

Abstract
Here comprehensive analysis was performed on the molecular and clinical features of colorectal carcinoma (CRC) harboring chromosome 20q amplification. Tumor and normal DNA from patients with advanced CRC underwent next generation sequencing (NGS) via MSK-IMPACT and a subset of case samples were subjected to high resolution microarray (Oncoscan). Relationships between genomic copy number and transcript expression were assessed with The Cancer Genome Atlas (TCGA) CRC data. Of the CRC patients sequenced (n=401) with MSK-IMPACT, 148 (37%) had 20q gain, and 30 (7%) had 20q amplification. In both the MSK-IMPACT and TCGA datasets, BCL2L1 was the most frequently amplified 20q oncogene. However, SRC was the only recognized 20q oncogene with a significant inverse relationship between mRNA upregulation and RAS/RAF mutation (OR: -0.4 +/- 0.2, p=0.02). In comparison to 20q diploid CRC, 20q gain/amplification was associated with wild-type (WT) KRAS (p<0.001) and BRAF (p=0.01), microsatellite stability (MSS) (p<0.001), distal primary tumors (p<0.001), and mutant TP53 (p<0.001), but not stage. On multi-variate analysis, longer overall survival from date of metastasis was observed with chromosome 20q gain (p=0.02) or amplification (p=0.04) compared to diploid 20q.
Keywords: colorectal carcinoma, Chromosome, 20q, AURKA, BCL2L1, SRC
INTRODUCTION
Gains and amplifications of chromosome arm 20q, which harbors multiple potential oncogenes including BCL2L1, AURKA and SRC, have been implicated as an important oncogenic event in colorectal carcinoma (CRC).(1-12) Despite multiple functional studies of various 20q genes, 20q gain in CRC remains under-investigated relation to other key molecular features of CRC,(2) and clinical features including prognosis and response to treatment. In this study, we aimed to further define the molecular and clinical features of CRC harboring 20q gain.
METHODS
Case Selection
After approval by our institutional review board, the molecular results and electronic medical records from all patients with CRC who underwent MSK-IMPACT molecular testing from January 1st, 2014 to May 31st, 2016 were reviewed. Patients with CRC whose tumors are submitted for MSK-IMPACT testing at our institution generally have advanced (distant metastatic) disease and are being considered for anti-EGFR therapies that require wild type KRAS, NRAS, and BRAF tumor status. Only one sample per patient was used for analyses in the manuscript. Minimum required tumor purity was 10% for MSK-IMPACT cases.
Molecular testing
MSK-IMPACT is a hybrid capture based next generation sequencing assay that interrogates the entire coding region and select non-coding regions of 410 genes to determine somatic mutations, copy number alterations, and structural variants from tumor and matched normal samples.(13) We calculated the Tumor/Normal log2 ratio (lr) for all 20q genes in the panel and then stratified cases into three groups: Amplification (lr • 0.95), Gain (lr: 0.45 to 0.95), and Diploid (lr:<0.45 ). Hotspot mutations in KRAS (G12, G13, Q61, K117, A146, K147), NRAS (G12, G13, Q61), or BRAF (V600E) were recorded; along with any coding mutations in all other genes within the panel. Microsatellite instability (MSI) status was assessed via MSIsensor, a program that assesses variations in repeat areas throughout the genome comparing the tumor to the normal.(14) Samples with MSIsensor scores greater than 10 are considered MSI-H. This program has been clinically validated for the evaluation of microsatellite instability-high (MSI-H) versus microsatellite stable (MSS) in MSK-IMPACT data. A select group of cases with the highest level of chromosome 20q amplification was assessed by genome-wide SNP microarray, Oncoscan, to allow a higher resolution, allele-specific analysis of the amplified region.
TCGA data analysis
To further investigate the prevalence and potential drivers of chromosome 20q amplification in colon cancer, we obtained Level 3 data available from The Cancer Genome Atlas (TCGA ) colorectal adenocarcinoma (COADREAD) cohort.(15) Somatic mutations and RNASeqV2 normalized expression data were obtained using the R/Bioconductor package TCGAbiolinks.(16) GISTIC2.0 gene and arm level copy number data were obtained from The Broad GDAC Firehose.(17,18) Samples were stratified by chromosome arm 20q absolute copy number (acn) into the following groups: Amplified (acn: >=4), Gain (acn: 2.5 to 4), Diploid (acn:1.5 to 2.5), and Loss (acn:<1.5). Gene level copy number calls were pulled directly from GISTIC2.0 output. Since RNA-Seq control samples are limited in TCGA data, the gene expression values (e) for each sample were normalized by calculating the mean (μ) and variance (σ) of the expression values for samples in which the gene was copy number diploid. Sample level gene expression (zscore) was then calculated as (e -μ) / . Sample zscore values of > 2 and < -2 were used as thresholds for gene upregulation and downregulation, respectively.
Statistical Analysis
Associations with clinical and molecular data were assessed by Fisher’s test with multiple hypothesis testing correction (Benjamini–Hochberg, alpha=0.05). To assess survival, a Cox proportional hazard model was fitted to the data. Here, the covariates of chromosome arm 20q copy number status, RAS/BRAF mutation, MSI status, age at diagnosis, and pathologic stage were each assessed through both univariate and multivariate Cox regressions. This analysis was repeated using the available molecular and clinical data available from TCGA. For all chromosome arm 20q genes, Pearson correlations were calculated between log2 copy number and log2 transformed normalized expected counts (RSEM) from RNA-Seq experiments, also available through the TCGA.
RESULTS
Incidence and Clinical Characteristics of Chromosome 20q Gain/ Amplification
We screened for chromosome arm 20q copy number alterations in 413 prospectively sequenced patient samples (401 patients). Of 401 consecutive cases of advanced CRC undergoing MSK-IMPACT testing results, 148 (37%) had 20q gain, and 30 (7%) had 20q arm level amplification. Ten patients had MSK-IMPACT testing on both primary and metastasis, and the concordance for 20q copy number status between paired samples was 100% (2 patients with gain of chromosome 20q and 8 patients with diploid chromosome 20q).
Further, we selected fourteen cases with high levels of 20q identified with MSK-IMPACT and analyzed them by Oncoscan to confirm MSK-IMPACT results and to assess whether DNA levels on 20q were preferentially higher at specific cytogenetic loci or genes. Oncoscan analysis revealed that 20q gains and amplifications are broad, without focal changes or discontinuity. Additionally, the ‘major clones’ rather than subclonal populations harbored 20q amplification in the cases studied, suggesting that 20q amplification may take place relatively early in carcinogenesis. The gains/ amplifications started between 20p11.2 and20q11.2 and included all of the long arm of chromosome 20 through qter (Suppl Figure 1, Suppl Table 1).
The distribution of right sided to left sided (right sided: left-sided) CRC was 1:9 (n= 30) in CRC with chromosome 20q amplification, 1:4.5 (n= 148) in CRC with 20q gain, and 1:1.1 (n=218) in patients with diploid chromosome 20q (Fisher’s P value < 0.001) (Table 1). There was no significant difference in stage distribution at diagnosis of CRC in patients with 20q amplified versus 20q diploid tumors in either MSK-IMPACT and TCGA data (Suppl Table 2).
Table 1.
Molecular signature according to chromosome 20q status in MSK-IMPACT dataset.
| Feature | diploid 20q | gain of 20q | amplification of 20q | P value*, OR | |||
|---|---|---|---|---|---|---|---|
| Mutated | Wild Type/ Negative | Mutated | Wild Type/ Negative | Mutated | Wild Type/ Negative | ||
| KRAS | 137 | 86 | 55 | 93 | 3 | 27 | <0.001, 0.33 |
| NRAS | 5 | 218 | 8 | 140 | 1 | 29 | 0.407, 2.32 |
| BRAF | 26 | 197 | 4 | 144 | 0 | 30 | 0.012, 0.17 |
| MSI-H | 30 | 193 | 1 | 147 | 0 | 30 | <0.001, 0.037 |
| Right sided tumor | 105 | 118 | 27 | 121 | 3 | 27 | <0.001, 0.126 |
| TP53 | 137 | 86 | 124 | 24 | 28 | 2 | <0.001, 3.82 |
| APC | 152 | 71 | 121 | 27 | 23 | 7 | 0.043, 1.98 |
P values and Odds Ratio (OR) calculated for gain or amplification of 20q versus diploid 20q.
Molecular Signature of CRC with 20q Gain/ Amplification
Since chromosome 20q amplifications are often implicated as an oncogenic event, we looked for patterns of co-mutation and mutual exclusivity of 20q alterations with other known driver events. KRAS and BRAF mutations were significantly associated with diploid chromosome 20q (p<0.0001 and p=0.01 for KRAS and BRAF, respectively) in the MSK-IMPACT data. Further, the relationships between RAS/ RAF mutation and 20q in MSK-IMPACT showed a higher mutual exclusivity with increased copy number of 20q indicating a dose dependency (5% of CRC with 20q amplification had RAS/ RAF mutations, 18% of CRC with 20q gain had RAS/ RAF mutations, and 34% of CRC with diploid 20q had RAS/ RAF mutation) (Table 1, Figure 1). Hotspot mutations in KRAS (G12, G13, Q61, K117, A146, K147), NRAS (G12, G13, Q61), or BRAF (V600E) were present in 10% of CRC with 20q amplification, 45% of CRC with 20q gain, and 72% of CRC with diploid 20q (Table 1).
Figure 1.
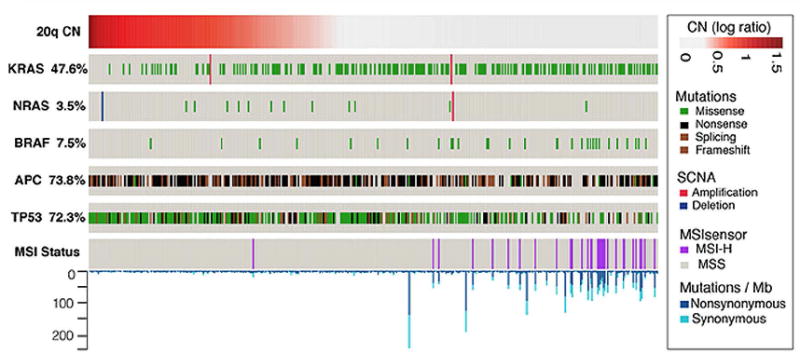
Molecular correlates of 20q amplified or gained MSK-IMPACT CRC. Increased copy number of chromosome 20q showed a mutually exclusive relationship with KRAS, NRAS and BRAF mutations, microsatellite instability- high status, and a higher incidence of TP53 and APC mutations.
Further analysis of the MSK-IMPACT data revealed relationships with MSI status, APC mutations, and TP53 mutations. MSI-H status was significantly associated with diploid chromosome 20q (p<0.0001). MSI-H status was present in none of the 30 cases with 20q amplification, 1 (<1%) of 148 cases with 20q gain, and 30 (13%) of the 223 cases with diploid 20q. APC mutations were associated with gain or amplification of chromosome 20q (p=0.04). APC mutations were present in 152 (68%) of CRC with diploid 20q, 121 (82%) of CRC with gain of 20q, and 23 (77%) of CRC with amplification of 20q. TP53 mutations were significantly associated with 20q gain or amplification (p<0.0001). TP53 mutations were present in 136 (61%) of CRC with diploid 20q, 124 (84%) of CRC with gain of 20q, and 28 (93%) of CRC with amplification of 20q. These findings are summarized in Table 1.
Overall Survival of Patients with Advanced CRC by 20q Copy Number Status
There were 354 CRC patients with MSK-IMPACT testing with distant metastases and available clinical follow up for survival analysis. This included 187 patients with diploid 20q, 139 patients with 20q gain, and 28 patients with amplified 20q.Chromosome 20q copy number status, RAS/RAF mutation, MSI status, pathologic stage, and age at diagnosis were included as covariates to a Cox proportional hazard model. On multivariate analysis, chromosome 20q gain (p=0.015; CI = 0.36 - 0.90) or amplification (p=0.039; CI= 0.11 - 0.94) was associated with longer overall survival (Figure 2) and RAS/ RAF mutation was associated with shorter overall survival that did not reach statistical significance (p=0.064; CI= 0.97 - 2.46).
Figure 2.
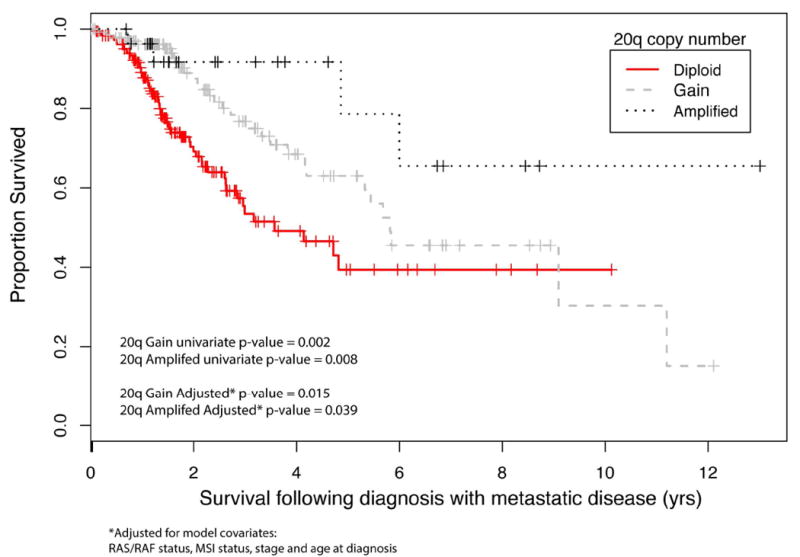
Overall survival from date of diagnosis with metastasis stratified by chromosome 20q status. Chromosome 20q gain or amplification was associated with longer overall survival (p=0.004; CI:0.34-0.82).
Of the 366 CRC patients analyzed by the TCGA, only stage IV disease at diagnosis was associated with worse survival (HR:4.8, 95% CI: 1.73-12.80) on multivariate analysis. RAS/ RAF mutation, MSI status, and 20q status were not signicantly associated with survival in TCGA data. However, only 47 patients in the TCGA data had metastatic disease at diagnosis (stage IV), while 240 patients with MSK-IMPACT data had metastatic disease at diagnosis (stage IV), reflecting the very different criteria for patient selection in these two datasets.
Response to anti-EGFR Therapy of Patients with 20q Gain/ Amplification
Nineteen patients with metastatic CRC and 20q gain/ amplification on MSK-IMPACT were treated with EGFR inhibitors (cetuximab/ panitumumab). Of these patients, 11 had adequate follow up and received EGFR inhibitors either alone or in combinations where response to the EGFR inhibitor could be assessed (e.g., combination with chemotherapy after progression to that chemotherapy) (Suppl Table 3). Review of radiology records suggested that of these 11 patients, 3 patients had decreased disease, 1 patient had unchanged disease, 1 patient had mixed response, and 6 patients had increased disease after cetuximab or panitumumab therapy.
Correlation between TCGA Chromosome 20q Oncogene DNA copies and mRNA Levels
In an effort to identify a potential driver gene within chromosome arm 20q, we integrated the somatic copy number and whole transcriptome RNAseq data from TCGA. MSK-IMPACT genes on 20q that showed amplification (in order of frequency) included BCL2L1, ASXL1, SRC, DNMT3B, GNAS, TOP1, AURKA, PTPRT, and NCOA3. A similar pattern of amplification frequency was observed in TCGA data (Suppl Table 4), with slightly higher percentages for CRC showing amplification due to higher tumor purity in TCGA data (Suppl Figure 2)
The correlation coefficient (r) values for TCGA data between DNA copy number and mRNA levels of potential oncogenes on chromosome 20q was 0.51 for TPX2, 0.65 for BCL2L1, 0.59 for SRC, 0.53 for AURKA, and 0.42 for GNAS. Given that our analysis showed a highly statistically significant mutually exclusive relationship between 20q amplification and RAS/RAF mutations suggesting that this alteration may play a similar driver role in CRC, we next examined which 20q amplified genes were most highly expressed in the absence of these well known driver mutations. This analysis showed that SRC mRNA upregulation had the strongest mutually exclusive relationship with KRAS/ NRAS/ BRAF hot spot mutations (odds ratio: -0.4+/- 0.2, p=0.02); while the relationships of mRNA upregulation versus KRAS/ NRAS/ BRAF for other potential 20q oncogenes did not reach statistical significance. These findings are illustrated in Figure 3.
Figure 3.

Correlation of copy number and mRNA expression of potential chromosome arm 20q oncogenes in TCGA COADREAD cohort and log odds ratio of RNA expression up-regulation and KRAS/NRAS/BRAF mutant. SRC mRNA upregulation was significantly associated with wild type RAS/RAF (p=0.02, 95% CI: -0.36+/-0.24).
DISCUSSION
In this study, we show that gain or amplification of chromosome 20q11-13.3 occurs in approximately 37% and 7% of advanced CRC, respectively; that chromosome 20q gain/ amplification is associated with molecular and clinical findings such as a 20q “dose-dependent” RAS/ RAF wild type status, left sided primary tumors, microsatellite stability, a higher incidence of mutations in TP53 and APC, and longer overall survival in patients with metastatic disease. These findings support the role of chromosome 20q amplification as a driver in colorectal carcinoma.
Previous studies have observed that chromosome 20q is recurrently amplified in CRC(1-12). One study of 133 CRC patients identified a statistically significant inverse correlation between ‘PI3K’ pathway mutations, which included PIK3CA as well as KRAS and BRAF, versus 20q amplification.(2) Some studies have reported that 20q amplification is consistent between primary and metastatic samples from the same patient(4,10) and occurs as an early event in invasive CRC.(3) Other studies have found that 20q amplification is more common in CRC patients with liver metastases,(2,11) lung metastases,(4) or within metastatic foci than in the primary tumor.(12) Chromosome 20q gain/ amplification is highly concordant between primary and metastasis in both our data and the above cited papers, and its positive selection in primary and metastatic samples argues for its role in the development of metastases to different organs.
We have found complete concordance for 20q DNA copy number in cases where both the primary and metastasis were analyzed. Additionally, allele-specific copy number analysis using the Oncoscan SNP array platform showed that 20q amplification occurs as a clonal rather than subclonal event, much like hot spot driver mutations in KRAS, NRAS, or BRAF; and that there is no difference in the proportion of cases with 20q amplification between different diagnostic stages. Further, a higher degree of mutual exclusivity between RAS/ RAF mutation and 20q level was identified (that is, the higher the level of 20q amplification, the less likely a tumor was to have a KRAS, NRAS, or BRAF mutation). These data suggest that 20q amplification occurs early rather than late in CRC as a ‘dose-dependent’ driving event.
A ‘driver’ mutation has been defined as a mutation that gives a growth advantage to the cell(s) harboring it and is selected for as a cancer evolves.(19) In the broad sense of the term, an individual cancer may have 5-20 ‘driver’ events, representing functionally significant mutations. These include both tumor suppressor gene mutations such as TP53 and APC as seen in the majority of CRC, as well as oncogene events that co-occur with tumor suppressor alterations. Strong mitogenic drivers (usually RTK/ RAS/ MAPK pathway) are usually mutually exclusive; and 20q amplification follows this pattern. The facts that 20q is amplified with high concordance between primary and metastasis and that it shows a dose-dependent inverse relationship with RAS/ RAF mutations suggest that amplification of certain gene(s) on 20q is important for the process of metastasis and may have growth advantages that overlap with those provided by RAS/ RAF mutations.
Although chromosome 20q gain/ amplification is an important recurrent alteration in CRC, attempts to identify the “driving” oncogene(s) within the amplified segment by standard cancer genomic approaches such as definition of a minimal common region of amplification and correlations of copy number and expression have been unsuccessful.(1) Both in our data and published studies, the amplified segment is consistently broad and amplification does not correlate well with RNA upregulation for many genes on 20q(20,21), with R values ranging from 0.42 - 0.65 for DNA amplification vs. RNA upregulation in our analysis of TCGA data for common oncogenes (Figure 2). We therefore used a different analytical approach, reasoning that, if 20q amplification is functioning as a CRC driver similar to RAS/RAF mutations and other kinase alterations recently described in CRC(22), then the 20q amplified genes with the strongest inverse expression correlation with RAS/RAF status may be the most likely drivers. We therefore examined how 20q amplified genes were expressed in relation to the presence or absence of RAS/RAF mutation and found that SRC had the strongest, and only statistically significant, mutually exclusive relationship between mRNA upregulation and RAS/ RAF mutation in the TCGA data. This suggests that SRC amplification may serve as a mechanism for SRC upregulation, and that SRC may be an important driving oncogene in the subset of CRC with 20q amplification. SRC encodes a non-receptor protein kinase that has been linked to cancer progression and metastatic disease. It is downstream of EGFR, yet upstream of both the PI3K and MAPK pathways. Several drugs targeting SRC are available, including dasatinib(23,24), and functional studies of CRC cell lines or patient-derived xenogratfs with 20q amplification are needed to assess whether the group of patients in this study may benefit from anti-SRC therapy.
Other recognized oncogenes on chromosome 20q include AURKA and TPX2. AURKA overexpression, located at 20q13.13, has been observed in some CRC.(25) It has also been shown that TPX2, located at 20q11.21 near BCL2L1, interacts with AURKA.(19) It has been hypothesized that these genes act on the same pathway of tumor progression and may have a gene-dosage effect in which amplification of both genes has a larger effect than amplification of just one or the other. AURKA and TPX2 have been shown to induce anchorage-independent growth (an in vitro measure of metastatic potential) and to co-regulate MYC; specifically, AURKA and TPX2 overexpression stabilizes and induces MYC.(26)
The limitations of this study include the fact that the effect of 20q amplification on the response to specific chemotherapies could not be assessed in patients due to multiple concomitant lines of chemotherapy given and pre-treatment. Only 11 patients who received anti-EGFR therapy had clinical courses that could be analyzed. Separately, we did not have expression data for MSK-IMPACT cases as material is often limited for biopsy specimens, so DNA copy number vs. RNA transcription levels could not be compared. Additionally, tumor purity requirements were lower for MSK-IMPACT (•10%) compared to TCGA (•50%) cases. This difference in tumor purity would result in an underestimate of 20q gain/ amplification due to dilution with the surrounding non-tumor DNA.
In summary, we show that chromosome 20q gain/ amplification drives and defines a subset of CRC with distinct clinical and molecular findings in a dosage dependent fashion. Oncogenes on chromosome 20q such as SRC and AURKA may serve as potential targetable alterations with available selective inhibitors.
Supplementary Material
Implications.
20q amplification defines a subset of colorectal cancer patients with better overall survival from date of metastasis, and further studies are warranted to assess whether inhibition of 20q oncogenes, such as SRC, may benefit this subset of patients.
Acknowledgments
This study was funded by the National Cancer Institute (NCI) under the MSK Cancer Center Support Grant/Core Grant (P30 CA008748).
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Sillars-Hardebol AH, Carvalho B, Beliën JA, de Wit M, Delis-van Diemen PM, Tijssen M, et al. BCL2L1 has a functional role in colorectal cancer and its protein expression is associated with chromosome 20q gain. J Pathol. 2012;226:442–50. doi: 10.1002/path.2983. [DOI] [PubMed] [Google Scholar]
- 2.Bruin SC, He Y, Mikolajewska-Hanclich I, Liefers GJ, Klijn C, Vincent A, Verwaal VJ, et al. Molecular alterations associated with liver metastases development in colorectal cancer patients. Br J Cancer. 2011;105(2):281–7. doi: 10.1038/bjc.2011.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diep CB, Kleivi K, Ribeiro FR, Teixeira MR, Lindgjaerde OC, Lothe RA. The order of genetic events associated with colorectal cancer progression inferred from meta-analysis of copy number changes. Genes Chromosomes Cancer. 2006;45(1):31–41. doi: 10.1002/gcc.20261. [DOI] [PubMed] [Google Scholar]
- 4.Danner BC, Gerdes JS, Jung K, Sander B, Enders C, Liersch T, et al. Comparison of chromosomal aberrations in primary colorectal carcinomas to their pulmonary metastases. Cancer Genet. 2011;204(3):122–8. doi: 10.1016/j.cancergen.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 5.Carvalho B, Postma C, Mongera S, Hopmans E, Diskin S, van de Wiel MA, et al. Multiple putative oncogenes at the chromosome 20q amplicon contribute to colorectal adenoma to carcinoma progression. Gut. 2009;58(1):79–89. doi: 10.1136/gut.2007.143065. [DOI] [PubMed] [Google Scholar]
- 6.Brenner BM, Rosenberg D. High-throughput SNP/CGH approaches for the analysis of genomic instability in colorectal cancer. Mutat Res. 2010;693(1-2):46–52. doi: 10.1016/j.mrfmmm.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 7.De Angelis PM, Stokke T, Beigi M, Mjåland O, Clausen OP. Prognostic significance of recurrent chromosomal aberrations detected by comparative genomic hybridization in sporadic colorectal cancer. Int J Colorectal Dis. 2001;16(1):38–45. doi: 10.1007/s003840000275. [DOI] [PubMed] [Google Scholar]
- 8.Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318(5853):1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 9.Schlegel J, Stumm G, Scherthan H, Bocker T, Zirngibl H, Rüschoff J, et al. Comparative genomic in situ hybridization of colon carcinomas with replication error. Cancer Res. 1995;55:6002–5. [PubMed] [Google Scholar]
- 10.Paredes-Zaglul A, Kang JJ, Essig YP, Mao W, Irby R, Wloch M, et al. Analysis of colorectal cancer by comparative genomic hybridization: evidence for induction of the metastatic phenotype by loss of tumor suppressor genes. Clin Cancer Res. 1998;4:879–86. [PubMed] [Google Scholar]
- 11.Hidaka S, Yasutake T, Takeshita H, Kondo M, Tsuji T, Nanashima A, et al. Differences in 20q13.2 copy number between colorectal cancers with and without liver metastasis. Clin Cancer Res. 2000;6:2712–7. [PubMed] [Google Scholar]
- 12.Parada LA, Marañon A, Hallén M, Tranberg KG, Stenram U, Bardi G, et al. Cytogenetic analyses of secondary liver tumors reveal significant differences in genomic imbalances between primary and metastatic colon carcinomas. Clin Exp Metastasis. 1999;17:471–9. doi: 10.1023/a:1006646901556. [DOI] [PubMed] [Google Scholar]
- 13.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17:251–64. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics. 2014;30:1015–6. doi: 10.1093/bioinformatics/btt755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Research. 2015;44:e71. doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Broad Institute TCGA Genome Data Analysis Center Firebrowse Portal. https://gdac.broadinstitute.org/
- 18.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biology. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–24. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Platzer P, Upender MB, Wilson K, Willis J, Lutterbaugh J, Nosrati A, et al. Silence of chromosomal amplifications in colon cancer. Cancer Res. 2002;62:1134–8. [PubMed] [Google Scholar]
- 21.Sillars-Hardebol AH, Carvalho B, Tijssen M, Beliën JA, de Wit M, Delis-van Diemen PM, et al. TPX2 and AURKA promote 20q amplicon-driven colorectal adenoma to carcinoma progression. Gut. 2012;61:1568–75. doi: 10.1136/gutjnl-2011-301153. [DOI] [PubMed] [Google Scholar]
- 22.Hechtman JF, Zehir A, Yaeger R, Wang L, Middha S, Zheng T, et al. Identification of targetable kinase alterations in patients with colorectal carcinoma that are preferentially associated with wild-type RAS/RAF. Mol Cancer Res. 2016;14:296–301. doi: 10.1158/1541-7786.MCR-15-0392-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perez M, Lucena-Cacace A, Marín-Gómez LM, Padillo-Ruiz J, Robles-Frias MJ, Saez C, et al. Dasatinib, a Src inhibitor, sensitizes liver metastatic colorectal carcinoma to oxaliplatin in tumors with high levels of phospho-Src. Oncotarget. 2016;7:33111–24. doi: 10.18632/oncotarget.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopetz S, Lesslie DP, Dallas NA, Park SI, Johnson M, Parikh NU, et al. Synergistic activity of the SRC family kinase inhibitor dasatinib and oxaliplatin in colon carcinoma cells is mediated by oxidative stress. Cancer Res. 2009;69:3842–9. doi: 10.1158/0008-5472.CAN-08-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chuang TP, Wang JY, Jao SW, Wu CC, Chen JH, Hsiao KH, et al. Over-expression of AURKA, SKA3 and DSN1 contributes to colorectal adenoma to carcinoma progression. Oncotarget. 2016;7(29):45803–18. doi: 10.18632/oncotarget.9960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi Y, Sheridan P, Niida A, Sawada G, Uchi R, Mizuno H, et al. The AURKA/TPX2 axis drives colon tumorigenesis cooperatively with MYC. Ann Oncol. 2015;26:935–942. doi: 10.1093/annonc/mdv034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.