

Abstract
Objective
Despite modern therapies, pulmonary arterial hypertension (PAH) harbors a high mortality. Vascular remodeling is a hallmark of the disease. Recent clinical studies revealed that antiremodeling approaches with tyrosine–kinase inhibitors such as imatinib are effective, but its applicability is limited by significant side effects. Although imatinib has multiple targets, expression analyses support a role for platelet-derived growth factor (PDGF) in the pathobiology of the disease. However, its precise role and downstream signaling events have not been established.
Approach and Results
Patients with PAH exhibit enhanced expression and phosphorylation of β PDGF receptor (βPDGFR) in remodeled pulmonary arterioles, particularly at the binding sites for phophatidyl-inositol-3-kinase and PLCγ at tyrosine residues 751 and 1021, respectively. These signaling molecules were identified as critical downstream mediators of βPDGFR-mediated proliferation and migration of pulmonary arterial smooth muscle cells. We, therefore, investigated mice expressing a mutated βPDGFR that is unable to recruit phophatidyl-inositol-3-kinase and PLCγ (βPDGFRF3/F3). PDGF-dependent Erk1/2 and Akt phosphorylation, cyclin D1 induction, and proliferation, migration, and protection against apoptosis were abolished in βPDGFRF3/F3 pulmonary arterial smooth muscle cells. On exposure to chronic hypoxia, vascular remodeling of pulmonary arteries was blunted in βPDGFRF3/F3 mice compared with wild-type littermates. These alterations led to protection from hypoxia-induced PAH and right ventricular hypertrophy.
Conclusions
By means of a genetic approach, our data provide definite evidence that the activated βPDGFR is a key contributor to pulmonary vascular remodeling and PAH. Selective disruption of PDGF-dependent phophatidyl-inositol-3-kinase and PLCγ activity is sufficient to abolish these pathogenic responses in vivo, identifying these signaling events as valuable targets for antiremodeling strategies in PAH.
Keywords: platelet-derived growth factor, pulmonary hypertension, vascular endothelial growth factor, vascular remodeling
Pulmonary arterial hypertension (PAH) is a devastating disease still harboring a poor prognosis. Although modern therapies have led to an improved outcome, mortality remains unacceptably high, with annual mortality rates at ≈10%.1–3 Although the current PAH therapies target on abnormal pulmonary vasoconstriction, PAH is nowadays recognized as a proliferative disease, which is mainly caused by pulmonary vascular remodeling. A variety of cell types contribute to this process, including fibroblasts, endothelial cells (ECs), inflammatory cells and vascular smooth muscle cells (VSMCs).4,5 Excessive proliferation and migration of ECs and VSMC result in medial hypertrophy of pulmonary resistance vessels, as well as muscularization—and the formation of obliterative lesions in small, normally nonmuscularized segments of the pulmonary vasculature. Consequently, pulmonary vascular resistance rises, causing a progressive increase of right ventricular afterload, which eventually leads to right ventricular failure and death.6,7
Peptide growth factors, such as platelet-derived growth factor (PDGF), which elicit their signals via highly selective receptor tyrosine kinases, seem to play a prominent role in pulmonary vascular remodeling. PDGF is regarded as the most potent mitogen for VSMC,8,9 critically involving the tyrosine kinase activity of the β PDGF receptor (βPDGFR) subtype.10,11 Data from atherosclerosis and restenosis models support an important role of PDGF in vivo.12,13 In the context of PAH, several studies reporting expression analyses and pharmacological interventions suggest a role for PDGF in experimental and human disease,14–20 but its precise role and downstream signaling remain to be established.
Inhibition of PDGFR signaling may be achieved by tyrosine kinase inhibitors, such as imatinib mesylate, which was developed for the treatment of chronic myeloid leukemia and targets the Bcr/Abl oncogene, c-kit, and the α and βPDGFR subtypes.21 Importantly, imatinib was sufficient to reverse established pulmonary vascular remodeling and PAH in experimental models.20 Case reports and a phase II study provided evidence that it may be effective in human PAH.22–24 Recently, a randomized, placebo-controlled phase III trial (IMPRES) has shown that imatinib profoundly improved exercise capacity and pulmonary hemodynamics (mean pulmonary artery pressure, cardiac output, and pulmonary vascular resistance) in patients with severe PAH who were already on combination therapy with approved PAH drugs.25 However, this was accompanied by severe adverse events, including poor tolerability and the unexpected occurrence of subdural hematomas in a significant number of patients that occurred in conjunction with therapeutic anticoagulation, which is recommended in PAH.26 These serious safety concerns preclude the routine clinical use of imatinib in PAH, despite proof of efficacy. It is thus paramount to precisely define the pathways involved in both the efficacy and safety aspects of imatinib in PAH, as such knowledge will provide the basis to develop more targeted compounds for this disease.25,27
Two target kinases of imatinib, PDGFR and c-kit, have been implicated in the pathobiology of PAH. The stem cell factor c-kit may be important because targeting of c-kit in circulating progenitor cells inhibited the formation of PAH in a mouse model,28 so that the effect of imatinib may—at least partially—be explained by interference with these cells.29 Consequently, the hypothesis that PDGF signaling is important in PAH is currently based on expression data and experimental evidence with nonspecific therapeutic interventions,14,18,20 whereas direct proof from genetic models is lacking. It thus seems of key importance to precisely define the role of PDGF for pulmonary vascular remodeling and to identify the signaling pathways, by which the βPDGFR may mediate cellular responses that contribute to disease progression. Because the presence of null alleles for the βPDGFR or PDGF-B in mice results in embryonic lethality, no studies on defective PDGF signaling in PH are available. We used an alternate genetic approach to address 3 important questions, such as (1) what are the signaling pathways by which the activated βPDGFR mediates proliferation, migration, and cell survival of pulmonary arterial smooth muscle cells (PASMCs)? (2) is βPDGFR signaling really important for pulmonary vascular remodeling in vivo? and (3) can the disruption of specific signaling events prevent pulmonary vascular remodeling and PAH?
On ligand binding, the βPDGFR is autophosphorylated at tyrosine residues that serve as specific docking sites for receptor-associated signaling molecules. The activated βPDGFR recruits and activates Src family kinases, phosphatidylinositol 3′-kinase (PI3K), the GTPase-activating protein of Ras, the protein tyrosine phosphatase SHP-2, and phospholipase C-γ1 (PLCγ).30 Recently, we systematically characterized signal relay by the βPDGFR in VSMCs by the use of 2 panels of chimeric CSF1R/βPDGFR mutants in the context of neointima formation.13 On the basis of our previous findings and on site-specific βPDGFR phosphorylation analyses in humans, we now used a mouse model defective of PDGF-dependent PI3K and PLCγ signaling (βPDGFRF3/F3 mice) to definitively analyze the contribution of PDGF-initiated signals to the pathobiology of PAH. These studies revealed that PI3K and PLCγ are the key signaling enzymes mediating the βPDGFR’s mitogenic, chemotactic, and antiapoptotic signals, and that these events are critical for pulmonary vascular remodeling and PAH in vivo.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Activation of PDGF-Dependent PI3K and PLCγ Signaling in Human PAH
To monitor PDGF-dependent signaling events in human idiopathic PAH (IPAH) in vivo, we used site-selective, phospho-specific antibodies directed against the tyrosine residues, which are required for the recruitment of PI3K (Y751) and PLCγ (Y1021) to the activated βPDGFR. These analyses revealed that patients with IPAH exhibit enhanced expression and activation of βPDGFR in remodeled pulmonary arterioles as assessed by immunostaining and Western blot analyses (Figure 1A–C). In particular, we found increased phosphorylation of the βPDGFR’s binding sites for PI3K and PLCγ at tyrosine residues 751 and 1021, respectively, indicating that PDGF-dependent PI3K and PLCγ signaling are activated in human IPAH.
Figure 1.

Expression and site-specific activation of the β platelet-derived growth factor receptor (βPDGFR) in human pulmonary arterial hypertension. Expression of the βPDGFR and phosphorylation of tyrosine residues required for binding of phophatidyl-inositol-3-kinase (Y751) and phospholipase C-γ1 (Y1021) in lung samples of patients with idiopathic pulmonary arterial hypertension (IPAH) and patients without pulmonary hypertension (control). Shown are representative immunostainings (A), representative Western blot analysis (B), and the quantification of Western blot analyses by densitometry (C). Shown is the protein expression/phosphorylation normalized for actin. Data in (C) represent mean values±SEM. From 5 patients in each group. AU indicates arbitrary units. *P<0.05.
PI3K and PLCγ Mediate PDGF-Induced PASMC Proliferation and Migration
To assess the signal relay mechanisms downstream of the βPDGFR that are relevant for vascular remodeling in mPASMC, we first used a pharmacological approach. To rule out effects in ECs, we assessed βPDGFR expression and activation in these cells. We found that the βPDGFR is not expressed and activated on PDGF stimulation in EC, whereas vascular endothelial growth factor–dependent activation of the vascular endothelial growth factor receptors was restricted to EC (Figure 2A). Thus, we mainly focused on VSMCs. On the basis of previous data pointing toward the importance of PI3K and PLCγ for PDGF-BB–mediated responses,13,18 we mainly focused on these signaling molecules. Hence, we performed BrdU incorporation and chemotaxis analyses using pharmacological inhibitors of PI3K (LY294002) and PLCγ (U73122). As shown in Figure 2B, inhibition of PI3K or PLCγ completely blocked PDGF-BB–mediated proliferation. In contrast, the MEKK-inhibitor PD98059 had no effect. Similarly, PDGF-BB–mediated chemotaxis, as assessed by modified Boyden chamber assays, was also abolished by inhibition of PI3K or PLCγ, whereas MEKK inhibition had no effect (Figure 2C). These data indicate that PI3K and PLCγ activity is required for the mitogenic and chemotactic responses to PDGF-BB in mPASMC, and this finding is consistent with data obtained in human PASMC.18 In conjunction with βPDGFR upregulation and the phosphorylation state of the βPDGFR’s docking sites for PI3K and PLCγ in human IPAH at Y751 and Y1021, respectively, our data point toward a pathogenic role of these pathways in pulmonary vascular remodeling and PAH.
Figure 2.
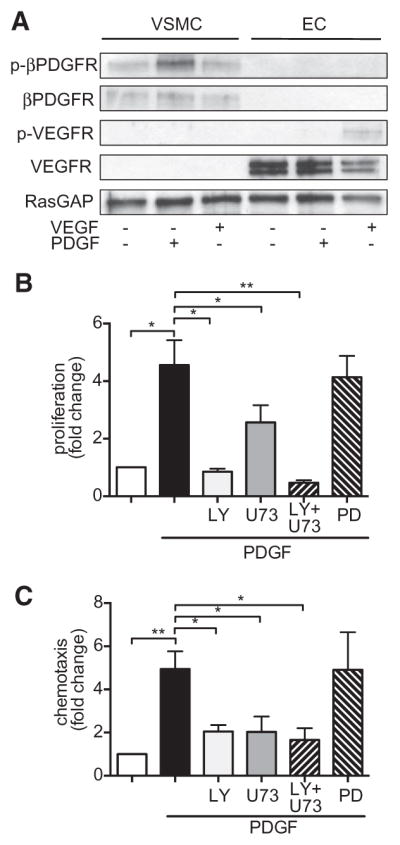
Critical role of platelet-derived growth factor receptor (PDGFR)–dependent activation of phophatidyl-inositol-3-kinase (PI3K) and phospholipase C-γ1 (PLCγ) in pulmonary hypertension. A, Expression of βPDGFR and vascular endothelial growth factor receptor (VEGFR) in vascular smooth muscle cells (VSMCs) and endothelial cells (EC), as well as activation (phosphorylation) of these receptors by platelet-derived growth factor (PDGF; 50 ng/mL) or VEGF (50 ng/mL) were assessed by western blot analyses. B and C, PDGF-dependent proliferation (B) and migration (C) of murine pulmonary arterial smooth muscle cells were assessed as BrdU incorporation and by modified Boyden chamber assays, respectively. To assess the role of PI3K and PLCγ, cells were incubated with pharmacological inhibitors: The PI3K inhibitor LY294002 (LY, 10 mmol/L), the PLCγ inhibitor U73122 (U73, 10 mmol/L) or (as a control) the MEK kinase inhibitor PD98059 (PD, 5 mmol/L) 30 minutes before PDGF treatment. Furthermore, cells were treated with both PI3K and PLCγ inhibitors (LY+U73) followed by addition of PDGF. RasGAP indicates GTPase-activating protein of Ras. *P<0.05, **P<0.01.
Functionality of the F3 Mutation and Characterization of PASMC From βPDGFRF3/F3 Mice
On the basis of the above findings, a genetic mouse model with defective binding of these 2 signal relay molecules to the βPDGFR would be expected to display impaired or abolished PDGF-dependent proliferation/migration in vivo. The F3-mouse model was generated by introducing point mutations into the coding sequence of the βPDGFR that led to the modification of amino acid residues 739 and 750 from tyrosine to phenylalanine (PI3K-binding site), and residue 1020 from tyrosine to isoleucine (PLCγbinding site), thus preventing binding of PI3K and PLCγ to the activated βPDGFR31 (Figure 3A). This genetic model would be expected to be associated with completely abolished PDGF-dependent proliferation and migration.13 Thus, it may be more valuable than a single mutation of the PI3K or the PLCγ-binding site because it enables us to definitely analyze the role of PDGF-dependent signaling events in pulmonary vascular remodeling in the context of PH. βPDGFRF3/F3 mice had no overt phenotype at baseline31 and did not show changes in body weight (21.6±1.4 g versus 21.9±0.9 g) or metabolic parameters as compared with wild-type (WT mice; not shown).
Figure 3.

Characterization of pulmonary arterial smooth muscle cells (PASMCs) isolated from wild-type (WT) and homozygous β platelet-derived growth factor receptor (βPDGFR)F3/F3 mice. A, Schematic diagram of the intracellular part of the βPDGFR F3 mutant showing lack of the tyrosine phosphorylation sites required for binding of phophatidyl-inositol-3-kinase (PI3K; Tyr-739/750) and phospholipase C-γ1 (PLCγ; Tyr-1020). B, Western blot analysis demonstrating equal protein expression levels of the βPDGFR in PASMC isolated from WT or βPDGFRF3/F3 mice (GTPase-activating protein of Ras [RasGAP] served as loading control). C, PASMC from WT or βPDGFRF3/F3 mice was treated with PDGF-BB or vehicle for 5 minutes. Phosphorylation of tyrosines Y1020 and Y750 was assessed by Western blot analysis using phospho- and site-specific antibodies. D, On treatment with PDGF-BB or vehicle, the βPDGFR was immunoprecipitated followed by Western blot analysis to detect total receptor phosphorylation (P-Y) or binding of associated signaling molecules (p85: regulatory subunit of PI3K, PLCγ, RasGAP, and SHP-2). E, To detect PDGF-dependent association of Src with the activated βPDGFR, Src was immunoprecipitated followed by detection of associated βPDGFR, cells treated as in (D). F and G, On stimulation with PDGF-BB or vehicle for the indicated time points, phosphorylation of Akt and Erk 1/2, as well as expression of the cell cycle protein cyclin D1 were assessed. Shown are a representative Western blot analysis (F) and the quantification by densitometry (G). AU indicates arbitrary units. *P<0.05.
To evaluate the consequences of the genetic modification on the cellular level, we isolated PASMC from mice that were homozygous for the F3 allele, and from WT litter-mates. First, we assessed the protein levels of the βPDGFR in F3/F3- and WT-PASMC to rule out that the mutation interfered with normal expression of the receptor. As shown in Figure 3B, the βPDGFR is expressed at similar levels in F3/F3- and WT-PASMC. Next, we evaluated the ligand-induced phosphorylation of the binding sites of PI3K- and PLCγ at tyrosine residues 739/750 and 1020, respectively, by the use of phospho- and site-specific antibodies.18 Figure 3C demonstrates that PDGF-BB–dependent phosphorylation of these sites was absent in F3-PASMC. Next, we sought to ensure the impaired recruitment of PI3K and PLCγ to the activated F3 receptor. To this end, we performed coimmunoprecipitation experiments to monitor the association of receptor-associated signal relay enzymes with the activated βPDGFR. The receptor was immunoprecipitated on stimulation with PDGF-BB (30 ng/mL; 5 minutes), followed by Western blot analyses for receptor-associated signaling molecules, including GTPase-activating protein of Ras, Src, SHP-2, PI3K, and PLCγ. As shown in Figure 3D, both the WT and F3 receptor were phosphorylated on PDGF stimulation (P-Y), indicating proper activation of the mutated receptor. As expected, there was no ligand-induced recruitment of PI3K and PLCγ to the activated F3 receptor, whereas binding of these 2 signaling molecules to the WT receptor was efficient, ruling out unspecific effects induced by insufficient antibody binding. Overall, phosphorylation of the F3 receptor was slightly decreased, most probably reflecting the lack of the 2 important phosphorylation sites. Furthermore, binding of the other signaling molecules that associate with the activated βPDGFR whose binding sites were not mutated (GTPase-activating protein of Ras, SHP-2, Figure 3D, Src, Figure 3E) was unaffected by the F3 mutation. Hence, PDGF-dependent signaling via PI3K and PLCγ is selectively abolished in F3-mPASMC. To assess whether these modifications translate into impaired downstream signaling, we monitored the phosphorylation state of Erk1/2 and Akt, as well as the expression of the cell cycle protein cyclin D1. Figure 3F and 3G demonstrates that the PDGF-induced phosphorylation of Erk1/2 and Akt, representing critical downstream mediators, and the induction of cyclin D1 were profoundly impaired in F3-mPASMC as compared with WT cells. Taken together, these data confirm the presence of the functional defect of the F3 mutation and show that this genetic modification results in altered downstream signaling.
PDGF-Induced Proliferation, Migration, and Cell Survival are Abolished in F3-mPASMC
As mentioned above, proliferation and migration of PASMC are critical during the onset and progression of pulmonary vascular remodeling. To assess whether these responses were altered in F3-mPASMC, we performed BrdU incorporation and chemotaxis assays. As demonstrated in Figure 4A, PDGF-BB induced a robust increase of BrdU incorporation in WT-PASMC, whereas this response was essentially completely absent in F3-PASMC. Furthermore, PDGF-BB- and thrombospondin-1–mediated chemotaxis were also blunted in F3-PASMC, when compared with WT cells (Figure 4B and 4C). Because chronic hypoxia is an important pathogenic factor in PH, we assessed PDGF-dependent proliferation after exposure of cells to hypoxia. As shown in Figure 4D, PDGF-dependent proliferation of F3 cells was also blocked under hypoxic conditions. This observation was reproduced in vivo, as shown by PCNA staining of lung tissue of WT- and βPDGFRF3/F3 mice that were exposed to chronic hypoxia for 3 weeks (Figure 4E and 4F). As PDGF was also shown to protect against apoptosis, an effect exclusively mediated by PI3K,32 we hypothesized that this protective effect was lost in F3-PASMC. Consequently, we performed an FACS-based assay and stained caspase-3-positive cells after induction of apoptosis by serum deprivation in the presence or absence of PDGF-BB. As shown in Figure 4G and 4H, PDGF-BB offered significant protection from apoptosis in WT-PASMC, whereas this effect was completely abolished in F3 cells. In summary, F3-PASMC lost their ability to proliferate and migrate on PDGF treatment, and PDGF-mediated protection from apoptosis is also lost in these cells. Although F3-PASMC are hence resistant to the critical PDGF-mediated events that relate to vascular remodeling in vitro, we next sought to analyze the extent of vascular remodeling and pulmonary hypertension in WT and βPDGFR-F3 mice in vivo.
Figure 4.

Abolished platelet-derived growth factor (PDGF)–dependent proliferation and migration and enhanced susceptibility to apoptosis in F3/F3 pulmonary arterial smooth muscle cells (PASMCs). PDGF-BB–dependent proliferation (A) and PDGF-BB or thrombospondin (TSP-1)–induced migration (B and C) of PASMC from wild-type (WT) or β platelet-derived growth factor receptor (βPDGFR)F3/F3 mice were assessed by BrdU incorporation or with modified Boyden chamber assays, respectively. C, PDGF-dependent proliferation (BrdU incorporation) of PASMC was analyzed after exposure of cells to hypoxia (1% O2) or normoxia. D and E, Lung sections of WT and βPDGFRF3/F3 mice subjected to normoxia (21% O2) or hypoxia (10% O2) for 3 weeks stained for PCNA, counterstain with methylgreen. Representative experiment (D) and quantification of 3 independent experiments (E). F and G, Detection of apoptosis by flow cytometric staining of active caspase-3-positive cells. Representative experiment (F) and quantification of 3 independent experiments (G). AU indicates arbitrary units. *P<0.05, **P<0.01.
Disruption of PDGF-Dependent PI3K and PLCγ Activity Protects From Hypoxia-Induced Pulmonary Hypertension
To assess, whether defective PDGF signaling in the F3 model results in an altered susceptibility to PH, we took advantage of the established mouse model of hypoxia-induced pulmonary hypertension. Because partial deficiency of PDGF signaling via PI3K and PLCγ may be sufficient to cause an altered response, heterozygous βPDGFRF3/+ mice (apart from homozygous βPDGFR βPDGFRF3/F3 and WT littermate mice) were also included in the analysis. Exposure of mice to chronic hypoxia (10% O2 for 3 weeks) led to the development of pulmonary hypertension in WT mice, as indicated by an increase of right ventricular systolic pressure from 24.3±0.5 mm Hg to 33.5±1.5 mm Hg (P<0.001; Figure 5A). This adaptive response to hypoxia was significantly impaired in heterozygous βPDGFRF3/+ mice (29.4±1.3 mm Hg; P<0.01), as well as in homozygous βPDGFRF3/F3 mice (28.6±1.2 mm Hg; P<0.01).
Figure 5.

Hypoxia-induced pulmonary hypertension and right ventricular hypertrophy are abrogated in β platelet-derived growth factor receptor (βPDGFR)F3/F3 mice. A and B, Right ventricular systolic pressure (RVPsyst) and right ventricular hypertrophy, presented as the ratio of right ventricle (RV) to left ventricle (LV) plus septum (S) weight (RV/LV+S) in mice (genotypes: wild-type, βPDGFRF3/+, and βPDGFRF3/F3) subjected to normoxia (21% O2) or hypoxia (10% O2) for 3 weeks. C and D, Systolic and diastolic blood pressure (BP) in the various genotypes treated as in (A) and (B) as assessed via cannulation of the carotid artery. E, Heart rate of mice treated as in (A) and (B). n ≥ 6 in each group; *P<0.05, **P<0.01, ***P<0.001.
Chronic PH causes right ventricular adaptation as indicated by the development of right ventricular hypertrophy. Figure 5B demonstrates that pulmonary hypertension in WT mice was associated with an increase of the right ventricle to left ventricle plus septum weight ratio (right ventricle/left ventricle+septum) from 0.21±0.01 to 0.35±0.03 (P<0.001). However, right ventricular hypertrophy was significantly reduced in both the heterozygous βPDGFRF3/+ (0.29±0.01; P<0.05) and homozygous βPDGFRF3/F3 mice (0.28±0.01; P<0.05). These results indicate that lack or impairment of PDGF-induced PI3K and PLCγ signaling leads to protection from hypoxia-induced PH and, consequently, to decreased hypertrophy of the right ventricle.
To rule out differences in systemic arterial pressures between genotypes, arterial blood pressure was recorded with a catheter placed in the carotid artery. As shown in Figure 5C and 5D, systolic and diastolic arterial blood pressure were similar between groups. Furthermore, we did not detect differences in heart rate between genotypes (Figure 5E).
βPDGFRF3/F3 Mice are Protected From Hypoxic Pulmonary Vascular Remodeling
On the basis of the blunted PDGF-dependent proliferation and migration of F3-mPASMC, the finding that F3 mice are protected from hypoxia-induced PH is most likely because of reduced pulmonary vascular remodeling in these mice. To assess the effect of the F3 mutation on hypoxia-induced remodeling processes in the pulmonary vasculature, morphometric analyses of lung tissues were performed in the various genotypes (Figure 6A–6D), representative stainings are presented in Figure 6E and 6F. To assess the degree of muscularization, we performed morphometric analyses in small pulmonary arteries with a diameter <70 μm, and medium-sized vessels (70–150 μm). As shown in Figure 6A, hypoxia led to a significant increase of the percentage of muscularized small pulmonary arteries in WT mice (P<0.01), whereas the number of nonmuscularized arteries significantly decreased (P<0.01) in these mice (arterial diameter <70 μm). In contrast, βPDGFRF3/F3 mice had significantly less fully (P<0.001) and partially (P<0.05) muscularized small pulmonary arteries (<70 μm) under these conditions, whereas the percentage of nonmuscularized arteries was significantly higher (P<0.001). Even among larger vessels with a diameter of 70 to 150 μm, there were significantly more nonmuscularized vessels (P<0.05) and a trend toward less fully muscularized arteries (P=0.08) in βPDGFRF3/F3 mice compared with WT animals (Figure 6B). As shown in Figure 6C and 6D, hypoxia induced a significant increase of medial wall thickness in small pulmonary arteries with a diameter <50 μm and in medium-sized arteries (50–100 μm) in WT mice, confirming the presence of pulmonary vascular remodeling. However, this response was significantly reduced in βPDGFRF3/+ mice and completely blunted in βPDGFRF3/F3 mice, in line with resistance to hypoxia-induced pulmonary vascular remodeling in these mice. In conclusion, ablation of mitogenic βPDGFR signaling protects from hypoxic pulmonary vascular remodeling.
Figure 6.

β platelet-derived growth factor receptor (βPDGFR)F3/F3 mice are protected from hypoxic pulmonary vascular remodeling. A and B, Mice (genotypes: wild-type (WT), βPDGFRF3/+, and βPDGFRF3/F3) were exposed to normoxia (21% O2) or hypoxia (10% O2) for 3 weeks. Shown are morphometric analyses of fully muscularized (open bars), partially muscularized (dashed bars), and nonmuscularized (closed bars) small (A: diameter <70 μm) and medium-sized (B: diameter 70–150 μm) pulmonary arteries. C and D, Medial wall thickness in small (C: diameter <50 μm) and medium-sized (D: diameter 50–150 μm) pulmonary arteries of mice treated as in (A) and (B). E and F, Van Gieson staining (E) and double staining for α smooth muscle actin (purple) and the endothelial marker von Willebrand factor (brown; F) in lung sections from mice treated as in (A) and (B). n ≥ 6 in each group; *P<0.05, **P<0.01, ***P<0.001.
Discussion
In this study, we analyzed the contribution of the activated βPDGFR and its associated signaling molecules to cellular responses of PASMCs in vitro and pulmonary hypertension in vivo. The data presented herein provide direct proof from a genetic model and allow 2 important conclusions, such as (1) signaling by the activated βPDGFR (particularly through PI3K and PLCγ) is crucial for pulmonary vascular remodeling and hypoxia-induced pulmonary hypertension, and (2) selective disruption of specific signaling pathways is sufficient to attenuate the onset and progression of a pathogenic vascular disease, pulmonary hypertension, and consecutive right ventricular remodeling in vivo. These results have potential clinical significance and clarify the role of PDGF signaling in PAH.
Although previous studies that provided expression analyses or applied pharmacological approaches, such as tyrosine–kinase inhibitors have indicated that PDGF is an important contributor to pulmonary vascular remodeling and pulmonary hypertension,14,20 these studies did not provide direct proof for a significant role of PDGF. For instance, although the TKI imatinib—targeting the PDGFR and the Bcr/Abl kinase—reversed established pulmonary vascular remodeling and experimental PH,20 this compound also inhibits c-kit which was recently shown to contribute to the pathobiology of PH.28,29 It may thus not be concluded from available data that inhibition of PDGF-dependent pathways is responsible for the therapeutic effects of TKIs such as imatinib. Furthermore, the role of PDGF isoforms and receptor subtypes has not been investigated. To directly prove the importance of βPDGFR-initiated signals for the pathobiology of pulmonary hypertension (independently of more or less specific pharmacological inhibitors) and to identify the crucial downstream signaling pathways, we took advantage of a targeted genetic modification. In mice that are homozygous for the F3 mutation, the βPDGFR is normally expressed, but is unable to recruit and activate PI3K and PLCγ on ligand binding. Ablation of these PDGF-dependent events did not lead to general defects of βPDGFR signaling, as overall receptor activation and binding of other signaling molecules were not impaired (not shown13,31).
The rationale to investigate pulmonary vascular remodeling and pulmonary hypertension in βPDGFRF3/F3 mice was based on a systematic in vitro approach using chimeric CSF1R/βPDGFR mutants that allowed the detailed characterization of βPDGFR signaling in aortic VSMCs.13,32 These studies revealed that (1) multiple signaling enzymes, including PI3K, PLCγ, and Src contribute to PDGF-dependent chemotaxis, (2) PI3K and PLCγ are essential for the βPDGFR’s mitogenic signal, and (3) antiapoptotic PDGF signaling is exclusively mediated via the PI3K/Akt pathway,13,32 thus identifying PI3K and PLCγ as the critical mediators of relevant cellular responses. The finding that PDGF-dependent chemotaxis is completely blocked in F3 cells, despite effective binding of Src to the activated βPDGFR further emphasizes the importance of PI3K and PLCγ for PDGF signaling. In the context of hypoxia, we further demonstrated that the ligand-mediated phosphorylation of the βPDGFR was enhanced under hypoxic conditions in PASMC, causing augmented proliferation and migration of PASMC in response to PDGF.18 Although receptor expression was not affected by hypoxia, the tyrosine residues representing the binding sites of PI3K and PLCγ were also hyperphosphorylated in this setting, indicating increased signaling via these two molecules. On the molecular level, hyperactivation of the βPDGFR correlated with hypoxia-inducible factor (HIF)-1α–dependent downregulation of protein tyrosine phosphatases, which dephosphorylate and thus inactivate the receptor. Taken together, these data highlight the critical role of PDGF-dependent PI3K and PLCγ signaling, particularly under hypoxic conditions.
Based on previous studies demonstrating that the presence of null alleles for the βPDGFR or PDGF-B in mice result in embryonic lethality, which is primarily because of vascular defects,33–36 it seems surprising that βPDGFRF3/F3 mice are viable and have no obvious phenotype.31 However, chimeric analysis revealed that when F3-receptor–expressing cells competed with WT βPDGFR-expressing cells in the same animals, there was a strong selection against βPDGFRF3/F3 cells in the layer of smooth muscle cells surrounding the vasculature.31 This finding, which indicates that the F3 mutant receptor does not transmit signals as efficiently as the WT receptor in vivo, prompted us to hypothesize that the F3 mutation may become relevant under pathological conditions. Indeed, when βPDGFRF3/F3 mice were exposed to chronic hypoxia, the remodeling process in the pulmonary vasculature was profoundly reduced when compared with WT mice. The presented data demonstrate for the first time that genetic ablation of essential elements of βPDGFR signaling abolishes experimental PH. The data are strengthened by the fact that heterozygous βPDGFRF3/+ mice were still significantly protected from PH and right ventricle hypertrophy and also exhibited significantly less hypoxia-induced medial hypertrophy. However, vascular changes in heterozygous βPDGFRF3/+ mice in the morphometric analyses (Figure 6A–6B) were not significant. This probably reflects the way the data are analyzed. In morphometric analyses, vessels are classified arbitrarily into nonmuscularized (<5% muscularization), partially muscularized (5%–70%), and fully muscularized (>70%). Thus, because of the large group of partial muscularization, smaller differences between genotypes may have been masked in this analysis. In contrast, analysis of medial wall thickness, which is independent of this arbitrary classification, revealed significant protection from hypoxia-induced vascular remodeling in heterozygous βPDGFRF3/+ mice, which was more pronounced in homozygous βPDGFRF3/F3 mice. The protection from PH in βPDGFRF3/F3 mice resulted from the complete absence of hypoxia-induced pulmonary vascular remodeling, and—on the cellular level—from impaired PDGF-dependent proliferation and migration of PASMC, as well as blunting of antiapoptotic PDGF signaling. These responses were assessed in VSMC, and may translate to other cell types, such as fibroblasts, as well. However, despite the prominent role of this cell type in PH biology, the observed effects are most probably independent of alterations in EC, as we did not detect expression of the βPDGFR in these cells and, furthermore, PDGF did not induce receptor activation.
With regard to the role of PI3K and PLCγ, our genetic approach bears 1 potential caveat: it cannot be ruled out completely that proteins other than the known binding partners of the mutated tyrosine residues also associate with these sites and thereby contribute to βPDGFR-mediated cellular responses. This seems, however, unlikely because pharmacological inhibition of PI3K or PLCγ also abrogated PDGF-induced PASMC proliferation and migration in vitro. In addition to mouse PASMCs, the respective binding sites were found to be in an active state in human disease, and we found that pharmacological inhibition of PI3K and PLCγ also attenuated PDGF-induced chemotaxis and proliferation of human PASMCs,18 suggesting species independency. Finally, our results are in line with previous observations demonstrating the relevance of these PDGF-dependent signaling pathways in a mouse model of neointima formation after carotid artery balloon injury.13
The findings obtained in βPDGFRF3/F3 mice are of potential clinical significance, as they address important mechanistic insights that evolved in recent clinical trials. Although the concept of tyrosine–kinase inhibition by imatinib has proven effective in human PAH when applied in addition to established therapies,24,25 it is currently not feasible because of poor tolerability in this population and the unexpected occurrence of subdural hematomas in patients on anticoagulation. In addition, cardiotoxic effects of imatinib have been described.37 Although the therapeutic efficacy of imatinib in PAH is most likely via inhibition of PDGF signaling, the mechanisms that are responsible for adverse events, such as subdural hematomas are completely unknown, or—in the case of cardiotoxicity—are independent of PDGF.37,38 Therefore, it is critical to resolve these issues and to better define both the efficacy and safety aspects of tyrosine–kinase inhibition in PAH. In that sense, the precise identification of the signal relay mechanisms driving disease onset and progression as provided in this study is of great value. Importantly, phosphorylation of the PI3K- and PLCγ-binding sites in the βPDGFR at tyrosine residues 751 and 1021, respectively, was also found in human tissue of patients with IPAH, indicating that these pathways are activated in human disease and thus represent a selective therapeutic target. With regard to translational aspects, our data may be helpful and relevant in 2 ways, such as (1) we provide the first evidence from a genetic loss of function model to demonstrate that PDGF and not other target kinases of imatinib is important, suggesting to design future TKIs particularly against PDGF and (2) we identify important pathways downstream of the PDGFR, which may enable more specific therapeutic strategies in the future (potentially limiting adverse events and improving safety and tolerability), and which may also be relevant for other growth factor receptors. Hence, targeting these downstream pathways potentially inhibits the responses of other growth factor receptors, as well and may, therefore, be effective in nonresponders to imatinib, in whom the disease may be driven primarily by other growth factors. Our findings thus set the ground for future studies exploring the role of specific PDGF inhibition on one hand, as well as targeting common downstream mediators (ie, PI3K and PLCγ) with small-molecule inhibitors that are currently being developed, which may represent a promising alternative strategy.
In summary, our data indicate that the lack of PI3K and PLCγ activity downstream of the βPDGFR prevents the proliferation, migration, and survival of PASMCs and provide direct proof that these signaling pathways are essential for pulmonary vascular remodeling and pulmonary hypertension in vivo. The identification of these signal relay mechanisms provides the basis for the development of selective novel therapeutic strategies that target on the morphological alterations in the pulmonary vasculature and may help to overcome current tolerability and safety issues with antiproliferative approaches.
Supplementary Material
Significance.
We found that selective disruption of PDGF-dependent phophatidyl-inositol-3-kinase and phospholipase C-γ1 activity in a genetic mouse model (β platelet-derived growth factor receptorF3/F3) is sufficient to abolish pulmonary vascular remodeling and pulmonary hypertension. In vitro, PDGF-induced proliferation, migration, and protection from apoptosis were blocked in vascular smooth muscle cells isolated from β platelet-derived growth factor receptorF3/F3 mice. Furthermore, patients with pulmonary arterial hypertension exhibit enhanced expression and phosphorylation of β platelet-derived growth factor receptor in remodeled pulmonary arterioles, particularly at the binding sites for phophatidyl-inositol-3-kinase and phospholipase C-γ1 at tyrosine residues 751 and 1021, respectively. These data highlight the critical importance of PDGF signaling in pulmonary hypertension and suggest that these signaling events probably represent valuable targets for antiremodeling strategies in pulmonary arterial hypertension.
Acknowledgments
We thank A. Kazlauskas (Harvard Medical School) for generously providing antibodies against the βPDGFR (97A) and GTPase-activating protein of Ras (69.3). The technical assistance by F. Oberhäuser and M. Becker is greatly appreciated. This article contains parts of a doctoral thesis by M. Leuchs.
Sources of Funding
This work was supported, in part, by the Deutsche Forschungsgemeinschaft (SFB612-B10 to S. Rosenkranz), the Deutsche Stiftung für Herzforschung (F/11/12 to H.t. Freyhaus), the Center for Molecular Medicine Cologne (project A6 to S. Rosenkranz), and by a research grant from Novartis Pharma (Basel, Switzerland).
Nonstandard Abbreviations and Acronyms
- EC
endothelial cell
- IPAH
idiopathic PAH
- PAP
pulmonary arterial pressure
- PAH
pulmonary arterial hypertension
- PASMC
pulmonary arterial smooth muscle cell
- PDGF
platelet-derived growth factor
- PDGFR
platelet-derived growth factor receptor
- PI3K
phophatidyl-inositol-3-kinase
- PLCγ
phospholipase C-γ1
- VSMC
vascular smooth muscle cell
- WT
wild-type
Footnotes
Disclosures
H.t. Freyhaus received honoraria for lectures. R.T. Schermuly received honoraria for lectures and consulting fees, research grants from Novartis. S. Rosenkranz received honoraria for lectures and consulting fees, research grants from Novartis. The other authors report no conflicts.
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.114.304864/-/DC1.
References
- 1.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) Circulation. 2010;122:164–172. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 2.Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–163. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 3.Hoeper MM, Huscher D, Ghofrani HA, et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: results from the COMPERA registry. Int J Cardiol. 2013;168:871–880. doi: 10.1016/j.ijcard.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 4.Hassoun PM, Mouthon L, Barberà JA, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009;54(suppl 1):S10–S19. doi: 10.1016/j.jacc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 5.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, Yuan JX, Weir EK. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(suppl 1):S20–S31. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haworth SG. The cell and molecular biology of right ventricular dysfunction in pulmonary hypertension. Eur Heart J Suppl. 2007;9(H):H10–H16. doi: 10.1093/eurheartj/sum025. [DOI] [Google Scholar]
- 7.van Wolferen SA, Marcus JT, Boonstra A, Marques KM, Bronzwaer JG, Spreeuwenberg MD, Postmus PE, Vonk-Noordegraaf A. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J. 2007;28:1250–1257. doi: 10.1093/eurheartj/ehl477. [DOI] [PubMed] [Google Scholar]
- 8.Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz SM. Perspectives series: cell adhesion in vascular Smooth muscle migration in atherosclerosis and restenosis. J Clin Invest. 1997;99:2814–2816. doi: 10.1172/JCI119472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raines EW, Ross R. Multiple growth factors are associated with lesions of atherosclerosis: specificity or redundancy? Bioessays. 1996;18:271–282. doi: 10.1002/bies.950180405. [DOI] [PubMed] [Google Scholar]
- 11.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 12.Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. LRP: role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–332. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
- 13.Caglayan E, Vantler M, Leppänen O, Gerhardt F, Mustafov L, Ten Freyhaus H, Kappert K, Odenthal M, Zimmermann WH, Tallquist MD, Rosenkranz S. Disruption of platelet-derived growth factor-dependent phosphatidylinositol 3-kinase and phospholipase Cγ 1 activity abolishes vascular smooth muscle cell proliferation and migration and attenuates neointima formation in vivo. J Am Coll Cardiol. 2011;57:2527–2538. doi: 10.1016/j.jacc.2011.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perros F, Montani D, Dorfmüller P, et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:81–88. doi: 10.1164/rccm.200707-1037OC. [DOI] [PubMed] [Google Scholar]
- 15.Katayose D, Ohe M, Yamauchi K, Ogata M, Shirato K, Fujita H, Shibahara S, Takishima T. Increased expression of PDGF A- and B-chain genes in rat lungs with hypoxic pulmonary hypertension. Am J Physiol. 1993;264(2 pt 1):L100–L106. doi: 10.1152/ajplung.1993.264.2.L100. [DOI] [PubMed] [Google Scholar]
- 16.Humbert M, Monti G, Fartoukh M, Magnan A, Brenot F, Rain B, Capron F, Galanaud P, Duroux P, Simonneau G, Emilie D. Platelet-derived growth factor expression in primary pulmonary hypertension: comparison of HIV seropositive and HIV seronegative patients. Eur Respir J. 1998;11:554–559. [PubMed] [Google Scholar]
- 17.Lannér MC, Raper M, Pratt WM, Rhoades RA. Heterotrimeric G proteins and the platelet-derived growth factor receptor-beta contribute to hypoxic proliferation of smooth muscle cells. Am J Respir Cell Mol Biol. 2005;33:412–419. doi: 10.1165/rcmb.2005-0004OC. [DOI] [PubMed] [Google Scholar]
- 18.ten Freyhaus H, Dagnell M, Leuchs M, Vantler M, Berghausen EM, Caglayan E, Weissmann N, Dahal BK, Schermuly RT, Ostman A, Kappert K, Rosenkranz S. Hypoxia enhances platelet-derived growth factor signaling in the pulmonary vasculature by down-regulation of protein tyrosine phosphatases. Am J Respir Crit Care Med. 2011;183:1092–1102. doi: 10.1164/rccm.200911-1663OC. [DOI] [PubMed] [Google Scholar]
- 19.Balasubramaniam V, Le Cras TD, Ivy DD, Grover TR, Kinsella JP, Abman SH. Role of platelet-derived growth factor in vascular remodeling during pulmonary hypertension in the ovine fetus. Am J Physiol Lung Cell Mol Physiol. 2003;284:L826–L833. doi: 10.1152/ajplung.00199.2002. [DOI] [PubMed] [Google Scholar]
- 20.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115:2811–2821. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.ten Freyhaus H, Dumitrescu D, Berghausen E, Vantler M, Caglayan E, Rosenkranz S. Imatinib mesylate for the treatment of pulmonary arterial hypertension. Expert Opin Investig Drugs. 2012;21:119–134. doi: 10.1517/13543784.2012.632408. [DOI] [PubMed] [Google Scholar]
- 22.Ghofrani HA, Seeger W, Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med. 2005;353:1412–1413. doi: 10.1056/NEJMc051946. [DOI] [PubMed] [Google Scholar]
- 23.ten Freyhaus H, Dumitrescu D, Bovenschulte H, Erdmann E, Rosenkranz S. Significant improvement of right ventricular function by imatinib. mesylate in scleroderma-associated pulmonary arterial hypertension. Clin Res Cardiol. 2009;98:265–267. doi: 10.1007/s00392-009-0752-3. [DOI] [PubMed] [Google Scholar]
- 24.Ghofrani HA, Morrell NW, Hoeper MM, Olschewski H, Peacock AJ, Barst RJ, Shapiro S, Golpon H, Toshner M, Grimminger F, Pascoe S. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am J Respir Crit Care Med. 2010;182:1171–1177. doi: 10.1164/rccm.201001-0123OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoeper MM, Barst RJ, Bourge RC, et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation. 2013;127:1128–1138. doi: 10.1161/CIRCULATIONAHA.112.000765. [DOI] [PubMed] [Google Scholar]
- 26.Galiè N, Hoeper MM, Humbert M, et al. ESC Committee for Practice Guidelines (CPG) Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT) Eur Heart J. 2009;30:2493–2537. doi: 10.1093/eurheartj/ehp297. [DOI] [PubMed] [Google Scholar]
- 27.Humbert M. Impression, sunset. Circulation. 2013;127:1098–1100. doi: 10.1161/CIRCULATIONAHA.113.001460. [DOI] [PubMed] [Google Scholar]
- 28.Gambaryan N, Perros F, Montani D, Cohen-Kaminsky S, Mazmanian M, Renaud JF, Simonneau G, Lombet A, Humbert M. Targeting of c-kit+ haematopoietic progenitor cells prevents hypoxic pulmonary hypertension. Eur Respir J. 2011;37:1392–1399. doi: 10.1183/09031936.00045710. [DOI] [PubMed] [Google Scholar]
- 29.Gambaryan N, Perros F, Montani D, Cohen-Kaminsky S, Mazmanian GM, Humbert M. Imatinib inhibits bone marrow-derived c-kit+ cell mobilisation in hypoxic pulmonary hypertension. Eur Respir J. 2010;36:1209–1211. doi: 10.1183/09031936.00052210. [DOI] [PubMed] [Google Scholar]
- 30.Rosenkranz S, Kazlauskas A. Evidence for distinct signaling properties and biological responses induced by the PDGF receptor alpha and beta subtypes. Growth Factors. 1999;16:201–216. doi: 10.3109/08977199909002130. [DOI] [PubMed] [Google Scholar]
- 31.Tallquist MD, Klinghoffer RA, Heuchel R, Mueting-Nelsen PF, Corrin PD, Heldin CH, Johnson RJ, Soriano P. Retention of PDGFR-beta function in mice in the absence of phosphatidylinositol 3′-kinase and phospholipase Cgamma signaling pathways. Genes Dev. 2000;14:3179–3190. doi: 10.1101/gad.844700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vantler M, Caglayan E, Zimmermann WH, Bäumer AT, Rosenkranz S. Systematic evaluation of anti-apoptotic growth factor signaling in vascular smooth muscle cells. Only phosphatidylinositol 3′-kinase is important. J Biol Chem. 2005;280:14168–14176. doi: 10.1074/jbc.M413310200. [DOI] [PubMed] [Google Scholar]
- 33.Soriano P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994;8:1888–1896. doi: 10.1101/gad.8.16.1888. [DOI] [PubMed] [Google Scholar]
- 34.Levéen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994;8:1875–1887. doi: 10.1101/gad.8.16.1875. [DOI] [PubMed] [Google Scholar]
- 35.Lindahl P, Johansson BR, Levéen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 36.Mellgren AM, Smith CL, Olsen GS, Eskiocak B, Zhou B, Kazi MN, Ruiz FR, Pu WT, Tallquist MD. Platelet-derived growth factor receptor beta signaling is required for efficient epicardial cell migration and development of two distinct coronary vascular smooth muscle cell populations. Circ Res. 2008;103:1393–1401. doi: 10.1161/CIRCRESAHA.108.176768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kerkelä R, Grazette L, Yacobi R, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. 2006;12:908–916. doi: 10.1038/nm1446. [DOI] [PubMed] [Google Scholar]
- 38.Cheng H, Force T. Molecular mechanisms of cardiovascular toxicity of targeted cancer therapeutics. Circ Res. 2010;106:21–34. doi: 10.1161/CIRCRESAHA.109.206920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.