

Xu et al. show that Abro1 localizes to stalled replication forks and protects stalled replication fork stability in response to replication stress by inhibiting DNA2 nuclease/WRN helicase-mediated degradation of stalled forks.
Keywords: Abro1, BRCA2, DNA2, MRE11, RAD51, stalled replication fork stability
Abstract
Protection of the stalled replication fork is crucial for responding to replication stress and minimizing its impact on chromosome instability, thus preventing diseases, including cancer. We found a new component, Abro1, in the protection of stalled replication fork integrity. Abro1 deficiency results in increased chromosome instability, and Abro1-null mice are tumor-prone. We show that Abro1 protects stalled replication fork stability by inhibiting DNA2 nuclease/WRN helicase-mediated degradation of stalled forks. Depletion of RAD51 prevents the DNA2/WRN-dependent degradation of stalled forks in Abro1-deficient cells. This mechanism is distinct from the BRCA2-dependent fork protection pathway, in which stable RAD51 filament formation prevents MRE11-dependent degradation of the newly synthesized DNA at stalled forks. Thus, our data reveal a new aspect of regulated protection of stalled replication forks that involves Abro1.
Replication stress occurs when a subset of DNA replication forks stalls due to endogenous or exogenous sources, such as depletion of nucleotides or collision of the replisome with DNA lesions, or at difficult to replicate genomic regions. Defects in cellular responses to replication stress often lead to genomic instability and human diseases, including cancer (Zeman and Cimprich 2014; Gaillard et al. 2015). One of the first consequences of replication stress is decreased stability of stalled replication forks. Stalled replication forks are susceptible to degradation by nucleases that are capable of processing stalled replication intermediates upon replication stress (Zeman and Cimprich 2014; Berti and Vindigni 2016). Cellular mechanisms have evolved to protect against degradation and maintain fork stability. Protection of stalled replication forks is crucial for responding to replication stress, thereby minimizing the impact of this stress on chromosome instability, which can lead to tumorigenesis. The breast and ovarian tumor suppressor proteins BRCA1 and BRCA2 and Fanconi anemia protein FANCD2 were implicated in the stabilization of replication forks (Schlacher et al. 2011, 2012; Ying et al. 2012). In the absence of BRCA1 or BRCA2, nascent DNA strands at stalled forks undergo MRE11 nuclease-dependent degradation, which contributes to the genetic instability observed in BRCA mutated cells (Schlacher et al. 2011, 2012). BRCA2 stabilizes RAD51 filaments at stalled replication forks, thereby protecting nascent strands from extensive MRE11-dependent degradation (Hashimoto et al. 2010; Schlacher et al. 2011, 2012). Recently, several additional new factors were also identified as playing a role in stalled replication fork protection (Pefani et al. 2014; Higgs et al. 2015). Thus, multiple factors and mechanisms exist to ensure fork protection.
Abro1 is a paralog of a BRCA1-interacting protein, Abraxas (Wang et al. 2007). It does not interact with Abraxas or BRCA1 (Wang and Elledge 2007; Wang et al. 2007; Hu et al. 2011) but forms a BRISC (BRCC36 isopeptidase complex) in a manner similar to Abraxas forming the BRCA1-A complex. In the Abro1/BRISC complex, Abro1 interacts with NBA1/MERIT40, BRE, and BRCC36, which are common components of the two complexes (Wang et al. 2009; Cooper et al. 2010; Feng et al. 2010; Patterson-Fortin et al. 2010; Hu et al. 2011). Abro1 contains an MPN domain at the N terminus and a coiled-coil domain at the central region of the protein. The MPN domain mediates the interaction of Abro1 with NBA1/MERIT40 and BRE, and the coiled-coiled domain is required for the interaction with BRCC36. While Abraxas plays a critical role in double-strand break (DSB) repair by recruiting BRCA1 to DSBs and is important for tumor suppression (Hu et al. 2012; Wang 2012; Castillo et al. 2014), the role of Abro1 in genome maintenance and tumor suppression is not clear.
In this study, by analyzing Abro1-null mice and cells, we revealed a role of Abro1 in protecting stalled replication forks for the maintenance of genomic stability. We demonstrated that Abro1 protects stalled replication forks from uncontrolled DNA2/WRN-dependent resection such that Abro1-null cells exhibited increased ssDNA accumulation and shortened newly synthesized DNA at stalled replication forks. We also show that RAD51 facilitates DNA2/WRN-dependent degradation in Abro1-deficient cells and that Abro1 protects stalled replication forks distinctively from the BRCA2-dependent pathway that stabilizes RAD51 filaments for protection against MRE11-dependent degradation. Thus, our data established that Abro1 is a critical factor in the intricate mechanisms for protection of stalled replication forks.
Results
Abro1 knockout mice displayed decreased survival and increased tumor development
To explore the function of Abro1 in vivo, we generated a conditional Abro1 knockout mouse model. We made a gene targeting construct containing exon 5 of the Abro1 genomic sequence flanked by two loxP sites and a selection marker, NEO (neomycin resistance) gene, flanked by two FRT sites inserted into the intron region between exons 4 and 5 of the Abro1 gene (Fig. 1A). The construct was introduced into murine embryonic stem (ES) cells. Properly targeted ES cells were identified by Southern blot and used for the generation of chimeric mice (Supplemental Fig. S1A). The chimeras were then crossed with C56BL/6 mice to identify germline transmission and generation of Abro1Neo/+ mice. Abro1+/Δ animals were generated after Cre recombinase-mediated deletion of exon 5. Upon deletion of exon 5, a frameshift occurs, resulting in a null allele of the Abro1 gene. The loss of Abro1 full-length protein was confirmed by immunoblotting with Abro1 antibodies (Supplemental Fig. S1B). Abro1−/− (Abro1Δ/Δ) mice did not exhibit any gross developmental defects, were born at expected Mendelian ratios, and were indistinguishable from their wild-type littermates after birth.
Figure 1.

Abro1-deficient mice exhibit decreased survival and increased susceptibility to tumor formation. (A) Generation of Abro1-deficient mice. A diagram of mouse Abro1 alleles generated for deletion of exon 5. (NEO) Neomycin resistance gene. (B) Overall disease-free survival analyzed by the Kaplan-Meier method. P < 0.001. A matched cohort of 19 Abro1+/+, 19 Abro1+/−, and 34 Abro1−/− mice was monitored over 26 mo. (C) Tumor-free survival analyzed by the Kaplan-Meier method. P < 0.02. Spontaneous tumor incidence in Abro1+/+, Abro1+/−, and Abro1−/− mice was monitored. From the mice that we analyzed, zero out of 15 Abro1+/+ mice (0%); two out of 13 Abro1+/− mice (15%), including lymphoma (one) and lung adenocarcinoma (one); and six out of 28 mice (21%) developed tumors, including lymphoma (four), lung adenocarcinoma (one), and sebaceous adnexa tumor (one). (D) Representative histological images (H&E staining) of detected tumors are shown. (E) Abro1-deficient mice are not sensitive to ionizing radiation (IR). A cohort of Abro1+/+ (n = 12) and Abro1−/− (n = 15) mice was treated with 7.5 Gy of IR. Overall survival was monitored for up to 1 mo and analyzed by the Kaplan-Meier method. (F) Abro1−/− mouse embryonic fibroblast cells are sensitive to mitomycin C (MMC) and ultraviolet radiation (UV) but not IR as measured by clonogenic survival assay. Colonies were counted and normalized to untreated samples to calculate the percentage of survival. Error bars represent standard deviation across triplicates. Three independent experiments were performed.
Compared with wild-type mice, Abro1+/− and Abro1−/− mice had a significantly reduced life span and exhibited decreased disease-free survival (Fig. 1B). We also analyzed tumor-free survival in the mouse cohort and observed an increase in tumor incidence in Abro1−/− mice, although the tumor frequency is low (20%) and latency is long (Fig. 1C). Abro1 nullizygous mice developed cancer, including lymphoma and lung adenocarcinoma (Fig. 1D). A possible explanation of the high incidence of death and relative low incidence of tumor formation in Abro1-null mice is that the Abro1−/− mice die from other defects before they develop tumors. Indeed, many of the Abro1−/− (36%) and Abro1+/− (31%) mice in our analyses showed an enlarged spleen (splenomegaly) and inflammatory lymphoid infiltration in major organs suggestive of infection or lymphoid proliferative diseases (Supplemental Fig. S1C,D).
Since Abro1's paralog protein, Abraxas, is required for cellular resistance to ionizing radiation (IR) (Castillo et al. 2014), we examined whether Abro1-deficient mice are sensitive to IR. We found that, unlike Abraxas-null mice, Abro1-null mice did not appear to be sensitive to acute treatment with 7.5 Gy of IR (Fig. 1E). Thus, Abro1 is less likely to play a direct role in DSB repair in response to IR. Consistent with this result, Abro1-null mouse embryonic fibroblasts (MEFs) did not show increased cellular sensitivity to IR. Interestingly, Abro1-null MEFs exhibited hypersensitivity to the DNA cross-linking agent mitomycin C (MMC) and ultraviolet radiation (UV) (Fig. 1F).
Abro1 maintains genome stability and plays a role in the response to replication stress
We found that Abro1−/− cells exhibited nuclear bridges between segregating chromosomes during anaphase (Fig. 2A). Using immunofluorescence staining with BLM antibody, we observed that >20% of the Abro1−/− cells contained ultrafine bridges formed in mitosis (Fig. 2B). Ultrafine bridges, to which BLM, FancD2, and PICH proteins localize, are thought to be due to the failure to resolve incomplete replication and are associated with chromosome instability (Chan et al. 2007; Chan and Hickson 2011; Biebricher et al. 2013). Micronuclei are the outcome of mitotic defects of cells under replication stress that fail to complete timely DNA replication, resulting in entry into mitosis of an underreplicated DNA region (Hoffelder et al. 2004; Chan et al. 2009). We observed that Abro1-null MEFs showed significantly more nuclear abnormalities, such as micronuclei and nuclear budding, than wild-type MEFs, consistent with a role of Abro1 in resolving replication stress (Fig. 2C). We also investigated the formation of 53BP1 bodies in G1 cells, which has been associated with fragile sites and difficult to replicate regions of the genome (Lukas et al. 2011). Abro1 deficiency led to an increase in both the frequency of cells containing 53BP1 nuclear bodies and the number of 53BP1 bodies per cell (Fig. 2D). Together, these data suggest that Abro1 is likely to play a role in resolving DNA replication stress in order to maintain genome stability.
Figure 2.

Abro1 deficiency leads to genomic instability. (A) Formation of anaphase bridges in Abro1−/− cells. MEFs were stained with DAPI, and the percentage of mitotic cells with anaphase bridges was quantitated. “#” represents 0. (B) An ultrafine bridge (UFB) formed in Abro1−/− cells. Representative images of immunostaining of wild-type and Abro1−/− MEFs with BLM antibody and DAPI are shown. The percentage of mitotic cells containing an ultrafine bridge was calculated. (C) Increased micronuclei in Abro1−/− cells. MEFs were stained with DAPI, and the percentage of cells with abnormal nuclear morphology was calculated. (D) Increased 53BP1 nuclear body formation in Abro1−/− cells. MEFs were analyzed by immunofluorescence with 53BP1 antibody. The percentage of cells containing 53BP1 nuclear bodies (NB) was calculated. P-value was determined by Student's t-test.
To test for a role of Abro1 in DNA replication stress response, we used the DNA fiber assay, a technique that allows analysis of DNA replication at the single-molecule level (Jackson and Pombo 1998; Zeman and Cimprich 2014). To monitor fork progression, we pulse-labeled Abro1 wild-type and Abro1-null MEFs in unperturbed S phase with nucleotide analog CldU (5-chloro-2′-deoxyuridien) (Fig. 3A, red) for 30 min followed by labeling with a second nucleotide analog, IdU (5-iodo-2′-deoxyuridine) (Fig. 3A, green), for 30 min (Fig. 3A). The speed of DNA replication in the Abro1-null cells was similar to that of the wild-type MEFs (Fig. 3B), indicating that the overall fork progression and rate are not affected in Abro1-null cells. However, when we treated the Abro1-null cells with a low dose of a DNA polymerase inhibitor, aphidicolin (APH), to mimic the physiological barriers that the DNA replication machinery approaches during DNA synthesis, the Abro1-null cells exhibited a marked decrease in fork progression when compared with the control (Fig. 3B), indicating a role of Abro1 in response to replication stress. Consistent with such a role, the percentage of cells containing 53BP1 nuclear bodies and the number of 53BP1 bodies per cell greatly increased when Abro1−/− cells were treated with APH (Fig. 2D). Furthermore, primary Abro1-null MEFs showed increased chromosome aberrations when compared with wild-type MEFs, and treatment with APH greatly increased chromosome aberrations in Abro1−/− cells (Fig. 3C). Thus, Abro1 is crucial for maintaining genome stability in response to replication stress.
Figure 3.
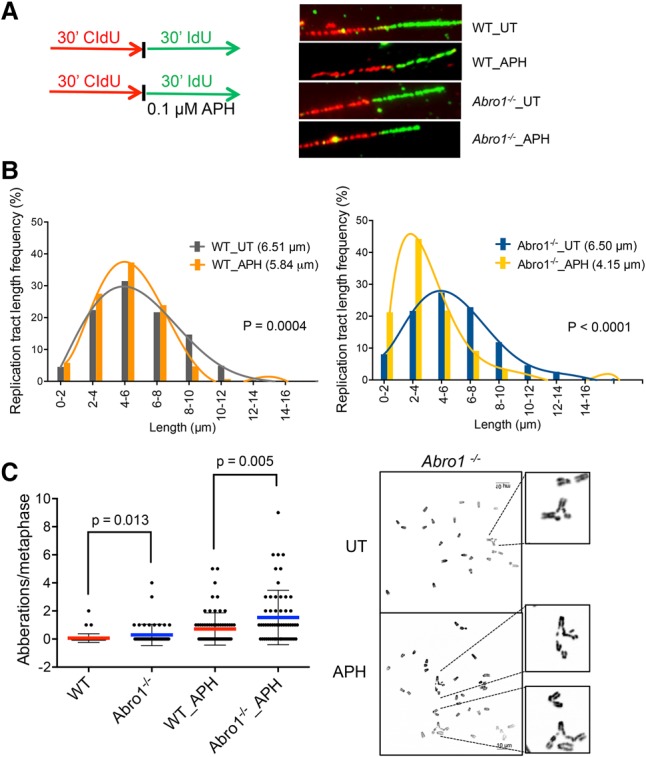
Abro1 plays a role in the response to replication stress. (A) Schematic for labeling cells with CIdU and IdU and representative images of DNA fibers. (B) Fork progression in cells untreated or treated with APH. Primary MEFs are labeled as indicated. More than 300 fibers were measured for each sample. Experiments were repeated twice. P-value was determined by two-tailed Student's t-test. (C) Abro1−/− cells displayed increased chromosome aberrations. Metaphase spreads were prepared from primary wild-type and Abro1−/− MEFs. Cells were treated with 0.3 µM APH for 16 h. The number of aberrations per metaphase was calculated. At least 50 metaphases were analyzed per experimental group. Significance was determined by Mann-Whitney test.
Abro1 suppresses uncontrolled resection
To determine the role of Abro1 in the replication stress response, we examined how Abro1-null cells respond to replication stress. Upon treatment of Abro1-null cells with hydroxyurea (HU), which depletes nucleotide pools and stalls replication forks, we observed a marked increase in RPA32 phosphorylation on S4/S8 (a well-established marker of DNA resection) that continued for a period of time after release from the treatment (Fig. 4A; Supplemental Fig. S2A). This suggests that there is increased resection at stalled replication forks and accumulation of ssDNA. Consistent with this, immunofluorescence staining with an RPA32pS4/8 antibody showed an increased percentage of Abro1-null cells containing RPA32pS4/8 foci when compared with the control (Fig. 4B). In addition, the percentage of cells containing pan-nuclear staining of γ-H2AX in Abro1−/− cells increased immensely, indicating increased replication stalling (Fig. 4C). Increased γ-H2AX phosphorylation was also detected by Western blot of the chromatin fraction and the total lysates of Abro1-null cells (Fig. 4A). A neutral comet assay showed no significant increase of DSB formation in Abro1−/− cells treated with HU (Fig. 4D), indicating that the increased RPA32 phosphorylation was not due to DSB formation. In addition, Chk1 was phosphorylated in Abro1-null cells in a manner similar to that in wild-type controls (Fig. 4A), indicating that the ATR signaling pathway to activate Chk1 in response to replication stress was largely unaffected by Abro1 deficiency.
Figure 4.

Abro1 suppresses uncontrolled resection. (A) Wild-type or Abro1−/− MEFs were either untreated (Un) or treated with 4 mM HU for 4 h (Tr) followed by release into fresh medium for 30 min (R30′) or 60 min (R60′). Western blot was carried out with the indicated antibodies on chromatin fraction or total lysate samples. (B) Increased RPA32pS4/8 in Abro1−/− cells. Overlaid images of immunofluorescence with RPA32pS4/8 antibody and DAPI staining are shown. (C) Increased γH2AX in Abro1−/− cells. Overlaid images of immunofluorescence with γH2AX antibody and DAPI staining are shown. (D) Neutral comet assay for cells untreated or treated with 4 mM HU for 4 h. Wild-type cells treated with 1 µM camptothecin (CPT) for 1 h were used as a control. Tail moment was measured, and P-value was determined by Student's t-test. (E) Increased ssDNA in Abro1−/− cells. Wild-type or Abro1−/− cells were labeled with BrdU overnight before treatment with 4 mM HU for 4 h. Immunofluorescence using BrdU antibody was carried out under native conditions. Relative BrdU labeling intensity (a.u.) was analyzed, and a cutoff value was used for scoring BrdU-positive cells. The percentage of cells with BrdU-positive (BrdU+) staining was determined. P-value was determined by Student's t-test. (F) Abro1 is present at unperturbed and stalled replication forks using isolation of proteins on nascent DNA (iPOND) analysis. Cells were pulse-labeled with EdU for 10 min (EdU), labeled with EdU followed by treatment with 2 mM HU for 2 h (HU), or labeled with EdU followed by thymidine chase for 2 h (Thd). Control (Ctrl) is the “no Click” sample. EdU-labeled newly synthesized DNA was captured, and bound proteins were analyzed by Western blot.
We then examined ssDNA generation at stalled forks in Abro1-null cells by detecting ssDNA through native BrdU immunofluorescence. Treatment with HU resulted in very little BrdU staining in wild-type cells but induced robust BrdU staining in Abro1-null cells, indicative of extensive ssDNA formation at stalled forks (Fig. 4E). We also pulse-labeled cells with BrdU immediately before stalling forks with HU and examined ssDNA generation from nascent strand DNA at stalled replication forks. Similarly, the BrdU staining in the pulse-labeled Abro1-null cells increased when compared with the control, indicating that nascent strand DNA at stalled forks also was resected to generate ssDNA (Supplemental Fig. S2B).
With an evident role for Abro1 in the response to fork stalling, we next examined whether Abro1 is present at stalled replication forks. The isolation of proteins on nascent DNA (iPOND) uses click chemistry to conjugate biotin to a nucleoside analog (EdU) incorporated in newly synthesized DNA, allowing analysis of proteins bound to DNA replication forks (Sirbu et al. 2011, 2013). As a control, we detected PCNA dissociation from and FANCD2 accumulation at stalled forks in HU-treated cells, consistent with previous reports (Sirbu et al. 2011; Lossaint et al. 2013). In addition, we detected a reproducible Abro1 association with replication forks at both unperturbed forks stalled and those stalled by HU treatment (Fig. 4F; Supplemental Fig. S2C) as well as a decreased association of Abro1 with replication forks after thymidine chase in a fashion similar to MCM2, suggesting a role of Abro1 in DNA replication near replication forks. These data indicate that Abro1 plays a role at replication forks to suppress extensive resection at stalled forks.
Abro1 maintains fork stability and promotes fork recovery in response to replication stress
We used the DNA fiber assay to analyze fork stability in cells exposed to sequential 30-min pulses with CIdU (Fig. 5A, red) and IdU (Fig. 5A, green) followed by 4 h of incubation with HU to induce DNA replication stalling (Fig. 5A). We found that Abro1 is necessary for the stability of nascent DNA at stalled replication forks, as measured by the ratio of the lengths of adjacent IdU (Fig. 5A, green) and CIdU (Fig. 5A, red) tracts. Upon HU treatment, stalled replication forks in wild-type cells are stable, showing an IdU/CIdU tract ratio close to 1. Degradation of newly synthesized DNA was observed in Abro1-null cells treated with HU, indicated by a reduced IdU/CIdU ratio due to shortening of the IdU tract (Fig. 5B). The shortening of newly synthesized DNA tracts at stalled replication forks was rescued by introduction of wild-type mouse Abro1 (mAbro1) gene expression in Abro1−/− cells but not by empty vector (Fig. 5C; Supplementary Fig. 3A). A similar defect in protection of nascent DNA at stalled replication forks was also observed in human U2OS cells after deletion of Abro1 by CRISPR–Cas9, and the defect could be rescued by reintroduction of GFP-tagged full-length human Abro1 (Supplemental Fig. S3B). We then examined whether Abro1 is involved in the recovery of DNA synthesis after fork stalling induced by a transient replication block. We prelabeled cells with CldU before HU treatment followed by IdU (Supplemental Fig. S3C). The frequency of forks that could not restart and collapsed (Supplemental Fig. S3D, red tracks only) during the HU treatment increased dramatically in Abro1-null cells when compared with the wild-type control (Supplemental Fig. S3D). This defect also could be rescued by complementation of expression of mAbro1 (Supplemental Fig. S3D). For the forks that resumed DNA synthesis, the IdU-labeled tracts were significantly shorter after HU treatment, indicating that fork progression was significantly reduced (Supplemental Fig. S3E). The decreased fork progression after stalling could be rescued by expression of mAbro1 in Abro1−/− cells (Supplemental Fig. S3F). Thus, Abro1 plays an important role in protecting stalled replication fork integrity and restarting replication forks after replication stress.
Figure 5.

Abro1 maintains fork stability and promotes fork recovery in response to replication stress. (A) Schematic for labeling cells with CIdU and IdU and representative images of DNA fibers. (B) Nascent DNA degradation at stalled replication forks in Abro1-null cells treated with HU. (C) Shortened nascent DNA tracts were rescued by re-expression of the mouse Abro1 gene in Abro1-null MEFs. Expression of mAbro1 in Abro1−/− cells is shown in Supplemental Figure S3. (D) A schematic view of mouse Abro1 and mutants. (E) The MPN domain, but not the coiled-coil domain, is required for protection of newly synthesized DNA at stalled forks. Expression of mAbro1 wild type and mutants in Abro1−/− cells is shown in Supplemental Figure S3. (F) NBA1 and BRE, but not BRCC36, are required for protection of newly synthesized DNA at stalled forks. The experiments were repeated multiple times. In each experiment, 170–200 fibers were measured for each sample. Line and bars represent the median IdU/CldU ratio and interquartile range. Significance was determined by Mann-Whitney test. (n.s.) Not significant; (****) P < 0.0001.
As Abro1 is known to interact with BRCC36, NBA1/MERIT40, and BRE, forming the BRISC deubiquitinating complex (Cooper et al. 2010; Feng et al. 2010; Patterson-Fortin et al. 2010; Hu et al. 2011), we asked whether the function of Abro1 in fork protection requires the Abro1 domains that are needed to form an intact BRISC. We found that deletion of the MPN domain but not the coiled-coil domain led to failure of Abro1 in the protection of stalled replication forks (Fig. 5D,E; Supplemental Fig. S3G). The coiled-coil domain is required for Abro1 interaction with BRCC36 (Hu et al. 2011). The fact that the coiled-coil deletion mutant was able to rescue the deficiency of Abro1 indicates that Abro1–BRCC36 interaction is dispensable for the role of Abro1 in fork protection. The MPN domain mediates Abro1 interaction with NBA1/MERIT40 and BRE in the BRISC. Since the MPN domain deletion mutant as well as a point mutation (W103E) in the MPN domain that abolishes the binding of Abro1 to NBA1/MERIT40 and BRE failed to rescue the deficiency of Abro1-null cells in protecting stalled replication fork stability, it indicates that the interaction of Abro1 with NBA1/MERIT40 and BRE is important for Abro1's function in the replication stress response. When we examined the effect of depletion of NBA1/MERIT40, BRE, and BRCC36, we found that knockdown of NBA1 and BRE, but not BRCC36, in wild-type MEFs resulted in deficiency in fork protection (Fig. 5F). Thus, Abro1, together with NBA1/MERIT40 and BRE but not BRCC36, is important for maintaining fork stability in response to replication stress.
DNA2, but not MRE11, is responsible for nascent strand shortening at stalled forks in Abro1-null cells
Next, we asked how Abro1 protects stalled fork stability. It has been demonstrated that loss of BRCA1, BRCA2, or FANCD2 protein leads to MRE11-dependent nucleolytic degradation of stalled replication forks (Schlacher et al. 2011, 2012). Therefore, we examined whether MRE11 is responsible for the observed nascent strand shortening at stalled forks in Abro1-null cells. As a control, we treated FANCD2-deficient cells with Mirin, a chemical inhibitor of MRE11 (Dupre et al. 2008). This reversed the degradation of stalled replication forks seen in FANCD2-deficient cells, as was reported previously (Supplemental Fig. S4A). However, treatment of Abro1−/− cells with Mirin was unable to rescue fork degradation (Fig. 6A). Knockdown of MRE11 using siRNAs also did not rescue the fork degradation in Abro1-null cells (Supplemental Fig. S4B). Instead, knockdown of DNA2 in Abro1−/− cells rescued the degradation of stalled forks (Fig. 6B; Supplemental Fig. S4C), indicating that DNA2 is responsible for the degradation of stalled forks in the absence of Abro1. In addition, the increased RPA32 phosphorylation observed in Abro1−/− cells was greatly reduced by knockdown of DNA2 (Fig. 6C). The increased ssDNA detected by native BrdU staining in Abro1-null cells also was reduced when cells were treated with shRNAs or siRNAs against DNA2 (Fig. 6D; Supplemental Fig. S4D).
Figure 6.

Degradation of nascent DNA strands at stalled forks in Abro1−/− cells is dependent on DNA2. (A) Mirin does not inhibit nascent strand DNA degradation in Abro1-null cells. Cells were labeled sequentially with CIdU and IdU as shown in Figure 5A, and left untreated (UT) or treated with 4 mM HU, 4 mM HU in the presence of 100 µM mirin, or DMSO/4 mM HU for 4 h. Line and bars represent the median IdU/CldU ratio and interquartile range. (n.s.) Not significant; (****) P < 0.0001, Mann-Whitney test. n > 170 fibers for each condition. Experiments were completed in duplicate. (B) DNA2 knockdown rescues end protection deficiency in Abro1-null MEFs in response to HU-induced replication fork stalling. Cells were infected with virus containing control or shRNAs to DNA2 and treated as described in Figure 5A. Western blot showing knockdown efficiency is in Supplemental Figure S4. Significance was determined as above. (C) DNA2 knockdown decreased RPA32pS4/8 phosphorylation in Abro1−/− cells after HU treatment. Wild-type (+/+) or Abro1-null (−/−) cells were transfected with siRNAs to DNA2 and treated as described in Figure 5A. Chromatin fraction or total cell lysate was analyzed by Western blot. (D) Expression of human DNA2 (hDNA2) wild type and mutants in Abro1−/− cells with knockdown of DNA2. Cells were labeled with BrdU overnight before treatment with 4 mM HU for 4 h. Immunofluorescence using BrdU antibody was carried out under native conditions. The percentage of cells with BrdU-positive (BrdU+) staining was determined. P-value was determined by Student's t-test. (n.s.) Not significant; (****) P < 0.0001. (E) Expression of hDNA2 D294A in Abro1-null cells with knockdown of DNA2 did not restore increased RPA32pS4/8 phosphorylation in Abro1−/− cells after HU treatment. Chromatin fractions from cells were analyzed by Western blot. Expression level of hDNA2 is shown in Supplemental Figure S4. (F) Expression of hDNA2 D294A in Abro1-null cells with knockdown of DNA2 failed to restore the end protection deficiency in Abro1-null cells after HU treatment. Abro1−/− MEFs were transfected with siRNAs targeting mouse DNA2 followed by transient transfection with hDNA2 wild type or mutants and then treated as described in Figure 5A for DNA fiber analysis. (G) Knockdown of WRN, but not BLM or EXO1, rescued end protection deficiency in Abro1-null MEFs in response to HU-induced replication fork stalling. (H) Knockdown of WRN reduced RPA2 hyperphosphorylation in Abro1−/− cells. (I) Knockdown of WRN reduced ssDNA accumulation in Abro1−/− cells.
As DNA2 is a nuclease/helicase, we tested whether its nuclease or helicase activities or both are responsible for the shortening of the nascent DNA tract in Abro1−/− cells. We expressed human DNA2 (hDNA2) wild type, nuclease-deficient mutant D294A, or the helicase-deficient mutant K671E (Lin et al. 2013) in Abro1−/− cells with endogenous DNA2 knocked down by siRNAs. Whereas expression of hDNA2 wild type and K671E was able to complement the effect of DNA2 knockdown on Abro1−/− cell and restored ssDNA and RPA32 phosphorylation to the increased levels seen in Abro1−/− cells, expression of D294A mutant did not have an obvious effect (Fig. 6D,E; Supplemental Fig. S4E). The D294A mutant, unlike wild type or the K671E mutant, was also unable to reinstate the shortening of nascent DNA at stalled forks in Abro1−/− cells depleted of endogenous DNA2 (Fig. 6F). Together, these results indicate that the nuclease activity, but not the helicase activity, of DNA2 is responsible for the fork degradation in HU-treated Abro1-null cells.
DNA2 has been shown to function in conjunction with BLM (Imamura and Campbell 2003; Nimonkar et al. 2011) and WRN (Thangavel et al. 2015) helicases in end processing. We found that WRN depletion, but not BLM depletion, mimicked the effect of DNA2 knockdown in rescuing the nascent strand degradation in Abro1-null cells, indicating that WRN helicase cooperates with DNA2 in shortening the nascent strand DNA at stalled forks in Abro1-null cells (Fig. 6G; Supplemental Fig. S4F). We also examined exonuclease I (EXO1), with which DNA2 plays a partially redundant role in DNA end resection during DSB repair (Zhu et al. 2008). Unlike knockdown of DNA2, knockdown of EXO1 did not rescue the degradation of nascent DNA in Abro1-null cells (Fig. 6G). Depletion of WRN, but not BLM or EXO1, also led to decreased RPA32 phosphorylation and reduced ssDNA accumulation in Abro1-null cells (Fig. 6H,I). Thus, Abro1 protects stalled replication forks from DNA2/WRN-dependent degradation of newly synthesized DNA.
Resistance to degradation of newly synthesized DNA in Abro1-null cells in the absence of RAD51
RAD51 filament stabilization plays an important role in maintaining nascent DNA strand integrity in BRCA1- or BRCA2-deficient cells (Schlacher et al. 2011, 2012). To determine whether this is the case for Abro1-null cells, we expressed a RAD51 K133R mutant in Abro1-null cells (Supplemental Fig. S5A). This mutant is devoid of ATPase activity and forms stable filaments (Morrison et al. 1999). We found that although stabilized RAD51 filaments rescued the degradation of stalled forks in FANCD2-deficient cells as indicated previously (Supplemental Fig. S5B), it did not rescue the degradation of stalled forks in Abro1−/− cells exposed to HU (Fig. 7A). Interestingly, expression of a BRCA2 BRC4 peptide fragment (Saeki et al. 2006), which suppresses DNA binding of RAD51, thus perturbing RAD51 filaments (Hashimoto et al. 2010), rendered IdU tracts resistant to degradation. This indicates that disruption of RAD51 filament formation suppresses the degradation of nascent strand DNA in Abro1-null cells (Fig. 7B; Supplemental Fig. S5C). As a control, expression of the BRC4 peptide in wild-type MEFs led to substantially shorter IdU tracts upon HU treatment, as reported previously (Schlacher et al. 2011). In addition, overexpression of BRC4 decreased the phosphorylation of RPA32 (Fig. 7C) and the overall amount of ssDNA in Abro1−/− cells (Supplemental Fig. S5D). Knockdown of RAD51 also rescued the degradation of nascent strand DNA in Abro1−/− cells, indicating that the fork instability caused by Abro1 deficiency can be compensated for by RAD51 deficiency. This is consistent with the previous finding that RAD51 knockdown largely prevents DNA2 nucleolytic processing of nascent DNA at stalled forks (Thangavel et al. 2015).
Figure 7.

Abro1 protects stalled replication fork integrity distinctively from BRCA2-dependent RAD51 filament stabilization. (A) Expression of the RAD51K133R mutant does not rescue the defect of Abro1−/− cells in protecting newly synthesized DNA. MEFs were transfected with either empty vector or the RAD51K133R expression construct and then treated as described in Figure 5A. Expression of RAD51K133R is shown in Supplemental Figure S5. (B) Expression of the BRC4 peptide rescues degradation of nascent strand DNA in Abro1−/− cells. MEFs were transfected with either empty vector or the BRC4 expression construct and then treated as described in Figure 5A. Expression of Flag-BRC4 is shown in Supplemental Figure S5. (C) Expression of BRC4 peptide decreased RPA32pS4/8 phosphorylation in Abro1−/− cells upon HU treatment. Chromatin fraction and total cell lysates were analyzed by Western blot with the indicated antibodies. (D) Knockdown of Rad51 rescued degradation of nascent strand DNA in Abro1−/− cells. (E) Knockdown of Abro1 in BRCA2-deficient MEFs further increased degradation of nascent strand DNA. Brca2fl/fl primary MEFs were transfected with control or siRNAs against Abro1 and infected with adenovirus or adenovirus expressing Cre. (F) Knockdown of Abro1 in CAPAN-1 cells led to increased degradation of nascent strand DNA that can be rescued by mirin and depletion of DNA2. CAPAN-1 cells or CAPAN-1 cells infected with control shRNA (empty space) or shRNA to Abro1 (labeled with a filled circle) were either untreated or treated with HU as described in Figure 5A. Cells were treated with mirin or siRNAs to DNA2 as indicated (samples not treated with siDNA2 were treated with control siRNA). (G) A proposed model showing that Abro1 protects stalled replication fork integrity in a mechanism distinct from the BRCA2-dependent pathway. For DNA fiber assay, >170 fibers were counted for each sample. Line and bars represent the median IdU/CldU ratio and interquartile range. Significance was determined by Mann-Whitney test. (n.s.) Not significant; (***) P < 0.001; (****) P < 0.0001.
Abro1 protects stalled replication forks independently of BRCA2
Since it appears that Abro1 protects stalled replication fork stability in a manner different from the BRCA1/BRCA2-dependent fork protection pathway, in which stable RAD51 filament formation prevents MRE11-dependent degradation of the newly synthesized DNA at stalled forks, we examined whether Abro1 deficiency further decreases the defect of BRCA2-deficient cells in the protection of stalled fork integrity. We knocked down mAbro1 by siRNAs in Brca2flox/flox MEFs (Jonkers et al. 2001), with Brca2 deleted upon Cre recombinase infection. As expected, deletion of Brca2 upon Cre infection resulted in degradation of nascent DNA tracts (Fig. 7E). Depletion of Abro1 by siRNA in Brca2fl/fl cells infected with Cre further increased degradation of newly synthesized DNA (Fig. 7E; Supplemental Fig. S5F), indicating that Abro1 protects stalled replication fork integrity in the absence of BRCA2. This was confirmed in human CAPAN-1 cells, which express a truncated BRCA2 and are defective in maintaining nascent DNA tracts at stalled forks (Schlacher et al. 2011). Knockdown of Abro1 by shRNAs (Supplemental Fig. S5G) further shortened the nascent DNA tract length at forks stalled by HU (Fig. 7F, lane 7) when compared with HU-treated CAPAN-1 cells (Fig. 7F, lane 2). Use of Mirin (Fig. 7F, lanes 3,5), but not treatment with siRNAs to DNA2 (Fig. 7F, lane 4), rescued the stalled fork degradation in CAPAN-1 cells. Importantly, the degree of degradation of nascent strand DNA in Abro1- and BRCA2-double-deficient cells (Fig. 7F, lane 7) was partially rescued by Mirin (Fig. 7F, lane 8) or depletion of DNA2 (Fig. 7F, lane 9) but fully rescued by a combination of treatment of Mirin and depletion of DNA2 (Fig. 7F, lane 10). Together, these results indicate that Abro1 maintains fork stability by limiting DNA2-dependent degradation of stalled forks, a mechanism that is distinct from the BRCA1/BRCA2-dependent pathway that inhibits MRE11-mediated fork degradation.
Discussion
The protection of stalled fork integrity is critical in the cellular mechanisms that resolve replication stress and maintain genome stability. It has been shown that in the BRCA1/BRCA2/FANCD2-dependent pathway, BRCA2 stabilizes RAD51 filaments at stalled forks, thereby preventing nucleolytic degradation of nascent strands by MRE11 nuclease (Schlacher et al. 2011, 2012). In this study, we identified Abro1 as a new component that protects stalled replication forks by limiting DNA2/WRN-mediated fork degradation in a manner distinct from the BRCA1/BRCA2/FANCD2-dependent pathway that protects stalled forks from MRE11-mediated degradation (Fig. 7G).
In the absence of Abro1, the DNA in the vicinity of stalled forks becomes susceptible to nuclease resection, generating an increased amount of ssDNA and hyperphosphorylation of RPA (Fig. 4). Nascent strand DNA at stalled forks is also shortened in Abro1-null cells (Fig. 5). Knocking down DNA2 nuclease reverses the degradation of stalled forks and generation of ssDNA in Abro1-null cells (Fig. 6). The effect of DNA2 knockdown was complemented for by both wild-type DNA2 and a helicase-deficient mutant but not by a nuclease-deficient mutant of DNA2. Therefore, we propose that Abro1 protects stalled replication forks by limiting uncontrolled DNA2 nuclease activity at stalled forks. Although the unwinding activity of DNA2 is dispensable, a DNA2-associated helicase, WRN, appears to be important for this process. Thus, DNA2 and WRN cooperate to degrade stalled forks in Abro1-null cells.
The Abro1-dependent fork protection mechanism is distinct from the BRCA2-dependent pathway of protecting stalled fork stability. In BRCA2-deficienct cells, nascent DNA strands are shortened by MRE11 endonuclease-dependent degradation, and inhibition of MRE11 restored fork integrity (Schlacher et al. 2011, 2012; Ying et al. 2012). However, nascent strand DNA at stalled replication forks in Abro1-null cells is susceptible to the degradation of DNA2/WRN but not MRE11 nuclease (Fig. 6). Furthermore, whereas the nascent strand instability in cells deficient for BRCA2 is rescued by stabilized RAD51 filament formation (Schlacher et al. 2011), the degradation of nascent DNA strands in Abro1−/− cells is not rescued by RAD51 filament stabilization (Fig. 7). Consistent with a role of Abro1 in a distinct mechanism that acts to protect stalled replication fork stability, concomitant depletion of Abro1 and BRCA2 in MEF cells and knockdown of Abro1 in BRCA2-deficient CAPAN-1 cells results in increased levels of fork degradation (Fig. 7). Importantly, the fork degradation due to BRCA2/Abro1 double deficiency is only partially rescued by inhibition of MRE 11 or inhibition of DNA2 but is completely reversed when both nucleases are inhibited. Thus, we propose that the Abro1-regulated pathway represents a new aspect in the intricate mechanisms that ensure fork stability independently of BRCA2.
RAD51 appears to play a facilitating role in the DNA2/WRN-mediated degradation of stalled forks in Abro1−/− cells, since RAD51 deficiency alleviates the degradation of nascent strand DNA in Abro1-null cells (Fig. 7). Why? It has been indicated that the DNA2 nuclease and WRN helicase act downstream from RAD51-mediated fork reversal to degrade reversed replication forks following HU treatment, promoting proper restart of the reversed fork (Thangavel et al. 2015; Zellweger et al. 2015). A recent study investigating a Fanconi anemia-associated RAD51 T131P mutant also indicates a role for RAD51 in DNA2/WRN-mediated degradation of interstrand cross-link repair intermediates (Wang et al. 2015). Thus, RAD51-mediated fork reversal is also likely to play a role in the extensive DNA2/WRN-dependent degradation of newly synthesized DNA in Abro1-null cells. In Abro1−/− cells, uncontrolled DNA2 activity acts on the reversed forks, leading to increased resection and shortening of nascent strand DNA. Depletion of RAD51 diminishes fork reversal and thus alleviates the degradation of nascent strand DNA in Abro1−/− cells. It is also possible that Abro1 deficiency leads to a change of DNA configuration at stalled forks that increases RAD51-mediated fork reversal and hence DNA2-dependent degradation. It remains to be seen whether reversed fork structures form in Abro1-null cells and whether they accumulate in Abro1-null cells in response to replication stress. The different roles of RAD51 in maintaining replication fork stability and promoting fork reversal during replication fork stalling may also explain why RAD51 deficiency eliminates fork degradation in Abro1−/− cells, while BRCA2 deficiency, which perturbs stable RAD51 filament formation for fork protection, instead increases fork degradation in Abro1-deficient cells (Fig. 7E,F). It is likely that, while BRCA2-dependent RAD51 filament stabilization protects fork integrity, BRCA2 does not have a direct effect on the role of RAD51 in promoting fork reversal.
It is important to note that RAD51-mediated fork reversal and controlled DNA2 activity-dependent processing of reversed forks appear to be important for the restart of the reversed fork (Thangavel et al. 2015; Zellweger et al. 2015). However, extended degradation of stalled forks by DNA2 in Abro1-null cells largely decreased the ability of the stalled fork to resume progression or fork restart (Supplemental Fig. S3). This suggests that regulated DNA2 activity is critical both for the protection of stalled forks and for a stalled fork to resume progression or restart. Interestingly, in addition to BRCA1/BRCA2/FANCD2/RAD51-dependent inhibition of MRE11, recent studies indicate that multiple components, including PTIP, MLL3/4, CHD4, and PARP1, are involved in promoting MRE11 activity at stalled forks (Ray Chaudhuri et al. 2016). Thus, nuclease activities at stalled forks are tightly controlled to ensure fork integrity.
The role of Abro1 in responding to replication stress is critical for its ability to maintain genome stability. Abro1-null cells exhibited chromosomal instability that is associated with the failure to resolve replication stress, such as increased ultrafine bridges during anaphase and increased 53BP1 G1 nuclear bodies, which are associated with underreplicated regions (Fig. 2). Due to its critical role in maintaining chromosome stability, it is not surprising that Abro1 knockout mice are prone to tumor development (Fig. 1). Abro1 forms the BRISC by interacting with BRCC36, NBA1/MERIT40, and BRE and is implicated in facilitating the deubiquitylating enzyme (DUB) activity of BRCC36 (Cooper et al. 2010; Feng et al. 2010; Patterson-Fortin et al. 2010; Hu et al. 2011). Previously, it has been shown that Abro1/BRISC complex DUB activity is involved in deubiquitinating substrates that regulate inflammsome activity, interferon response, and mitotic spindle assembly (Py et al. 2013; Zheng et al. 2013; Yan et al. 2015). Our study indicates that the function of Abro1 in the protection of stalled fork stability is independent of BRCC36, since the coiled-coil deletion mutant of Abro1 that does not interact with BRCC36 retains the ability to protect stalled replication forks, and knockdown of BRCC36 does not lead to a defect in fork protection (Fig. 5). Although the majority of Abro1 protein appears to localize in the cytoplasm of the cells (Feng et al. 2010; Hu et al. 2011), we show that Abro1 is present at both undisturbed and stalled replication forks in the nucleus (Fig. 4). In addition, we did not observe translocation of Abro1 in response to DNA-damaging agents IR, UV, or HU (data not shown). It is likely that Abro1 forms different subcomplexes to carry out distinct functions in the nucleus or cytoplasm. We speculate that Abro1, NBA1/MERIT40, and BRE may form a subcomplex independent of the BRISC in nucleus at the replication fork and that the DUB activity of BRCC36 is not required.
In summary, our study reveals a new aspect to the mechanisms of protection of stalled replication forks that involves Abro1, which has critical implications for the maintenance of genomic integrity and tumor suppression.
Materials and methods
Generation and analysis of Abro1-deficient mice
Detailed methods are described in the Supplemental Material. Age-matched wild type mice (+/+), heterogeneous mice (+/−), and null mice (−/−) were monitored daily for survival and tumor formation. All mouse experiments were approved by the M.D. Anderson Cancer Center Institutional Animal Care and Use Committee, protocol RN01-00001247, and followed the guidelines of the United States Animal Welfare Act and the National Institutes of Health.
For the whole-animal radiosensitivity assay, age-matched 4- to 6-wk-old wild-type and Abro1-null mice were treated with 7.5 Gy of IR and monitored daily for survival. The survival curve was analyzed by the Kaplan-Meier method. The log-rank test was used for statistical analysis.
Cell lines, cell culture, antibodies, and chemicals
MEF cells were isolated from embryonic day 13.5 (E13.5) embryos as described previously (Castillo et al. 2014). Primary MEFs were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin antibiotics and maintained in 3% O2/5% CO2 in an incubator at 37°C. Immortalized MEFs were generated by spontaneous immortalization following the 3T3 protocol as described previously (Castillo et al. 2014). PD20 and PD20/FANCD2 cells were maintained in RPMI1640 medium with 15% FBS. U2OS cells were grown in McCoy's 5A medium with 10% FBS. CAPAN-1 cells were cultured in Iscove's modified Dulbecco's medium (IMDM) (Hyclone, Thermo Scientific) with 20% FBS. Antibodies used in this study included Abro1 (Hu et al. 2011), BRCC36 (Abcam), BRE (Hu et al. 2011), NBA1 (Hu et al. 2011), 53BP1 (Novus Biologicals), BLM (Bethyl Laboratories), BrdU (BD Bioscience), RAD51 (Calbiochem), RPA32pS4/8 (Bethyl Laboratories), p-CHK1 (Cell Signaling), CHK1 (Santa Cruz Biotechnolgoy), γH2AX (Ser139; Upstate Biotechnology), Flag (Sigma), BRCA2 (Abcam, ab27976), γ-tubulin (Sigma), DNA2 (Proteintech, 18727-1-AP), FANCD2 (Santa Cruz Biotechnology, sc-20022), and H3 (Abcam). Chemicals used in this study included BrdU (Sigma), APH (Sigma), HU (Sigma), MMC (Sigma), and camptothecin (CPT) (Sigma).
Plasmids, siRNAs, and shRNAs
Retroviral constructs expressing mouse Abro1 and mutants were generated using the MSCV vector. Flag-BRC4 and RAD51 K133R expression constructs were described previously (Schlacher et al. 2011). hDNA2 wild-type, D294A, and K671E mutant expression plasmids were described previously (Lin et al. 2013). The shRNAs used in this study were mouse DNA2 shRNA (5′-TTGGACAGCATGGCTTCTG-3′ and 5′-ACTGTGTTAACCTCAACCA-3′), human Abro1 shRNA (5′-CGTAGTTTCTGTTCTTTAA-3′ and 5′-CTGATTAGAACAAATCAGA-3′), mouse shNBA1(5′-ATTTACAGTGGACAGAGTG-3′), and mouse BRE (5′-TTCATTTCCATCCCATCTG-3′ and 5′-AGCCTTGAATCACATACTG-3′). The siGENOME SMARTpool siRNAs targeting mouse Dna2 (M-062864-01-0005), mouse Mre11 (M-051344-00-0005), mouse Blm (M-061987-01-0005), mouse Wrn (M-058494-01-0005), mouse Exo1 (M-060591-01-0005), mouse Rad51 (M-062730-01-0005), and mouse Abro1 (M-041233-01-0005) were purchased from Dharmacon. siRNAs targeting mouse BRCC36 (ID:167166 and 167177) and negative control siRNA (#12935110) were purchased from Invitrogen.
Generation of Abro1/BRCA2-deficient MEFs
BRCA2flox/flox MEFs were transfected with SMARTpool siRNA against mouse Abro1 by Lipofectamine RNAiMAX reagent. Twenty-four hours later, cells were infected with either Cre expression adenovirus or control adenovirus. Cre-mediated recombination efficiency was detected by PCR genotype of cells 48 h after virus infection using primers specific for the BRCA2 flox allele (5′-GGCTGTCTTAGAACTTAGGCTG-3′ and 5′-CTCCACACATACATCATGTGTC-3′).
CAPAN-1 cells were infected with virus containing either control shRNA or shRNA targeting Abro1, and stable cell lines were selected. Cells were then transfected with control siRNA or siRNA targeting DNA2 for 48 h or treated with 4 mM HU or 100 µM mirin as indicated.
DNA fiber assay
DNA fiber assay was carried out as described previously (Castillo et al. 2014). Briefly, cells were pulse-labeled with 100 µM CldU and 250 µM IdU for 30 min. After labeling and treatment, cells were harvested, lysed, and spread on microscope slides. Immunofluorescence was carried out using α-IdU/α-BrdU (BD Biosciences) and α-CldU/α-BrdU (Abcam) and secondary antibodies, including Alexa fluor 488 (green) and Alexa fluor 555 (Invitrogen). Images were taken using a Nikon 80 microscope with a 60× lens and analyzed using ImageJ software. Statistics were calculated using Prism software.
Clonogenic survival assay
For colony survival studies, cells were seeded at low density and treated with the indicated doses of UV or IR and then left for 10–14 d of incubation at 37°C as described previously (Wu et al. 2016). For MMC treatment, cells were treated with the indicated doses of MMC for 1 h followed by two PBS washes and then left for incubation. Colonies were fixed and stained with 2% methylene blue and 50% ethanol. Colonies with ≥50 cells were counted as positive. Colonies were counted and normalized to untreated samples to calculate the percentage of survival. Error bars represent standard deviation across triplicates.
iPOND
iPOND was performed according to the protocol described by Sirbu et al. (2012) with slight modification. HEK293T cells were pulse-labeled with 10 µM EdU (Invitrogen) for 10 min followed by treatment with 2 mM HU for 2 h or thymidine chase for 2 h. Cells were fixed in 1% formaldehyde for 20 min at room temperature, quenched by adding glycine to a final concentration of 0.125 M, and washed three times in PBS. Cell pellets were collected and frozen at −80°C. Cell permeablization was carried out by resuspending cells in ice-cold 0.25% Triton-X/PBS at a concentration of 1 × 107 cells per milliliter followed by incubation for 30 min at room temperature. Samples were then washed once in 0.5% BSA/PBS and once in PBS. Click reaction was performed by incubating the sample for 1 h at room temperature in Click reaction buffer containing 10 µM biotin-azide (Invitrogen), 10 mM sodium ascorbate, and 2 mM CuSO4 in PBS. The control was a “no Click” reaction using DMSO instead of biotin-azide. Following the Click reaction, cells were washed once in 0.5% BSA/PBS and once in PBS, resuspended in lysis buffer (50 mM Tris–HCl at pH 8.0, 1% SDS) containing protease inhibitors, and then sonicated using a microtip sonicator with a power setting of 13–16 W for 20-sec pulses and 40-sec pauses, repeated five times. After centrifugation, the supernatant was diluted 1:3 with NTN buffer (100 mM NaCl, 20 mM Tris at pH 7.4, 0.05% NP40) containing protease inhibitors. Streptavidin-agarose beads (Novagen) were washed with NTN buffer before being added to the sample. One-hundred microliters of bead slurry was used per 1 × 108 cells. The sample mixture was then incubated overnight at 4°C. The beads were then washed four times with NTN buffer. Cross-links were reversed by incubated samples in 2× SDS sample loading buffer for 12 min at 95°C.
Immunofluorescence
Immunofluorescence was carried out as described previously (Hu et al. 2011). Briefly, cells were fixed with fresh 3% paraformaldehyde/2% sucrose solution for 10 min at room temperature followed by permeablization with 0.5% Triton X-100 solution for 5 min on ice. For RPA32pS4/8 immunostaining, cells were pre-extracted using pre-extraction buffer (10 mM PIPES at pH 6.8, 100 mM sodium chloride, 300 mM sucrose, 3 mM magnesium chloride, 1 mM EGTA, 0.2% Triton-X100) for 5 min on ice before fixation and permeabilization. Cells were then incubated with the indicated primary antibody for 1 h at 37°C followed by secondary antibody, including Alexa 488 or Alexa 555, for 1 h at 37°C. Coverslips were then washed with PBS and mounted using DAPI containing anti-fade solution (Invitrogen). Images were captured by a Nikon 80i upright microscope. For native BrdU staining, cells were grown on glass coverslips and labeled with 10 µM BrdU for 36 h or pulse-labeled with 10 µM BrdU for 10 min before treatment with 4 mM HU for 4 h. Cells were pre-extracted on ice using pre-extraction buffer for 5 min, washed with PBS, and fixed with freshly prepared 3% paraformaldehyde/2% sucrose solution for 10 min at room temperature. After washes with PBS, cells were then fixed with cold methanol (−20°C) for 20 min, washed with PBS, and treated with cold acetone (4°C) for 30 sec. Cells were then washed three times with PBS and blocked in PBST containing 2% BSA for 1 h at room temperature. Immunofluorescence was then carried out with anti-BrdU primary antibody and Alexa 488-conjugated secondary antibody. Images were captured using a Nikon 80i upright microscope. Native BrdU intensities were measured using NIS Elements AR software (Nikon).
Cell lysis and chromatin fractionation
Cells were lysed using NETN buffer (50 mM Tris-HCl at pH 8.0, 150 mM sodium chloride, 1 mM EDTA, 0.5% nonidet P-40, 1 mM dithiothreitol [DTT], 1 mM PMSF, 5 mM NaF, 1 mM Na3VO4, 50 mM β-glyceral, protease inhibitor cocktails [Roche]) and centrifuged at 15,000 rpm for 10 min at 4°C. The supernatants were used as total cell lysate. For cell fractionation, 1 × 106 cells were incubated with 100 µL of cold solution A (10 mM HEPES at pH 7.9, 10 mM KCL, 1.5 mM MgCL2, 0.34 M sucrose, 10% glycerol, 5 mM NaF, 1 mM Na3VO4, 1 mM DTT, protease cocktails, 0.1% Triton X-100) for 5 min on ice. After centrifugation at 4000 rpm for 5 min at 4°C, cell pellets were collected and washed with cold solution A. Cell pellets were then incubated with cold solution B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, protease inhibitor cocktails) for 15 min on ice. After centrifugation, chromatin pellets were collected, suspended in NETN buffer, and sonicated before use.
Neutral comet assay
Comet assay was performed as described previously (Castillo et al. 2014). Briefly, cells were trypsinized, pelleted, and gently resuspended in 1 mL of PBS. Ten microliters of cell suspension (10,000 cells) was mixed with low-melting agarose gel and used for cell lysis and electrophoresis in neutral electrophoresis buffer (Trevigen). Nuclear DNA was visualized by staining with SYBR Gold solution, and images were captured using a Nikon 80i microscope. The tail moment of each cell was analyzed by OpenComet software.
Metaphase analysis
MEFs were grown to reach 60%–70% confluence and incubated in medium containing 0.1 µg/mL colcemid for 4 h. For cells treated with APH, cells were incubated in 0.3 µM APH for 16 h before treatment with colcemid. Cells were harvested by trypsinization, incubated in 0.075 M KCl hypotonic solution, and fixed in 3:1 methanol:acetic acid. Metaphase spreads were dropped and dried on glass slides. Slides were stained with DAPI and examined for chromosomal breaks and structural aberrations. At least 50 metaphases per condition were analyzed.
Supplementary Material
Acknowledgments
We thank Henry P. Adams and Dr. Adriana Paulucci-Holthauzen for providing technical support, assistance with the use of the microscope, and analysis of images, and Dr. Binghui Shen for hDNA2 wild-type, D294A, and K671E expression constructs. E.A. is a recipient of the Cancer Prevention Research Institute of Texas (CPRIT) Graduate Scholar Award. M.J.Y. is supported by 1R01CA164346, 1R01CA200703, and CPRIT RP140402. This work was supported by the National Institutes of Health (CA155025 to B.W.), the Mel Klein Family Fund, and funds from the University of Texas M.D. Anderson Cancer Center (Institutional Research Grant Program). S.X., X.W., and B.W. designed the study. S.X., X.W., L.W., A.C., J.L., E.A., A.P., D.S., and B.W. performed the experiments. K.S. and Y.K. provided critical reagents. M.J.Y. performed pathologic analysis. B.W. supervised the project. S.X., X.W., and B.W. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.299172.117.
References
- Berti M, Vindigni A. 2016. Replication stress: getting back on track. Nat Struct Mol Biol 23: 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biebricher A, Hirano S, Enzlin JH, Wiechens N, Streicher WW, Huttner D, Wang LH, Nigg EA, Owen-Hughes T, Liu Y, et al. 2013. PICH: a DNA translocase specially adapted for processing anaphase bridge DNA. Mol Cell 51: 691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo A, Paul A, Sun B, Huang TH, Wang Y, Yazinski SA, Tyler J, Li L, You MJ, Zou L, et al. 2014. The BRCA1-interacting protein Abraxas is required for genomic stability and tumor suppression. Cell Rep 8: 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KL, Hickson ID. 2011. New insights into the formation and resolution of ultra-fine anaphase bridges. Semin Cell Dev Biol 22: 906–912. [DOI] [PubMed] [Google Scholar]
- Chan KL, North PS, Hickson ID. 2007. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J 26: 3397–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KL, Palmai-Pallag T, Ying S, Hickson ID. 2009. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 11: 753–760. [DOI] [PubMed] [Google Scholar]
- Cooper EM, Boeke JD, Cohen RE. 2010. Specificity of the BRISC deubiquitinating enzyme is not due to selective binding to Lys63-linked polyubiquitin. J Biol Chem 285: 10344–10352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT, et al. 2008. A forward chemical genetic screen reveals an inhibitor of the Mre11–Rad50–Nbs1 complex. Nat Chem Biol 4: 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Wang J, Chen J. 2010. The Lys63-specific deubiquitinating enzyme BRCC36 is regulated by two scaffold proteins localizing in different subcellular compartments. J Biol Chem 285: 30982–30988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard H, Garcia-Muse T, Aguilera A. 2015. Replication stress and cancer. Nat Rev Cancer 15: 276–289. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V. 2010. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17: 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs MR, Reynolds JJ, Winczura A, Blackford AN, Borel V, Miller ES, Zlatanou A, Nieminuszczy J, Ryan EL, Davies NJ, et al. 2015. BOD1L is required to suppress deleterious resection of stressed replication forks. Mol Cell 59: 462–477. [DOI] [PubMed] [Google Scholar]
- Hoffelder DR, Luo L, Burke NA, Watkins SC, Gollin SM, Saunders WS. 2004. Resolution of anaphase bridges in cancer cells. Chromosoma 112: 389–397. [DOI] [PubMed] [Google Scholar]
- Hu X, Kim JA, Castillo A, Huang M, Liu J, Wang B. 2011. NBA1/MERIT40 and BRE interaction is required for the integrity of two distinct deubiquitinating enzyme BRCC36-containing complexes. J Biol Chem 286: 11734–11745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Paul A, Wang B. 2012. Rap80 protein recruitment to DNA double-strand breaks requires binding to both small ubiquitin-like modifier (SUMO) and ubiquitin conjugates. J Biol Chem 287: 25510–25519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura O, Campbell JL. 2003. The human Bloom syndrome gene suppresses the DNA replication and repair defects of yeast dna2 mutants. Proc Natl Acad Sci 100: 8193–8198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DA, Pombo A. 1998. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 140: 1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. 2001. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 29: 418–425. [DOI] [PubMed] [Google Scholar]
- Lin W, Sampathi S, Dai H, Liu C, Zhou M, Hu J, Huang Q, Campbell J, Shin-Ya K, Zheng L, et al. 2013. Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J 32: 1425–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Decaillet C, Gari K, Constantinou A. 2013. FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol Cell 51: 678–690. [DOI] [PubMed] [Google Scholar]
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, et al. 2011. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 13: 243–253. [DOI] [PubMed] [Google Scholar]
- Morrison C, Shinohara A, Sonoda E, Yamaguchi-Iwai Y, Takata M, Weichselbaum RR, Takeda S. 1999. The essential functions of human Rad51 are independent of ATP hydrolysis. Mol Cell Biol 19: 6891–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC. 2011. BLM–DNA2–RPA–MRN and EXO1–BLM–RPA–MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 25: 350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson-Fortin J, Shao G, Bretscher H, Messick TE, Greenberg RA. 2010. Differential regulation of JAMM domain deubiquitinating enzyme activity within the RAP80 complex. J Biol Chem 285: 30971–30981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pefani DE, Latusek R, Pires I, Grawenda AM, Yee KS, Hamilton G, van der Weyden L, Esashi F, Hammond EM, O'Neill E. 2014. RASSF1A-LATS1 signalling stabilizes replication forks by restricting CDK2-mediated phosphorylation of BRCA2. Nat Cell Biol 16: 962–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. 2013. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 49: 331–338. [DOI] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. 2016. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535: 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki H, Siaud N, Christ N, Wiegant WW, van Buul PP, Han M, Zdzienicka MZ, Stark JM, Jasin M. 2006. Suppression of the DNA repair defects of BRCA2-deficient cells with heterologous protein fusions. Proc Natl Acad Sci 103: 8768–8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. 2011. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145: 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M. 2012. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22: 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. 2011. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25: 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Cortez D. 2012. Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat Protoc 7: 594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, Cortez D. 2013. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J Biol Chem 288: 31458–31467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, et al. 2015. DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 208: 545–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B. 2012. BRCA1 tumor suppressor network: focusing on its tail. Cell Biosci 2: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Elledge SJ. 2007. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci 104: 20759–20763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. 2007. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316: 1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Hurov K, Hofmann K, Elledge SJ. 2009. NBA1, a new player in the Brca1 A complex, is required for DNA damage resistance and checkpoint control. Genes Dev 23: 729–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AT, Kim T, Wagner JE, Conti BA, Lach FP, Huang AL, Molina H, Sanborn EM, Zierhut H, Cornes BK, et al. 2015. A dominant mutation in human RAD51 reveals its function in DNA interstrand crosslink repair independent of homologous recombination. Mol Cell 59: 478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Paul A, Su D, Mehmood S, Foo TK, Ochi T, Bunting EL, Xia B, Robinson CV, Wang B, et al. 2016. Structure of BRCA1–BRCT/Abraxas complex reveals phosphorylation-dependent BRCT dimerization at DNA damage sites. Mol Cell 61: 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan K, Li L, Wang X, Hong R, Zhang Y, Yang H, Lin M, Zhang S, He Q, Zheng D, et al. 2015. The deubiquitinating enzyme complex BRISC is required for proper mitotic spindle assembly in mammalian cells. J Cell Biol 210: 209–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Hamdy FC, Helleday T. 2012. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 72: 2814–2821. [DOI] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M. 2015. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208: 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. 2014. Causes and consequences of replication stress. Nat Cell Biol 16: 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Gupta V, Patterson-Fortin J, Bhattacharya S, Katlinski K, Wu J, Varghese B, Carbone CJ, Aressy B, Fuchs SY, et al. 2013. A BRISC–SHMT complex deubiquitinates IFNAR1 and regulates interferon responses. Cell Rep 5: 180–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. 2008. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134: 981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.