

Abstract
The EBNA1 protein of Epstein–Barr virus (EBV) activates latent-phase DNA replication by an unknown mechanism that involves binding to four recognition sites in the dyad symmetry (DS) element of the viral latent origin of DNA replication. Since EBV episomes are assembled into nucleosomes, we have examined the ability of Epstein–Barr virus nuclear antigen 1 (EBNA1) to interact with the DS element when it is assembled into a nucleosome core particle. EBNA1 bound to its recognition sites within this nucleosome, forming a ternary complex, and displaced the histone octamer upon competitor DNA challenge. The DNA binding and dimerization region of EBNA1 was sufficient for nucleosome binding and destabilization. Although EBNA1 was able to bind to nucleosomes containing two recognition sites from the DS element positioned at the edge of the nucleosome, nucleosome destabilization was only observed when all four sites of the DS element were present. Our results indicate that the presence of a nucleosome at the viral origin will not prevent EBNA1 binding to its recognition sites. In addition, since four EBNA1 recognition sites are required for both nucleosome destabilization and efficient origin activation, our findings also suggest that nucleosome destabilization by EBNA1 is important for origin activation.
INTRODUCTION
Epstein–Barr virus (EBV) genomes are maintained in latently infected human cells as double-stranded, circular DNA episomes in the host cell nucleus (1). The viral episomes are replicated once per cell cycle and are efficiently partitioned to daughter cells during cell division (reviewed in 2). One viral protein, Epstein–Barr nuclear antigen 1 (EBNA1), and one cis-acting viral DNA fragment, oriP, are required for the replication and partitioning of the viral episomes. OriP includes two essential elements termed the family of repeats (FR) and the dyad symmetry (DS) element, that contain twenty and four EBNA1 binding sites respectively. The DS element is the site of initiation of bidirectional DNA replication and EBNA1 binding to the DS is essential for origin activation. The binding of EBNA1 to the FR activates the expression of other EBV latent genes and mediates the segregation of EBV episomes.
Sequence-specific DNA binding is essential for all of the functions of EBNA1, and the mechanism of the EBNA1–DNA interaction is reasonably well understood. EBNA1 dimers recognize an 18 bp palindromic sequence and assemble cooperatively on the multiple sites of the DS element (3–6). EBNA1 complexes formed on the FR and DS elements of oriP interact at a distance, causing the looping out of the DNA separating the two elements and the linking of multiple oriP molecules (7–9). The region of EBNA1 responsible for DNA binding and dimerization has been localized to the C-terminal portion of the protein (amino acids 459–607) and the crystal structure of this EBNA1 fragment has been solved in complex with the DNA recognition site (6,10,11). The EBNA1 DNA binding region is comprised of two domains, termed the core and flanking domains, both of which directly contribute to sequence-specific DNA recognition (11–13). The core domain, which is structurally homologous to the DNA binding domain of the papillomavirus E2 protein, contains the dimerization interface as well as an α-helix that contacts the major groove of the DNA (12,13). Flanking domain base contacts are made by an α-helix oriented perpendicular to the DNA and an extended chain that tunnels along the minor groove (11).
The mechanism by which EBNA1 governs the partitioning of EBV episomes involves the tethering of the episomes to the host cell mitotic chromosomes (14,15). Recent evidence indicates that tethering occurs through the attachment of FR-bound EBNA1 to the cellular EBP2 component of the mitotic chromosomes (16–18). The mechanisms by which EBNA1 activates DNA replication and transcription, however, are not yet known. Unlike other characterized viral origin binding proteins, EBNA1 does not melt the origin DNA nor does it have intrinsic DNA helicase activity; thus cellular factors with these activities must be utilized (19). The contribution of EBNA1 to DNA replication and transcription may be in the recruitment of cellular replication/transcription factors and/or in the alteration of the EBV chromatin structure.
Like cellular DNA, EBV episomes have been shown to be assembled into nucleosomes with a spacing similar to that of cellular chromatin (20). The close association of DNA with histone proteins can serve as a mechanism for the regulation of DNA replication and transcription, as the packaging of DNA into chromatin inhibits the interaction of many DNA binding proteins with their recognition sites (21,22). Some sequence-specific DNA binding proteins, however, have been shown to access their recognition sites when folded into nucleosome core particles (23–28). For two of these proteins, namely GAL4 and Fos/Jun, this interaction results in nucleosome disruption, which may facilitate the assembly of transcription complexes.
The possibility that the displacement of nucleosomes from the EBV replication origin is important for the activation of DNA replication is based on several pieces of evidence from other replication systems. First, DNA binding by several transcriptional activators has been shown to increase the efficiency of replication initiation from neighboring Simian Virus 40 (SV40) or papillomavirus origins by a mechanism that appears to involve the disruption of the nucleosome structure at the origin (29–31). Secondly, replication from a yeast ARS element has been shown to decrease when it is positioned within the central region of a nucleosome core particle (32), likely due to the inability of the origin recognition complex (ORC) to access its DNA recognition site in the nucleosomes (33). Thirdly, the human ORC1 subunit interacts with a histone acetyltransferase suggesting a role for this initiation protein in chromatin remodeling (34). Fourthly, the assembly of core histones on SV40 DNA inhibits its replication in vitro and this inhibition can be reduced by binding the origin binding protein (T antigen) to the origin prior to nucleosome assembly or by adding the chromatin accessibility complex (CHRAC) to the assembled SV40 chromatin (35,36).
Although the interaction of EBNA1 with naked DNA has been extensively studied, the interaction of EBNA1 with chromatin has not been investigated. Given its role in the activation of DNA replication and transcription and the importance of nucleosome disruption in these processes, we investigated the possible role of EBNA1 in chromatin remodeling. We began by examining the interaction of EBNA1 with the DS element of the latent origin of DNA replication containing an assembled nucleosome. Here we show that EBNA1 forms a ternary complex with the DS-nucleosome and that this interaction results in nucleosome destabilization.
MATERIALS AND METHODS
Preparation of DNA fragments
Nucleosomes were assembled on DNA fragments that contained either the DS element of oriP (EBV nucleotides 9019–9137) or EBNA1 binding sites 1 and 2 (plus intervening sequence) from the DS element. The 179 bp DS DNA fragment was generated by PCR amplification of the DS element and flanking sequences from pGEMoriP (19). Primers were designed so that the 118 bp DS element was centered within the fragment (Fig. 1A). The DNA fragments containing EBNA1 binding sites 1 and 2 from the DS element either centered in a 174 bp fragment (Fig. 1B) or positioned 40 bp from one end of a 179 bp fragment (Fig. 1C) were generated by PCR using pGEMs1+2 as a template. This plasmid contains EBNA1 binding sites 1 and 2 and flanking EBV sequences (EBV nucleotides 9076–9164) between the SacI and HindIII sites of pGEM2. The PCR products were separated by agarose gel electrophoresis, excised and purified using Qiaex beads (Qiagen). The purified DNA fragments were cleaved at one end (within one of the PCR primers) with XbaI, to generate a 5′ overhang, then repurified by agarose gel electrophoresis and Qiaex treatment. For DS DNA fragments to be used in footprinting, DNA fragments with 5′ overhangs at the opposite end were also generated by cleavage with NgoMI (in the other PCR primer) in order to enable labeling of the opposite DNA strand. The DNA fragments were labeled by filling in the 3′ recessed ends with DNA polymerase Klenow in the presence of a [α-32P]dCTP and purified from unincorporated nucleotides using a G-25 Sephadex spun column (Boehringer Mannheim).
Figure 1.

The DNA fragments used to reconstitute nucleosomes. A schematic representation of the DNA fragments assembled into nucleosome core particles showing the positioning of EBNA1 binding sites 1, 2, 3 and 4. (A) The DS DNA fragment showing the location of the 118 bp DS element (DS). (B) Centered site 1+2 DNA fragment. (C) Site 1+2 DNA fragment with the same site positioning as in the DS DNA fragment.
Nucleosome reconstitution with chicken histone octamers
To purify chicken histone octamers, erythrocytes from chicken blood were lysed and pellets were treated with micrococcal nuclease as previously described (37), except that butyrate was omitted from the buffers. The chromatin fragments were then applied to a hydroxylapatite column in 100 mM potassium phosphate, pH 6.7, 630 mM NaCl, and all four core histones were eluted by increasing the salt to 2 M NaCl (38). Fractions containing the core histones were pooled and stored at –20°C in 50% glycerol. Purified histone octamers were assembled on the end-labeled DNA fragments by salt dilution. To this end, 40 µg of purified chicken histone octamers were incubated with 100 ng of end-labeled DNA fragment and 40 µg of herring sperm DNA in 40 µl reaction containing 10 mM Tris–HCl, pH 8.0, 1 M NaCl, 1 mM EDTA. After 10 min at 37°C, the reaction was sequentially diluted to 800, 600 and 300 mM NaCl by the addition of 10 mM Tris–HCl pH 8.0, 1 mM EDTA, with a 10 min incubation at 37°C at each dilution step. Mock assembled DNA fragments were also prepared where the end-labeled DNA fragments were subjected to the gradual salt dilution treatments in the absence of histone octamers.
Nucleosome reconstitution by transfer of human histone octamers
Oligonucleosomes were prepared from 1 l of HeLa cells at 1.0 × 106 cells/ml as described by Cote et al. (39). Briefly, nuclei were sequentially extracted in buffer containing 300 mM KCl, 400 mM NaCl and 650 mM NaCl plus 340 mM sucrose. After dialysis, the extraction supernatant was digested with micrococcal nuclease then applied to a Sepharose CL-6B gel filtration column. Column fractions containing the four core histones but lacking H1 were pooled, and dialyzed against buffer A (20 mM HEPES pH 7.5, 1 mM EDTA, 1 mM β-mercaptoethanol, 0.5 mM PMSF). Oligonucleosomes were concentrated by dialysis against solid sucrose, dialyzed against buffer A, further concentrated on a Centriprep-10 (Amicon), aliquoted, and stored at –70°C. The histone octamers from the HeLa oligonucleosomes were transferred to the end-labeled DS DNA fragment as according to Cote et al. (39). HeLa oligonucleosome cores (10 µg) were incubated with 25 ng of DNA fragments at 37°C for 20 min in a 20 µl reaction containing 1 M NaCl in dilution buffer (50 mM HEPES, pH 7.5, 1 mM EDTA, 5 mM DTT, 0.5 mM PMSF). The reaction was sequentially diluted to 850, 650, 500 and 300 mM NaCl by the addition of dilution buffer and incubated at 30°C for 30 min at each dilution. The assembly reaction was then diluted to 100 mM NaCl by the addition of 10 mM Tris–HCl pH 7.5, 1 mM EDTA, 5 mM DTT, 0.5 mM PMSF, 20% glycerol, 100 µg/ml bovine serum albumin and incubated at 30°C for 30 min. Mock assembled DNA fragments were also prepared where the end-labeled DNA fragments were subjected to the gradual salt dilution treatments in the absence of HeLa oligonucleosomes.
Sucrose gradient purification of reconstituted nucleosomes
Mononucleosomes assembled on end-labeled DNA fragments with either the chicken or human histone octamers were purified away from unbound DNA, unbound histones and oligonucleosomes by sedimentation on a continuous sucrose gradient. Nucleosome reconstitution reactions were layered on top of 4 ml gradients containing 5–25% sucrose in 100 mM HEPES pH 7.5, 10 mM EGTA, 0.1% NP-40, 0.1 mM PMSF. After centrifugation for 17 h at 32 500 r.p.m. in a SW60 rotor, 115 µl fractions were collected and 5 µl of each was analyzed by electrophoresis on a 5% polyacrylamide gel. Labeled DNA was visualized by autoradiography and fractions containing nucleosome core particles were pooled and stored at 4°C.
Electrophoretic mobility shift assays (EMSAs)
EBNA1 proteins were purified as described in Frappier and O’Donnell (19) for functional EBNA1 or in Barwell et al. (40) for EBNA452–641 and EBNA459–607. Purified EBNA1 proteins were titrated onto 2 fmol of either mock-assembled DNA fragments or reconstituted nucleosome cores in a 6 µl reaction containing 10 mM HEPES, pH 7.0, 5 mM MgCl2, 150 mM NaCl. After 10 min at room temperature, DNA complexes were separated by electrophoresis on a 5% polyacrylamide gel and visualized by autoradiography. For competition assays, ternary complexes were formed between the EBNA1 proteins and nucleosomes as described above, using an amount of the EBNA1 protein sufficient for complete binding of the nucleosome. After a 10 min incubation at room temperature, unlabeled competitor DNA (either the DS DNA fragment or pGEM2 plasmid) was added, while maintaining the 150 mM NaCl concentration. Samples were incubated for 20 min at room temperature, then analyzed by polyacrylamide gel electrophoresis and autoradiography. The percentage of labeled DNA migrating as unbound, nucleosome cores, EBNA1–DNA and EBNA1–nucleosomes complexes was quantified by phosphorimager analysis using Imagequant software.
DNase I footprinting
For footprint analysis, purified EBNA452–641 was titrated with end-labeled DS DNA or reconstituted DS-nucleosomes as described for EMSAs. DNase I digestion and subsequent preparation of the DNA fragments for denaturing polyacrylamide gel electrophoresis was performed according to Cote et al. (39). DNA fragments were separated by electrophoresis on an 8% polyacrylamide/50% urea gel and visualized by autoradiography.
RESULTS
Characterization of EBNA452–641 ternary complexes
We first asked whether EBNA1 could bind to nucleosome core particles containing EBNA1 binding sites. For these experiments we generated a 179 bp DNA fragment containing the 118 bp DS element of oriP centered within the fragment (Fig. 1A). The DNA fragment was designed such that it could form a mononucleosome centered over the DS element. The DS-containing fragments, which contain four EBNA1 binding sites, were end-labeled and reconstituted into nucleosome core particles by gradual salt dilution in the presence of histone octamers purified from chicken erythrocytes. Reconstituted nucleosomes were purified away from free DNA via a continuous sucrose gradient. The efficient formation of nucleosome cores was verified by EMSAs, as indicated by their reduced mobility in a native polyacrylamide gel (Fig. 2A, lane 2) as compared to DS DNA fragments subjected to the salt dilution treatment in the absence of histones (mock-assembled DNA; Fig. 2A, lane 1).
Figure 2.
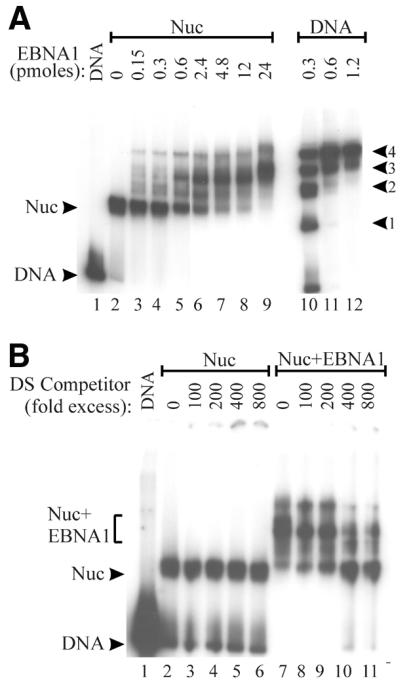
EBNA452–641 forms a ternary complex with the DS-nucleosome. (A) EBNA452–641 was titrated with DS-nucleosomes (Nuc) or with naked DS DNA fragments (DNA), and complexes were analyzed by EMSAs. The intermediates formed during the filling of the four EBNA1 sites in the naked DS DNA are labeled according to the number of sites bound (1–4). (B) DS-nucleosomes (Nuc) or ternary complexes formed between EBNA452–641 and DS-nucleosomes (Nuc+EBNA1) were challenged by the addition of increasing amounts of unlabeled DS DNA competitor prior to acrylamide gel electrophoresis. DS competitor added is shown as fold excess over the labeled DS DNA fragments.
The ability of EBNA1 to interact with the DS-nucleosome particles was assessed by EMSAs using EBNA452–641. EBNA452–641, a truncation mutant of EBNA1, contains the DNA binding and dimerization domains (amino acids 459–607) and the C-terminal acidic tail (amino acids 619–641). Although EBNA452–641 is unable to mediate the replication, segregation and transcriptional activation functions of EBNA1, wild-type affinity for EBNA1 recognition sites is maintained (41). The binding of EBNA452–641 to mock-assembled DS templates produced a series of shifted complexes representing binding to one to four of the EBNA1 recognition sites and saturation of the sites occurred at 0.6–1.2 pmol protein (Fig. 2A, lanes 10–12). The titration of EBNA452–641 onto the DS-nucleosome core particles revealed an interaction between this protein and the core particles that resulted in a shifted complex migrating at a position distinct from that of EBNA452–641 bound to naked DNA (Fig. 2A, lanes 3–9). The small amount of DNA that migrates at the position of naked DNA bound by four EBNA452–641 dimers is likely generated from EBNA452–641 binding to the residual amount of naked DS fragment that is present in the assembled DS-nucleosome. Comparisons of complete titrations of EBNA452–641 with DS-nucleosomes and mock-assembled DS DNA fragments indicate that nucleosome formation inhibits EBNA1 binding ~10-fold (data not shown). The interaction of EBNA452–641 with DS-nucleosome particles appeared to involve EBNA1 recognition sites, as binding was not observed to nucleosome core particles assembled with DNA lacking EBNA1 binding sites (data not shown).
The decreased mobility of the DS-nucleosome upon addition of EBNA452–641 suggested that EBNA452–641 was forming a ternary complex with these core particles. To assess the composition of this complex, we took the following two approaches. First, we used a DNA competition assay to determine if the complexes contained histones. This assay was previously used to demonstrate the presence of histones in ternary complexes formed between GAL4 dimers and nucleosome core particles and involves using specific recognition sites to remove the sequence-specific DNA binding protein from the ternary complex (42). To this end, increasing amounts of unlabeled DS DNA fragments were added to the ternary complexes formed by EBNA452–641 and the DS-nucleosome and, after a 20 min incubation, complexes were separated on a native polyacrylamide gel. As shown in Figure 2B, the addition of the DS competitor DNA altered the mobility of the ternary complexes containing EBNA452–641, presumably by removing EBNA452–641, but did not affect that of the DS-nucleosome. After removal of EBNA452–641 from the ternary complexes, the residual complex migrated at a position consistent with the nucleosome core particle. We conclude that the histones were not displaced when EBNA452–641 bound to the DS-nucleosome and that the ternary complex formed contained the DS DNA fragment, histones and EBNA452–641.
The second approach that we took to analyse the complex formed between EBNA452–641 and the DS-nucleosome was DNase I footprinting (Fig. 3). Cleavage of the DS-nucleosomes with DNase I produced the 10 bp periodicity characteristic of a rotationally phased nucleosome (Fig. 3A and B, lane 3). This cleavage pattern was most prominent in regions farthest from the radiolabeled end of the DS fragment and was not observed with naked DS DNA. When EBNA452–641 was titrated onto the DS-nucleosomes, protection of some of the nucleotides within each of the four EBNA1 binding sites from DNase I cleavage was observed, indicating that this protein occupies all four sites of the nucleosome core particle. EBNA452–641 binding was also accompanied by the induction of a DNase I hypersensitive site adjacent to EBNA1 binding site 4 on one DNA strand (Fig. 3A) and adjacent to binding site 1 on the other DNA strand (Fig. 3B). The EBNA452–641 protection pattern on the nucleosome was different from that observed on the naked DS DNA, where protection of the entire length of the EBNA1 recognition sites was observed. EBNA452–641 binding did not completely disrupt the 10 bp cleavage pattern of the core particle, suggesting that the histones were still present on the DNA. Protection of all four of the recognition sites occurred at the same concentration of EBNA452–641, despite the fact that, when taken individually, EBNA1 has a higher affinity for the outer sites (sites 1 and 4) than the inner sites (sites 2 and 3) (3,6). The simultaneous filling of the four sites in the DS-nucleosomes suggests that the cooperative assembly of EBNA1 on these four sites, which has been well documented on naked DS DNA, still occurs in the presence of the histone octamer (6,43).
Figure 3.

DNase I footprints of ternary complexes. Nucleosomes containing DS DNA fragments (Nuc) were incubated with increasing amounts of EBNA452–641 to form ternary complexes, then subjected to DNase I footprint analysis. Bands protected by EBNA452–641 binding are indicated (arrows and brackets) as are DNase I-hypersensitive bands induced by EBNA452–641 (*). The DNase I digestion patterns of the naked DS DNA fragment (lane 2) and EBNA452–641 bound to the naked DS (lane 1) are also shown. The positions of EBNA1 binding sites 1–4 are indicated. (A) and (B) are footprints of opposite DNA strands.
Functional EBNA1 also forms a ternary complex with the DS-nucleosome
To ensure that the ability of EBNA452–641 to bind nucleosome core particles reflected the properties of the full-length EBNA1 protein, we assessed the nucleosome interactions of an EBNA1 protein that is functional for replication, segregation and transcriptional activation. Functional EBNA1 was not used in the initial study because of the DNA linking activity of this protein, which causes the aggregation of bound DNA molecules containing EBNA1 recognition sites (7–9). These EBNA1-linked DNA complexes remain in the wells during native polyacrylamide gel electrophoresis, making the assessment of their contents difficult.
When the ability of EBNA1 to interact with the DS-nucleosome was examined by EMSAs, EBNA1 was found to bind the naked DS and the core particles as indicated by the shift of the labeled DNA to the wells of the gel (Fig. 4, lanes 4 and 6). In order to determine whether the histones were still present in the supershifted core particles, the competitor assay was used as described above for EBNA452–641. When excess DS DNA fragments were added to the supershifted DS-nucleosome, EBNA1 was removed from some of these complexes and the released DNA migrated at the position of the core particle (Fig. 4, lane 5). Thus, like EBNA452–641, EBNA1 forms a ternary complex with the DS-nucleosome and does not displace the histone octamer. The decreased ability of the competitor DS DNA to disrupt the ternary complex, as compared to that formed with EBNA452–641, indicates that EBNA1 is more stably bound to the DS-nucleosome than EBNA452–641. This is in keeping with previous reports in which the linking activity associated with EBNA1 has been shown to stabilize the binding of EBNA1 to its recognition sites in the DS element (9,44).
Figure 4.
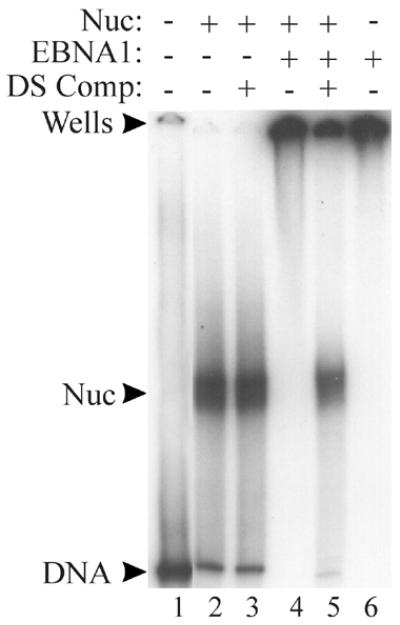
Functional EBNA1 forms a ternary complex with the DS-nucleosome. The DS-nucleosome (Nuc) was incubated with sufficient EBNA1 (7 pmol) to form a ternary complex (lane 4) then this complex was challenged by the addition of a 1000-fold excess of unlabeled DS DNA (DS Comp; lane 5). Complexes were analyzed by EMSA. The positions of the labeled DS DNA (lane 1), the DS-nucleosome (lane 2) and EBNA1 bound to naked DS DNA (lane 6) are also shown. Lanes 1 and 6 contain labeled DS DNA and lanes 2–5 contain the labeled DS-nucleosome.
The nature of the ternary complex formed between EBNA1 and the DS-nucleosome was also examined by DNase I footprinting and the footprints were found to be indistinguishable from those obtained with ternary complexes containing EBNA452–641. As was observed for EBNA452–641, EBNA1 protected nucleotides in all four of the recognition sites from DNase I cleavage but did not completely disrupt the 10 bp cleavage pattern of the nucleosome (data not shown). We conclude that the ternary complex formed between EBNA452–641 and the DS-nucleosome accurately reflects complex formation by functional EBNA1.
EBNA452–641 destabilizes the DS-nucleosome
We next examined whether the interaction of EBNA452–641 with the nucleosome formed on the DS element destabilized the interaction of the histone octamer with the DNA. Destabilized nucleosomes can be detected by the addition of non-specific competitor DNA (25,28). While this treatment has no effect on assembled nucleosomes, it caused the release of histone octamers from ~40% of the ternary complexes formed between GAL4 and nucleosomes containing GAL4 binding sites, leaving GAL4 bound to the recognition sites in the naked DNA (28).
To determine if EBNA452–641 destabilized nucleosomes formed at the DS element, ternary complexes were challenged with an excess of non-specific competitor DNA, then separated on a native polyacrylamide gel. The addition of competitor DNA led to a change in the mobility of the labeled DS DNA, from the position of the ternary complex to that of naked DNA containing four EBNA452–641 dimers (Fig. 5A, lanes 9–13). As expected, the addition of competitor DNA had no effect on the stability of DS-nucleosome core particles in the absence of EBNA452–641 (Fig. 5A, lanes 2–7). Quantification of these results revealed that the histones were released from ~60% of the ternary complexes after addition of the competitor DNA. The results indicate that EBNA452–641 displaced the histone octamers from the DS element in the presence of competitor DNA. In some instances we also observed that, during the formation of the ternary complex, more EBNA1–DS complexes were generated than could be accounted for by EBNA1 binding to the residual naked DS DNA present in the assembled DS-nucleosome (compare ‘DNA’ band in lane 2 to ‘DNA+EBNA1’ band in lane 8). This suggests that EBNA1 causes histones to be displaced from a proportion of the ternary complexes prior to the addition of non-specific competitor DNA. For the ternary complexes that remain, however, no further dissociation is observed with time (up to 2 h) in the absence of competitor DNA (data not shown); only in the presence of non-specific competitor DNA is efficient displacement of the histones observed.
Figure 5.
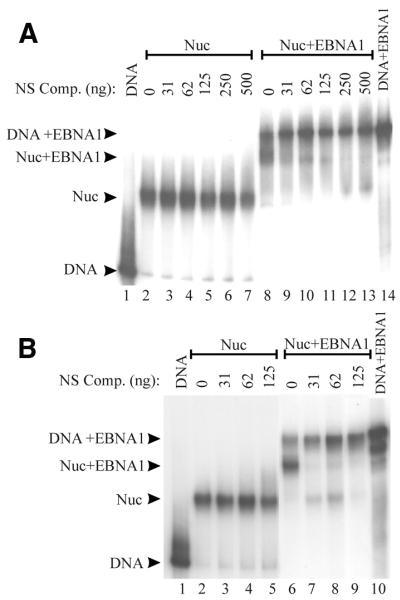
Histones are displaced from ternary complexes upon non-specific competitor DNA challenge. Nucleosomes containing DS DNA fragments were incubated with 12 pmol of EBNA452–641 to form ternary complexes (Nuc+EBNA1) then were challenged by the addition of increasing amounts of plasmid competitor DNA. Nucleosomes that had not been incubated with EBNA1 (Nuc) were also challenged with plasmid competitor. The positions of the naked DS DNA (DNA) and DS DNA bound by EBNA452–641 (DNA+EBNA1) are indicated. (A) Nucleosomes were formed from purified chicken histone octamers by salt dialysis. (B) Nucleosomes were formed from HeLa oligonucleosomes by octamer transfer.
In the above experiment, nucleosome core particles were assembled from purified chicken histone octamers using the salt dialysis method. To ensure that the destabilization of the nucleosome observed with EBNA452–641 was not particular to the source of the histone octamers or the method used to assemble the nucleosome, we repeated the assay using DS-nucleosomes assembled from unacetylated HeLa oligonucleosomes by the octamer transfer method. The ternary complex formed between these nucleosomes and EBNA452–641 was similar in its mobility to that formed with the chicken histones (Fig. 5B, lane 6). The addition of non-specific competitor DNA disrupted the ternary complex, predominantly resulting in EBNA452–641 bound to naked DNA (Fig. 5B, lanes 7–9). A small amount of nucleosome lacking EBNA1 was also generated indicating that the non-specific competitor DNA can also promote dissociation of EBNA1 from the ternary complexes. Similar effects of non-specific competitor DNA on the dissociation of GAL4 from ternary complexes have been reported (28). We conclude that EBNA452–641 can destabilize nucleosomes resulting in the displacement of histone octamers upon competitor DNA challenge.
The EBNA1 DNA binding and dimerization domain is sufficient for nucleosome destabilization
EBNA452–641 contains the DNA binding and dimerization domains of EBNA1 (amino acids 459–607) as well as a highly acidic C-terminal region (amino acids 619–641) of unknown function. Since the mechanism by which some transcription factors alleviate nucleosome repression involves acidic domains (30,31), we tested the possibility that the acidic C-terminus of EBNA1 contributed to the destabilization of nucleosomes. To this end, the nucleosome binding and destabilization experiments were repeated with an EBNA1 fragment that contained only the DNA binding and dimerization domains, EBNA459–607. EBNA459–607 was added to purified nucleosome core particles containing the end-labeled DS fragments and chicken histone octamers and complexes were analysed by EMSAs (Fig. 6). EBNA459–607 bound to the nucleosomes, forming a ternary complex that shifted to the gel wells (Fig. 6, lane 3), likely due to aggregation between the ternary complexes. As was observed with EBNA452–641, the addition of non-specific DNA to ternary complexes resulted in the release of the histone octamer, leaving EBNA459–607 bound to naked DNA (Fig. 6, lane 4). Thus the DNA binding and dimerization region of EBNA1 is sufficient to bind and destabilize DS-nucleosomes.
Figure 6.
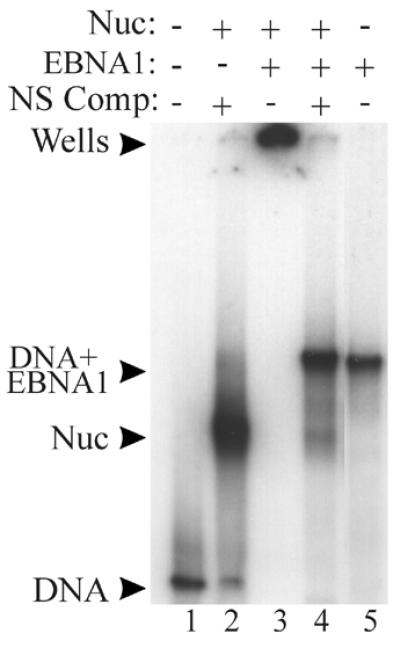
The DNA binding and dimerization domain of EBNA1 is sufficient for nucleosome disruption. EBNA459–607 (15 pmol) was incubated with a DS-nucleosomes (lane 2) to form a ternary complex (lane 3), which was challenged by the addition of plasmid competitor DNA (lane 4). The positions of naked DS DNA fragments, either unbound (lane 1) or bound by EBNA459–607 (lane 5) are also indicated.
EBNA1 recognition site requirement for nucleosome destabilization
It has been shown that the affinity of GAL4 dimers for nucleosome core particles is affected by the number and the positioning of the GAL4 binding sites in the nucleosomes (45). We have evaluated EBNA1 binding to nucleosomes containing the four EBNA1 recognition sites of the latent origin of DNA replication. These four sites are organized into two sets of two sites with conserved spacing (sites 1+2 and 3+4). To investigate the number of recognition sites required for EBNA1 to bind and destablize nucleosomes, we generated DNA fragments containing two EBNA1 binding sites (sites 1 and 2) positioned either in the center or 40 bp from one end of the DNA fragment (Fig. 1B and C). Nucleosomes were reconstituted from these end-labeled DNA fragments and chicken histone octamers, and the purified nucleosomes were tested for binding to EBNA452–641. No binding of EBNA452–641 was detected to the nucleosome containing the centered sites with up to 24 pmol of EBNA1 (data not shown). However, binding was detected to the nucleosomes when sites 1 and 2 were placed at the edge of the nucleosome, the same positioning as occurs in the DS DNA fragment. EBNA452–641 bound to this nucleosome, producing a ternary complex that migrated to a position distinct from that of EBNA452–641 bound to naked DNA (Fig. 7A, lanes 4–7). To determine if the binding of EBNA452–641 to the nucleosome containing sites 1 and 2 destabilizes the nucleosome, we challenged these complexes with an excess of non-specific competitor DNA. The addition of competitor DNA did not alter the migration of the ternary complex, even at competitor DNA concentrations >15-fold higher than that needed to visualize the disruption of the DS-nucleosome bound by EBNA452–641 (Fig. 7B, compare lanes 4 and 5). The results indicate that EBNA1 can bind to nucleosomes containing two recognition sites if these sites are positioned near the edge of the nucleosome, but that binding to these two sites is insufficient for nucleosome destabilization.
Figure 7.
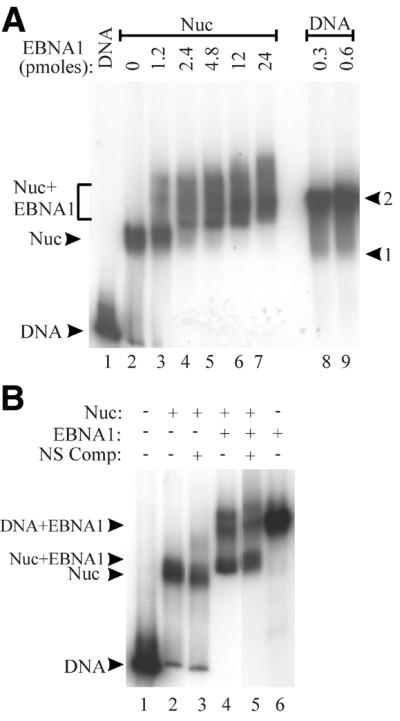
Two EBNA1 recognition sites are insufficient for nucleosome destabilization. (A) A DNA fragment containing sites 1 and 2 with the same positioning as in the DS DNA fragment (Fig. 1C) was assembled into a nucleosome (lane 2) then titrated with EBNA452–641 to form a ternary complex (lanes 3–7; Nuc+EBNA1). EBNA452–641 bound to the naked DNA fragment (lanes 8 and 9) is shown and positions of shifted complexes representing EBNA452–641 binding to one or two of these sites is indicated. (B) Ternary complexes from (A) (lane 4) were challenged with 500 ng of plasmid competitor DNA (lane 5), as were nucleosomes that lacked EBNA452–641 (lane 3). The positions of the naked DNA fragments, unbound (DNA) or bound by EBNA452–641 (DNA+EBNA1) are shown. Note that while two bands are observed in the vicinity of the DNA+EBNA1 arrow after addition of EBNA452–641 to the nucleosomes (lanes 4 and 5), only the lower band corresponds to EBNA452–641 bound to naked DNA. The composition of the upper band is not known.
DISCUSSION
Latent EBV episomes are assembled into nucleosomes and therefore mechanisms must exist to facilitate access to important regulatory sequences, enabling the activation of DNA replication and transcription. EBNA1 plays an essential role in both latent EBV replication and transcription and we have begun to investigate its ability to interact with and alter chromatin. We have shown that EBNA1 retains the ability to bind its recognition sites within a nucleosome core particle containing the latent origin of replication, the DS element of oriP. This interaction results in the formation of a ternary complex in which the nucleosome is destabilized. Our observations suggest that, in vivo, EBNA1 would be able to access its recognition sites at the origin, even if the sites were assembled into a nucleosome, and could then facilitate the access of other proteins to origin sequences.
An EBNA1 fragment containing only the DNA binding and dimerization region (EBNA459–607) was sufficient to bind and destabilize nucleosomes formed at the replication origin. This implies that it is the act of DNA binding that destabilizes the nucleosome. These results are consistent with those of GAL4 and Fos/Jun, where the DNA binding domains of these proteins were also found to be sufficient for nucleosome disruption (25,28). The DNA binding and dimerization region of EBNA1 comprises two domains, termed the core and flanking domains, both of which make sequence-specific DNA contacts that are important for the assembly of EBNA1 on naked DNA templates containing EBNA1 recognition sites (11–13). The core domain can bind DNA independent of the flanking domain and, in the context of the complete DNA binding region, likely makes the first DNA contacts (13). It is not yet clear whether the binding of EBNA1 to its recognition site in the context of a nucleosome involves one or both of these domains. The binding of EBNA459–607 to a single recognition site causes DNA bending and localized regions of helical over- and under-winding (11). The assembly of EBNA459–607 on the adjacent closely-spaced sites from the DS element is predicted to be accompanied by additional structural changes in the DNA, including unwinding (11). The nucleosome destabilization induced by EBNA1 suggests that EBNA1 binding alters the shape of or the histone–DNA contacts within the nucleosome core particle. These alterations may be due, at least in part, to the DNA structural changes caused by EBNA1.
The effect of EBNA1 binding on assembled nucleosomes was very similar to that observed for the yeast GAL4 transcriptional activator. Like EBNA1, GAL4 has been shown to form a ternary complex with nucleosome core particles containing multiple recognition sites and to destabilize the nucleosome (28). For both proteins, nucleosome formation is associated with an ~10-fold decrease in the ability of EBNA1 and GAL4 to bind to multiple recognition sites (45). In GAL4, an order of filling of the sites in the nucleosomes has been observed, with the outer sites becoming bound before the inner sites (46). Such a sequential order of filling was not obvious for EBNA1, rather all four sites became bound simultaneously. This simultaneous filling of the sites in the DS, which is also observed in naked DNA, indicates the cooperative assembly of EBNA1 because the recognition sites, when taken individually, have 10-fold different affinities for EBNA1 (i.e. EBNA1 has a 10-fold higher affinity for sites 1 and 4 than sites 2 and 3) (3,6,43,47). Effects of recognition site positioning within the nucleosome were observed however; DNA fragments containing two recognition sites were only bound when the sites were positioned at the edge, rather than the center of the nucleosome. Like EBNA1, GAL4 assembles on its recognition sites in nucleosome core particles in a cooperative manner (45). For GAL4, this cooperativity requires the N-terminal histone tails (46).
We have studied the interaction of EBNA1 with a nucleosome formed from the DS element of the latent origin of DNA replication, oriP. A previous study suggested that the DS element was unfavorable to nucleosome reconstitution, because half as many nucleosomes were observed to assemble on this element than on flanking DNA sequences when histone octamers were incubated with oriP DNA under high salt (1.5 M NaCl) conditions (48). Using conditions that are more conducive to nucleosome assembly, however, we have found that a nucleosome with the expected stability to salt, DNA competitors and sucrose gradient sedimentation forms at the DS element. Therefore, we expect that nucleosomes could also form at the DS in vivo.
There are several instances during EBV latent infection where the ability of EBNA1 to access its recognition sites within the DS-nucleosome would be important for the replication of the EBV genomes. First, this ability may be important during the establishment of the latent infection. During initial EBV infection, EBNA1 expression begins ~20 h post-infection and reaches the level seen in EBV-infected lymphoblastoid cell lines only after 46 h (49). Thus the expression of EBNA1 likely occurs after the EBV genomes are assembled into chromatin. Secondly, in established EBV latency, it appears that resting B-cells that harbor EBV genomes but do not express EBNA1 can progress to proliferating cells that express EBNA1 (50). When this occurs, EBNA1 must gain access to oriP in order for the EBV genomes to replicate and be maintained in the dividing cells. Thirdly, in replicating cells expressing EBNA1, EBNA1 may have to reassemble on oriP at each cell cycle. Although in vivo footprints of oriP have suggested that EBNA1 occupies its recognition site throughout most of the cell cycle (51,52), EBNA1 may dissociate from its sites during the replication process and have to compete with nucleosomes to reaccess the sites after replication is complete.
The four EBNA1 recognition sites of the DS element are organized into two sets of two sites (sites 1+2 and sites 3+4) where each set of sites (1+2 or 3+4) has been shown to have some capacity to initiate DNA replication in recombinant plasmids (43,53). The efficiency with which a two-site origin element functions appears to vary in different cell lines, but in the only B-cells examined (the natural host for EBV latent replication), replication efficiency was greatly increased by the presence of all four EBNA1 binding sites (53). Our data indicate that increasing the number of EBNA1 binding sites from two to four profoundly affects the ability of EBNA1 to destabilize nucleosome core particles. EBNA1 can bind to nucleosome core particles containing two sites only if the sites are positioned near the edge of the nucleosome, and destabilization of the nucleosome was not observed. We suggest that the increased replication efficiency observed from origins containing four, as opposed to two, EBNA1 binding sites reflects, at least in part, the ability of EBNA1 to destabilize nucleosomes at these sequences. It is also possible that, in the context of the viral DNA (as opposed to the recombinant plasmids used in the replication assays), there may be an absolute requirement for four EBNA1 binding sites due to more efficient occupancy of DS by histone octamers. Indeed the importance of at least four recognition sites is suggested by the finding that the DS element in the closely related Herpesviruses Papio contains five recognition sites for its EBNA1-like origin binding protein (54).
While EBNA1 is the only viral protein required for DNA replication from oriP, it is not sufficient for origin activation. This process relies heavily on the host cell replication proteins and results in one round of EBV DNA replication per cell cycle. The cellular factors and mechanisms involved in the assembly of the preinitiation complex on the DS element and the melting of the origin DNA are not yet clear, but it is likely that these processes require the interaction of one or more cellular proteins with the origin sequences. One of the contributions that EBNA1 makes to DNA replication may be to facilitate the access of these cellular proteins to origin sequences through the destabilization of nucleosomes at the origin. This EBNA1-facilitated access to origin sequences might involve the sliding of the histone octamer away from the origin or simply the loosening of the histone–DNA contacts within the nucleosome. Studies to determine where nucleosomes are positioned within the complete origin of replication and how EBNA1 binding affects their positioning will be necessary to assess these possibilities.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Jennifer Cruickshank for EBNA452–641 and Cinzia Commisso-Cappelli for assistance with chicken histone purification. This work was supported by a grant from the National Cancer Institute of Canada (NCIC), which receives funds from the Canadian Cancer Society. T.M.A.-H. is a research student of the NCIC, supported with funds provided by the Terry Fox run. L.F. is a Medical Research Council of Canada Scientist.
References
- 1.Kieff E. (1996) Epstein–Barr virus and its replication. In Fields,B.N., Knipe,D.M. and Howley,P.M. (eds), Fields Virology, 3rd edition. Lippincott-Raven Publishers, PA, pp. 2343–2396. [Google Scholar]
- 2.Yates J.L. (1996) Epstein–Barr virus DNA replication. In DePamphilis,M.L. (ed.), DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 751–773.
- 3.Ambinder R.F., Shah,W.A., Rawlins,D.R., Hayward,G.S. and Hayward,S.D. (1990) Definition of the sequence requirements for binding of the EBNA-1 protein to its palindromic target sites in Epstein-Barr virus DNA. J. Virol., 64, 2369–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hearing J., Mulhaupt,Y. and Harper,S. (1992) Interaction of Epstein-Barr virus nuclear antigen 1 with the viral latent origin of replication. J. Virol., 66, 694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rawlins D.R., Milman,G., Hayward,S.D. and Hayward,G.S. (1985) Sequence-specific DNA binding of the Epstein-Barr virus nuclear antigen (EBNA1) to clustered sites in the plasmid maintenance region. Cell, 42, 859–868. [DOI] [PubMed] [Google Scholar]
- 6.Summers H., Barwell,J.A., Pfuetzner,R.A., Edwards,A.M. and Frappier,L. (1996) Cooperative assembly of EBNA1 on the Epstein-Barr virus latent origin of replication. J. Virol., 70, 1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frappier L. and O’Donnell,M. (1991) Epstein-Barr nuclear antigen 1 mediates a DNA loop within the latent replication origin of Epstein-Barr virus. Proc. Natl Acad. Sci. USA, 88, 10875–10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldsmith K., Bendell,L. and Frappier,L. (1993) Identification of EBNA1 amino acid sequences required for the interaction of the functional elements of the Epstein-Barr virus latent origin of DNA replication. J. Virol., 67, 3418–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su W., Middleton,T., Sugden,B. and Echols,H. (1991) DNA looping between the origin of replication of Epstein-Barr virus and its enhancer site: stabilization of an origin complex with Epstein-Barr nuclear antigen 1. Proc. Natl Acad. Sci. USA, 88, 10870–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ambinder R.F., Mullen,M., Chang,Y., Hayward,G.S. and Hayward,S.D. (1991) Functional domains of Epstein-Barr nuclear antigen EBNA-1. J. Virol., 65, 1466–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bochkarev A., Barwell,J., Pfuetzner,R., Bochkareva,E., Frappier,L. and Edwards,A.M. (1996) Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin binding protein, EBNA1, bound to DNA. Cell, 84, 791–800. [DOI] [PubMed] [Google Scholar]
- 12.Bochkarev A., Barwell,J., Pfuetzner,R., Furey,W., Edwards,A. and Frappier,L. (1995) Crystal structure of the DNA binding domain of the Epstein-Barr virus origin binding protein EBNA1. Cell, 83, 39–46. [DOI] [PubMed] [Google Scholar]
- 13.Cruickshank J., Davidson,A., Edwards,A.M. and Frappier,L. (2000) Two domains of the Epstein-Barr virus origin DNA binding protein, EBNA1, orchestrate sequence-specific DNA binding. J. Biol. Chem., 275, 22273–22277. [DOI] [PubMed] [Google Scholar]
- 14.Calos M.P. (1998) Stability without a centromere. Proc. Natl Acad. Sci. USA, 95, 4084–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hung S.C., Kang,M.-S. and Kieff,E. (2001) Maintenance of Epstein-Barr virus (EBV) oriP-based episomes requires EBV-encoded nuclear antigen-1 chromosome-binding domains, which can be replaced by high-mobility group-I or histone H1. Proc. Natl Acad. Sci. USA, 98, 1865–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shire K., Ceccarelli,D.F.J., Avolio-Hunter,T.M. and Frappier,L. (1999) EBP2, a human protein that interacts with sequences of the Epstein-Barr nuclear antigen 1 important for plasmid maintenance. J. Virol., 73, 2587–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu H., Ceccarelli,D.F.J. and Frappier,L. (2000) The DNA segregation mechanism of the Epstein-Barr virus EBNA1 protein. EMBO Rep., 1, 140–144. [DOI] [PMC free article] [PubMed]
- 18.Kapoor P., Shire,K. and Frappier,L. (2001) Reconstitution of Epstein-Barr virus-based plasmid partitioning in budding yeast. EMBO J., 20, 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frappier L. and O’Donnell,M. (1991) Overproduction, purification and characterization of EBNA1, the origin binding protein of Epstein-Barr virus. J. Biol. Chem., 266, 7819–7826. [PubMed] [Google Scholar]
- 20.Shaw J., Levinger,L. and Carter,C. (1979) Nucleosomal structure of Epstein-Barr virus DNA in transformed cell lines. J. Virol., 29, 657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Workman J.L. and Kingston,R.E. (1998) Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu. Rev. Biochem., 67, 545–579. [DOI] [PubMed] [Google Scholar]
- 22.Felsenfeld G. (1992) Chromatin as an essential part of the transcriptional mechanism. Nature, 355, 219–223. [DOI] [PubMed] [Google Scholar]
- 23.Li B., Adams,C.C. and Workman,J.L. (1994) Nucleosome binding by the constitutive transcription factor Sp1. J. Biol. Chem., 269, 7756–7763. [PubMed] [Google Scholar]
- 24.Lee D.Y., Hayes,J.J., Pruss,D. and Wolffe,A.P. (1993) A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell, 72, 73–84. [DOI] [PubMed] [Google Scholar]
- 25.Ng K.W., Ridgway,P., Cohen,D.R. and Tremethick,D.J. (1997) The binding of a Fos/Jun heterodimer can completely disrupt the structure of a nucleosome. EMBO J., 16, 2072–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perlmann T. and Wrange,O. (1988) Specific glucocorticoid receptor binding to DNA reconstituted in a nucleosome. EMBO J., 7, 3073–3079. [DOI] [PMC free article] [PubMed]
- 27.Wechsler D.S., Papoulas,O., Dang,C.V. and Kingston,R.E. (1994) Differential binding of c-Myc and Max to nucleosomal DNA. Mol. Cell. Biol., 14, 4097–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Workman J.L. and Kingston,R.E. (1992) Nucleosome core displacement in vitro via a metastable transcription factor-nucleosome complex. Science, 258, 1780–1784. [DOI] [PubMed] [Google Scholar]
- 29.Cheng L. and Kelly,T. (1989) Transcriptional activator nuclear factor I stimulates the replication of SV40 minichromosomes in vivo and in vitro. Cell, 59, 541–551. [DOI] [PubMed] [Google Scholar]
- 30.Cheng L., Workman,J., Kingston,R. and Kelly,T. (1992) Regulation of DNA replication in vitro by the transcriptional activation domain of GAL4-VP16. Proc. Natl Acad. Sci. USA, 89, 589–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li R. and Botchan,M.R. (1994) Acidic transcription factors alleviate nucleosome-mediated repression of DNA replication of bovine papillomavirus type 1. Proc. Natl Acad. Sci. USA, 91, 7051–7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simpson R.T. (1990) Nucleosome positioning can affect the function of a cis-acting DNA element in vivo. Nature, 343, 387–389. [DOI] [PubMed] [Google Scholar]
- 33.Lipford J.R. and Bell,S.P. (2001) Nucleosome positioned by ORC facilitate the initiation of DNA replication. Mol. Cell, 7, 21–30. [DOI] [PubMed] [Google Scholar]
- 34.Iizuka M. and Stillman,B. (1999) Histone acetyltransferase HBO1 interacts with the ORC1 subunit of the human initiator protein. J. Biol. Chem., 274, 23027–23034. [DOI] [PubMed] [Google Scholar]
- 35.Alexiadis V., Varga-Weisz,P.D., Bonte,E., Becker,P.B. and Gruss,C. (1998) In vitro chromatin remodelling by chromatin accessibility complex (CHRAC) at the SV40 origin of DNA replication. EMBO J., 17, 3428–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishimi Y. (1992) Preincubation of T antigen with DNA overcomes repression of SV40 DNA replication by nucleosome assembly. J. Biol. Chem., 267, 10910–10913. [PubMed] [Google Scholar]
- 37.Chan S., Attisano,L. and Lewis,P.N. (1988) Histone H3 thiol reactivity and acetyltransferases in chicken erythrocyte nuclei. J. Biol. Chem., 263, 15643–15651. [PubMed] [Google Scholar]
- 38.Simon R.H. and Felsenfeld,G. (1979) A new procedure for purifying histone pairs H2A+H2B and H3+H4 from chromatin using hydroxylapatite. Nucleic Acids Res., 6, 689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cote J., Utley,R.T. and Workman,J.L. (1995) Basic analysis of transcription factor binding to nucleosomes. Methods Mol. Genet., 6, 108–127. [DOI] [PubMed] [Google Scholar]
- 40.Barwell J., Bochkarev,A., Pfuetzner,R., Tong,H., Yang,D., Frappier,L. and Edwards,A. (1995) Purification and crystallization of the DNA-binding and dimerization domain of the Epstein-Barr virus nuclear antigen 1. J. Biol. Chem., 270, 20556–20559. [DOI] [PubMed] [Google Scholar]
- 41.Ceccarelli D.F.J. and Frappier,L. (2000) Functional analyses of the EBNA1 origin DNA binding protein of Epstein-Barr virus. J. Virol., 74, 4939–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walter P.P., Owen-Hughes,T.A., Cote,J. and Workman,J.L. (1995) Stimulation of transcription factor binding and histone displacement by nucleosome assembly protein 1 and nucleoplasmin requires disruption of the histone octamer. Mol. Cell. Biol., 15, 6178–6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harrison S., Fisenne,K. and Hearing,J. (1994) Sequence requirements of the Epstein-Barr Virus latent origin of DNA replication. J. Virol., 68, 1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frappier L., Goldsmith,K. and Bendell,L. (1994) Stabilization of the EBNA1 protein on the Epstein-Barr virus latent origin of DNA replication by a DNA looping mechanism. J. Biol. Chem., 269, 1057–1062. [PubMed] [Google Scholar]
- 45.Taylor I.C.A., Workman,J.L., Schuetz,T.J. and Kingston,R.E. (1991) Facilitated binding of GAL4 and heat shock factor to nucleosomal templates: differential function of DNA binding domains. Genes Dev., 5, 1285–1298. [DOI] [PubMed] [Google Scholar]
- 46.Vettese-Dadey M., Walter,P., Chen,H., Juan,L. and Workman,J.L. (1994) Role of the histone amino termini in facilitated binding of a transcription factor, GAL4-AH, to nucleosome cores. Mol. Cell. Biol., 14, 970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frappier L. and O’Donnell,M. (1992) EBNA1 distorts oriP, the Epstein-Barr virus latent replication origin. J. Virol., 66, 1786–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sexton C.J. and Pagano,J.S. (1989) Analysis of the Epstein-Barr virus origin of plasmid replication (oriP) reveals an area of nucleosome sparing that spans the 3′ dyad. J. Virol., 63, 5505–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alfieri C., Birkenbach,M. and Kieff,E. (1991) Early events in Epstein-Barr virus infection of human B lymphocytes. Virology, 181, 595–608. [DOI] [PubMed]
- 50.Babcock G.J., Hochberg,D. and Thorley-Lawson,D.A. (2000) The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity, 13, 497–506. [DOI] [PubMed] [Google Scholar]
- 51.Niller H.H., Glaser,G., Knuchel,R. and Wolf,H. (1995) Nucleoprotein complexes and DNA 5′-ends at oriP of Epstein-Barr virus. J. Biol. Chem., 270, 12864–12868. [DOI] [PubMed] [Google Scholar]
- 52.Hsieh D.-J., Camiolo,S.M. and Yates,J.L. (1993) Constitutive binding of EBNA1 protein to the Epstein-Barr virus replication origin, oriP, with distortion of DNA structure during latent infection. EMBO J., 12, 4933–4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yates J.L., Camiolo,S.M. and Bashaw,J.M. (2000) The minimal replicator of Epstein-Barr virus oriP. J. Virol., 74, 4512–4522. [DOI] [PMC free article] [PubMed]
- 54.Loeb D.D., Sung,N.S., Pesano,R.L., Sexton,C.J., Hutchison,C.,III and Pagano,J.S. (1990) Plasmid origin of replication of herpesvirus papio: DNA sequence and enhancer function. J. Virol., 64, 2876–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]