

Abstract
Anti-tumor efficacy of T cells engineered to express chimeric antigen receptors (CARs) is dependent on their specificity, survival, and in vivo expansion following adoptive transfer. Toll-like receptor (TLR) and CD40 signaling in T cells can improve persistence and drive proliferation of antigen-specific CD4+ and CD8+ T cells following pathogen challenge or in graft-versus-host disease (GvHD) settings, suggesting that these costimulatory pathways may be co-opted to improve CAR-T cell persistence and function. Here, we present a novel strategy to activate TLR and CD40 signaling in human T cells using inducible MyD88/CD40 (iMC), which can be triggered in vivo via the synthetic dimerizing ligand, rimiducid, to provide potent costimulation to CAR-modified T cells. Importantly, the concurrent activation of iMC (with rimiducid) and CAR (by antigen recognition) is required for interleukin (IL)-2 production and robust CAR-T cell expansion and may provide a user-controlled mechanism to amplify CAR-T cell levels in vivo and augment anti-tumor efficacy.
Keywords: T cell, chimeric antigen receptor, costimulation, dimerizer, MyD88, CD40, inducible
Graphical Abstract
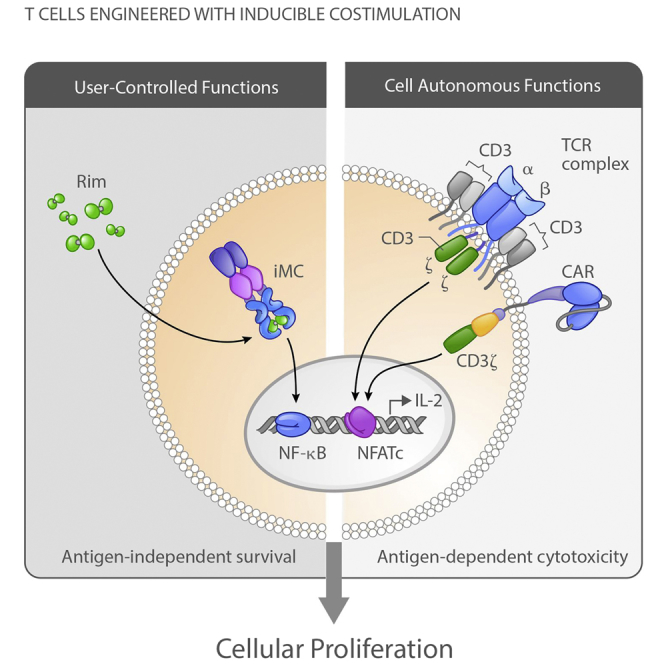
Foster et al. describe the use of inducible MyD88/CD40 (iMC) to provide costimulation for CAR-T cells to drive their proliferation, survival, and anti-tumor efficacy. iMC can be activated in vivo using systemic administration of a small molecule ligand, rimiducid, to provide physician-controlled amplification of CAR-T cell therapies.
Introduction
Immunotherapy with T cells genetically engineered with chimeric antigen receptors (CARs) has shown remarkable efficacy for the treatment of a number of distinct CD19+ lymphoid malignancies.1 In these patients, objective response rates are closely correlated with in vivo expansion of CAR-T cells following adoptive transfer. However, in solid tumors, efficacy has been more muted,2, 3 possibly due to reduced costimulation along with a more profoundly inhibitory tumor microenvironment, which suppresses T cell activation. To increase T cell expansion, preparative “conditioning” regimens and systemic administration of the T cell growth factor, interleukin (IL)-2, have been used to deplete host regulatory immune cells and promote T cell proliferation, respectively.4 However, these approaches are not devoid of side effects and may not be applicable in all patients with cancer, including patients with higher tumor burdens or other comorbidities that limit their use.5 Therefore, novel approaches to predictably amplify CAR-T cells in vivo to increase efficacy are needed.
Mechanistic studies aimed at understanding the effect of lymphodepleting regimens (chemotherapy and total body irradiation [TBI]) on T cell proliferation have shown that Toll-like receptor (TLR) activation via lipopolysaccharide (LPS) liberated from gut flora contributes to the expansion of T cells following adoptive transfer.6, 7 TLRs are widely expressed on most, if not all, nucleated cells, including T cells,8, 9 and engagement of TLRs by agonists induces costimulatory pathways in T cells that decrease the T cell receptor (TCR) activation threshold, increases cytokine production and cytotoxicity, promotes T cell memory formation, and increases expression of pro-survival proteins (e.g., Bcl-2 and Bcl-xL).10, 11, 12 In addition, using tumor-specific T cells derived from MyD88-deficient mice, Geng et al.12 demonstrated that signaling through the TLR adaptor protein, MyD88, was essential for tumor control by tumor-specific T cells following TLR2 agonist treatment, indicating that T cell-intrinsic TLR activation can provide direct costimulation. These studies further support that TLR signaling pathways might be useful to improve persistence of gene-modified T cells.
We previously developed a novel molecular switch, inducible MyD88/CD40 (iMC), to activate downstream TLR and CD40 signaling pathways following dimerization using a small molecule ligand, rimiducid (Rim; formerly AP1903).13 iMC was originally developed as an inducible adjuvant for a gene-modified dendritic cell (DC) vaccine, where exposure to Rim results in DC maturation and robust IL-12 production. Because TLR signaling also provides costimulation to T cells, and due to the highly conserved nature of the signaling pathways triggered by iMC in DCs (e.g., nuclear factor κB [NF-κB], Akt, mitogen-activated protein kinase [MAPK], and c-Jun N-terminal kinase [JNK]),13 we hypothesized that iMC might be useful as a drug-dependent, inducible costimulatory module to enhance survival and proliferation of CAR-T cells. While iMC activation alone is not sufficient to drive expansion of transduced primary human T cells, iMC signaling induces a network of pro-survival cellular pathways. Further, when T cells are coactivated through their endogenous TCR, or by antigen recognition with a “first-generation” CAR that engages CD3ζ signaling, iMC provides the necessary costimulatory signals to drive T cell proliferation and enhance anti-tumor activity. Thus, iMC represents a potent genetic tool to control CAR-T cells in vivo using a highly specific, clinically validated,14, 15 small molecule dimerizing ligand.
Results
Rim-Dependent iMC Costimulation of T Cells
The iMC molecule is a cell membrane-targeted signaling molecule composed of truncated MyD88 (lacking the C-terminal Toll/IL-1 homology domain [TIR] domain) and truncated CD40 (lacking the transmembrane and extracellular domains) fused in frame to tandem FKBP12v36 (FKBP) domains, which bind to the lipid-permeable dimerizing ligand, Rim, at high affinity (Kd ∼0.1 nM) (Figure 1A).13 To test iMC function in T cells, we generated retroviral vectors encoding FKBP, iMyD88, iCD40, or iMC (Figure 1B). After transduction was confirmed (Figure 1C), T cells were incubated with 10 nM Rim, which induced significant IL-6 production by iMC-modified T cells (Rim = 498 ± 3.9 pg/mL; media = 94.5 ± 3.5 pg/mL; p < 0.001), without affecting control construct-modified cells (Figure 1D). A multiplex cytokine array subsequently showed that iMC activation induced a broad spectrum of cytokines and chemokines, including interferon (IFN)-γ, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-5, IL-8, IL-13, tumor necrosis factor (TNF)-α, and IP-10 (Table S1). Interestingly, iMC activation was not sufficient for secretion of IL-2, a key T cell growth and survival factor, which typically requires TCR-mediated induction of Ca2+-dependent transcription factor, NFATc, in concert with NF-κB activation, primarily through costimulatory pathways. Since iMC induces NF-κB in DCs, we hypothesized that this molecule might also provide costimulation to T cells and facilitate IL-2 production. Indeed, iMC-modified T cells stimulated with both a TCR cross-linking antibody (plate-bound OKT3) and Rim secreted significantly more IL-2 (683 ± 63 pg/mL) than iMC-modified T cells receiving only Rim (26 ± 8 pg/mL) or OKT3 activation (120 ± 22 pg/mL) (p = 0.0001) (Figures 1E and 1F). Like IL-6 secretion, FKBP-, iCD40- and iMyD88-modified T cells produced significantly less IL-2 than iMC-expressing T cells, suggesting that MyD88 and CD40 provide synergistic costimulation. Importantly, even sub-nanomolar Rim levels (half-maximum activation concentration [EC50] = ∼0.12 nM) were sufficient for IFN-γ induction (Figure 1G).
Figure 1.

Transduction of Human T Cells with iMC Allows Rimiducid-Dependent Activation
(A) Schematic representation of iMC dimerization following exposure to the lipid-permeable ligand, rimiducid (Rim). (B) Retroviral constructs encoding the control (FKBP, iMyD88, and iCD40) and the iMC vectors. The constructs consist of the following: 5′ and 3′ long terminal repeats (LTRs) (white), ψ packaging signal, membrane-targeting myristoylation peptide (purple), MyD88 (blue), CD40 (green), tandem FKBP12v36 (red), Thoseaasigna virus 2A skipping polypeptide (T2A; gray), and truncated CD19 (ΔCD19; yellow). (C) Transduction levels were determined by flow cytometry staining for CD3 and CD19. (D) Non-transduced (NT) or human T cells modified with either FKBP only (n = 3), iMyD88, iCD40, or iMC were stimulated with 10 nM Rim, and IL-6 production was measured after 48 hr by ELISA (see also Tables S1). (E) Schematic of integrated signaling in T cells, where activation of the endogenous TCR via MHC/peptide recognition or by antibody cross-linking provides TCR signal one and Rim-dependent dimerization provides costimulatory signal two. (F) NT or gene-modified T cells (n = 3) were cultured in media alone or stimulated with either 10 nM Rim, 50 ng/mL anti-CD3 antibody (OKT3), or both Rim and anti-CD3 antibody. IL-2 secretion was measured 48 hr after stimulation by ELISA. (G) iMC-modified T cells (n = 3) were stimulated with log titrations of Rim (0.001–100 nM) and IFN-γ production was measured after 48 hr. The half-maximum activation concentration (EC50) was calculated using GraphPad Prism curve fitting. *p < 0.05, **p < 0.01.
Further analysis examining the downstream signaling effects of iMC activation showed that iMC rapidly induced (within 15 min of Rim exposure) the phosphorylation of a broad network of signaling pathways, including JNK, Akt, extracellular signal-regulated kinase (ERK), RelA, and MAPK (Figures 2A and 2B). In addition, Rim-dependent iMC activation upregulated cytokine-related genes (e.g., IFIT1, IFIT3, CXCL10, and CXCL11), genes downstream of MyD88-dependent TLR signaling (e.g., IL-5, IL-6, and IL-13), and CD40-associated genes that regulate apoptosis (e.g., BCL2L1) (Figure 2C). Pathway analysis showed significant upregulation of genes associated with NF-κB activation (p = 4.4 × 10−15), as well as a significant overlap with gene expression profiling in DCs activated by agonists of TLRs 4, 7, and 8 (p = 6.1 × 10−40) (Tables S2–S5). Together, this analysis indicates that iMC activates a broad, pro-survival signaling network (Figure 2E).
Figure 2.

iMC Activates Multiple Signaling Pathways and Upregulates Gene Expression in T Cells
(A) T cells were transduced with either control (FKBP) or iMC retroviral constructs and treated with 10 nM Rim for 15 or 60 min. PMA/ionomycin (PI) was used as a positive control. An immunoblot was then probed with antibodies specific for phosphorylated JNK, AKT, RelA, ERK, and p38. β-tublin was included as a protein-loading control. (B) Time-course multiplex assay for phosphorylated JNK, RelA, and p38 at 0, 15, 30, 60, 120, and 240 min post-rimiducid exposure. Relative phosphorylation was calculated by normalizing values to untreated gene-modified T cells. (C) Gene expression analysis was performed on FKBP- and iMC-transduced T cells generated from three healthy donors 48 hr after treatment with 10 nM Rim and compared to non-stimulated, gene-modified T cells. Hierarchical clustering for genes at >4-fold difference is displayed. (D) Cytokine production from iMC-transduced T cells for select upregulated genes in the gene expression array (e.g., IL-6, IL-5, IL-13, and GM-CSF). (E) Using differentially expressed genes (>2-fold change), ConsensusPathDB-human analysis was performed to identify potential network nodes that are affected by iMC activation, which include NF-κB (red), TNF family member (green), and anti-apoptotic (blue) pathway networks (see also Tables S2–S5).
Augmentation of a First-Generation CAR with Inducible iMC Costimulation
As iMC multimerization appeared to synergize with TCR activation, resulting in high-level IL-2 production, we reasoned that iMC might similarly provide costimulation to CD3ζ endodomain-encoding CARs. To discover the relative contributions of iMC and target antigen in regulating key T cell functions (e.g., cytotoxicity, survival, cytokine production, and proliferation), we used two distinct retroviruses to generate T cells expressing FKBP, iMC, and/or a first-generation prostate stem cell antigen (PSCA)-specific CAR (PSCA.ζ) (Figure 3A). Consistent with molecular profiling, iMC activation by Rim provided a potent pro-survival signal, which promoted antigen- and exogenous IL-2-independent CAR-T cell survival (Figure 3B). In short-term coculture assays (24 hr), cytotoxic potential was unaffected by iMC costimulation; however, longer culture periods (7 days) revealed that combined CAR and iMC activation increased tumor killing, enhanced outgrowth of T cells coexpressing iMC and CAR, and promoted T cell proliferation that was associated with CD25 upregulation and iMC/CAR-dependent IL-2 production, suggesting a concomitant nuclear factor of activated T cells (NFAT) and NF-κB signaling requirement (Figures 3C–3H).
Figure 3.

iMC Provides Rimiducid-Dependent Costimulation for CAR-T Cells
(A) Using two retroviral vectors, T cells were transduced with different virus combinations to test the ability of iMC to provide Rim-dependent costimulation to CAR-T cells: (a) non-transduced (NT), (b) FKBP only, (c) iMC only, (d) FKBP + PSCA.ζ, and (e) iMC plus PSCA.ζ. Below the schematic diagram are predicted T cell functions endowed by either the costimulatory or CAR construct. (B) NT (not shown) or T cells transduced (n = 3) with iMC plus PSCA.ζ were cultured in media without exogenous IL-2 and stimulated with 10 nM Rim on a weekly basis, where indicated. T cells were counted weekly; on day 21, they were harvested and measured for viability and expression of iMC (CD19+) and CAR by flow cytometry (shown for a single donor). Gates are set on an isotype control (not shown). (C) Coculture assays were performed to measure cytotoxicity (24 hr), cytokine production (48 hr), and T cell or tumor cell proliferation (7 days) with or without Rim addition. (D) Specific lysis of NT and T cells modified with either FKBP plus PSCA.ζ or iMC plus PSCA.ζ were performed over 24 hr using DELFIA-labeled Capan-1 tumor cells with or without 10 nM Rim. (E) NT and T cells modified with either FKBP plus PSCA.ζ or iMC plus PSCA.ζ were cultured at a 1:1 ratio with Capan-1-GFP tumor cells with or without 10 nM Rim on day 0. After 7 days, cells were harvested and analyzed for tumor killing by flow cytometry using anti-CD3 antibody and GFP (top panel) or for FKBP/iMC (CD19) or CAR expression. (F) T cell proliferation was determined in coculture assays by cell enumeration followed by flow cytometry for CD3+ (T cells) and GFP cells (tumor cells) (n = 4). (G) Rim exposure upregulated the IL-2 receptor (CD25) on iMC- and CAR-modified T cells (n = 4). (H) IL-2 production was measured by ELISA after 48 hr with each of the vector combinations and with or without tumor and/or Rim (n = 2). *p < 0.05, **p < 0.01.
We subsequently generated a bicistronic vector encoding both iMC and PSCA.ζ, using the affinity-matured, humanized A11 single-chain variable fragment (scFv) (iMC-PSCA.ζ)16 (Figure 4A). The myristoylation-targeting peptide used to tether the iMC molecule to the cell membrane in the original vaccine design13 increased basal activity. Therefore, we subsequently removed the sequence without a loss of inducibility (not shown). T cells were efficiently transduced (>70%) with the unified vector (Figure 4B). As shown previously, Rim exposure in the absence of exogenous IL-2 increased antigen-independent survival without inducing CAR-T proliferation (Figure 4C). In coculture assays with red fluorescent protein (RFP)-modified HPAC (HPAC-RFP) tumor cells at a 1:10 T cell to tumor cell ratio, cytokine secretion (IL-6 and IL-2), tumor elimination, and CAR-T proliferation were significantly enhanced by activating iMC with Rim (Figures 4D and E). Further dilution of iMC-PSCA.ζ-modified T cells (1:20 effector-to-tumor [E:T] ratio) showed efficient and Rim-dependent killing of HPAC-RFP tumor cells under conditions where T cell proliferation was required to control tumor growth over 9 days (Figures 4F and 4G). Together, these data indicate that the unified, bicistronic vector, encoding both iMC and PSCA.ζ, retains in culture both CAR cytotoxicity and Rim-dependent costimulatory activity.
Figure 4.

Design and Validation of a Unified Vector Encoding iMC and PSCA.ζ CAR
(A) Retroviral transgene design encoding MyD88, CD40, tandem FKBP12v36, T2A, signal peptide, A11 scFv variable light (VL) and variable heavy (VH) domains, the minimal CD34 epitope, CD8α stalk and transmembrane region, and the CD3ζ signaling domain. (B) Flow cytometric analysis to determine transduction efficiency using anti-CD3 and anti-CD34 antibodies compared to non-transduced (NT) T cells. (C) iMC-PSCA.ζ-modified T cells only proliferate in the presence of exogenous IL-2 (100 U/mL) when stimulated with 10 nM rimiducid (Rim), but they demonstrate antigen-independent survival in the presence of IL-2 or with weekly iMC activation over 5 weeks (two individual donors shown). (D) T cells were transduced with EGFPluc or with both EGFPluc and iMC-PSCA.ζ-encoding-retrovirus and subsequently cultured with HPAC-RFP tumor cells at a 1:10 effector-to-tumor (E:T) cell ratio for 48 hr with variable Rim concentrations (0–250 nM) (n = 3). IL-6 and IL-2 production was measured by ELISA. (E) Control T cells (EGFPluc) or iMC-PSCA.ζ/EGFPluc-modified T cells were cultured at a 1:10 E:T ratio with HPAC-RFP cells at variable Rim concentrations (0–250 nM) for 7 days. Tumor cell killing (RFP; red) and T cell (EGFP; green) proliferation were measured by live-cell imaging using an IncuCyte imager. (F and G) EGFPluc and iMC-PSCA.ζ/EGFPluc-modified T cells were cultured with HPAC-RFP tumor cells at an E:T ratio of 1:20 and imaged over 7 days (n = 2). T cells transduced with iMC-PSCA.ζ/EGFPluc were stimulated with or without 2 nM Rim on day 0 of culture initiation.
iMC Activation Allows User-Controlled CAR-T Cell Survival and Proliferation In Vivo
As iMC activation enhances survival in the absence of antigenic activation (i.e., TCR or CAR ligation) (Figures 3B and 4C), it could enhance CAR-T cell engraftment prior to tumor encounter or sustain CAR-T cell persistence after tumor elimination. To examine this possibility, T cells were cotransduced with iMC-PSCA.ζ and with a vector encoding an EGFP-firefly luciferase fusion protein (EGFPluc), subsequently injected into tumor-free NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice and analyzed for persistence using in vivo bioluminescent imaging (BLI). While the bioluminescent signal rapidly decreased in mice injected with iMC-PSCA.ζ/EGFPluc only, the addition of a single Rim dose (day +1; 5 mg/kg intraperitoneally [i.p.]) increased the BLI area under the curve (AUC) by 10-fold (1.2 × 105 versus 1.2 × 106 p/s/cm2/sr, respectively; p = 0.008) (Figures 5A and 5B). To further determine whether iMC could enhance CAR-T proliferation and anti-tumor efficacy, HPAC-EGFPluc-bearing mice were injected with a single intravenous (i.v.) dose of non-transduced or iMC-PSCA.ζ-modified T cells, where one group also received weekly i.p. Rim (5 mg/kg). Using a relatively low CAR-T cell dose (1.0 × 106), Rim-dependent iMC activation was required to enhance tumor control following a single T cell injection (day 50 tumor BLI: 3.6 × 105 ± 5.2 × 105 versus 1.8 × 108 ± 2.3 × 108, p = 0.007 for Rim or vehicle, respectively) (Figures 5D and 5E). Importantly, in a separate experiment, qPCR analysis 21 days post-T cell injection showed that the vector copy number (VCN) was markedly increased in mice treated with Rim compared to mice receiving iMC-PSCA.ζ-modified T cells only, in which VCN was undetectable (Figure 5F). Thus, iMC provides an inducible mechanism to enhance CAR-T cell persistence and amplification in vivo.
Figure 5.

iMC Activation Enhances In Vivo CAR-T Engraftment, Proliferation, and Anti-tumor Efficacy
(A and B) 2.5 × 106 T cells were cotransduced with iMC-PSCA.ζ and EGFPluc and injected i.v. into tumor-free NSG mice. Mice (n = 5 per group) were either left untreated or treated with a single i.p. injection of Rim at 5 mg/kg 24 hr post-T cell injection. Radiance was calculated from in vivo BLI measurements on days +1, +8, and +30 post-T cell injection (LoD or limit of detection for luminescence). (C) NSG mice (n = 5 per group) were engrafted with HPAC-EGFPluc and treated with 1.0 × 106 iMC-PSCA.ζ-modified T cells or NT cells after 7 days. Mice subsequently received weekly i.p. vehicle (Veh) or Rim injections of 5 mg/kg. (D) Tumor BLI was assessed weekly (dotted lines indicate individual animals and full circles show group average). These data are representative of three independent experiments. (E) To quantitate CAR-T cell persistence, a separate experiment was performed where after 21 days, mice were euthanized and tissues were isolated for vector copy number (VCN) analysis by qPCR. *p < 0.05.
iMC Enhances Function of Additional Antigen-Specific CAR Molecules
To investigate the broad applicability of iMC in CAR-T cells, we replaced the PSCA scFv with a binding domain targeting CD123. As with the PSCA-targeting vector, transduction with iMC-CD123.ζ retrovirus was highly efficient (>90% CD3+CD34+) (Figure 6A). Among the four leukemia/lymphoma cell lines we examined for CD123 expression, KG-1, MOLM-13 and THP-1 expressed CD123, while U-937 did not (Figure 6B). The cell lines were subsequently modified to express EGFPluc and used in coculture assays at a 1:1 E:T ratio with unmodified or iMC-CD123.ζ-transduced T cells. At this E:T ratio, CAR-T cells demonstrated Rim- and proliferation-independent, specific elimination of CD123+ cell lines (KG-1, MOLM-13, and THP-1), consistent with the cytolytic activity of the CD123.ζ CAR molecule (Figure 6C). However, Rim stimulated robust T cell proliferation in response to CD123 antigenic stimulation (14-fold, 20-fold, and 39-fold increases over the starting T cell number for KG-1, MOLM-13, and THP-1, respectively) and corresponding IL-2 production (Figures 6D and 6E). To determine whether these results translated into improved efficacy in vivo, NSG mice were challenged with THP-1-EGFPluc tumor cells and treated with 2.5 × 106 non-transduced or iMC-CD123.ζ-modified T cells. Additionally, on days 1 and 15 post-T cell injection, groups received vehicle or Rim. THP-1 leukemic cells rapidly proliferated in mice receiving non-transduced T cells, or CAR-T cells without inducible costimulation, but were effectively controlled following Rim administration (Figures 6F and 6G). As observed with iMC-PSCA.ζ, Rim treatment increased overall CAR-T levels by 3.5-fold (836 ± 411 versus 2894 ± 527 copies/μg genomic DNA [gDNA] for vehicle- and Rim-treated groups, respectively; p = 0.02) (Figure 6I), which also corresponded to an increased frequency of CAR-T cells by flow cytometry (Figure S1). These data support a broadly applicable format by which iMC can be induced to augment a broad variety of CAR-T cells.
Figure 6.

iMC Enhances Proliferation and Anti-tumor Efficacy of CD123-Targeted CARs against Leukemia
(A) Transduction efficiency as measured by CD3+CD34+ expression in non-transduced and iMC-CD123.ζ-modified T cells. (B) Flow cytometric analysis of CD123 expression on four leukemia and lymphoma cell lines (KG-1, MOLM-13, THP-1, and U-937). (C) Non-transduced (NT) and iMC-CD123.ζ-modified T cells were cultured together with CD123+ (KG-1, MOLM-13, or THP-1) or CD123− (U-937) cancer cell lines for 7 days at an E:T ratio of 1:1 effector-to-target cells and evaluated for outgrowth of GFP+ tumor cells. (D) T cell proliferation of NT and iMC-CD123.ζ-modified T cells following coculture with KG-1, MOLM-13, THP-1, and U-937 cell lines was calculated as the fold increase following costimulation with 1 nM Rim. (E) IL-2 production was measured by ELISA following 48 hr of coculture of NT or iMC-CD123.ζ-modified T cells with THP-1 cells in the presence or absence of 1 nM Rim. (F) NSG mice (n = 5 per group) were challenged with 1 × 106 THP-1-EGFPluc tumor cells via i.v. injection, followed by i.v. treatment with 2.5 × 106 NT or iMC-CD123.ζ-modified T cells 7 days after THP-1 administration. Mice subsequently received vehicle only or 1 mg/kg Rim by i.p. injection on days +1 and +15 post-T cell infusion. (G) Animals were imaged on a weekly basis for tumor burden using BLI. (H) On day 28, animals were euthanized and spleens of the mice treated with gene-modified T cells, with and without Rim-induced costimulation, were isolated for vector copy number (VCN) analysis (points indicate individual animals) (see also Figure S1). *p < 0.05, **p < 0.01.
iMC Improves GD2-Specific CAR Potency Compared to Conventional Costimulatory Domains
To compare the activity of iMC-based costimulation relative to more widely used costimulatory domains, we generated first-, second-, and third-generation CAR vectors encoding CD28, 4-1BB, or OX40 endodomains as well as an iMC-GD2.ζ construct (Figure 7A). Healthy donor T cells (n = 4) were transduced and compared for CAR expression levels by flow cytometry, showing equivalent transduction among the vectors (Figure 7B). GD2-specific CAR-T cells were subsequently stress tested for anti-tumor activity using 7-day IncuCyte coculture assays at low E:T (1:10 and 1:100 E:T ratios). At a 1:10 ratio, all vectors demonstrated killing of GD2+ HT144 tumor cells, with GD2.28.BB.ζ CARs showing modestly less activity (Figure 7C). However, at a very high 1:100 E:T ratio, HT144 tumor outgrowth was apparent in the presence of T cells transduced with all CAR-T cell vectors except Rim-treated iMC-GD2.ζ-modified T cells (p < 0.05) (Figure 7D). iMC also enhanced IL-2 production 8- to 16-fold (p < 0.001) compared to conventional GD2 CAR vectors (Figure 7E). Together, these data indicate that iMC provides more potent costimulatory activity compared to CD28, 4-1BB, OX40, or combinations thereof.
Figure 7.

iMC Enhances Potency of GD2-Targeted CARs Compared to Conventional Costimulatory Molecules
(A) Vector design for GD2 CAR-T cells using various costimulatory signaling domains. (B) Transduction efficiency of GD2 CAR-T cells (n = 4) as measured by CD3+CD34+ expression by flow cytometry. (C and D) IncuCyte coculture of CAR-modified T cells with GD2+ HT144-EGFPluc tumor cells using IncuCyte live-cell imaging at effector-to-tumor (E:T) ratios of 1:10 (C) and 1:100 (D), where EGFPluc cell count was measured on day 7. Cocultures with iMC-GD2.ζ were stimulated with and without 10 nM Rim. (E) IL-2 production was measured from 1:100 E:T coculture supernatants after 48 hr of culture initiation (see also Figure S2).
Discussion
Herein, we report on a novel CAR-T cell platform that utilizes ligand-dependent costimulation to modulate T cell proliferation and survival. The iMC-CAR vector was designed to complement a first-generation CAR molecule (PSCA.ζ), which can initiate PSCA-dependent target cell lysis but lacks the requisite accessory signaling for T cell persistence and expansion. In contrast, iMC can be used to activate costimulatory pathways (e.g., NF-κB, JNK, ERK, Akt, MAPK) to assist CAR-dependent signaling (e.g., NFAT) by multimerization of the chimeric MyD88/CD40 protein using Rim, a class of synthetic dimerizing ligands that are only bioactive in the presence of their cognate binding partners. Activation of both iMC and the CAR completes the T cell signaling circuit, inducing IL-2 production and T cell proliferation.
Both MyD88 and CD40 play important, cell-intrinsic roles in T cell survival, differentiation, and effector function. CD40 signaling has been shown to contribute to memory formation and can rescue T cells from exhaustion.17, 18 TLRs are expressed in lymphocytes and ligation by TLR agonists [e.g., poly(I:C) and CpG oligodeoxynucleotides] can directly costimulate multiple T cell subsets, enhancing survival and effector functions and promoting proliferation following TCR cross-linking.19, 20, 21, 22, 23, 24 Using MyD88-deficient mice, previous reports have shown that MyD88 provides T cell-intrinsic anti-apoptotic signals in both CD4+25, 26 and CD8+ T cells27 that support survival and accumulation of antigen-specific T cells during clonal expansion. Our data concur with these previous studies, where activation of iMC alone (i.e., without TCR or CAR cross-linking) in gene-modified T cells allows antigen-independent survival, but not proliferation, and in tumor-free animals, promotes CAR-T cell engraftment.
We assessed the impact of iMC activation in primary human T cells using both gene expression analysis and phosphorylation of signaling molecules. The synergistic activity of MyD88 and CD40 suggests that the pathways induced following oligomerization are complementary and provide a potentially unique and potent signal in transduced T cells. Using GD2-targeted CARs constructed with conventional costimulatory domains (e.g., CD28, 4-1BB, and OX40), we performed a side-by-side stress test comparison (using limiting numbers of T cells). Here, we observed that activation of iMC by Rim induced higher levels of IL-2 production, as well as other cytokines (i.e., IL-6, IFN-γ, and TNF-α) (Figure S2), resulting in enhanced anti-tumor effects. As an adaptor molecule, MyD88 is an essential signaling component for TLRs 1, 2, 4, 6, 7, 8, and 9, which can impact NF-κB, IRF3, and IRF7.28, 29. Previous studies have shown that IRF7/IFN-β enhances the anti-tumor activity of CAR-T cells,30 suggesting that activating these IFN-related pathways with MyD88 may further improve CAR-T function. In addition, CD40 seems to augment this activity through a variety of TNF receptor associated factor (TRAF)-mediated pathways that can overlap with (i.e., NF-κB) or complement MyD88 signaling (i.e., p38, ERK, JNK).31 Indeed, iMC activation in concert with CAR-T ligation enhances secretion of a broad range of cytokines, suggesting that the molecule is a broad enhancer of T cell activity (Figure S3). While an in vivo comparison between iMC and standard costimulatory domains needs to be assessed, the increased potency on T cell survival and proliferation may make iMC a suitable technology for targeting solid tumors, where the inhibitory “hostile” tumor microenvironment can suppress T cell function.
Enhancing potency of CAR-T cells carries a risk of increasing toxicity. Synthetic biology is now being widely employed to design safety and control mechanisms to address acute and off-tumor toxicities.32 Regulatable switches, like iMC, have the potential to provide essential costimulation for CAR-T persistence when the dimerizing ligand is provided but may also allow a reduction or elimination of CAR-T cells if drug is withheld. For antigenic targets that are shared on normal cells (e.g., CD19), these types of control systems may be important in minimizing long-term side effects (e.g., B cell aplasia) if an acceptable anti-tumor response has been reached. It is worth noting that as iMC functions as an independent costimulatory module, it may be employed to augment other T cell therapies apart from CARs, including antigen-specific cytotoxic T lymphocytes (CTLs), tumor-infiltrating lymphocytes (TILs), or TCRαβ gene-modified T cells, providing a flexible control system for maintaining and amplifying targeted T cell therapies.
Overall, this study demonstrates that the chimeric MyD88/CD40 protein can provide a potent costimulatory signal that enhances T cell survival and augments T cell proliferation in the context of TCR or CAR signaling. Additionally, this costimulatory pathway can be activated in vivo using a highly specific, synthetic, small molecule dimerizing ligand, Rim. The separation of the cytolytic signal 1 (CD3ζ) domain from costimulatory signal 2 (iMC) provides a potential mechanism by which T cells may be expanded in response to an administered ligand and tumor antigen, or potentially contracted by ligand withdrawal, where insufficient costimulation could result in T cell anergy. Studies are currently underway to better understand how iMC can be used to control CAR-T cell therapies and maximize therapeutic efficacy.
Materials and Methods
Mice
NSG mice were obtained from Jackson Laboratory and maintained at the Bellicum Pharmaceuticals vivarium. These studies were approved by Bellicum Pharmaceuticals’ Institutional Animal Care and Use Committee (IACUC), comprising both internal and external reviewers from Baylor College of Medicine and the University of Texas Health Science Center.
Cell Lines, Media, and Reagents
HEK293T, Capan-1, HPAC, HT144, KG-1, THP-1, U-937 (all from ATCC), and MOLM-13 (from DSMZ) were cultured in media per the suppliers’ recommendation. T cells were generated from peripheral blood mononuclear cells (PBMCs) obtained from the Gulf Coast Blood Bank and were cultured in 45% RPMI 1640 and 45% Click’s media (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 2 mM GlutaMAX (T cell media; TCM), and 100 U/mL IL-2. Clinical-grade Rim was diluted in ethanol as a 100-μM working solution for in vitro assays and further into 0.9% saline for animal studies.
Retroviral and Plasmid Constructs
iMC comprising the myristoylation-targeting sequence (M) from v-Src,13 a TIR domain-deleted version of the TLR adaptor molecule, MyD88, the CD40 cytoplasmic region, and two tandem ligand-binding FKBP domains were cloned in frame with truncated CD19 (ΔCD19) in the SFG retroviral backbone using Gibson assembly (New England Biolabs) to generate SFG-iMC-2A-ΔCD19 (iMC). Similarly, a control vector was generated containing only the myristoylation sequence and tandem FKBP domains. Additional retroviral vectors were constructed using a synthetic DNA approach (Integrated DNA Technologies) to generate MyD88- or CD40-only constructs, termed iMyD88 or iCD40, respectively. A first-generation PSCA CAR was synthesized containing the murine bm2B3 scFv, the immunoglobulin G1 (IgG1) CH2CH3 spacer region, the CD28 transmembrane domain, and the CD3ζ cytoplasmic domain (PSCA.ζ), as previously described.33 Subsequently, to reduce basal iMC activity, bicistronic vectors were generated encoding a non-myristoylated iMC and the anti-PSCA CAR. Here, the CAR comprises the antigen-specific scFv (A11).16 the QBEnd-10 minimal epitope (CD34 epitope),34 a CD8α stalk and transmembrane domain (CD8stm), and the cytoplasmic CD3ζ domain (iMC-PSCA.ζ). An additional iMC-enabled CAR targeting CD123 was generated by replacing the variable light (VL) and variable heavy (VH) domains with the 32716 scFv.35 GD2 vectors encoding CD28, 4-1BB, and OX40 endodomains were constructed with the 14 g2a scFv.36 Tumor cells were transduced with the SFG-EGFPluciferase (EGFPluc) retrovirus or transfected with expression plasmids expressing GFP or RFP. For in vivo bioluminescence studies, either tumor cells or CAR-T cells were transduced with the EGFPluc retrovirus.
Transduction of T Cells
Retrovirus was produced by transient transfection of 293T cells using GeneJuice (EMD Millipore) with Moloney murine leukemia virus gag and pol polyproteins (MoMLV gag-pol; PegPam3-e plasmid), an RD114 envelop (RDF plasmid), and the retroviral vector, and the supernatant was collected after 48–72 hr. To transduce T cells, 5 × 105 PBMCs were stimulated with 0.5 μg/mL each of anti-CD3 and anti-CD28 antibodies (Miltenyi Biotec) bound to non-tissue culture-treated plates in the presence of 100 U/mL IL-2. Activated T cells were subsequently transduced using the Retronectin (Takara Bio) and spinfection technique and were expanded for 10–14 days post-transduction unless otherwise stated. For transductions with multiple vectors, the protocol was identical to the above except the wells were coated with equal amounts of each retroviral supernatant.
Immunophenotyping
Gene-modified T cells were analyzed for iMC transgene expression 10–14 days post-transduction by using anti-CD3-PerCP.Cy5 and anti-CD19-phycoerythrin (PE) antibodies (BioLegend). To detect CAR-transduced cells, T cells were also stained with an Fc-specific allophycocyanin (APC)-conjugated monoclonal antibody (Jackson ImmunoResearch Laboratories), which recognizes the IgG1 CH2CH3 component of the receptor. T cells were also analyzed for CD4, CD8, and CD25 (BioLegend) following activation with 10 nM Rim and Capan-1 tumor cells. T cells modified with the bicistronic iMC-CAR.ζ constructs were analyzed with PE-conjugated anti-CD34 QBEnd-10 antibody (Abnova) to determine transduction efficiency. Phenotypic analysis of T cells isolated from animals was assessed by nine-color flow cytometry using the following antibody panel: anti-murine CD45-BV510, anti-CD3-APC/Cy7, anti-CD4-PerCP-Cy5.5, anti-CD8-BV711, anti-CD34-PE, anti-CD45RA-fluorescein isothiocyanate (FITC), anti-CD62L-APC, anti-CD95-ECD, and anti-PD-1-BV421 (BD Biosciences). Flow cytometry was performed using LSRII (Becton Dickinson) or Gallios (Beckman Coulter) flow cytometers and the data were analyzed using Kaluza (Beckman Coulter) software.
Cytokine Production
Production of IFN-γ, IL-2, and IL-6 by T cells modified with iMC or control vectors was analyzed by ELISA or cytometric bead array as recommended (eBioscience or Becton Dickinson). In some experiments, cytokines were analyzed using a 27-cytokine/chemokine (Milliplex MAP; Millipore) multiplex array system with a multiplex reader (Bio-Plex MAGPIX; Bio-Rad).
Immunoblotting and Milliplex MAP Assay
Primary human T cells transduced with either FKBP or iMC were cultured for the indicated time points in a 37°C water bath with either 10 nM Rim, 250 nM each of phorbol 12-myristate 13-acetate (PMA) and ionomycin, or media alone. Western blots were performed by probing with antibodies specific for p-RelA (S536), p-Akt (S473), p-p38 (T180/Y182), p-JNK (T183/Y185), p-ERK1/2(T202/Y204) (Cell Signaling Technology), or total β-tubulin (Santa Cruz Biotechnology). Bound antibodies were detected by horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG antibody (Pierce), followed by enhanced chemiluminescence (ECL) (Pierce) and detection on a Gel Logic 6000 Pro imaging system (Carestream Health). Additionally, cell lysates were processed and analyzed per the manufacturer’s protocol using a BioPlex MAGPIX Multiplex Reader (Bio-Rad) to detect phosphorylation of the signaling molecules described above (custom Milliplex Magnetic Bead MAPmate kit; Millipore). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a control for total protein content. The fold increase in phosphorylation was calculated by the “net mean fluorescence intensity (MFI)” of Rim treatment at a given time point divided by the net MFI of the corresponding non-stimulated time point.
Gene Expression Analysis
Non-transduced or FKBP- or iMC-modified T cells were derived from PBMCs isolated from three healthy donors using the T cell generation protocol described above. T cells were stimulated with and without 10 nM Rim for 24 hr and were then harvested for mRNA extraction (by RNeasy; QIAGEN) and hybridization on Human Genome U133 Plus 2.0 arrays at the Baylor College of Medicine Genomics and RNA Profiling Core. Signaling data were derived from the image files using ArrayStar software (version 12.0.0). Genes that showed a Benjamini-Hochberg false discovery rate (FDR) of <0.05 and a >2-fold increase between datasets were used to perform gene ontology using Molecular Signatures Database (MSigDB)37 using C2 CP:KEGG gene sets, C3 transcription factor targets, and C7 immunological targets gene sets. Induced network module analysis was performed using the ConsensusPathDB-human database.38
Cytotoxicity and Coculture Assays
Cytotoxicity against PSCA+ tumor cells was measured in the 24-hr dissociation-enhanced lanthanide fluorescence immunoassay (DELFIA) assay (PerkinElmer) as recommended (Clontech Laboratories). Coculture assays were performed with GFP-modified tumor cells at various E:T with or without 10 nM Rim and in the absence of exogenous IL-2. After 7 days, all residual cells were harvested from the well, counted, and stained with CD3-, CD19-, and CAR-specific antibodies and analyzed by flow cytometry. Additional coculture assays were performed using real-time fluorescent microscopy (IncuCyte; Essen Biosciences).
In Vivo Studies
To evaluate the efficacy of iMC-modified CAR-T cells in vivo against PSCA+ tumors, NSG mice were engrafted with 2 × 106 HPAC tumor cells resuspended in Matrigel (BD Biosciences) and injected subcutaneously (s.c.). After 7–10 days, mice were injected i.v. with a single dose of 1 × 106 T cells per animal. Rim was administered via i.p. injections at a dose of 5 mg/kg. Tumor size was measured both by calipers and by BLI using an In Vivo Imaging System (IVIS; PerkinElmer). To assess CAR-T cell persistence and expansion in vivo, CAR-T cells were cotransduced with EGFPluc and measured using in vivo BLI, as above. Additional experiments were performed using a CD123-targeted CAR and EGFPluc-modified CD123+ THP-1 cells. Here, 1 × 106 tumor cells were injected i.v. and after 7 days, the mice were treated with 2.5 × 106 non-transduced or CAR-T cells via tail vein injection. Some groups also received an i.p. injection of Rim at 1 mg/kg (adjusted to more closely match Rim pharmacokinetics in humans) on day +1 and +15 after T cell injection. BLI was measured weekly; on day 28, mice were euthanized and splenocytes were assessed for CAR-T frequency by qPCR.
VCN Assay
CAR-T cell biodistribution was determined by isolating tissues and blood from treated mice and performing VCN analysis by qPCR. gDNA was isolated from each tissue specimen (<25 mg) using DNeasy Blood and Tissue Kits (QIAGEN). TaqMan PCR primers and fluorescent probe were designed to distinguish between MyD88 and CD40 (forward: GGGCATCACCACACTTGAT; reverse: GCCTTATTGGTTGGCTTCTTG; 6-carboxyfluoroscein (FAM) probe: FAM/ATATGCCTG/ZEN/AGCGTTTCGATGCCT). The retrovirus parent plasmid was used as a standard by serially diluting into gDNA isolated from non-transduced T cells.
Statistics
Data are presented as means ± SEM. Data were analyzed using Mann-Whitney statistical comparisons to determine significant differences between groups. One-way ANOVA followed by Bonferroni’s multiple-comparisons test was used to compare multiple treatment groups. Two-way ANOVA followed by Bonferroni’s test was used to assess statistical significance of differences in tumor growth between multiple treatment groups at different time points. Survival was recorded by Kaplan-Meier graphs, with significance determined by the log-rank test. Data were analyzed using GraphPad Prism software (version 5.0; GraphPad Software).
Author Contributions
A.E.F., K.M.S., and D.M.S. conceived this study and designed the research. A.E.F., A.M., N.P.S., W-C.C., J.C., E.M., J.L.S., S.S., M.T.D., M.R.C.-P., and J.H.B. generated research material and performed the experiments. A.E.F. wrote the manuscript with input from all authors.
Conflicts of Interest
A.E.F., K.M.S., and D.M.S. are coinventors of patent PCT/US2014/026734 (“Methods for controlling T cell proliferation”) and A.E.F., M.R.C., K.M.S, and D.M.S. are coinventors of patent PCT/US2015/015829 (“Methods for activating T cells using an inducible chimeric polypeptide”) filed by Bellicum Pharmaceuticals. K.M.S. and D.M.S. are cofounders of Bellicum, and all other authors are employees of Bellicum and have financial interests in the company.
Footnotes
Supplemental Information includes three figures and five tables and can be found with this article online at http://dx.doi.org/10.1016/j.ymthe.2017.06.014.
Contributor Information
Aaron E. Foster, Email: afoster@bellicum.com.
David M. Spencer, Email: dspencer@bellicum.com.
Supplemental Information
References
- 1.Park J.H., Geyer M.B., Brentjens R.J. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. 2016;127:3312–3320. doi: 10.1182/blood-2016-02-629063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pule M.A., Savoldo B., Myers G.D., Rossig C., Russell H.V., Dotti G., Huls M.H., Liu E., Gee A.P., Mei Z. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed N., Brawley V.S., Hegde M., Robertson C., Ghazi A., Gerken C., Liu E., Dakhova O., Ashoori A., Corder A. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J. Clin. Oncol. 2015;33:1688–1696. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gattinoni L., Finkelstein S.E., Klebanoff C.A., Antony P.A., Palmer D.C., Spiess P.J., Hwang L.N., Yu Z., Wrzesinski C., Heimann D.M. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goff S.L., Dudley M.E., Citrin D.E., Somerville R.P., Wunderlich J.R., Danforth D.N., Zlott D.A., Yang J.C., Sherry R.M., Kammula U.S. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J. Clin. Oncol. 2016;34:2389–2397. doi: 10.1200/JCO.2016.66.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paulos C.M., Wrzesinski C., Kaiser A., Hinrichs C.S., Chieppa M., Cassard L., Palmer D.C., Boni A., Muranski P., Yu Z. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J. Clin. Invest. 2007;117:2197–2204. doi: 10.1172/JCI32205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson M.H., Bowers J.S., Bailey S.R., Diven M.A., Fugle C.W., Kaiser A.D., Wrzesinski C., Liu B., Restifo N.P., Paulos C.M. Toll-like receptor agonist therapy can profoundly augment the antitumor activity of adoptively transferred CD8(+) T cells without host preconditioning. J. Immunother. Cancer. 2016;4:6. doi: 10.1186/s40425-016-0110-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muzio M., Bosisio D., Polentarutti N., D’amico G., Stoppacciaro A., Mancinelli R., van’t Veer C., Penton-Rol G., Ruco L.P., Allavena P., Mantovani A. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J. Immunol. 2000;164:5998–6004. doi: 10.4049/jimmunol.164.11.5998. [DOI] [PubMed] [Google Scholar]
- 9.Hornung V., Rothenfusser S., Britsch S., Krug A., Jahrsdörfer B., Giese T., Endres S., Hartmann G. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 2002;168:4531–4537. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 10.Hervas-Stubbs S., Olivier A., Boisgerault F., Thieblemont N., Leclerc C. TLR3 ligand stimulates fully functional memory CD8+ T cells in the absence of CD4+ T-cell help. Blood. 2007;109:5318–5326. doi: 10.1182/blood-2006-10-053256. [DOI] [PubMed] [Google Scholar]
- 11.Geng D., Zheng L., Srivastava R., Asprodites N., Velasco-Gonzalez C., Davila E. When Toll-like receptor and T-cell receptor signals collide: a mechanism for enhanced CD8 T-cell effector function. Blood. 2010;116:3494–3504. doi: 10.1182/blood-2010-02-268169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geng D., Zheng L., Srivastava R., Velasco-Gonzalez C., Riker A., Markovic S.N., Davila E. Amplifying TLR-MyD88 signals within tumor-specific T cells enhances antitumor activity to suboptimal levels of weakly immunogenic tumor antigens. Cancer Res. 2010;70:7442–7454. doi: 10.1158/0008-5472.CAN-10-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narayanan P., Lapteva N., Seethammagari M., Levitt J.M., Slawin K.M., Spencer D.M. A composite MyD88/CD40 switch synergistically activates mouse and human dendritic cells for enhanced antitumor efficacy. J. Clin. Invest. 2011;121:1524–1534. doi: 10.1172/JCI44327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iuliucci J.D., Oliver S.D., Morley S., Ward C., Ward J., Dalgarno D., Clackson T., Berger H.J. Intravenous safety and pharmacokinetics of a novel dimerizer drug, AP1903, in healthy volunteers. J. Clin. Pharmacol. 2001;41:870–879. doi: 10.1177/00912700122010771. [DOI] [PubMed] [Google Scholar]
- 15.Di Stasi A., Tey S.K., Dotti G., Fujita Y., Kennedy-Nasser A., Martinez C., Straathof K., Liu E., Durett A.G., Grilley B. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lepin E.J., Leyton J.V., Zhou Y., Olafsen T., Salazar F.B., McCabe K.E., Hahm S., Marks J.D., Reiter R.E., Wu A.M. An affinity matured minibody for PET imaging of prostate stem cell antigen (PSCA)-expressing tumors. Eur. J. Nucl. Med. Mol. Imaging. 2010;37:1529–1538. doi: 10.1007/s00259-010-1433-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bourgeois C., Rocha B., Tanchot C. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science. 2002;297:2060–2063. doi: 10.1126/science.1072615. [DOI] [PubMed] [Google Scholar]
- 18.Bhadra R., Gigley J.P., Khan I.A. Cutting edge: CD40-CD40 ligand pathway plays a critical CD8-intrinsic and -extrinsic role during rescue of exhausted CD8 T cells. J. Immunol. 2011;187:4421–4425. doi: 10.4049/jimmunol.1102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bendigs S., Salzer U., Lipford G.B., Wagner H., Heeg K. CpG-oligodeoxynucleotides co-stimulate primary T cells in the absence of antigen-presenting cells. Eur. J. Immunol. 1999;29:1209–1218. doi: 10.1002/(SICI)1521-4141(199904)29:04<1209::AID-IMMU1209>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 20.Gelman A.E., Zhang J., Choi Y., Turka L.A. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J. Immunol. 2004;172:6065–6073. doi: 10.4049/jimmunol.172.10.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caron G., Duluc D., Frémaux I., Jeannin P., David C., Gascan H., Delneste Y. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J. Immunol. 2005;175:1551–1557. doi: 10.4049/jimmunol.175.3.1551. [DOI] [PubMed] [Google Scholar]
- 22.Gelman A.E., LaRosa D.F., Zhang J., Walsh P.T., Choi Y., Sunyer J.O., Turka L.A. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity. 2006;25:783–793. doi: 10.1016/j.immuni.2006.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cottalorda A., Verschelde C., Marçais A., Tomkowiak M., Musette P., Uematsu S., Akira S., Marvel J., Bonnefoy-Berard N. TLR2 engagement on CD8 T cells lowers the threshold for optimal antigen-induced T cell activation. Eur. J. Immunol. 2006;36:1684–1693. doi: 10.1002/eji.200636181. [DOI] [PubMed] [Google Scholar]
- 24.Imanishi T., Hara H., Suzuki S., Suzuki N., Akira S., Saito T. Cutting edge: TLR2 directly triggers Th1 effector functions. J. Immunol. 2007;178:6715–6719. doi: 10.4049/jimmunol.178.11.6715. [DOI] [PubMed] [Google Scholar]
- 25.Fukata M., Breglio K., Chen A., Vamadevan A.S., Goo T., Hsu D., Conduah D., Xu R., Abreu M.T. The myeloid differentiation factor 88 (MyD88) is required for CD4+ T cell effector function in a murine model of inflammatory bowel disease. J. Immunol. 2008;180:1886–1894. doi: 10.4049/jimmunol.180.3.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomita T., Kanai T., Fujii T., Nemoto Y., Okamoto R., Tsuchiya K., Totsuka T., Sakamoto N., Akira S., Watanabe M. MyD88-dependent pathway in T cells directly modulates the expansion of colitogenic CD4+ T cells in chronic colitis. J. Immunol. 2008;180:5291–5299. doi: 10.4049/jimmunol.180.8.5291. [DOI] [PubMed] [Google Scholar]
- 27.Rahman A.H., Cui W., Larosa D.F., Taylor D.K., Zhang J., Goldstein D.R., Wherry E.J., Kaech S.M., Turka L.A. MyD88 plays a critical T cell-intrinsic role in supporting CD8 T cell expansion during acute lymphocytic choriomeningitis virus infection. J. Immunol. 2008;181:3804–3810. doi: 10.4049/jimmunol.181.6.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barton G.M., Kagan J.C. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat. Rev. Immunol. 2009;9:535–542. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X., Jiang S., Tapping R.I. Toll-like receptor signaling in cell proliferation and survival. Cytokine. 2010;49:1–9. doi: 10.1016/j.cyto.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Z., Condomines M., van der Stegen S.J., Perna F., Kloss C.C., Gunset G., Plotkin J., Sadelain M. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28:415–428. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown K.D., Hostager B.S., Bishop G.A. Differential signaling and tumor necrosis factor receptor-associated factor (TRAF) degradation mediated by CD40 and the Epstein-Barr virus oncoprotein latent membrane protein 1 (LMP1) J. Exp. Med. 2001;193:943–954. doi: 10.1084/jem.193.8.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roybal K.T., Lim W.A. Synthetic immunology: hacking immune cells to expand their therapeutic capabilities. Annu. Rev. Immunol. 2017;35:229–253. doi: 10.1146/annurev-immunol-051116-052302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katari U.L., Keirnan J.M., Worth A.C., Hodges S.E., Leen A.M., Fisher W.E., Vera J.F. Engineered T cells for pancreatic cancer treatment. HPB (Oxford) 2011;13:643–650. doi: 10.1111/j.1477-2574.2011.00344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Philip B., Kokalaki E., Mekkaoui L., Thomas S., Straathof K., Flutter B., Marin V., Marafioti T., Chakraverty R., Linch D. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. 2014;124:1277–1287. doi: 10.1182/blood-2014-01-545020. [DOI] [PubMed] [Google Scholar]
- 35.Mardiros A., Dos Santos C., McDonald T., Brown C.E., Wang X., Budde L.E., Hoffman L., Aguilar B., Chang W.C., Bretzlaff W. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122:3138–3148. doi: 10.1182/blood-2012-12-474056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rossig C., Bollard C.M., Nuchtern J.G., Merchant D.A., Brenner M.K. Targeting of G(D2)-positive tumor cells by human T lymphocytes engineered to express chimeric T-cell receptor genes. Int. J. Cancer. 2001;94:228–236. doi: 10.1002/ijc.1457. [DOI] [PubMed] [Google Scholar]
- 37.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamburov A., Wierling C., Lehrach H., Herwig R. ConsensusPathDB--a database for integrating human functional interaction networks. Nucleic Acids Res. 2009;37:D623–D628. doi: 10.1093/nar/gkn698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.