


Abstract
In the Myb family, as in other families of transcription factors sharing similar DNA-binding domains (DBDs), diversity of function is believed to rely mainly on the less conserved parts of the proteins and on their distinct patterns of expression. However, small conserved differences between DBDs of individual members could play a role in fine-tuning their function. We have compared the highly conserved DBDs of the three vertebrate Myb proteins (A-, B- and c-Myb) and found distinct functional differences. While A- and c-Myb behaved virtually identically in a variety of DNA-binding assays, B-Myb formed complexes of comparatively lower stability, rapidly dissociating under competitive conditions and showing less tolerance to binding site variations. The three protein domains also differed as substrates for protein kinases. Whereas PKA in theory should target the DBDs of A- and c-Myb, but not B-Myb, only c-Myb was phosphorylated by PKA. CK2 phosphorylated all three proteins, although on different sites in the N-terminal region. Finally, B-Myb was remarkably sensitive to cysteine-directed oxidation compared to the other Myb proteins. Our data suggest that the small differences that have evolved between individual Myb family members lead to clear differences in DBD properties even if their sequence recognition remains the same.
INTRODUCTION
Eukaryotic transcription factors are grouped in families classified according to common types of DNA-binding domains (DBDs). Diversity of function is usually held to rely more on other parts of the proteins than on their DBDs since members within a family often recognise the same DNA sequence. This is the case with the Myb family of transcription factors where the three vertebrate members A-, B- and c-Myb share DBDs with closely similar amino acid sequences that all bind the same recognition sequence (1–4). In the regions outside their DBDs these proteins are much more divergent contributing to their functional diversity. However, the small differences that exist between their DBDs are conserved between species and could play a role in fine-tuning their function.
The diversity of function of the three vertebrate Myb proteins is to some extent caused by their distinct patterns of expression. The founder member of the family, c-myb, is expressed mainly in immature hematopoietic cells, but also in other immature cell lineages of rapidly proliferating tissues such as hair follicles and gastrointestinal crypt epithelial cells (5–7). A-myb is likewise a gene expressed in specific cell types, mainly found in the developing central nervous system, in germinal centre B-lymphocytes, in mammary gland ductal epithelium and in testis (8–10). In contrast, the third member, B-myb, is a ubiquitously expressed gene (reviewed in 11,12). These patterns of expression are also reflected in the distinct phenotypes observed after myb gene inactivation in mice. The abrogation of fetal liver hematopoiesis in mice with a c-mybnull mutation confirmed a vital role for c-Myb in the development of hematopoietic cells (13). The phenotype of mice with an A-mybnull mutation demonstrated a critical role in spermatogenesis and mammary gland development (10). Finally, homozygous B-myb-deficient mice died at an early stage of development, and blastocyst culture experiments suggested a role for B-Myb in inner cell mass formation (14).
All three proteins act as transactivators. A- and c-Myb have related transactivation domains, while the same domain in B-Myb shows a minor degree of similarity. Earlier conflicting reports on whether B-Myb is a transactivator or not was probably due to cell type-specific effects as well as to B-Myb being present in a self-repressed conformation (11,12,15,16). In all three proteins the transactivation capacity is modified by a more C-terminal negative regulatory domain (NRD) (reviewed in 3,4). This arrangement of domains allows regulation of the transactivation potential through modification of the NRD. Accordingly, A- and B-Myb have been found to be direct targets for cyclin A/Cdk2 leading to activation of the proteins (reviewed in 11,12). Cyclin A does not seem to affect c-Myb (17), while the NRD in c-Myb is targeted by other modifications (18–20).
The DBDs of the vertebrate Myb proteins are composed of three imperfect repeats of 52–53 amino acids, each with a characteristic tryptophan-rich signature motif. The R1, R2 and R3 repeats each contain three α-helices of which the last two form a helix–turn–helix (HTH)-related motif (21–23). R2 plus R3 (designated R2R3) constitutes a minimal DBD sufficient for sequence-specific DNA binding, while R1 appears to have no specific interactions with DNA (23), and its role remains to be elucidated (reviewed in 24). Despite the close similarity between the repeats, they seem to have dynamic differences. While R1 and R3 are tightly folded in solution (25,26), several lines of evidence suggest that R2 is less ordered and does not generate a full HTH motif until it interacts with DNA (27–30). Evidence for a disordered region in R2 was reported for chicken and human c-Myb (28) as well as for B-Myb (31,32), but not for mouse c-Myb (26). However, the latter R2 was found to exhibit slow conformational exchange fluctuations owing to a cavity in the hydrophobic core of the repeat (33,34).
The Myb recognition element (MRE) was identified by several approaches and defined by deduced consensus sequences that are all related to YAACNGHH (35–39). This sequence is recognised through the double HTH-related motif using two recognition helices, one in R2 and one in R3, which in the DNA-bound form are closely packed in the major groove (23). Key residues making direct contacts with bases include K128 (R2), N183 (R3) and K182 (R3). As a result the first half-site, YAAC, is recognised mainly by R3 and the second half-site, NGHH, is recognised mainly by R2 (23). Also, in a functional sense, the MRE seems to be bipartite. While the first half-site is dominant and absolutely required for binding, the loosely defined second half-site is only modulatory and mainly affects the half life of the complex (40).
The A- and c-Myb DBDs are more closely related to each other than they are to the B-Myb DBD (Fig. 1) (1,2). However, A- and B-Myb have been found to bind the same DNA sequence as c-Myb (37,41–43). The residues that in c-Myb have been found to interact directly with exposed bases in the recognition site are all conserved in the three proteins. Sequence recognition is also preserved between members more distantly related than A-, B- and c-Myb. A Myb protein from the slime mould Dictyostelium discoideum was capable of binding to the same DNA sequence as the vertebrate and Drosophila Myb proteins (44).
Figure 1.

The aligned amino acid sequences of the DBDs (NR123) of human A-, B- and c-Myb. Identical amino acids are marked with black boxes and similar amino acids are marked with grey boxes. The locations of the α-helices determined by NMR (28) are indicated as grey barrels and the flexible region (nascent helix) in R2 is indicated by a dotted barrel. The locations of kinase phosphorylation sites and redox-sensitive cysteines are indicated.
The DBD in a transcription factor obviously has its main function in recognising a specific DNA sequence, but many DBDs have turned out to have additional functions. Hence, the simplistic view of DBDs as plain DNA attachment tools has gradually been replaced by a more complex picture of DBDs as multifunctional domains. Several lines of evidence suggest that the Myb-family DBDs serve functions beyond sequence-specific DNA binding. The DBD of AMV v-Myb has the same recognition specificity as c-Myb, but still contains point mutations that are important for the ability of v-Myb to transform cells (45). Additional capacities of Myb-family DBDs include protein–protein interaction functions, where both inter- and intramolecular interactions have been reported for the c-Myb DBD (reviewed in 46). This DBD is also a regulatory domain modified by phosphorylation and responsible for its redox sensitivity. Both protein kinase CK2 and cAMP-dependent protein kinase (PKA) have been found to phosphorylate the c-Myb DBD. CK2 phosphorylates S11 and S12 leading to inhibition of DNA binding (47,48). As for redox regulation, the invariant C130 (49) has been shown to be highly sensitive to oxidative conditions, turning specific DNA binding off by controlling the DNA-induced conformational change in R2 (27). This sensitivity is altered in the oncogenic v-Myb protein (50,51). With respect to modifications of the DBD and functions beyond DNA binding, limited information is available for A- and B-Myb.
In the present work we have performed a comparative study to investigate whether the highly similar DBDs of A-, B- and c-Myb are identical in a functional sense and whether small differences could play a differential role in fine-tuning their function. Our results show that the latter is indeed the case. Detailed studies of DNA binding revealed that A- and c-Myb have very similar DNA-binding properties. In contrast, B-Myb interacts with DNA in a different fashion forming complexes of significantly lower stability both in vitro and in vivo, as well as having a lower tolerance to MRE sequence variations. The three DBDs also behaved differently as substrates for protein kinases. All three were phosphorylated by CK2, while only c-Myb was phosphorylated by PKA despite the fact that very similar PKA sites were predicted in A- and c-Myb. The redox sensitivity of A-Myb was comparable to c-Myb, whereas B-Myb was much more sensitive to oxidative conditions than the other two proteins.
MATERIALS AND METHODS
Expression and purification of Myb proteins
All three DBDs from human A-, B- and c-Myb were expressed in Escherichia coli [strain BL21 (DE3) LysS] using the T7 system (52). The soluble N-terminal regions expressed spanned the DBD of the three proteins and included residues 1–187 for A-Myb, 1–183 for B-Myb and 1–192 for c-Myb. These domains were designated NR123 (N-terminal + repeats 1, 2 and 3) to be distinguished from minimal DBD proteins designated R2R3 (repeats 2 and 3). Recombinant E.coli was grown at 37°C to an optical density of 0.6–0.9 and then induced by addition of isopropyl-β-d-thiogalactopyranoside (IPTG; final concentration 0.4 mM). The cells were harvested after 2 h of induction, washed once in cold TEN buffer (10 mM Tris–HCl, 1 mM EDTA and 100 mM NaCl), and resuspended on ice in buffer A [20 mM Tris–HCl pH 8.0, 1 mM EDTA, 10% glycerol and 1 mM dithiothreitol (DTT)] containing 500 mM NaCl. Phenylmethylsulfonyl fluoride (PMSF; 1 mM) and Triton X-100 (0.1%) was added to the suspension on ice. After lysis (5–10 min), the viscous suspension was centrifuged at 40 000 r.p.m. for 2 h in a TFT 65.13 rotor at 4°C. The non-viscous part of the supernatant was collected, diluted with 1 vol buffer A and the overexpressed protein was purified on a heparin–Sepharose (Amersham Pharmacia) affinity column using a 250–1500 mM NaCl gradient in buffer A containing 0.01% Triton X-100 and 0.1 mM PMSF. Peak fractions were pooled, diluted with 1 vol buffer A and further purified on an SP Sepharose Fast Flow (Amersham Pharmacia) ion exchange column using a 150–1500 mM NaCl gradient in buffer A supplemented as above. Typical protein yields from a 500 ml culture were 3–6 mg with a purity >95%.
Electrophoretic mobility shift assay (EMSA)
DNA binding was monitored by EMSA (53). Protein–DNA complexes were formed in 20 mM Tris–HCl pH 8.0, 0.1 mM EDTA, 10% glycerol, 0.1 mM DTT, 0.005% Triton X-100 and 50 mM NaCl (53). In the redox experiments, DTT was omitted. The following duplex oligonucleotides were used: MRE-mim1A, 5′-GCATTATAACGGTTTTTTAGCGC-3′; MRE-GG, 5′-GCATTATAACGGTCTTTTAGCGCCTGG-3′; MRE-TG, 5′-GCATTATAACTGTCTTTTAGCGCCTGG-3′; MRE-GT, 5′-GCATTATAACGTTCTTTTAGCGCCTGG-3′; MRE-TT, 5′-GCATTATAACTTTCTTTTAGCGCCTGG-3′. The sequence of MRE-mim1A is based on the A-site MRE in the upstream region of the mim-1 gene (54). The four derived variants with different configurations of Gs in positions 5 and 6 of the MRE consensus sequence (MRE-GG, -TG, -GT and -TT) were labelled to give identical specific activities as described (40). The latter oligos are longer and also contain a T8C alteration relative to the mim-1 sequence for reasons explained in (40). For redox studies, recombinant proteins were treated with azodicarboxylic acid bis(dimethylamide), abbreviated as diamide, as previously described (50).
Transactivation studies in yeast with Myb–VP16 fusion proteins
The MRE-lacZ reporter plasmids (pGL670 3×MRE) are described elsewhere (39). The effector plasmids were derived from the plasmid pDBD11 (55), a yeast centromeric plasmid designed to express DBDs fused with the herpes simplex virus VP16 transactivation domain under the control of the yeast GAL1 promoter. Into this vector were cloned inserts encoding the NR123 region from A-, B- or c-Myb. The yeast strain INVSc1 (Invitrogen) was transformed with different combinations of effector and reporter plasmids, using a high efficiency transformation protocol (56). Induced β-galactosidase (β-Gal) was measured as β-Gal units assayed with o-nitrophenyl-β-d-galactopyranoside (ONPG) as substrate (57).
Western blot
The yeast strain INVSc1 (Invitrogen) transformed with Myb expression plasmids and the 3×GG MRE reporter was grown in selective galactose media. Equal amounts of cells were lysed with glass beads and 5% trichloroacetic acid, run on a 15% SDS–polyacrylamide gel and blotted onto polyvinylidene difluoride membranes as previously described (58). NR123VP16 fusion proteins were detected by the murine monoclonal anti-VP16 antibody 2GV4 (59) and horseradish peroxidase-conjugated donkey anti-mouse antibody as previously described (58). Horseradish peroxidase activity was detected by ECL+, as described by the manufacturer (Amersham Pharmacia Biotech).
Phosphorylation of Myb proteins
Purified recombinant NR123 and R2R3 domains of A-, B- and c-Myb (100 pmol each) were phosphorylated with 4.6 U of bovine heart PKA catalytic subunit (Roche) and 0.25 µl [γ-32P]ATP (1.6 pmol, 2.5 µCi) in 10 mM MgAc in a final volume of 12 µl for 15 min at 30°C. Then 4 nmol cold ATP was added and the phosphorylation reactions placed on ice. 3× SDS sample buffer was added and the reactions were run out on a 15% SDS–polyacrylamide gel. The gel was autoradiographed for 2–10 min, and subsequently stained by Coomassie brilliant blue.
Phosphorylation by CK2 was performed by treatment of 1.5 µg Myb protein (71–73 pmol) with 96 ng CK2 in a buffer containing 0.1 mM ATP, 30 µCi [γ-32P]ATP, 50 mM Tris–HCl pH 7.5, 12 mM MgCl2 and 100 mM NaCl in a total volume of 30 µl. The samples were incubated at 30°C for 30 min. The reaction was stopped by the addition of 25 mM EDTA. Aliquots of 10 µl of each sample were analysed on a 15% SDS–polyacrylamide gel and the phosphorylation was detected by autoradiography.
RESULTS
Quantitative analysis of the DNA-binding properties of A-, B- and c-Myb
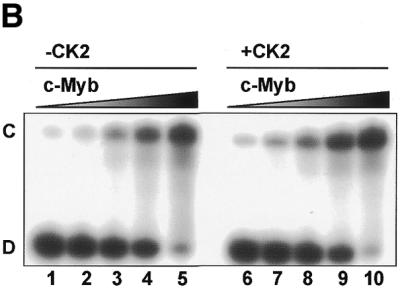
To address whether the three vertebrate members of the Myb family differ in their DNA-binding properties, we selected one common binding sequence (MRE-mim1A) and asked whether A-, B- and c-Myb bound the MRE probe with similar affinities and kinetics. We expressed the soluble N-terminal regions spanning the DBD of the three proteins in E.coli and purified them to near homogeneity. In a simple DNA-binding assay, without any competitor present, the NR123-domains of all three Myb proteins bound the MRE oligo with similar affinities (Fig. 2). Quantitative analysis of the data suggested dissociation constants in the range 0.2–0.5 nM. However, a clear difference in DNA-binding properties between the Myb family members was evident when specific or non-specific competitor oligo was added. B-Myb was slightly more affected by addition of increasing amounts of the non-specific competitor poly(dI-dC) than A- and c-Myb, suggesting a lower specific DNA-binding constant relative to the others (results not shown). The rate of dissociation from preformed protein–DNA complexes was evaluated by ‘decay-EMSA’. A large excess of specific competitor oligo was added to preformed Myb–DNA complexes and the time course of complex dissociation was monitored (Fig. 3A). The B-Myb DNA dissociation rate (lanes 6–10) was much higher than for c-Myb (lanes 11–15) or for A-Myb (lanes 1–5). While the half-lives for the A- and c-Myb complexes were ∼30 min under the experimental conditions used, the B-Myb–DNA complex was fully dissociated within 10 min of competition.
Figure 2.

Comparative analysis of DNA binding by recombinant A-, B- and c-Myb DBDs. The DNA binding of purified NR123-domains from A-Myb (lanes 1–5), B-Myb (lanes 6–10) and c-Myb (lanes 11–15) are compared. Increasing amounts of each of the proteins (10, 20, 40, 80 and 160 fmol) were incubated with 20 fmol MRE-mim1A probe at 25°C for 15 min and analysed by EMSA.
Figure 3.
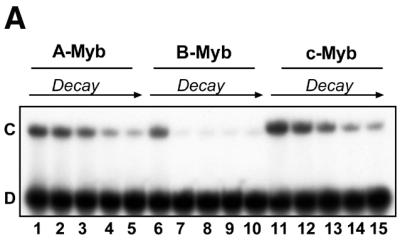
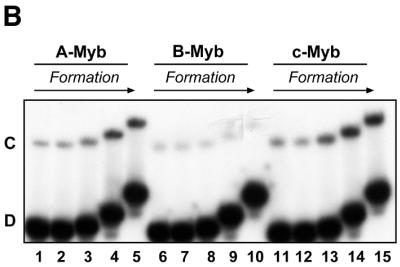
Myb–DNA complex formation and dissociation: comparative analysis of A-, B- and c-Myb DBDs. (A) Time course of complex dissociation upon competition. Myb–DNA complexes were generated using 40 fmol NR123 from A-Myb (lanes 1–5), B-Myb (lanes 6–10) and c-Myb (lanes 11–15) and 20 fmol MRE-mim1A probe and allowed to form for 15 min at 25°C. The complexes were then exposed to 750 fmol unlabeled MRE-mim1A probe for 0, 10, 20, 40 and 60 min before the samples were analysed by EMSA (as described in Materials and Methods). (B) Time course of complex formation. 40 fmol NR123 of A-Myb (lanes 1–5), B-Myb (lanes 6–10) and c-Myb (lanes 11–15) were incubated with 20 fmol MRE-mim1A for 0.5, 1, 5, 15 and 30 min in the presence of 0.1 µg poly(dI-dC). Then 750 fmol unlabeled MRE-mim1A was added and the samples were incubated for 5 min before loading on a running EMSA gel.
Evidence suggests that c-Myb–DNA complex formation proceeds by a two-step kinetic process (29). The first association step is rapid, leading to formation of an unstable protein–DNA complex, probably associated mainly through non-specific contacts to the phosphate backbone. After several minutes [rate constant estimated to 0.004 s–1 (29)], a more stable complex appears, presumably after a structural reorganisation where specific contacts to the bases are established. To study whether this slow formation of stable complexes was similar for all three Myb proteins, we used the following approach to follow the time course of stable complex formation. A protein–DNA complex was allowed to form in the presence of poly(dI-dC) to select for specific complexes. The binding reaction was quenched at different time points by addition of a large excess of unlabeled specific competitor followed by a brief incubation before loading on a running EMSA gel (Fig. 3B). For c-Myb we observed a slow formation of stable complexes exactly as predicted by the kinetic model of Zargarian et al. (29). Again A-Myb showed the same behaviour as c-Myb. In sharp contrast, B-Myb was unable to form a distinct and specific complex even after 30 min under these conditions. This could be explained by the quenching with cold oligo in this assay, since B-Myb was very sensitive to specific competition (Fig. 3A). It thus appears that B-Myb manifests features of specific DNA binding that are clearly distinct from those of A- and c-Myb. B-Myb DNA binding is characterised by low stability complexes that rapidly dissociate under competitive conditions.
In order to verify that this difference also operates in vivo, we used a yeast effector–reporter system designed to give a transcriptional read-out of DNA binding in vivo (39). This system allowed the conditional expression of either A-, B- or c-Myb DBDs fused to the strong VP16 transactivation domain. DNA binding in vivo was measured as induced β-Gal activity. A- and c-Myb bound very well to DNA as evidenced from their high and similar transactivation values (Fig. 4A). B-Myb, on the other hand, bound significantly weaker giving a reporter value only ∼20% of the two others. This in vivo analysis confirmed the weaker DNA binding of B-Myb relative to the two other Myb proteins. A western blot showed that all three Myb proteins were expressed at similar levels in the yeast cultures (Fig. 4B). The low transactivation obtained with B-Myb was not due to insufficient B-Myb levels since in fact B-Myb was expressed at a slightly higher level than the A- and c-Myb fusion proteins.
Figure 4.

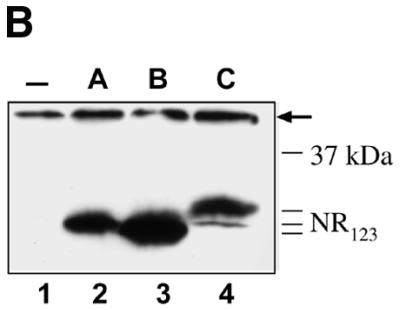
Comparative analysis of DNA binding in vivo by A-, B- and c-Myb DBDs. (A) DNA binding in vivo was estimated using a yeast effector–reporter system (39). An effector plasmid expressing one of the NR123VP-16 fusions A-Myb–VP16, B-Myb–VP16 or c-Myb–VP16 were cotransformed with a lacZ reporter plasmid containing three copies of the MRE-GG element. Transactivation was measured as β-Gal activity after induction by galactose of the effector plasmids expressing the fusion proteins. (B) A western blot shows the expression levels of Myb NR123VP16 fusion proteins as detected by the anti-VP16 antibody 2GV4 in yeast cells transformed with the lacZ reporter and the following expression plasmids as indicated: lane 1, empty vector pDBD11 (–); lane 2, pDBD11-hA-Myb-NR123 (A); lane 3, pDBD11-hB-Myb-NR123 (B); lane 4, pDBD11-hc-Myb-NR123 (C). A non-specific band, serving as loading control, is marked by an arrow.
MRE sequence preferences of A-, B- and c-Myb, in vitro and in vivo assays
We have shown in a previous analysis of DNA binding of c-Myb that the MRE is functionally bipartite. The first half-site, YAAC, was absolutely required for DNA binding, whereas the second half-site, NGHH, could adopt a range of sequences mainly affecting the half-life of the protein–DNA complex in vitro (40). The adaptable sequence requirement in the second half-site is presumably caused by the flexible structure of the second repeat R2, and was particularly striking when analysed with respect to configuration of Gs in positions 5 and 6 of the binding site (39,58). We performed similar experiments to investigate whether A- and B-Myb showed the same adaptive behaviour towards second half-site variants. The effect of base variations in positions 5 and 6 of the MRE was studied in vitro and in vivo.
In EMSA analysis, increasing amounts of the three different proteins were added to a fixed amount of the oligonucleotides MRE-GG, MRE-TG, MRE-GT and MRE-TT (Fig. 5A). Again A- and c-Myb showed very similar properties, binding most efficiently to the MRE-GG followed by MRE-TG and MRE-GT and with very weak binding to MRE-TT. The MRE-GG was also efficiently bound by B-Myb in the absence of competitor as already observed (Fig. 2A). However, the two single-G variants, MRE-GT and MRE-TG, were recognised by B-Myb with lower binding affinities than they were by A- and c-Myb.
Figure 5.


Binding of NR123-domains from A-, B- and c-Myb to different MRE variants in vitro and in vivo. (A) DNA binding in vitro. Increasing amounts of NR123 protein (10, 20, 40 and 80 fmol) of A-, B- and c-Myb were incubated for 15 min at 25°C with 20 fmol of one of the following MRE probes: MRE-GG, -TG, -GT or -TT. The samples were analysed by EMSA (as described in Materials and Methods). (B) DNA binding in vivo measured as Myb-dependent transactivation in a yeast effector–reporter system using different MRE variants. An effector-plasmid expressing one of the NR123VP-16 fusions A-Myb–VP16, B-Myb–VP16 or c-Myb–VP16 was cotransformed with one of four lacZ reporters containing triple copies of either the MRE-GG, MRE-TG, MRE-GT or MRE-TT. Transactivation was measured as described in the legend to Figure 4.
Studies in the yeast transactivation system (described above) to assay for DNA binding in vivo supported and complemented the results from the in vitro assay. Myb-dependent transactivation was measured using the three different Myb proteins and the four binding site variants (Fig. 5B). A- and c-Myb showed again almost indistinguishable activation profiles: efficient activation from the MRE-GG site, moderate activation from the MRE-GT and MRE-TG sites and no activation from the MRE-TT site. B-Myb showed overall weaker transactivation, causing a moderate level of activation with the MRE-GG site (as already discussed), but apparently unable to interact with sufficient strength with the single-G variants in a competitive in vivo situation to give transactivation above background level.
We conclude from these DNA-binding studies that A- and c-Myb are almost indistinguishable regarding both quantitative and qualitative aspects of DNA binding. B-Myb, on the other hand, interacts differently with MRE sequences and forms complexes of reduced stability compared to the other two Myb proteins. B-Myb also appears to have a lower tolerance to second half-site variations, since B-Myb–DNA complexes formed with single-G variants were too weak or unstable to be productive in vivo.
A possible explanation for the comparatively weak DNA binding of B-Myb could be that this protein has a more extended DBD than the others, and requires a region C-terminal to the truncation point for full activity, similar to what is the case for the yeast Myb-related protein Bas1p (60). However, when a B-Myb construct encoding a 44 residues larger protein (B-Myb[1–231]) was analysed in the yeast system, no significant increase in reporter activation was observed compared to the B-Myb[1–187] analysed above (results not shown). We therefore conclude that the B-Myb DBD interacts with consensus MREs less avidly than the DBDs of A- and c-Myb.
Competion between c- and B-Myb for a common binding site
A- and c-Myb proteins have restricted tissue-specific expression patterns. B-Myb on the other hand is ubiquitously expressed. Hence, cells that express A- or c-Myb also express B-Myb, and competition for common binding sites would be expected to occur. We therefore analysed how efficiently c- and B-Myb competed for the same MRE. In this EMSA the input of the two proteins (of unequal size to allow the complexes to be resolved) were adjusted to give similar complex intensities under conditions of MRE-probe excess. Then the DNA input was lowered to see which of the two proteins were preferentially bound under conditions of limiting DNA concentration. In both experiments shown (two combinations of small and large versions of DBDs), the results were the same. As expected from the less stable binding of B-Myb relative to c-Myb, c-Myb was preferentially bound to the MRE-GG oligo under limiting concentrations of DNA (Fig. 6).
Figure 6.

Competition between c- and B-Myb for the same MRE. Titration of protein–DNA complexes with decreasing amounts of DNA. Proteins of unequal size were used to allow the complex to be resolved. As a reference of migration, lanes 1 and 2 show c-Myb alone (4 fmol NR123 and 5 fmol R2R3) and B-Myb alone (40 fmol R2R3 and 30 fmol NR123), respectively, bound to 35 fmol MRE-mim1A probe. Complexes were allowed to form for 15 min at 25°C. After incubation the samples were analysed by EMSA (as described in Materials and Methods). (A) Lanes 3–9, complex formation in a mixture containing 4 fmol c-Myb NR123, 40 fmol B-Myb R2R3 and decreasing amounts of MRE-mim1A probe (35–2.5 fmol). Complexes were allowed to form for 15 min at 25°C. After incubation the samples were analysed by EMSA (as described in Materials and Methods). (B) A similar analysis was performed with 30 fmol B-Myb NR123, 5 fmol c-Myb R2R3 and decreasing amounts of MRE-mim1A probe (35–2.5 fmol).
Phosphorylation studies
Myb-family DBDs seem to have functions beyond sequence-specific DNA binding. The DBD of c-Myb has previously been shown to be a target for several types of posttranslational modifications, in particular phosphorylation and redox-related cysteine modification (47,48,50,51,61). The DBDs of A- and B-Myb are less studied in this respect. Since such modifications could have a regulatory role, we asked whether the three Myb proteins were similar as substrates for modifications or whether they had evolved differently and become distinct targets.
It has been reported that c-Myb could be a substrate for PKA (61) and the S116 residue in the second repeat was mapped as a critical PKA site in the DBD of chicken c-Myb (61; K.B.Andersson, E.Kowenz-Leutz, E.Brendeford, A.H.H.Tygsett, A.Leutz and O.S.Gabrielsen, manuscript in preparation). To analyse whether also A- and B-Myb DBDs were PKA substrates, we performed in vitro phosphorylation experiments with the three recombinant A-, B- and c-Myb DBDs and the catalytic subunit Cα of PKA. As shown in Figure 7, only c-Myb was efficiently phosphorylated. This was true for both the minimal (R2R3) and extended NR123 versions of the Myb DBDs. For B-Myb this result was expected since the PKA site is not conserved in B-Myb (KRWS in c-Myb replaced by KQWT). For A-Myb the very low efficiency of phosphorylation was a surprise since the amino acid sequence of the consensus phosphorylation site is identical between c- and A-Myb. Inspection of the sequence context of the putative PKA site in A-Myb (KRWS111L corresponding to KRWS116V in c-Myb), suggests that the failure of A-Myb to become similarly phosphorylated could be due to differences in the +1 residue (L versus V) since this position is known to influence substrate activity (62). However, we cannot exclude the possibility that the difference is due to unequal accessibility of the site caused by differences in local conformation of the two proteins.
Figure 7.
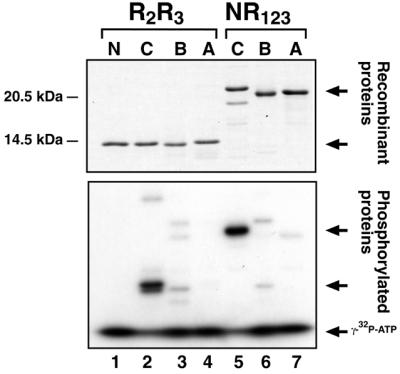
PKA phosphorylation of A-, B- and c-Myb DBDs. Recombinant NR123 and R2R3 domains for A-, B- and c-Myb were phosphorylated in vitro with [γ-32P]ATP and purified bovine PKA Cα catalytic subunit for 15 min before addition of SDS buffer, separation on a 15% SDS–polyacrylamide gel and autoradiography (as described in Materials and Methods). Top, Coomassie-stained gel; bottom, corresponding autoradiogram. N, phosphorylation reaction c-Myb R2R3 without PKA Cα added; C, B and A indicate c-, B- or A-Myb R2R3 or NR123 domains, respectively, as indicated. Proteins, phosphoproteins and free [γ-32P]ATP are marked with arrows.
In the N-terminal part of c-Myb there is a double site for protein kinase CK2 (S11 + S12 followed by an acidic stretch). Phosphorylation of this site has been shown to inhibit sequence-specific DNA binding of c-Myb (47,48,63). The sequence around the S11 site in c-Myb is conserved in A-Myb (a single S followed by an acidic stretch), but not in B-Myb, where a cysteine is located just upstream of the acidic stretch (Fig. 1). However, a putative B-Myb-specific CK2 site is found close by, in residue T18 in the sequence T18DSDV (Fig. 1). Hence we expected A-Myb to be phosphorylated by CK2 similarly to c-Myb, while it was less clear whether B-Myb would be a substrate for CK2. When incubating A-, B- and c-Myb with CK2 in vitro, all the three proteins became phosphorylated (Fig. 8A). The lack of phosphorylation of a c-Myb S11D/S12D mutant indicates that for c-Myb there are no additional CK2 sites. A- and B-Myb were readily phosphorylated, probably at the sites proposed (Fig. 1), although these sites were not further mapped.
Figure 8.
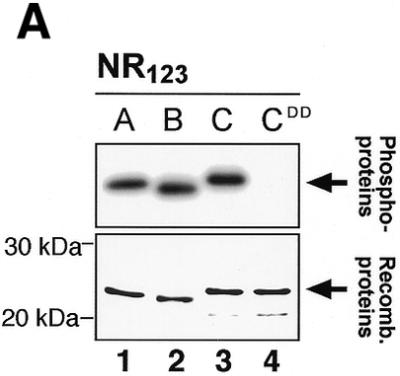
CK2 phosphorylation of A-, B- and c-Myb DBDs. (A) Recombinant NR123 domains of A-Myb (lane 1), B-Myb (lane 2), c-Myb (lane 3) and the S11D/S112D mutant of c-Myb (CDD; lane 4) were phosphorylated in vitro with [γ-32P]ATP and protein kinase CK2 at 30°C for 30 min before separation by SDS–PAGE and autoradiography (as described in Materials and Methods). The S11D/S12D mutant was constructed as previously described (22). The lower panel shows an SDS–PAGE analysis of the four NR123 domains from A-Myb (lane 1), B-Myb (lane 2), c-Myb (lane 3) and CDD (lane 4) (1.5 µg of each), purified as described, and visualized by staining with Coomassie brilliant blue. The theoretical molecular weights of the NR123 domains are 22.6, 22.0 and 23.1 kDa for A-, B- and c-Myb, respectively. (B) Recombinant NR123 domain of c-Myb (200 µg) was phosphorylated in vitro with 0.1 mM ATP (and trace amounts of [γ-32P]ATP) and protein kinase CK2 at 30°C for 30 min before separation on a SP Sepharose Fast Flow (Amersham Pharmacia) ion exchange column using a 150–1500 mM NaCl gradient. Equal amounts of non-phosphorylated NR123 domain of c-Myb and protein from the peak fraction corresponding to phosphorylated c-Myb were analysed by EMSA (as described in Materials and Methods). The degree of phosphorylation in the purified fraction was determined as described (75) and estimated to be 70%.
The mechanism of the inhibitory effect of CK2 phosphorylation on c-Myb DNA binding is not known. It could be a direct charge effect, an induced conformational change in the DBD or an effect that depends on intramolecular interactions in full-length c-Myb or on intermolecular interactions with other proteins. To study in vitro the effect of CK2 phosphorylation of an isolated c-Myb DBD, we compared DNA binding of wild-type c-Myb, CK2-phosphorylated c-Myb and a phosphate-mimicking mutant version where the two serines were replaced by two aspartates (S11D/S12D mutant). No difference in DNA binding between these forms could be detected in an EMSA (results not shown). We also tried to separate phosphorylated from non-phosphorylated c-Myb on an ion-exchange column to ensure analysis of a quantitatively modified protein, but again no difference in affinity for a MRE-GG probe was discernible in an EMSA with variable protein input (Fig. 8B). The level of phosphorylation in the CK2 treated fraction was estimated to be 70%. This suggests that the previously reported impaired DNA binding caused by phosphorylation of serines 11 or 12 is probably not a local effect involving only the DBD, but must imply a more complex mechanism involving intra- or intermolecular interactions.
Regulation studies by redox treatment
The DNA-binding activity of c-Myb R2R3 has previously been shown to be sensitive to the redox state of the protein (50,64), a feature conserved in Myb proteins from yeast to humans (49). This property is caused by a redox-sensitive cysteine (C130) in the recognition helix of R2 of c-Myb. The C130 residue has been proposed to function as a molecular redox sensor turning specific DNA binding on or off by controlling a DNA-induced conformational change in R2 (27). Since all three Myb proteins studied in this work contain a cysteine located at the same position in R2 (Fig. 1), we asked whether all three DBDs would behave in the same manner when exposed to conditions of oxidative stress. In all three proteins there is an additional cysteine in R1, located in a position corresponding to the one in R2 (Fig. 1). A-Myb contains only these two cysteines, but c-Myb contains a third cysteine in the N-terminal region, while B-Myb contains four more cysteines distributed throughout the DBD. We therefore expected B-Myb DBD to be more sensitive to oxidation than c- and A-Myb DBDs. This was indeed the case. When all three proteins were exposed to increasing amounts of the cysteine-specific oxidation agent diamide, we observed a general impairment of DNA binding (Fig. 9). A- and c-Myb showed similar sensitivity towards the oxidative reagent and gradually lost activity in the concentration range tested (lanes 1–5 and 11–15). B-Myb turned out to be highly sensitive (lanes 6–10). DNA binding of B-Myb was totally abolished by addition of only 30 µM diamide. We conclude that with respect to redox sensitivity, the DBDs of A- and c-Myb are similar, whereas the B-Myb DBD is exceptionally sensitive to oxidative inactivation.
Figure 9.
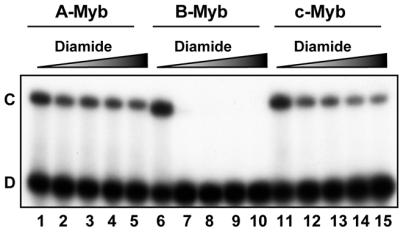
Cysteine-specific oxidation of A-, B- and c-Myb DBDs. 40 fmol of the NR123 domains of A-Myb (lanes 1–5), B-Myb (lanes 6–10) and c-Myb (lanes 11–15) were treated with 0, 30, 60, 90 and 120 µM diamide at 25°C for 10 min without a reducing agent present. MRE-mim1A probe (20 fmol) was then added and the samples were incubated at 25°C for 15 min before separation on an EMSA gel (as described in Materials and Methods).
DISCUSSION
In the present work we have compared the DBDs of three vertebrate members of the Myb family of transcription factors with respect to DNA binding and posttranslational modifications. The domains in all three proteins have remained very similar during evolution. In the minimal DBDs (R2R3) responsible for sequence-specific DNA binding A- and c-Myb are 96% identical, while they show 83 and 85% identity when compared individually to B-Myb. When the enlarged DBDs (NR123 as analysed in the present work) are compared, these figures fall to 77% identity between A- and c-Myb and to 65 and 70% identity when B-Myb is compared to A- and c-Myb, respectively. Because of this high level of conservation between the Myb-family members, these domains have been assumed to have closely similar functions. While this is certainly correct with respect to the main function in sequence recognition, our data show that the small differences that remain between the DBDs of A-, B- and c-Myb lead to distinct functional differences and thus cannot be regarded as results of silent divergence.
When DNA-binding properties were carefully analysed, no significant difference was observed between the DBDs of A- and c-Myb, consistent with their very close sequence similarity. The DBD of B-Myb, however, had properties that set it apart from the other two. Both in vitro and in vivo experiments supported that B-Myb forms DNA complexes of lower stability, with rapid dissociation under competitive conditions, and less tolerance to binding site variations than the A- and c-Myb cousins. Even after extended incubations with the best MRE probe we did not obtain complexes with B-Myb that were comparable in stability with A- and c-Myb. This weak binding also explains why it has been difficult to generate clear footprints in DNase I protection analysis of B-Myb DNA binding (J.Borrebæk and O.S.Gabrielsen, unpublished results). It might seem peculiar that we observe no difference between A-, B- and c-Myb in a direct EMSA without competition (Fig. 2), while a marked difference appears for B-Myb upon competition (Fig. 3A). This is probably due to the two-step mechanism of DNA binding found for c-Myb (29). In the first case, we expect the direct EMSA to monitor mainly the rapid first step association, while the competitive EMSA will reveal whether the complex has undergone the subsequent slower maturation to a more stable form. If this interpretation is correct, it means that B-Myb is either unable to perform this second step on the MRE variants used or that this docking step is extremely slow (requiring hours to complete). In a study where the DBDs of c-and B-Myb were swapped, the large difference in activity between the two factors was assigned to the transcriptional activation domain (TAD) since the chimerical proteins had activities most close to the Myb protein from which their TAD was derived (65). In addition, this study revealed that c-Myb lost activity after having its DBD replaced with that of B-Myb. Similarly, B-Myb slightly gained activity after having its DBD replaced with that of c-Myb. The latter observations fit with the present conclusions of differences in DNA-binding avidity between c- and B-Myb.
What would be a plausible biological explanation for the weak DNA binding of B-Myb? It seems to be important for the cell to carefully regulate the activity of B-Myb. During the cell cycle B-Myb expression is controlled by an E2F-mediated repression mechanism and B-Myb activity is regulated by cyclins: enhancement by cyclin A/Cdk2 and repression by the binding of cyclin D1 (11,12,15). In addition, the DNA-binding activity of B-Myb is negatively regulated by an intramolecular mechanism in Xenopus oocytes (66). Consistent with the idea of the tight B-Myb regulation, elevated B-Myb levels have been associated with several forms of cancer (67,68). It is possible that the weak DNA binding fits into the picture of a factor whose level and activity must be carefully controlled. B-Myb may be strictly dependent on the synergistic cooperation with other transcription factors to be able to activate target gene promoters. It is also possible that some intramolecular mechanism exists that upon certain signals induces a conformational change in B-Myb that results in a more efficient DBD. On the other hand, we cannot exclude the fact that an optimal recognition sequence for B-Myb has not yet been determined, since most reports have tested B-Myb DNA binding using c-Myb recognition sequences. To our knowledge, no direct selection of B-Myb recognition sequences has been reported. One study reported that B-Myb shows a more distinct preference for the base at position 7 than c-Myb (37). Others have reported that some sequences are only recognised by B-Myb and not by c-Myb (41), although this was not confirmed in an independent analysis (69). It is noteworthy that B-Myb has been suggested to have MRE-independent functions and is able to transactivate promoters that do not contain Myb binding sites (65,70–72). Others have shown that activation by B-Myb is dependent on the presence of Myb binding sites (73). In one report, the heat shock element (HSE) was shown to be specifically activated by B-Myb but not c-Myb (74). All these results taken together show that the transactivation activity of B- and c-Myb act differently in different promoter contexts and that the DNA-binding properties of the DBDs could account for some of these differences.
An increasing amount of functional data supports the notion of the c-Myb DBD being a multifunctional domain. We were interested in whether such additional features as being substrates for specific kinases had evolved independently in the three proteins or whether these features are conserved, tightly linked to the DNA-binding capacity. Two kinases, PKA and CK2, have previously been reported to phosphorylate the DBD of c-Myb. We showed that only c-Myb was a PKA substrate. This phosphorylation is exclusively on S116 in the conserved R2R3 region (K.B.Andersson, E.Kowenz-Leutz, E.Brendeford, A.H.H.Tygsett, A.Leutz and O.S.Gabrielsen manuscript in preparation). The difference between A- and c-Myb is particularly interesting. Apparently only a subtle difference in sequence between these two proteins was sufficient for c-Myb to become a PKA substrate but not A-Myb. This difference is located in the minimal DBD without having any effect on their DNA-binding properties in the non-phosphorylated state. In this way, one family member has evolved the capacity to be linked to a signalling pathway, while the others remain unaffected.
All three DBDs were phosphorylated by CK2 even though only A- and c-Myb contain the strong CK2 site previously investigated (47,48,63). We did not map the alternative CK2 site in B-Myb, but since none of the three minimal DBDs (R2R3 of A-, B- and c-Myb) were phosphorylated by this kinase (results not shown), the site must be located within R1 or the N-terminal part. Moreover, since the S11D/S12D mutant of c-Myb was not phosphorylated, it is unlikely that B-Myb is phosphorylated in potential sites that are conserved between the two proteins. We noticed, however, that B-Myb has evolved a distinct putative CK2 site a few residues away from the CK2 site in c-Myb. This site, residue T18 in the sequence T18DSDV (Fig. 1), is conserved between species in B-Myb proteins and is a good candidate for an alternative CK2 site in B-Myb.
Since the mechanism of the inhibitory effect of CK2 phosphorylation of c-Myb in S11 and S12 has remained elusive, we tested the effect on DNA binding of CK2 phosphorylation of recombinant c-Myb DBD. We found no effect, even after efforts to enrich for fully phosphorylated protein. Neither was the S11D/S12D mutant of c-Myb affected in DNA binding as measured by EMSA using the MRE-mim1A binding probe. We thus have to conclude that the reported effects of CK2 (47,48) cannot be the result of a local effect on the DBD alone or a simple charge effect acting negatively on DNA interaction, but must operate through a mechanism where other regions of the protein play a part.
When the three different DBDs were exposed to the cysteine specific oxidation agent diamide, A- and c-Myb again showed virtually identical behaviour. In contrast, B-Myb demonstrated a much higher sensitivity to this treatment than the other two proteins. Since these redox studies with enlarged DBDs (NR123) uncovered the same effects as the previous reported for c-Myb R2R3 (50), we hypothesise that mainly the second repeat cysteine in A- and c-Myb plays a major role in the redox modification of DNA binding. This is also consistent with a recent careful analysis of cysteine modification in the Myb-related Bas1p, concluding that the particular redox sensitivity of the second repeat cysteine is a highly conserved feature of the Myb family (49). Moreover, since neither the N-terminal nor the R1 repeat participate directly in DNA recognition, oxidation of cysteines in these regions should not have a major effect on DNA interaction. The enhanced sensitivity of B-Myb is probably caused by the presence of two additional cysteines in R3 that are not present is A- or c-Myb.
In summary, our comparative study of A-, B- and c-Myb illustrates that even highly similar DBDs that bind to the same recognition element still can hide differences with respect to quantitative aspects of DNA binding and in functions beyond DNA binding.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Per Arne Risøen for skillful assistance in parts of the work. We are grateful to Martino Introna for providing cDNA for human A- and B-Myb and to Lorenzo A. Pinna for providing CK2 enzyme. This work was supported by The Norwegian Research Council (S.B., O.S.G.), The Norwegian Cancer Society (O.S.G.), the Anders Jahres Foundation (O.S.G.) and the Deutsche Forschungsgemeinschaft, DFG (B.L.).
References
- 1.Lipsick J.S. (1996) One billion years of Myb. Oncogene, 13, 223–235. [PubMed] [Google Scholar]
- 2.Rosinski J.A. and Atchley,W.R. (1998) Molecular evolution of the Myb family of transcription factors: evidence for polyphyletic origin. J. Mol. Evol., 46, 74–83. [DOI] [PubMed] [Google Scholar]
- 3.Ganter B. and Lipsick,J.S. (1999) Myb and oncogenesis. Adv. Cancer Res., 76, 21–60. [DOI] [PubMed] [Google Scholar]
- 4.Oh I.H. and Reddy,E.P. (1999) The myb gene family in cell growth, differentiation and apoptosis. Oncogene, 18, 3017–3033. [DOI] [PubMed] [Google Scholar]
- 5.Kastan M.B., Slamon,D.J. and Civin,C.I. (1989) Expression of protooncogene c-myb in normal human hematopoietic cells. Blood, 73, 1444–1451. [PubMed] [Google Scholar]
- 6.Sitzmann J., Noben-Trauth,K. and Klempnauer,K.H. (1995) Expression of mouse c-myb during embryonic development. Oncogene, 11, 2273–2279. [PubMed] [Google Scholar]
- 7.Ess K.C., Witte,D.P., Bascomb,C.P. and Aronow,B.J. (1999) Diverse developing mouse lineages exhibit high-level c-Myb expression in immature cells and loss of expression upon differentiation. Oncogene, 18, 1103–1111. [DOI] [PubMed] [Google Scholar]
- 8.Mettus R.V., Litvin,J., Wali,A., Toscani,A., Latham,K., Hatton,K. and Reddy,E.P. (1994) Murine A-myb: evidence for differential splicing and tissue-specific expression. Oncogene, 9, 3077–3086. [PubMed] [Google Scholar]
- 9.Trauth K., Mutschler,B., Jenkins,N.A., Gilbert,D.J., Copeland,N.G. and Klempnauer,K.H. (1994) Mouse A-myb encodes a trans-activator and is expressed in mitotically active cells of the developing central nervous system, adult testis and B lymphocytes. EMBO J., 13, 5994–6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toscani A., Mettus,R.V., Coupland,R., Simpkins,H., Litvin,J., Orth,J., Hatton,K.S. and Reddy,E.P. (1997) Arrest of spermatogenesis and defective breast development in mice lacking A-myb. Nature, 386, 713–717. [DOI] [PubMed] [Google Scholar]
- 11.Saville M.K. and Watson,R.J. (1998) B-Myb: a key regulator of the cell cycle. Adv. Cancer Res., 72, 109–140. [DOI] [PubMed] [Google Scholar]
- 12.Sala A. and Watson,R. (1999) B-Myb protein in cellular proliferation, transcription control, and cancer: latest developments. J. Cell Physiol., 179, 245–250. [DOI] [PubMed] [Google Scholar]
- 13.Mucenski M.L., McLain,K., Kier,A.B., Swerdlow,S.H., Schreiner,C.M., Miller,T.A., Pietryga,D.W., Scott,W.J.,Jr and Potter,S.S. (1991) A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell, 65, 677–689. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka Y., Patestos,N.P., Maekawa,T. and Ishii,S. (1999) B-myb is required for inner cell mass formation at an early stage of development. J. Biol. Chem., 274, 28067–28070. [DOI] [PubMed] [Google Scholar]
- 15.Horstmann S., Ferrari,S. and Klempnauer,K.H. (2000) Regulation of B-Myb activity by cyclin D1. Oncogene, 19, 298–306. [DOI] [PubMed] [Google Scholar]
- 16.Ansieau S., Kowenz-Leutz,E., Dechend,R. and Leutz,A. (1997) B-Myb, a repressed trans-activating protein. J. Mol. Med., 75, 815–819. [DOI] [PubMed] [Google Scholar]
- 17.Ziebold U. and Klempnauer,K.H. (1997) Linking Myb to the cell cycle: cyclin-dependent phosphorylation and regulation of A-Myb activity. Oncogene, 15, 1011–1019. [DOI] [PubMed] [Google Scholar]
- 18.Aziz N., Miglarese,M.R., Hendrickson,R.C., Shabanowitz,J., Sturgill,T.W., Hunt,D.F. and Bender,T.P. (1995) Modulation of c-Myb-induced transcription activation by a phosphorylation site near the negative regulatory domain. Proc. Natl Acad. Sci. USA, 92, 6429–6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miglarese M.R., Richardson,A.F., Aziz,N. and Bender,T.P. (1996) Differential regulation of c-Myb-induced transcription activation by a phosphorylation site in the negative regulatory domain. J. Biol. Chem., 271, 22697–22705. [DOI] [PubMed] [Google Scholar]
- 20.Sano Y. and Ishii,S. (2000) Increased affinity of c-Myb for CBP after CBP-induced acetylation. J. Biol. Chem., 276, 3674–3682. [DOI] [PubMed] [Google Scholar]
- 21.Frampton J., Gibson,T.J., Ness,S.A., Doderlein,G. and Graf,T. (1991) Proposed structure for the DNA-binding domain of the Myb oncoprotein based on model building and mutational analysis. Protein Eng., 4, 891–901. [DOI] [PubMed] [Google Scholar]
- 22.Gabrielsen O.S., Sentenac,A. and Fromageot,P. (1991) Specific DNA binding by c-Myb: evidence for a double helix-turn-helix-related motif. Science, 253, 1140–1143. [DOI] [PubMed] [Google Scholar]
- 23.Ogata K., Morikawa,S., Nakamura,H., Sekikawa,A., Inoue,T., Kanai,H., Sarai,A., Ishii,S. and Nishimura,Y. (1994) Solution structure of a specific DNA complex of the Myb DNA-binding domain with cooperative recognition helices. Cell, 79, 639–648. [DOI] [PubMed] [Google Scholar]
- 24.Ness S.A. (1996) The Myb oncoprotein: regulating a regulator. Biochim. Biophys. Acta, 1288, F123–F139. [DOI] [PubMed] [Google Scholar]
- 25.Ogata K., Hojo,H., Aimoto,S., Nakai,T., Nakamura,H., Sarai,A., Ishii,S. and Nishimura,Y. (1992) Solution structure of a DNA-binding unit of Myb: a helix-turn-helix- related motif with conserved tryptophans forming a hydrophobic core. Proc. Natl Acad. Sci. USA, 89, 6428–6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogata K., Morikawa,S., Nakamura,H., Hojo,H., Yoshimura,S., Zhang,R., Aimoto,S., Ametani,Y., Hirata,Z., Sarai,A. et al. (1995) Comparison of the free and DNA-complexed forms of the DNA-binding domain from c-Myb. Nature Struct. Biol., 2, 309–320. [DOI] [PubMed] [Google Scholar]
- 27.Myrset A.H., Bostad,A., Jamin,N., Lirsac,P.N., Toma,F. and Gabrielsen,O.S. (1993) DNA and redox state induced conformational changes in the DNA-binding domain of the Myb oncoprotein. EMBO J., 12, 4625–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jamin N., Gabrielsen,O.S., Gilles,N., Lirsac,P.N. and Toma,F. (1993) Secondary structure of the DNA-binding domain of the c-Myb oncoprotein in solution. A multidimensional double and triple heteronuclear NMR study. Eur. J. Biochem., 216, 147–154. [DOI] [PubMed] [Google Scholar]
- 29.Zargarian L., Le Tilly,V., Jamin,N., Chaffotte,A., Gabrielsen,O.S., Toma,F. and Alpert,B. (1999) Myb-DNA recognition: role of tryptophan residues and structural changes of the minimal DNA binding domain of c-Myb. Biochemistry, 38, 1921–1929. [DOI] [PubMed] [Google Scholar]
- 30.Jamin N., Le Tilly,V., Zargarian,L., Bostad,A., Besançon-Yoshpe,I., Lirsac,P.N., Gabrielsen,O.S. and Toma,F. (1996) Preliminary investigation of the interaction between the R2R3 DNA-binding domain of the oncoprotein and DNA fragments. Int. J. Quant. Chem., 59, 333–341. [Google Scholar]
- 31.Carr M.D., Wollborn,U., McIntosh,P.B., Frenkiel,T.A., McCormick,J.E., Bauer,C.J., Klempnauer,K.H. and Feeney,J. (1996) Structure of the B-Myb DNA-binding domain in solution and evidence for multiple conformations in the region of repeat-2 involved in DNA binding: implications for sequence-specific DNA binding by Myb proteins. Eur. J. Biochem., 235, 721–735. [DOI] [PubMed] [Google Scholar]
- 32.McIntosh P.B., Frenkiel,T.A., Wollborn,U., McCormick,J.E., Klempnauer,K.H., Feeney,J. and Carr,M.D. (1998) Solution structure of the B-Myb DNA-binding domain: a possible role for conformational instability of the protein in DNA binding and control of gene expression. Biochemistry, 37, 9619–9629. [DOI] [PubMed] [Google Scholar]
- 33.Ogata K., Kanei-Ishii,C., Sasaki,M., Hatanaka,H., Nagadoi,A., Enari,M., Nakamura,H., Nishimura,Y., Ishii,S. and Sarai,A. (1996) The cavity in the hydrophobic core of Myb DNA-binding domain is reserved for DNA recognition and trans-activation. Nature Struct. Biol., 3, 178–187. [DOI] [PubMed] [Google Scholar]
- 34.Sasaki M., Ogata,K., Hatanaka,H. and Nishimura,Y. (2000) Backbone dynamics of the c-Myb DNA-binding domain complexed with a specific DNA. J. Biochem. (Tokyo), 127, 945–953. [DOI] [PubMed] [Google Scholar]
- 35.Biedenkapp H., Borgmeyer,U., Sippel,A.E. and Klempnauer,K.H. (1988) Viral myb oncogene encodes a sequence-specific DNA-binding activity. Nature, 335, 835–837. [DOI] [PubMed] [Google Scholar]
- 36.Nakagoshi H., Nagase,T., Kanei-Ishii,C., Ueno,Y. and Ishii,S. (1990) Binding of the c-myb proto-oncogene product to the simian virus 40 enhancer stimulates transcription. J. Biol. Chem., 265, 3479–3483. [PubMed] [Google Scholar]
- 37.Howe K.M. and Watson,R.J. (1991) Nucleotide preferences in sequence-specific recognition of DNA by c-myb protein. Nucleic Acids Res., 19, 3913–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weston K. (1992) Extension of the DNA binding consensus of the chicken c-Myb and v-Myb proteins. Nucleic Acids Res., 20, 3043–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ording E., Bergholtz,S., Brendeford,E.M., Jamin,N. and Gabrielsen,O.S. (1996) Flexibility in the second half-site sequence recognised by the c-Myb R2 domain—in vitro and in vivo analysis. Oncogene, 13, 1043–1051. [PubMed] [Google Scholar]
- 40.Ording E., Kvavik,W., Bostad,A. and Gabrielsen,O.S. (1994) Two functionally distinct half sites in the DNA-recognition sequence of the Myb oncoprotein. Eur. J. Biochem., 222, 113–120. [DOI] [PubMed] [Google Scholar]
- 41.Mizuguchi G., Nakagoshi,H., Nagase,T., Nomura,N., Date,T., Ueno,Y. and Ishii,S. (1990) DNA binding activity and transcriptional activator function of the human B-myb protein compared with c-MYB. J. Biol. Chem., 265, 9280–9284. [PubMed] [Google Scholar]
- 42.Foos G., Grimm,S. and Klempnauer,K.H. (1994) The chicken A-myb protein is a transcriptional activator. Oncogene, 9, 2481–2488. [PubMed] [Google Scholar]
- 43.Golay J., Loffarelli,L., Luppi,M., Castellano,M. and Introna,M. (1994) The human A-myb protein is a strong activator of transcription. Oncogene, 9, 2469–2479. [PubMed] [Google Scholar]
- 44.Stober-Grasser U., Brydolf,B., Bin,X., Grasser,F., Firtel,R.A. and Lipsick,J.S. (1992) The Myb DNA-binding domain is highly conserved in Dictyostelium discoideum. Oncogene, 7, 589–596. [PubMed] [Google Scholar]
- 45.Introna M., Golay,J., Frampton,J., Nakano,T., Ness,S.A. and Graf,T. (1990) Mutations in v-myb alter the differentiation of myelomonocytic cells transformed by the oncogene. Cell, 63, 1289–1297. [DOI] [PubMed] [Google Scholar]
- 46.Ness S.A. (1999) Myb binding proteins: regulators and cohorts in transformation. Oncogene, 18, 3039–3046. [DOI] [PubMed] [Google Scholar]
- 47.Lüscher B., Christenson,E., Litchfield,D.W., Krebs,E.G. and Eisenman,R.N. (1990) Myb DNA binding inhibited by phosphorylation at a site deleted during oncogenic activation. Nature, 344, 517–522. [DOI] [PubMed] [Google Scholar]
- 48.Oelgeschlager M., Krieg,J., Lüscher-Firzlaff,J.M. and Lüscher,B. (1995) Casein kinase II phosphorylation site mutations in c-Myb affect DNA binding and transcriptional cooperativity with NF-M. Mol. Cell. Biol., 15, 5966–5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinson B., Brendeford,E.M., Gabrielsen,O.S. and Daignan-Fornier,B. (2001) Highly conserved features of DNA binding between two divergent members of the myb family of transcription factors Nucleic Acids Res., 29, 527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brendeford E.M., Myrset,A.H., Hegvold,A.B., Lundin,M. and Gabrielsen,O.S. (1997) Oncogenic point mutations induce altered conformation, redox sensitivity, and DNA binding in the minimal DNA binding domain of avian myeloblastosis virus v-Myb. J. Biol. Chem., 272, 4436–4443. [DOI] [PubMed] [Google Scholar]
- 51.Brendeford E.M., Andersson,K.B. and Gabrielsen,O.S. (1998) Nitric oxide (NO) disrupts specific DNA binding of the transcription factor c-Myb in vitro. FEBS Lett., 425, 52–56. [DOI] [PubMed] [Google Scholar]
- 52.Studier F.W., Rosenberg,A.H., Dunn,J.J. and Dubendorff,J.W. (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol., 185, 60–89. [DOI] [PubMed] [Google Scholar]
- 53.Gabrielsen O.S., Matre,V. and Bergholtz,S. (2000) Protein–oligonucleotide interactions. In Meyers,R.A. (ed), Encyclopedia of Analytical Chemistry. John Wiley & Sons Ltd, Chichester, UK, pp. 5997–6017.
- 54.Ness S.A., Marknell,A. and Graf,T. (1989) The v-myb oncogene product binds to and activates the promyelocyte-specific mim-1 gene. Cell, 59, 1115–1125. [DOI] [PubMed] [Google Scholar]
- 55.Bonner J.J. (1991) Vectors for the expression and analysis of DNA-binding proteins in yeast. Gene, 104, 113–118. [DOI] [PubMed] [Google Scholar]
- 56.Gietz R.D., Schiestl,R.H., Willems,A.R. and Woods,R.A. (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast, 11, 355–360. [DOI] [PubMed] [Google Scholar]
- 57.Kippert F. (1995) A rapid permeabilization procedure for accurate quantitative determination of beta-galactosidase activity in yeast cells. FEMS Microbiol. Lett., 128, 201–206. [DOI] [PubMed] [Google Scholar]
- 58.Andersson K.B., Berge,T., Matre,V. and Gabrielsen,O.S. (1999) Sequence selectivity of c-Myb in vivo. Resolution of a DNA target specificity paradox. J. Biol. Chem., 274, 21986–21994. [DOI] [PubMed] [Google Scholar]
- 59.Tanaka Y., Nomura,T. and Ishii,S. (1997) Two regions in c-myb proto-oncogene product negatively regulating its DNA-binding activity. FEBS Lett., 413, 162–168. [DOI] [PubMed] [Google Scholar]
- 60.Høvring P.I., Bostad,A., Ording,E., Myrset,A.H. and Gabrielsen,O.S. (1994) DNA-binding domain and recognition sequence of the yeast BAS1 protein, a divergent member of the Myb family of transcription factors. J. Biol. Chem., 269, 17663–17669. [PubMed] [Google Scholar]
- 61.Ramsay R.G., Morrice,N., Van Eeden,P., Kanagasundaram,V., Nomura,T., De Blaquiere,J., Ishii,S. and Wettenhall,R. (1995) Regulation of c-Myb through protein phosphorylation and leucine zipper interactions. Oncogene, 11, 2113–2120. [PubMed] [Google Scholar]
- 62.Songyang Z., Blechner,S., Hoagland,N., Hoekstra,M.F., Piwnica-Worms,H. and Cantley,L.C. (1994) Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr. Biol., 4, 973–982. [DOI] [PubMed] [Google Scholar]
- 63.Bousset K., Oelgeschlager,M.H., Henriksson,M., Schreek,S., Burkhardt,H., Litchfield,D.W., Lüscher-Firzlaff,J.M. and Lüscher,B. (1994) Regulation of transcription factors c-Myc, Max, and c-Myb by casein kinase II. Cell. Mol. Biol. Res., 40, 501–511. [PubMed] [Google Scholar]
- 64.Myrset A.H., Bostad,A., Jamin,N., Lirsac,P.N., Toma,F. and Gabrielsen,O.S. (1993) DNA and redox state induced conformational changes in the DNA-binding domain of the Myb oncoprotein. EMBO J., 12, 4625–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Watson R.J., Robinson,C. and Lam,E.W. (1993) Transcription regulation by murine B-myb is distinct from that by c-myb. Nucleic Acids Res., 21, 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Humbert-Lan G. and Pieler,T. (1999) Regulation of DNA binding activity and nuclear transport of B-Myb in Xenopus oocytes. J. Biol. Chem., 274, 10293–10300. [DOI] [PubMed] [Google Scholar]
- 67.Hibi K., Liu,Q., Beaudry,G.A., Madden,S.L., Westra,W.H., Wehage,S.L., Yang,S.C., Heitmiller,R.F., Bertelsen,A.H., Sidransky,D. and Jen,J. (1998) Serial analysis of gene expression in non-small cell lung cancer. Cancer Res., 58, 5690–5694. [PubMed] [Google Scholar]
- 68.Khan J., Simon,R., Bittner,M., Chen,Y., Leighton,S.B., Pohida,T., Smith,P.D., Jiang,Y., Gooden,G.C., Trent,J.M. and Meltzer,P.S. (1998) Gene expression profiling of alveolar rhabdomyosarcoma with cDNA microarrays. Cancer Res., 58, 5009–5013. [PubMed] [Google Scholar]
- 69.Foos G., Grimm,S. and Klempnauer,K.H. (1992) Functional antagonism between members of the myb family: B-myb inhibits v-myb-induced gene activation. EMBO J., 11, 4619–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Foos G., Natour,S. and Klempnauer,K.H. (1993) TATA-box dependent trans-activation of the human HSP70 promoter by Myb proteins. Oncogene, 8, 1775–1782. [PubMed] [Google Scholar]
- 71.Nakagoshi H., Takemoto,Y. and Ishii,S. (1993) Functional domains of the human B-myb gene product. J. Biol. Chem., 268, 14161–14167. [PubMed] [Google Scholar]
- 72.Sala A., Kundu,M., Casella,I., Engelhard,A., Calabretta,B., Grasso,L., Paggi,M.G., Giordano,A., Watson,R.J., Khalili,K. and Peschle,C. (1997) Activation of human B-MYB by cyclins. Proc. Natl Acad. Sci. USA, 94, 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lane S., Farlie,P. and Watson,R. (1997) B-Myb function can be markedly enhanced by cyclin A-dependent kinase and protein truncation. Oncogene, 14, 2445–2453. [DOI] [PubMed] [Google Scholar]
- 74.Kamano H. and Klempnauer,K.H. (1997) B-Myb and cyclin D1 mediate heat shock element dependent activation of the human HSP70 promoter. Oncogene, 14, 1223–1229. [DOI] [PubMed] [Google Scholar]
- 75.Gatica M., Jacob,G., Allende,C.C. and Allende,J.E. (1995) DNA inhibits the catalytic activity of the alpha subunit of protein kinase CK2. Biochemistry, 34, 122–127. [DOI] [PubMed] [Google Scholar]