

Abstract
Lateral meningocele syndrome (LMS, OMIM%130720), also known as Lehman syndrome, is a very rare skeletal disorder with facial anomalies, hypotonia and meningocele-related neurologic dysfunction. The characteristic lateral meningoceles represent the severe end of the dural ectasia spectrum and are typically most severe in the lower spine. Facial features of LMS include hypertelorism and telecanthus, high arched eyebrows, ptosis, midfacial hypoplasia, micrognathia, high and narrow palate, low-set ears and a hypotonic appearance. Hyperextensibility, hernias and scoliosis reflect a connective tissue abnormality, and aortic dilation, a high-pitched nasal voice, wormian bones and osteolysis may be present. Lateral meningocele syndrome has phenotypic overlap with Hajdu–Cheney syndrome. We performed exome resequencing in five unrelated individuals with LMS and identified heterozygous truncating NOTCH3 mutations. In an additional unrelated individual Sanger sequencing revealed a deleterious variant in the same exon 33. In total, five novel de novo NOTCH3 mutations were identified in six unrelated patients. One had a 26 bp deletion (c.6461_6486del, p.G2154fsTer78), two carried the same single base pair insertion (c.6692_93insC, p.P2231fsTer11), and three individuals had a nonsense point mutation at c.6247A > T (pK2083*), c.6663C>G (p.Y2221*) or c.6732C >A, (p. Y2244*). All mutations cluster into the last coding exon, resulting in premature termination of the protein and truncation of the negative regulatory proline-glutamate-serine-threonine rich PEST domain. Our results suggest that mutant mRNA products escape nonsense mediated decay. The truncated NOTCH3 may cause gain-of-function through decreased clearance of the active intracellular product, resembling NOTCH2 mutations in the clinically related Hajdu–Cheney syndrome and contrasting the NOTCH3 missense mutations causing CADASIL.
Keywords: NOTCH3, lateral meningocele syndrome, Lehman syndrome, Hajdu-Cheney syndrome, PEST domain, dural ectasia
Introduction
Lateral meningocele syndrome (LMS; OMIM%130720) is a very rare condition with distinctive facial features, hyperextensibility, hypotonia, developmental delay and characteristic lateral meningoceles, which can result in neurologic complications such as bladder dysfunction and neuropathy. Lateral meningoceles comprise a protrusion of the arachnoid and dura through the spinal foramina and represent the severe end of the dural ectasia spectrum. They are typically most severe in the lower spine, presumably due to hydrostatic pressure. Additional more variable findings of LMS include cryptorchidism, cardiovascular anomalies, coarse hair, vertebral anomalies, wormian bones and connective tissue anomalies such as hernias and keloids. Since its first description by Lehman et al. [1977], fewer than 20 patients with LMS have been reported [Philip et al., 1995; Gripp et al., 1997; Chen et al., 2005; Avela et al., 2011; Alves et al., 2013; Castori et al., 2014]. Autosomal dominant inheritance [Lehman et al., 1977; Chen et al., 2005] and phenotypic overlap with Hajdu–Cheney syndrome (HCS, MIM%102500) [Avela et al., 2011; Gripp, 2011; Avela and Makitie, 2011] have been suggested. Despite apparent clinical overlap with connective tissue disorders and with HCS, candidate gene testing had not revealed an underlying cause for LMS [Alves et al., 2013; Castori et al., 2014].
Materials and Methods
In order to identify the genetic cause for LMS, we enrolled affected individuals and their parents into an IRB approved research study. We performed whole exome sequencing and/or Sanger sequencing on samples from individuals or trios as available. The identification numbers were assigned based on the enrollment into the research study for exome analysis, which contained individuals with phenotypes other than lateral meningocele.
Individuals
Individual 1
This male patient was described in detail as Patient 2 in Gripp et al. [1997] and his findings are summarized in Table I. At age 24 years, his height was 166cm (5–10th centile), weight 51.3 kg (<5th centile, 50th centile for 14 years) and OFC 58.8 cm (+2 SD). His headshape was dolichocephalic and he had a low posterior hairline. Facial findings included ptosis and a long and flat midface, a high and narrow palate and posteriorly angulated low-set ears (Fig. 1A,B). Stature appeared disproportionate with long and slender extremities (at age 17 years, arm span was 183.5 cm; corresponding height 166 cm). Fingers 2 and 3 showed pseudo-clubbing with short and widened tips (Fig. 1C,D). His muscle strength was grossly normal. He had well healed but widened scars on his back. His voice was relatively high-pitched and his interaction reflected his intellectual disability. At age 27 years his general health remained stable, he lived with his parents and worked in a sheltered workshop. He enjoyed music and playing the piano. No additional clinical, imaging or laboratory studies were available.
Table I. Overview of Clinical and Molecular Findings in Individuals With Lateral Meningocele Syndrome.
| Individual ID | 1 [Gripp et al., 1997] | 7 [Avela et al., 2011] | 15 [Gripp et al., 1997] | 20 [Chen et al., 2005] | 26 [Alves et al., 2013] | 28 |
|---|---|---|---|---|---|---|
| Gender | M | M | M | M | M | M |
| Age (years) at last evaluation | 24 | 10 | Died age 8 | 10 | 9 | 5 |
| Craniofacial features | ||||||
| Hypertelorism | − | + | + | + | + telecanthus | —telecanthus |
| Downslanting palpebral fissures | + | − | + | + | + | + |
| Ptosis | + | + | + | + | + | + |
| Low-set ears | + | + | + | + | + | + |
| Malar hypoplasia | + | + | + | + | + | + |
| High, narrow palate | + | + | + | cleft | + | − |
| Cleft palate | − | - | - | + | - | - |
| Micrognathia | + | + | + | + | + | + |
| Coarse hair | + | + | + | + | + | + |
| Cognitive and neurologic function | ||||||
| Developmental delay | + | + | + | + | + | + |
| Intellectual disability | + | − | N/A | - | − | − |
| Hypotonia | + | + | + | + | + | + |
| Decreased muscle bulk | + | + | + | N/A | − | N/A |
| Neurologic findings | Syringomyelia | Syringomyelia, Leg weakness | Chiari 1, Syringomyelia Hydrocephalus, Neurogenic bladder | Tethered cord, incontinence, neuropathy | ||
| Skeletal features | ||||||
| Short stature | − | − | − | + | − | + |
| Hypermobility | + | + | + | + | + | + |
| Scoliosis | + | + | + | + | + | − |
| Pectus | − | − | − | mild | + | + |
| Wormian bones | + | − | + | N/A | − | |
| Thickened calvaria | + | − | − | N/A | + | |
| Additional features | ||||||
| Cryptorchidism | − | - | + | + | + | + |
| Inguinal hernia | + | + | − | + | − | |
| Keloids | + | − | + | − | + | + |
| High nasal voice | + | + | N/A | high | + | + |
| Obstructive sleep apnea or sleep hypoxemia | N/A | + | + | − | + | N/A |
| Other | Spinal fusion, Pseudo-clubbing, Aortic dilation | Osteolysis treated with bisphosphonate | Malformed CI vertebra, Tracheostomy, PDA, VSD, structural vascular anomalies | PDA, VSD, Aortic dilation, Spinal fusion, Mixed hearing loss | Umbilical hernia, Bicuspid aortic valve, mandibular distraction osteogenesis, conductive hearing loss | Omphalocele Macrocytic anemia |
| Molecular studies | ||||||
| Technique | Exome/Sanger | Exome/Sanger | Sanger | Exome/Sanger | Exome/Sanger | Sanger/Exome |
| cDNA | C.G4G1 6486del | c.GG92 93insC | c.GG92 93insC | c.G732C>A | C.GGG30G | c.G247A>T |
| protein | p.G2154fsTer78 | p.P2231fsTerll | p.P2231fsTerll | p.Y2244* | p.Y2221* | p.K2083* |
| Inheritance | de novo | de novo | N/A | de novo | de novo | de novo |
+: present; − : absent ; M: Male; N/A: not available or not applicable; PDA, persistent ductus arteriosus; VSD, ventricular septal defect.
FIG. 1.
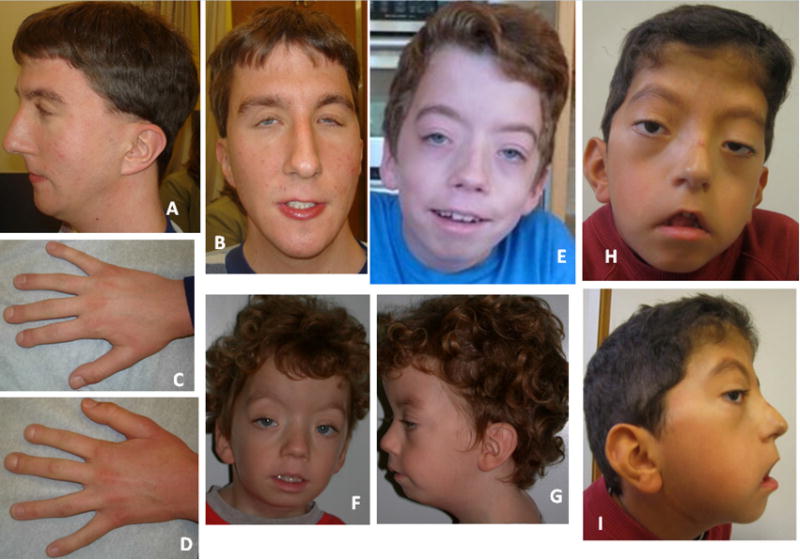
Photographs of Individual 1 at age 24 years showing face and head (A and B) with arched eye brows, ptosis, flat midface, thin upper lip, low-set and posteriorly angulated ears and low posterior hairline, and hands (C and D) with short and wide distal 2nd and 3rd fingers (pseudo-clubbing). Individual 20 ‘s facial features at age 13 years (E) are similar to those at age 4 years (F and G) with coarse and curly hair, tall forehead, high arched brows, ptosis, midfacial hypoplasia, long flat philtrum and thin upper lip, micrognathia and low-set ears. Facial photographs (H and I) of Individual 26 at age 6 years, showing a high forehead, shallow supraorbital ridges with arched eye brows, ptosis, flat midface, thin and tented upper lip, low-set and posteriorly angulated ears, low posterior hairline and a submandibular scar with keloid formation.
Individual 7
This male patient was reported in detail in Avela et al. [2011] and his findings are summarized in Table I. Osteolysis of the distal phalanges of his toes was treated with bisphophonate, resulting in decreased biochemical markers for bone absorption [Avela et al., 2011 for details]. He had a vertical expandable prosthetic titanium rib (VEPTR) implant placed for progressive kyphoscoliosis; this device was replaced at age 9 years and the surgery was complicated by dural spinal fluid leaks requiring three additional surgical procedures. Following these, he had leg pain and diminished muscle strength in his right leg. He received physical therapy. Obstructive sleep apnea was successfully treated with tonsillectomy at age 7 years. The patient continued orthodontic treatment for dental crowding and a very high and narrow palate. He was cognitively intact and his school performance in a typical classroom was excellent. He enjoyed reading books and was physically active.
At age 10 5/12 years his height was 140 cm (25th–50th centile), weight 27 kg (10th centile) and OFC 56.5 cm (+0.6 SD). Long and slender extremities with decreased muscle mass and a disproportionately short torso were notable. The family declined publication of recent photographic images. The kyphosis and the lateral meningoceles are shown in Figure 2A,B.
FIG. 2.
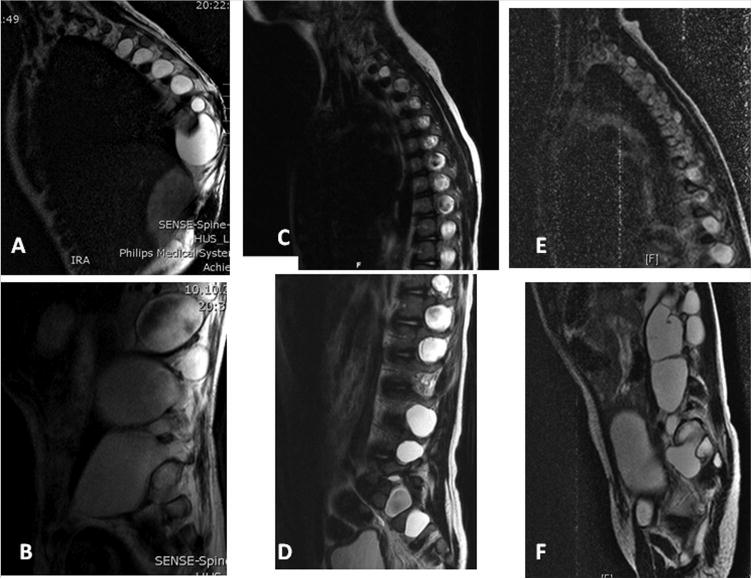
Sagittal T2-weighted MR images of the spine from Individual 7 at age 8 years showing numerous lateral meningoceles increasing in size from the thoracic (A) to the lumbar spine (B); from Individual 26 showing numerous lateral meningoceles increasing in size from the high thoracic (C) to the lumbar region (D); and similar findings seen on MR images obtained at age 5 years from Individual 28 (E, F).
Individual 15
This male patient was described in detail as Patient 1 in Gripp et al. [1997] and his findings are listed in Table I. Limited additional information is available because the patient died at age 8 years. He had a mitrofanoff procedure, presumably in order to allow for regular bladder catheterization due to neurogenic bladder. Subsequently he became febrile, developed a hyponatremic seizure and had circulatory failure despite multiple resuscitation attempts. At autopsy his length was 125cm (25–50th centile for standing height) and OFC 53cm (75th centile). Notable findings on autopsy included evidence of surgical repair of lateral meningoceles and shunting for hydrocephalus, chronic cerebellar herniation with sclerotic cerebellum and leptomeninges, and syrinx of the cervical spinal cord. The undescended testes were located intra-abdominally, in an unusually recessed retroperitoneal pouch. No additional information or images are available and parental samples were not available for molecular studies.
Individual 20
This male individual was described in detail as Patient 3 in Chen et al. [2005] and his findings are summarized in Table I. His facial features are shown in Figure 1E–G. His VSD closed spontaneously. His PDA was obliterated with a coil. By age 2.5 years, his aorta was dilated (maximum Z-score 3.64 at aortic sinuses). He began taking atenolol at age 5.5 years, when his ascending aorta Z-score rose to 5. Since then, his aortic dilation has been stable with Z-scores between 3 and 4.
His early-onset scoliosis was managed initially with a Risser cast, and at age 3.5 years, he had a growing rod construct placed, followed by multiple lengthening procedures, implant revision and removal, and Shilla construct placement, which was removed for deep infection. At his most recent evaluation he was Tanner stage 4 and a definitive spinal fusion procedure was considered. He had an unexplained precipitous drop in his BMI beginning at age 9 years. He had short stature, with normal growth hormone stimulation testing at age 11 years. His lower segment growth rate was appropriate for his Tanner stage, but his trunk growth rate was extremely slow, probably reflecting his severe scoliosis.
At age 13 years his height was 134.6 cm(<3rd centile, 50th centile for 9.5 years) and weight 25.9 kg (<3rd centile, 50th centile for 9.5 years). He attended regular education classes. His vision was 20/70. He had continued bilateral middle ear effusions with intermittent drainage. He used bone-anchored hearing aids for a moderate-tosevere mixed conductive and sensorineural hearing loss.
Individual 26
The male patient was first described clinically by Alves et al. [2013] and subsequently re-reported in Correia-Sa et al. [2013]. His findings are listed in Table I. His facial features and the keloid after mandibular distraction osteogenesis are shown in Figure 1H,I and the lateral meningoceles in Figure 2C,D. Sleep apnea was treated at age 2 years with tonsillectomy and adenoidectomy. At 9 years, his height was 133.5 cm (50th centile), weight 37 kg (90th centile) and OFC 57 cm (>97th centile). He was pre-pubertal. Facial features included hypertelorism, telecanthus, downslanting palpebral fissures and bilateral ptosis, arched eyebrows, posteriorly angulated low-set ears, long philtrum and tented upper lip, a high and narrow palate and dental crowding. He had pectus excavatum, mild scoliosis and increased intermammary distance. He had coarse hair, ligamentous laxity and keloid scaring after surgical interventions. Muscle strength was normal and hands had a normal shape. His voice was high-pitched and nasal. He was a good student and enjoyed good peer relationships.
Individual 28
This male patient was born at term to a 28-year-old mother and a 30-year-old father after an uneventful pregnancy without prenatal exposures. Both parents were of Caucasian ancestry, in good health and not consanguineous. A maternal half-brother and a full brother were in good health. His birth weight was 3.3 kg (25th– 50th centile). At age 3 weeks an “enlarged umbilical stump” was surgically repaired and reportedly considered an omphalocele by the surgeon. The patient had pneumonia at age 2 months and 12 months. He gained weight slowly. His motor development was delayed with sitting independently after age 8 months and walking at age 22 month. His speech and language development was age appropriate.
At age 13 months his length was 76.2 cm (25th–50th centile), weight 8 kg (<3rd centile, 50th centile for age 6 months) and OFC 45 cm (25–50th centile). Findings noted on physical examination included a hypotonic facial appearance (Fig. 3A–D) with epicanthal folds, a thin upper lip and micrognathia, high arched palate, pectus excavatum and “bell-shaped” chest. His anterior fontanel was open and “generous for age”. Generalized hypotonia was present.
FIG. 3.
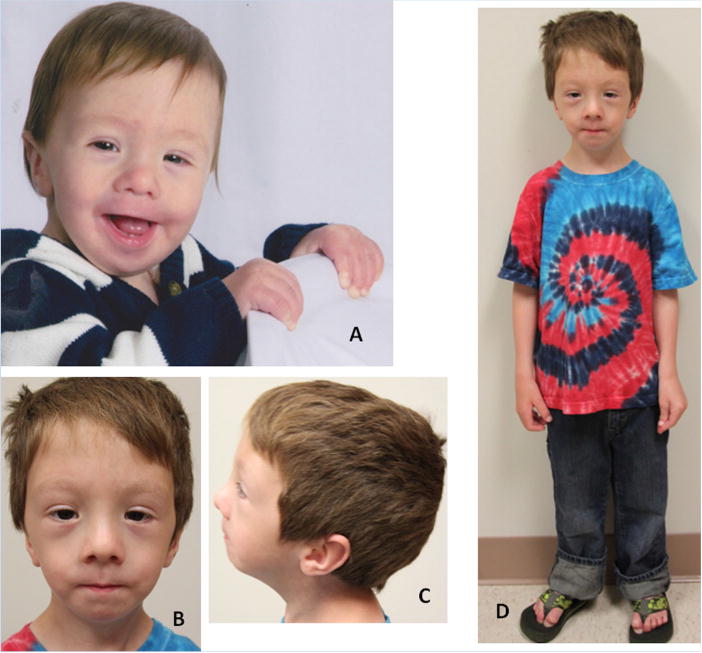
Photographs of Individual 28 at age 12 months (A) and 5 2/12 years (B–D), showing dolichocephaly, high forehead, shallow supraorbital ridges with high arched eyebrows, telecanthus and ptosis, a short nasal tip, thin upper lip, midfacial hypoplasia, low-set and posteriorly angulated ears and coarse hair.
At age 4 4/12 years his length was 95.1 cm (<3rd centile, 50th centile for 3 years), weight 11.6 kg (<3rd centile, 50th centile for 17 months) and OFC 49.6 cm (10th–25th centile). Newly identified physical findings included “wooly” hair, downslanting palpebral fissures, low-set and posteriorly angulated ears, mild ptosis, dental crowding, high-pitched nasal voice, unilateral cryptorchidism and ligamentous laxity. Abone age study performed due to short stature had a normal result (60 months at chronological age 52 months, normal range 46–62 months).
Acute onset low back pain, urinary incontinence and worsening stool incontinence occurred at age 4 9/12 years. His gait became irregular and he dragged one foot. A tethered cord was suspected and MR imaging of the spine revealed multiple lateral meningoceles (Fig. 2E,F). Exploratory surgery confirmed a tethered cord and release resulted in resolution of the gait abnormalities and urinary incontinence, with persistence of stool incontinence. Re-tethering resulted in a repeat surgical release, but urinary incontinence remained. Neuropathic pain developed in his legs and he fatigued easily with walking. At his most recent evaluation at age 5 5/12 years his height was 103.1 cm (<3rd centile, 50th centile for 4 3/12 years), weight 12.7 kg (<3rd centile, 50th centile for 2 years) and OFC 50.7 cm (25th–50th centile).
Molecular studies performed on a clinical basis with normal results included FISH for del22q11.2; chromosome microarray and panel testing for Noonan syndrome and other rasopathies.
Mutation Analysis
Genomic DNA was extracted from formalin fixed paraffin embedded tissue, buccal and/or peripheral white blood cells from the patients, parents and unaffected family members, using Puregene DNA Isolation Kit (Qiagen Inc., Valencia, CA; www.qiagen.com). RNA was isolated from peripheral white blood cells from Individual 28 and his parents. Biological relationships were confirmed as previously reported [Sol-Church et al., 2006] using microsatellite markers.
Whole Exome Sequencing
DNAs from the first four trios (1, 7, 20, and 26) were submitted to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG) through the online submission portal PhenoDB [Hamosh et al., 2013]. The Agilent SureSelect HumanAllExonV4_51Mb-Kit_S03723314 was used for exome capture. Libraries were sequenced on the HiSeq2500 platform with onboard clustering using 100 bp paired end runs and sequencing chemistry kits TruSeq Rapid PE Cluster Kit-HS and TruSeq Rapid SBS-HS. Data analysis was essentially as described in Hoover-Fong et al. [2014], with some notable changes. For patients 1 and 7, fastq files were aligned with BWA [Li and Durbin, 2009] version 0.5.10-tpx to the 1000 genomes phase 2 (GRCh37) human genome reference. Duplicate molecules were flagged with Picard version 1.74. Local realignment around indels and base call quality score recalibration were performed using the Genome Analysis Toolkit (GATK) version 2.3–9-ge5ebf34 [McKenna et al., 2010]. Unified Genotyper (GATK) was used for multi-sample calling of SNVs and indels using Reduced BAMs (GATK). Variant filtering was done using the Variant Quality Score Recalibration (VQSR) method [DePristo et al., 2011]. For patients 20 and 26, fastq files were aligned with BWA mem [Li, 2013] version 0.7.8 to the 1000 genomes phase 2 (GRCh37) human genome reference. Duplicate molecules were flagged with Picard version 1.109(1716). Local realignment around indels and base call quality score recalibration were performed using the Genome Analysis Toolkit (GATK) version v3.1–1-g07a4bf8 [McKenna et al., 2010]. GATK's reference confidence model workflow was used to perform joint sample genotyping. Briefly, this workflow entails: (i) Producing a gVCF (genomic VCF) for each sample individually using HaplotypeCaller (–emitRefConfidence GVCF) for all bait intervals to generate likelihoods that the sites are homozygote reference; and (ii) Joint genotyping the single sample gVCFs together with GenotypeGVCFs to produce a multi-sample VCF file. Variant filtering was done using the Variant Quality Score Recalibration (VQSR) method [DePristo et al., 2011]. A variant prioritization strategy was designed using the Analysis Tool of PhenoDB [Hamosh et al., 2013].
Sanger Sequencing
PCR primers were designed to amplify exon 33 of NOTCH3. Three of the DNA variants clustered within a 599 bp DNA fragment amplified using primer pair 5′-TCGCCCGTGGACTCGCTGGAC-3′ and 5′-GAGCTGGGAACAGACAAGGGAAGT-3′. The mutation in Individual 15 was sequenced from a 446 bp DNA fragment amplified using primer pair 5′-CAGCCCCCTGGAGGATGTGTA-3′ and 5′-GAGCTGGGAACAGACAAGGGAAGT-3′. For individual 28, a 685 bp product was amplified using primer pair 5′-CTCTGGCACT-TAGTAGGTGATGG-3′ and 5′-TGGCAGTGGCAGCTGCATAGG-3′. Sanger sequencing was performed using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Life Technologies).
Results
After resequencing the exomes of individuals 1, 7, 20, and 26 and their parents, we prioritized rare functional variants (missense, nonsense, splice site variants and indels) that were heterozygous, homozygous or compound heterozygous in each proband and excluded variants with a MAF > 0.01 in dbSNP 126, 129, and 131 or in the Exome Variant Server (release ESP6500SI-V2) or 1000 Genomes Project [1000 Genomes Project Consortium, 2012]. We generated a heterozygous, homozygous and a compound heterozygous variant list for each proband and merged them to identify genes mutated in three or four probands. All four individuals (1, 7, 20, and 26) had a novel mutation in exon 33 of NOTCH3 (Fig. 4). The variants described here were based on RefGene transcript NM_000435 and NCBI human genome assembly build 37. Individual 1 carried a 26 bp deletion c.6461_6486del predicted to result in a premature stop codon at p.G2154fsTer78. This variant was not present in the Unified Genotyper call set, but was detected following reanalysis with HaplotypeCaller. Individual 7 had a single base pair insertion at c.6692_93insC predicting a premature termination (p.P2231fsTer11). Patients 20 and 26 had a nonsense point mutation c.6732C > A (p.Y2244*), c.6663C > G p.Y2221*, respectively. PCR primers were designed to amplify this exon and Sanger sequencing was used to validate the candidate causative variants. Targeted Sanger sequencing was performed on two additional probands: Patient 15, who was deceased and DNA extracted from a formalin fixed paraffin embedded tissue sample, carried a single base pair insertion at c.6692_93insC resulting in premature termination (p.P2231fsTer11) and patient 28 had a nonsense mutation c.6247A >T; pK2083* (Fig. 4). These variants were not present in the available unaffected family members (Table I), the >6,000 individuals in the Exome Variant Server, nor in the 1092 individuals whose sequence is available from the 1000 Genomes Project [1000 Genomes Project Consortium, 2012]. The mutation identified by Sanger sequencing in Individual 28 was present in the subsequently completed exome analysis. Using RNA extracted from peripheral white blood cells of Individual 28, we found that NOTCH3 expression was low compared to expression in unrelated control fibroblasts. However, the mutant allele was expressed (data not shown).
FIG. 4.
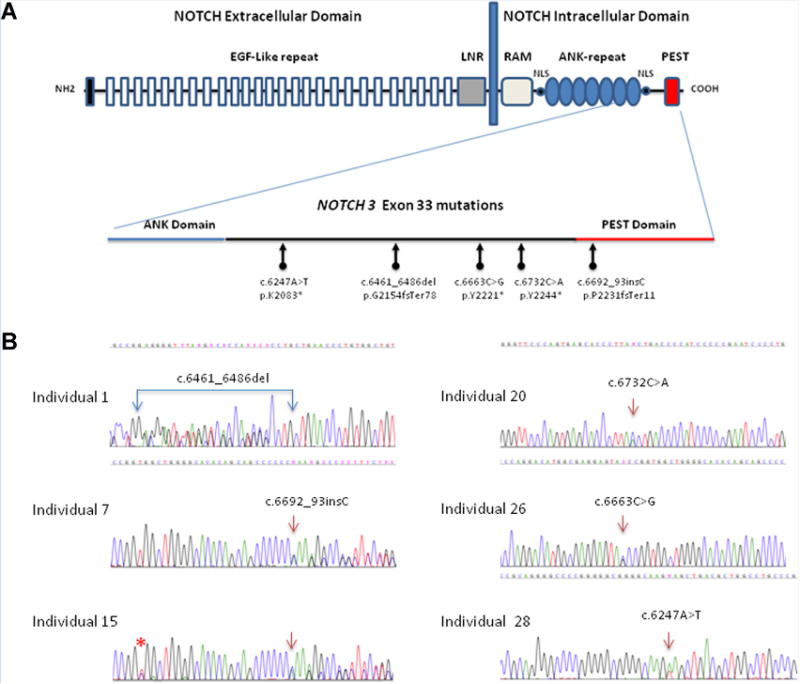
Diagram of NOTCH3 mutations in lateral meningocele syndrome, not to scale. (A) Structural organization of the NOTCH3 receptor: NOTCH3 is expressed on the cell surface as a heterodimer composed of a large extracellular domain non-covalently linked to the intracellular domain. The extracellular domain contains 34 epidermal growth factor (EGF-like) repeats and 3 cysteine-rich repeats (LNR). The intracellular domain includes a RAM23 domain; nuclear localizing signals (NLS); and ankyrin repeats (ANK) for protein protein interaction. At the carboxyl terminus, the PEST domain is a region rich in proline (P), glutamine (E), serine (S) and threonine (T) residues used for protein degradation. A close up of exon 33's structure indicates the location of the 5 different de novo DNA variants identified in this study. Panel (B) shows the sequencing chromatogram for each of the six individuals. All sequences are presented 5′ -> 3′. Double sequences indicate the frameshift mutations in patient 1, 7, and 15. Asterisk (*) indicates a common SNP (rs 1044009) identified in Individual 15.
While de novo mutations in other genes were suspected based on the exome analysis results, no mutation was found consistently in most or all patients' samples (see supplemental Table I for detail in supporting information online).
Discussion
We identified a heterozygous truncating mutation in exon 33 of NOTCH3 in every LMS individual available for testing, suggesting that such mutations are the cardinal cause of LMS. The mutations occurred de novo in each proband with parental samples available for testing. All mutations are predicted to result in premature termination of the protein product (Fig. 4) and while they may be subject to nonsense mediated decay, nonsense mutations in the last exon typically escape this effect[Nagy and Maquat, 1998; Khajavi et al., 2006]. The uniform location of the mutations identified here in the last coding exon suggests a dominant gain-of-function effect of the abnormal protein product, rather than haploinsufficiency resulting from nonsense mediated decay. We saw expression of the mutant NOTCH3 in RNA derived from peripheral white blood cells of one patient, additional studies were not feasible due lack of samples.
NOTCH3 Structure and Function
NOTCH3 encodes a transmembrane receptor of the highly conserved notch signaling pathway [for review see Bellavia et al., 2008; Penton et al., 2013]. NOTCH3 consists of an extracellular domain with 34 EGF-like repeats and 3 Lin-Notch repeats, a single transmembrane region and 4 conserved intracellular domains (Fig. 4). Upon ligand binding a series of cleavage events results in the intracellular release of the active soluble cytoplasmic domain (NICD, Fig. 5), which enters the nucleus and interacts with the CSL transcription factor and a transcriptional coactivator of the Mastermind-like family [Bellavia et al., 2008]. While the RAM and ANK domain serve this protein-protein interaction, the C-terminal PEST domain is involved in negative regulation of protein stability and promotes degradation [Bellavia et al., 2008]. The truncated NOTCH3 protein predicted to result from the mutations in LMS patients lacks a functional PEST domain, which may prolong the intracellular half-life of NICD and thus increase its signaling effects (Fig. 5). Overexpression of NOTCH1-derived NICD resulted in increased canonical signaling in a mouse model for osteosclerosis [Tao et al., 2010].
FIG. 5.
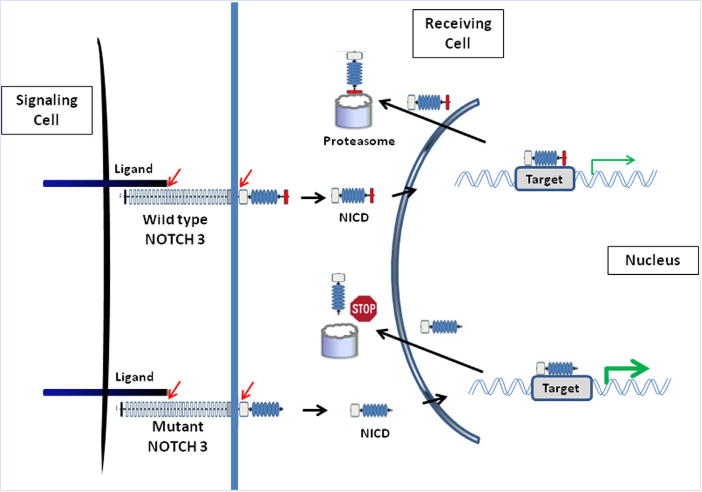
Diagram of normal and abnormal NOTCH signaling. Binding of a Notch ligand to the membrane bound NOTCH receptor triggers proteolytic cleavage (indicated by red arrows) and release of the Notch intracellular domain (NICD). NICD translocates to the nucleus, where it binds to specific transcription factors (not shown). This activated complex directs the transcription of target genes. In wildtype NOTCH3, the PEST sequence (red box) serves as a signal for protein degradation by the proteasome machinery and controls the intracellular half-life of NICD. In mutant NOTCH3, truncation of the C-terminal PEST domain presumably impairs proteasome degradation of NICD (Stop sign), resulting in an extended half-life of NICD and prolonged signaling (gain-of-function effect).
NOTCH Signaling and Human Disease
The NOTCH signaling pathway governs cell fate determination and thus allows for a secondary role in carcinogenesis and maintenance of cancers [Bellavia et al., 2008; Penton et al., 2013]. The four NOTCH genes (NOTCH 1–4) encode single–pass type 1 trans-membrane proteins binding at least 5 ligands, and germline mutations affecting this signaling pathway cause congenital anomaly syndromes and a progressive neurovascular disorder [Penton et al., 2013], in addition to somatic mutations found in T-cell leukemias and other malignancies. Missense mutations and deletions predicted to result in haploinsufficiency for NOTCH1 were found in individuals with congenital heart defects such as coarctation and bicuspid aortic valve, and similar NOTCH1 changes were found in individuals with Adams–Oliver syndrome (AOS), a developmental disorder characterized by aplasia cutis congenita and terminal transverse limb defects in combination with vascular anomalies and congenital heart defects [Stittrich et al., 2014]. Point mutations or whole gene deletions of the NOTCH ligand JAG1 or point mutations in NOTCH2 cause Alagille syndrome, an autosomal dominant multiple congenital anomaly syndrome with bile duct paucity, cardiac, eye and vertebral anomalies [Penton et al., 2013].
NOTCH2 and Hajdu-Cheney Syndrome
NOTCH2 mutations are associated with Hajdu-Cheney syndrome (HCS) [Isidor et al., 2011; Simpson et al., 2012], a skeletal disorder closely related to LMS [Gripp, 2011]. Similar to the NOTCH3 mutations in LMS reported here, all NOTCH2 mutations in HCS cluster to the last exon and are predicted to result in premature termination of the abnormal protein product. The effects of the truncated NOTCH2 are responsible for thickened occipital bone, wormian bones, hypoplastic midface, micrognathia, joint hyperlaxity and osteoporosis with acro-osteolysis in HCS. Similar findings occur in LMS, with acro-osteolysis resulting in a previous misdiagnosis of HCS in Patient 7 [Avela et al., 2011], and may be attributed to a similar effect of the abnormal NOTCH3 product on skeletal and connective tissue. These findings further support the importance of the NOTCH signaling pathway in bone maintenance.
NOTCH3 and CADASIL
Heterozygous NOTCH3 mutations cause the progressive neurodegenerative cerebral arteriopathy, autosomal dominant, with subcortical infarcts and leukencephalopathy (CADASIL) [Joutel et al., 1996]. CADASIL causing mutations located in exons 3–11 are typically missense changes and invariably affect the number of cysteine residues in the extracellular domain, presumably resulting in a neomorphic effect because neither a nonsense mutation nor an intragenic multi-exon deletion within this region resulted in the phenotype [Rutton et al., 2013]. In contrast, the pathogenicity of a missense mutation in exon 25 resulting in a predicted p.Leu1515Pro substitution, suspected to render the protein product hyperactive through destabilization of the heterodimer, remains questionable as the affected individual lacked the NOTCH3 deposits pathognomonic of CADASIL. Identification of another missense mutation (p.Leu1519Pro) in the NOTCH3 heterodimerization domain in a family segregating infantile myofibromatosis (IMF) [Martignetti et al., 2013] may support pathogenicity of missense changes in this domain, however, there is no phenotypic overlap between CADASIL and IMF, no additional patients with mutations in this region were reported, and IMF is caused by PDGFRB mutations in most cases. Haploinsufficiency for NOTCH3 does not result in an identifiable phenotype as evidenced by the variable and unspecific phenotype associated with whole gene deletions in the decipher database (http://decipher.sanger.ac.uk).
Phenotype of Individuals Reported Here
While the patients reported here were originally identified based on their lateral meningoceles, they share a recognizable phenotype of facial, developmental and physical findings. All patients in this series are male, however, it is unlikely that the phenotype is sex specific beyond the cryptorchidism seen in 4/6 individuals. Females with lateral meningocele have been reported [Lehman et al., 1977; Gripp et al., 1997; Chen et al., 2005; Castori et al., 2013], but DNA samples were unavailable for testing.
Characteristic facial findings include dolichocephaly with a high forehead, shallow supraorbital ridges, high arched eyebrows, ptosis, hyperteloric appearance with telecanthus, flat midface, short and upturned nasal tip in younger individuals, long and slightly flat philtrum, tented and thin upper lip, high and narrow palate or cleft palate, micro- or retrognathia, and low-set posteriorly angulated ears. Facial expression tends to by hypotonic. The posterior hairline is low and the hair has a coarse texture. Facial features may resemble those seen in Noonan syndrome, as evidenced by the rasopathies panel testing performed in Individual 28.
Hypotonia and hyperextensibility are likely causes for the developmental delay seen in all patients reported here. Intellectual disability was noted only in Individual 1.Neurologic complications arising from the meningoceles and secondary to surgical procedures include incontinence, neuropathic pain and muscle weakness. Syringomyelia, noted in 3/6 patients, may contribute to neuropathic problems. Chiari 1 malformation and tethered cord occurred in one patient each.
Structural bone abnormalities including wormian bones, thickened skull, micrognathia with a diminished mandibular angle and vertebral anomalies were seen and overlap with findings in HCS. Osteolysis, which is characteristic for HCS, was confirmed by increased biochemical markers for bone turnover and treated with bisphosphonates in Individual 7, and may be suspected based on the pseudo-clubbing of fingers 2 and 3 in Individual 1. Decreased muscle mass and scoliosis give rise to a dolichostenomelic appearance [see Fig. 14 in Gripp et al., 1997; and Fig. 2a in Avela et al., 2011]. In combination with midfacial hypoplasia, micrognathia and ligamentous laxity, the features may be considered “Marfan-like”, and thus patients with a milder phenotype may not be recognized as having LMS, but could be included in cohorts of “individuals with Marfan-like features but exclusion of mutations in the genes FBN1, TGFBR1 and TGFBR2” [Sheikhzadeh et al., 2011].
Conclusions
We identified heterozygous truncating mutations in the last coding exon of NOTCH3 in all tested LMS patients. As suggested by the phenotypic overlap of LMS and HCS, these data support a close biological relationship between truncating mutations in closely related genes. The effect of abnormal NOTCH signaling on bone metabolism and maintenance requires further study. The disease causing mechanism of the truncating NOTCH3 mutations reported here contrasts with the missense mutations affecting the cysteine residues in the extracellular domain causing CADASIL and highlights the phenotypic heterogeneity of NOTCH3 mutations.
Supplementary Material
Acknowledgments
Grant sponsor: Nemours Foundation Cluster; Grant numbers: P20GM103464, P20GM103446, 1U54HG006542.
We thank the families and patients for their participation. We thank Stacey Koletty and Corinne Boehm for administrative help with IRB approval and clinical coordination and Carolyn Schanen for discussion of the data. The views expressed are those of the author and do not reflect the official policy of the Department of the Army, the Department of Defense or the U.S. Government. This work was supported in part by the Nemours Foundation Cluster grant program; NIH-NIGMS grants P20GM103464 and grant P20GM103446 to KSC;and 1U54HG006542 to the Baylor Hopkins Center for Mendelian Genomics.
Footnotes
Supporting Information: Additional supporting information may be found in the online version of this article at the publisher's web-site.
Conflict of interest: none
References
- Abecasis GR. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avela K, Makitie O. Response to “Lateral Meningocele Syndrome and Hajdu–Cheney syndrome: Different disorders with overlapping phenotypes” by Gripp. Am J Med Genet Part A. 2011;155A:1775. doi: 10.1002/ajmg.a.34075. [DOI] [PubMed] [Google Scholar]
- Avela K, Valanne L, Helenius I, Makitie O. Hajdu–Cheney syndrome with severe dural ectasia. Am J Med Genet Part A. 2011;155A:595–598. doi: 10.1002/ajmg.a.33510. [DOI] [PubMed] [Google Scholar]
- Bellavia D, Checquolo S, Campese AF, Felli MP, Gulino A, Screpanti I. Notch3: From subtle structural differences to functional diversity. Oncogene. 2008;27:5092–5098. doi: 10.1038/onc.2008.230. [DOI] [PubMed] [Google Scholar]
- Castori M, Morlino S, Ritelli M, Brancati F, de Bernardo C, Colombi M, Grammatico P. Late diagnosis of lateral meningocele syndrome in 55-year-old woman with symptoms of joint instability and chronic musculoskeletal pain. Am J Med Genet Part A. 2014;164A:528–534. doi: 10.1002/ajmg.a.36301. [DOI] [PubMed] [Google Scholar]
- Chen KM, Bird L, Barnes P, Barth R, Hudgins L. Lateral meningocele syndrome: Vertical transmission and expansion of the phenotype. Am J Med Genet Part A. 2005;133A:115–121. doi: 10.1002/ajmg.a.30526. [DOI] [PubMed] [Google Scholar]
- Correia-Sa I, Horta R, Neto T, Amarante J, Marques M. Lehman syndrome: A new syndrome for Pierre Robin Sequence. Cleft palate-Craniofacial Journal. 2013:51. doi: 10.1597/13-063. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp KW. Lateral meningocele syndrome and Hajdu–Cheney syndrome: Different disorders with overlapping phenotypes. Am J Med Genet Part A. 2011;155:1773–1774. doi: 10.1002/ajmg.a.34075. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Scott CI, Jr, Hughes HE, Wallerstein R, Nicholson L, States L, Bason LD, Kaplan P, Zderic SA, Duhaime AC, Miller F, Magnusson MR, Zackai EH. The lateral meningocele syndrome: Three new patients and review of the literature. Am J Med Genet. 1997;70:229–239. [PubMed] [Google Scholar]
- Hamosh A, Sobreira N, Hoover-Fong J, Sutton VR, Boehm C, Schiettecatte F, Valle D. PhenoDB: a new web-based tool for the collection, storage, and analysis of phenotypic features. Hum Mutat. 2013;34:566–571. doi: 10.1002/humu.22283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover-Fong J, Sobreira N, Jurgens J, Modaff P, Blout C, Moser A, Kim OH, Cho TJ, Cho SY, Kim SJ, Jin DK, Kitoh H, Park WY, Ling H, Hetrick KN, Doheny KF, Valle D, Pauli RM. Mutations in PCYT1A, encoding a key regulator of phosphatidylcholine metabolism, cause spondylometaphyseal dysplasia with cone-rod dystrophy. Am J Hum Genet. 2014;94:105–112. doi: 10.1016/j.ajhg.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cecillion M, Marechal E, Maclazek J, Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier-Lasserve E. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- Isidor B, Lindenbaum P, Pichon O, Bézieau S, Dina C, Jacquemont S, Martin-Coignard D, Thauvin-Robinet C, LeMerrer M, Mandel JL, David A, Faivre L, Cormier-Daire V, Redon R, Le Caignec C. Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis. Nat Genet. 2011;43:306–308. doi: 10.1038/ng.778. [DOI] [PubMed] [Google Scholar]
- Khajavi M, Inoue K, Lupski JR. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Europ J Hum Genet. 2006;14:1074–1081. doi: 10.1038/sj.ejhg.5201649. [DOI] [PubMed] [Google Scholar]
- Lehman RA, Stears JC, Wesenberg RL, Nusbaum ED. Familial osteosclerosis with abnormalities of the nervous system and meninges. J Pediatr. 1977;90:49–54. doi: 10.1016/s0022-3476(77)80763-4. [DOI] [PubMed] [Google Scholar]
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013 arXiv:1303.3997v1 [q-bio.GN] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martignetti JA, Tian L, Li D, Ramirez MC, Camacho-Vanegas O, Camacho SC, Guo Y, Zand DJ, Bernstein AM, Masur SK, Kim CE, Otieno FG, Hou C, Abdel-Magid N, Tweddale B, Metry D, Fournet JC, Papp E, McPherson EW, Zabel C, Vaksmann G, Morisot C, Keating B, Sleiman PM, Cleveland JA, Everman DB, Zackai E, Hakonarson H. Mutations in PDGFRB cause autosomal-dominant infantile myofibromatosis. Am J Hum Genet. 2013;92:1001–1007. doi: 10.1016/j.ajhg.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem Sci. 1998;23:198–199. doi: 10.1016/s0968-0004(98)01208-0. [DOI] [PubMed] [Google Scholar]
- Penton AL, Leonard LD, Spinner NB. Notch signaling in human development and disease. J Med Genet. 2013;49:138–144. doi: 10.1016/j.semcdb.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip N, Andrac L, Moncla A, Sigaudy S, Zanon N, Lena G, Choux M. Multiple lateral meningoceles, distinctive facies and skeletal anomalies: A new case of Lehman syndrome. Clin Dysmorphol. 1995;4:347–351. doi: 10.1097/00019605-199510000-00011. [DOI] [PubMed] [Google Scholar]
- Rutten JW, Boon EM, Liem MK, Dauwerse JG, Pont MJ, Vollebregt E, Maat-Kievit AJ, Ginjaar HB, Lakeman P, van Duinen SG, Terwindt GM, Lesnik Oberstein SA. Hypomorphic NOTCH3 alleles do not cause CADASIL in humans. Hum Mutat. 2013;34:1486–1489. doi: 10.1002/humu.22432. [DOI] [PubMed] [Google Scholar]
- Sheikhzadeh S, Rybczynski M, Habermann CR, Bernhardt AMJ, Arslan-Kirchner M, Keyser B, Kaemmerer H, Mir TS, Staebler A, Oezdal N, Robinson PN, Berger J, Meinertz T, von Kodolitsch Y. Dural ectasia in individuals with Marfan-like features but exclusion of mutations in the genes FBN1. TGFBR1 and TGFBR2. Clin Genet. 2011;79:568–574. doi: 10.1111/j.1399-0004.2010.01494.x. [DOI] [PubMed] [Google Scholar]
- Simpson MA, Irving MD, Asilmaz E, Gray MJ, Dafou D, Elmslie FV, Mansour S, Holder SE, Brain CE, Burton BK, Kim KH, Pauli RM, Aftimos S, Stewart H, Kim CA, Holder-Espinasse M, Robertson SP, Drake WM, Trembath RC. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat Genet. 2011;43:303–305. doi: 10.1038/ng.779. [DOI] [PubMed] [Google Scholar]
- Sol-Church K, Stabley DL, Nicholson L, Gonzalez IL, Gripp KW. Paternal bias in parental origin of HRAS mutations in Costello syndrome. Hum Mutat. 2006;27:736–741. doi: 10.1002/humu.20381. [DOI] [PubMed] [Google Scholar]
- Stittrich AB, Lehman A, Bodian DL, Ashworth J, Zong Z, Li H, Lam P, Khromykh A, Iyer RK, Vockley JG, Baveja R, Silva ES, Dixon J, Leon EL, Solomon BD, Glusman G, Niederhuber JE, Roach JC, Patel MS. Mutations in NOTCH1 cause Adams-Oliver syndrome. Am J Hum Genet. 2014;95:275–284. doi: 10.1016/j.ajhg.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Chen S, Yang T, Dawson B, Munivez E, Bertin T, Lee B. Osteosclerosis owing to Notch gain of function is solely Rbpj-dependent. J Bone Miner Res. 2010;25:2175–2183. doi: 10.1002/jbmr.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.