

Abstract
Excessive endoplasmic reticulum (ER) stress leads to cell loss in many diseases, e.g., contributing to endothelial cell loss after spinal cord injury. Here, we determined whether ER stress-induced mitochondrial dysfunction could be explained by interruption of the focal adhesion kinase (FAK)-mitochondrial STAT3 pathway we recently discovered. ER stress was induced in brain-derived mouse bEnd5 endothelial cells by thapsigargin or tunicamycin and caused apoptotic cell death over a 72 h period. In concert, ER stress caused mitochondrial dysfunction as shown by reduced bioenergetic function, loss of mitochondrial membrane potential and increased mitophagy. ER stress caused a reduction in mitochondrial phosphorylated S727-STAT3, known to be important for maintaining mitochondrial function. Normal activation or phosphorylation of the upstream cytoplasmic FAK was also reduced, through mechanisms that involve tyrosine phosphatases and calcium signaling, as shown by pharmacological inhibitors, bisperoxovanadium (bpV) and 2-aminoethoxydiphenylborane (APB), respectively. APB mitigated the reduction in FAK and STAT3 phosphorylation, and improved endothelial cell survival caused by ER stress. Transfection of cells rendered null for STAT3 using CRISPR technology with STAT3 mutants confirmed the specific involvement of S727-STAT3 inhibition in ER stress-mediated cell loss. These data suggest that loss of FAK signaling during ER stress causes mitochondrial dysfunction by reducing the protective effects of mitochondrial STAT3, leading to endothelial cell death. We propose that stimulation of the FAK-STAT3 pathway is a novel therapeutic approach against pathological ER stress.
Keywords: cell death, endoplasmic reticulum stress, focal adhesion kinase, integrin, mitochondria
Graphical abstract
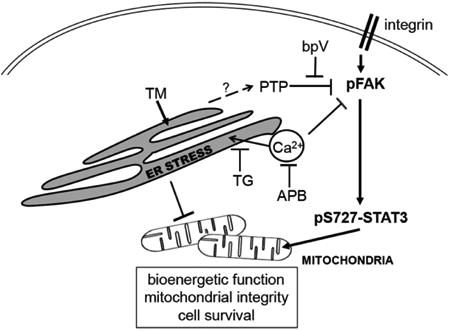
Proposed FAK-STAT pathway involvement in ER stress. Under physiological conditions, the integrin signaling effector FAK promotes phosphorylation of S727-STAT3 which leads to its mitochondrial translocation to promote mitochondrial bioenergetics and integrity, and cell survival. ER stress induced by TM or TG decreases pFAK through protein tyrosine phosphatase (PTP) activity (blocked by bpV) or high calcium levels (blocked by APB), leading to decreased mitochondrial pS727-STAT3 and subsequent dysfunction.
1. Introduction
The ER is responsible for synthesis and proper folding of cellular proteins. A variety of insults, such as inflammation, trauma and hypoxia disrupt ER function and cause accumulation of unfolded proteins. This can induce the unfolded protein or ER stress response, which can be beneficial, but when excessive or prolonged causes apoptosis. The CCAAT-enhancer-binding protein homologous protein (CHOP) transcription factor is up-regulated after ER stress [1] and can initiate apoptotic cell death [2, 3]. ER stress can be induced by thapsigargin (TG), which blocks the sarcoplasmic/ER calcium ATPase resulting in increased cytoplasmic calcium [4] and tunicamycin (TM), which interferes with protein glycosylation, causing an unfolded protein response [5, 6]. Endothelial cells undergo apoptosis in response to TG and TM which involves caspase-3 activation [7, 8]. Excessive ER stress and CHOP induction cause CNS microvascular endothelial cell death in vitro and in vivo after spinal cord injury [9, 10], which is responsible for ensuing secondary tissue loss [11, 12]. Unchecked ER stress also causes mitochondrial dysfunction and damage in a number of cell types through mitochondrial membrane pore formation, which is associated with ER-stress induced apoptosis [13]. ER stress-induced damage is mediated in part through excessive calcium influx [14] through the mitochondria-associated ER membrane contact sites [15, 16]. Both the mitochondria and the ER can take up calcium from the cytosol to maintain low cytosolic calcium levels. ER stress leads to a calcium imbalance by altering uptake/release of calcium from the ER [15].
Vascular endothelial cells are dependent on specific transmembrane integrin receptors binding to extracellular matrix molecules in the basement membrane, and their detachment leads to apoptosis, possibly through reduced FAK signaling [17, 18]. Integrins prevent apoptosis through FAK-AKT signaling [19] and inhibiting mitochondria-associated bit1 [20]. We discovered a selective integrin signaling pathway that inhibits CNTF expression, involving FAK and the S727 residue of the downstream STAT3 transcription factor [21]. STAT3, when phosphorylated on S727, translocates to the mitochondria instead of the nucleus and plays a major, non-transcriptional role in preserving mitochondrial function [22]. Mitochondrial localized STAT3 is known to reduce mitochondrial damage markers such as calcium overload, ROS production and mitochondrial permeability transition pore (mPTP) formation [23-25]. We have found that antagonists of integrins, FAK, and STAT3 rapidly and substantially reduce mitochondrial bioenergetics leading to endothelial cell loss [26].
FAK is a cytoplasmic non-receptor protein tyrosine kinase which depends on Y397 phosphorylation for its activation and subsequent downstream signaling [27]. It is dephosphorylated by tyrosine phosphatases such as SHP2 [28, 29], PTP-PEST/PTPN12 [30-32], and PTP 1B [29], which can be inhibited by tyrosine phosphatase inhibitors [33]. High calcium transients can also dephosphorylate FAK in cultured neurons [34].
In this study, we determined whether excessive ER stress caused by TG and TM can induce mitochondrial dysfunction, leading to endothelial cell loss in vitro and whether interrupted FAK-STAT3 signaling might contribute to this. We also tested the capability of calcium channel and tyrosine phosphatase inhibitors to maintain FAK signaling and endothelial cell survival under ER stress conditions.
2. Experimental Procedures
2.1. bEnd5 endothelial cell culture
The establishment of the bEnd5 endothelial cell line and its characterization was described previously [35-37]. bEnd5 cells were maintained at 37°C in a humidified incubator in an environment of 5% CO2 and 95% air in Dulbecco modified Eagle's medium (DMEM). Media was supplemented with 3 mM glutamine, 1% non-essential amino acids, 1 mM Na-pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin and 10% fetal bovine serum (FBS). All reagents were from Gibco. For cell culture experiments, bEnd5 cells were counted in a hemocytometer and seeded at a density of 0.5 × 106 cells per 100 mm plate. After 3 days (at ∼60% confluence), the cells were treated with 10 μg/ml tunicamycin (TM, cat# T7765, Sigma) or 300 nM thapsigargin (TG, cat# 9033, Sigma) for 24, 48 and 72 h to induce ER stress. For co-treatments, 2 μM stattic (cat# 2798, Tocris), 10 μM APB (cat# D9754, Sigma), or 3 μM bpV (potassium bisperoxo (1,10-phenanthroline) oxovanadate V) [bpV(phen)]; a gift from Dr. Alan Shaver [38], were added to the media for the same period of time as TM/TG. Afterwards, the cells were processed for mitochondrial fractionation, whole cell lysate preparation, or incubated with mitophagy or membrane potential markers (see below). The cell numbers were quantified using Image J software and images were captured with a 10X and 40X objective (Olympus Q Color 3, CKX41 microscope). Averages were calculated from five fields per well. To confirm cell death, cells were stained with propidium iodide (PI, 2 μg/ml; Cat#: P3566, Life Technologies) and counterstained with Hoechst 33342 (50 μM, Cat#: H1399, Molecular Probes) according to the manufacturer's protocol.
2.2. Western blotting
Proteins were extracted from whole cells using a freeze-thaw cycle with 1% RIPA lysis buffer (cat# R0278, Sigma) supplemented with protease and phosphatase inhibitors. The samples were mixed 5:1 (v:v) with Laemmli loading buffer plus 5% β-mercaptoethanol and heated for 5 min at 95°C. Then, 15-30 μg mitochondrial or 30 μg of whole cell protein lysate samples were loaded in 4-20% Criterion Tris-HCl precast gels (cat#3450033, Bio-rad Lab, Hercules, CA, USA) and run for 2-3 h (25mA, 100V). This was followed by overnight wet transfer to PVDF membranes using 30 volts. After blocking with 5% milk in TBST (TBS, 0.1% Tween-20) for 4 h, the membranes were incubated overnight at 4° C with primary antibodies diluted in TBST with 0.5% milk. The antibodies (Cell Signaling Technology) were against Parkin (1:1,000, cat#32282), PDH (1:5,000, cat#3205), pS727-STAT3 (1:400, cat#9134), total STAT3 (1:1,000, cat#12640), pFAKY397 (1:500, cat#3283), total FAK (1:1,000, cat#3285), cleaved caspase 3 (1:1000, cat#9661) and a-tubulin (1:10,000, cat#2125). LC3 antibody was from MBL (1:3,000, cat# M186-3). The next day, the membranes were washed 2X with TBST buffer and then incubated with species-appropriate HRP-conjugated secondary antibodies (1:2,000, cat#7074 and 7076) for 1.5 h. After the secondary antibody incubation, three washes with TBST were done, all at room temperature. Chemiluminescence substrate (ECL, cat#34080, ThermoFisher) was added and the bands were visualized and analyzed by the Li-Cor Odyssey FC (Lincoln, NE) quantitative western blotting system and Image Studio application software. Integrated optical densities of pFAK in each sample was normalized to the value of total FAK protein. Then the values in all groups were normalized to the average of controls at 24 hrs. Each time point included 4 or 5 independent experiments.
2.3. LDH release assay
LDH release is a marker of cell death [39] and was measured using a Promega kit according to the manufacturer's protocol (Cat# G1780). Briefly, 50 μl of cell culture medium was taken from each treatment condition, mixed with 50 μl substrate and maintained at room temperature for 30 min. Afterwards, stop solution (50 μl) was added and absorbance measured at 492 nm in a 96 well plate. Calculations were performed by subtracting the background (medium only) and then normalizing treatments to untreated controls and expressing the result as a percentage. Lysed cells were used as a positive control.
2.4. Mitochondrial membrane potential
Changes in mitochondrial membrane potential were determined by changes in mitochondrial permeability to the cationic red-orange dye tetramethylrhodamine methyl ester (TMRM, cat# T668, Invitrogen). TMRM accumulation within mitochondria is dependent on the mitochondrial transmembrane potential. Freshly prepared TMRM was added at 20 nM to the treated cells in a 96 well plate and incubated in a CO2 tissue culture incubator for 30 min. DAPI (4′,6-diamidino-2-phenylindole, cat# D3571, Invitrogen) was added at 0.2 μg/ml for the last 15 min of TMRM incubation. DAPI stains the nucleus and was used to normalize TMRM fluorescence to the cell number in each well. The intact cells were washed once with 200 μl HBSS followed by immediate measurement of fluorescence intensities (λex 540 nm, λem 575 nm for TMRM and λex 340 nm, λem 460 nm for DAPI) in a spectrofluorometer.
2.5. Mitochondrial bioenergetics
Mitochondrial bioenergetic function in bEnd5 cells was determined by measuring the oxygen consumption rate (OCR) with a Seahorse XF24 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA).The cells were seeded at 20,000 cells per XF24 culture plate well and next day were treated with 300 nM TG or 10 μg/ml TM for 24 h, all in regular culture medium (see above). Afterwards, the cells were washed once with XF serum-free base media supplemented with 2.5 mM glucose and 1 mM sodium pyruvate. This was followed by a 1 h incubation without CO2. The sensor cartridges were prepared according to the manufacturer's instructions. Baseline measurements of OCR were taken before sequential injection of 5 μM oligomycin (495455, Millipore), 1 μM FCCP (0453, Tocris), and 10 μM antimycin A (A8654, Sigma). The mitochondrial basal OCR, ATP production, maximum reserve capacity and maximum respiratory capacity were calculated as described [40].
2.6. Mitophagy markers: microscopy
The occurrence of mitophagy within the individual bEnd5 cells was determined by lysosomes associating with mitochondria, one of the defining steps of mitophagy. Cells were seeded at 105 cells per 35 mm clear bottom culture dishes (P35G-1.5-14-c, MarTek Corp, Ashland, MA) and treated with TG or TM for 24, 48 and 72 h, as above. Afterwards, the cells were washed twice with media and incubated with 200 nM mitotracker green (cat# M7514, Invitrogen) and 75 nM lysosomal red dye (cat# L7528, Invitrogen) in 500 μl of media for 20 min at 37° C. After washing 3 times with PBS, the cells were imaged with a Leica SP8 confocal microscope. Co-localization of green and red pixels was quantified in 3 dimensions in Z-stacks using Leica Application Suite X software.
2.7. Isolation of mitochondrial fraction from bEnd5 cells
Mitochondria were isolated as previously described [41] with minor modifications. All steps were performed at 4°C. Cells were lysed in mitochondrial isolation buffer (1 mM EGTA, 75 mM sucrose, 5 mM HEPES, 225 mM mannitol, 1 mg/ml, BSA, supplemented with a cocktail of protease and phosphatase inhibitors) and homogenized in a Dounce glass homogenizer with 40 strokes. After centrifugation at 1500 g for 10 min to remove the unbroken cells and nuclei, and decanting of the supernatant, the supernatant was then centrifuged at 15,000 g for 20 min. The pellet containing the mitochondrial fraction was collected and resuspended in isolation buffer without BSA.
2.8. CRISPR-Cas9 mediated STAT-3 KO in bEnd5 cells
STAT3 null bEnd5 cells were produced by CRISPR-Cas9 technology as described previously [26]. Briefly, a STAT3 gRNA-Cas9 plasmid with a puromycin resistance gene was transfected into bEnd5 cells. After 24 hours, cells were treated with 2 μg/ml puromycin for 4 days to remove non-transfected cells. Puromycin resistant cells were maintained for ∼14 days then screened by PCR and Western blotting to confirm STAT3 deletion [26]. Cells were passaged normally and used for reintroduction of STAT3 WT or mutant plasmids.
2.9. Generation of GFP tagged STAT3 wild type (WT) and mutant (S727A, S727D) plasmids
Wild type STAT3 was PCR amplified from bEnd5 cDNA, produced by reverse transcription using oligo dT primers and 2 μg of template RNA with NEB's ProtoScript II kit according to manufacturer's instructions (Cat # E6560S, NEB). All PCR reactions were performed with a high fidelity Taq polymerase, using 2 μl of cDNA as a template according to kit guidelines (Cat#KK2102, KAPA Biosystems). Primers for amplifying STAT3 were: FWD 5′GGCTCCGGACTCAGATCTAGAAGGATGGCTCAGTGGAACCAG, 3′ REV 5′ACGCCGGAATTC TTTTAATTTAAAGAGGAACCTCTGGG 3′. GFP was PCR amplified with the following primers: FWD 5′ CCC AAGCTT ACCATGGTGAGCAAGGGC, 3′ REV 5′TCTAGATCTGAGTCCGGAGCC CTTGTACAGCTCGTCCATG 3′ using plasmid DNA containing GFP as template. Reactions were gel purified and products used at a 1:1 molar ratio for subsequent fusion PCR reactions. Fusion PCR reactions were performed with primers FWD:5′ CCC AAGCTT ACCATGGTGAGCAAGGGC 3′ and REV 5′ ACGCCGGAATTC TTTTAATTTAAAGAGGAACCTCTGGG ‘3. The resulting fragment was gel purified, confirmed by restriction digest, and directionally cloned into pcDNA 3.1 (+) vector before being transformed into DH5α E.Coli according to standard protocol [26]. The constitutionally active, phosphomimetic, STAT3 S727D mutant was generated using the Q5 mutagenesis kit (Cat# E0554S, NEB) according to manufacturer's instructions with the following PCR primers 5′ CCTGCCGATGGACCCCCGCACTT 3′, and REV 5′ TCAATGGTATTGCTGCAGGTC 3′. The functionally inactive STAT3 S727A mutant was produced with the following PCR primers: FWD 5′ GCCGATGGCCCCCCGCACTT -3′ REV 5′ AGGTCAATGGTATTGCTGCAGGTCGTT -3′. GFP fused WT STAT3 cloned into pcDNA 3.1 (+) vector served as the template for Q5 mutagenesis. Successful mutagenesis was confirmed by sequencing. It was observed through sequencing that Serine-701 was not present in the wild type, S727D and S727A STAT3 clones. Plasmids were grown in competent DH5α E.coli and isolated using an endotoxin free midi-prep kit (Cat# D6915-03, Omega Biotek). bEnd5 cells were transfected using lipofectamine 3000 at a final concentration of 0.5% in penicillin-streptomycin free culture medium with 1 μg plasmid/well in a 24 well plate. Transfection was visualized by GFP fluorescence.
2.10. Transfection of GFP-fused plasmids
Due to a relatively low transfection efficiency in these cells (5-15%), we fusion tagged these STAT3 isoforms with an N-terminal GFP tag to visualize expression of STAT3 and to rule out cells that did not take up the plasmid. Transfections of GFP-pcDNA 3.1 (control) and GFP-STAT3-pcDNA 3.1 fused plasmids (WT-STAT3, S727A-STAT3, S727D-STAT3) were performed with lipofectamine 3000 as mentioned above. After 24 h of transfection, media was changed to normal bEnd5 media and treated with TM (10 μg/ml) for 72 h. At 24 h after transfection, GFP+ cells were quantified from 5 fields per well and normalized to total cell numbers (overlayed on brightfield) to determine transfection efficiency. We counted on average ∼70 cells/experiment from 5 fields/well over 4 independent experiments. To account for potential differences in transfection efficiency between groups and experiments, the number of transfected cells at 72 h was counted the same way and expressed as a percentage of GFP+ cells at 24 h. Images were taken using a fluorescent microscope (Olympus Q Color 3, CKX41 microscope).
2.11. Statistical Analysis
All data were expressed as mean ± SEM. Statistical significance was set at p<0.05 and calculated using Graphpad Prism software. For the experiments with one variable (drug treatment or time), a one-way ANOVA followed by Bonferroni post hoc multiple comparison was used. For the experiment with two variables (time and drug treatment), a two-way ANOVA followed by Tukey post hoc multiple comparison was used.
3. Results
3.1. Excessive ER stress causes death of endothelial cells
ER stress induced by TG and TM caused a loss of mouse brain-derived bEnd5 cells by 72 h (Fig. 1 A, B). The stressed cells which were adhered appeared swollen and more elongated and progressively more rounded, presumably dying, cells were observed (Fig. 1 A). The cell damage and loss seemed to be more severe with TM than with TG. ER stress was induced as early as 24 h and to a similar extent as at 48 and 72 h as shown by induction of CHOP protein (Fig. 1D), consistent with the idea that prolonged ER stress is needed to kill these cells. The extent of cell death was confirmed by measuring LDH release, which showed a clear increase at 72 h (Fig 1D), consistent with the cell numbers. TM values were in the same range as lysed cells, indicative of severe cell damage. Propidium Iodide (PI) staining, which occurs only in dying/dead cells, was observed in TG- and TM-treated cells (Fig. 1 E). Endothelial cells are known to undergo apoptosis involving caspase-3 activation in response to TG and TM [7, 8]. Here, ER stress also caused apoptosis as shown by fragmented and condensed nuclei (Fig. 1E) and the appearance of cleaved caspase-3 in TG and, much more so, TM-treated cells (Fig. 1F).
Figure 1. ER stress causes mitochondrial dysfunction and endothelial cell loss.
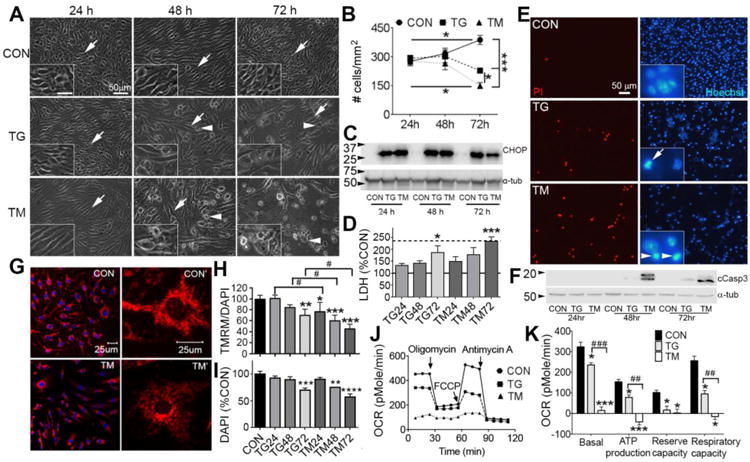
bEnd5 endothelial cells were treated with TM or TG for 24, 48 or 72 h to induce ER stress. A) Microscopic images show cell swelling from 48 h and a clear loss of cells by 72 h compared to control (CON) culture conditions. Arrows indicate adhered cells and arrowheads indicate rounded dying cells. TM treatment caused noticeably more damage. Insets show a 2 fold higher magnification of selected fields (scale bar = 25 μm). B) Quantification shows the progressive cell loss under ER stress conditions with TM causing more cell loss than TG. Data are mean ± SEM from three independent experiments for each group. *p<0.05, ***p<0.001. C) ER stress as shown by CHOP protein expression in a Western blot was evident already at 24 h and similar between TG and TM. Tub = α-tubulin loading control. D) In separate experiments, LDH release confirmed the loss of cells at 72 h of ER stress. The solid horizontal line indicate control values (100%) and the stippled line the positive control values of lysed cells. N=3 independent experiments. E) Fluorescence images of cells stained with PI for dead cells at 72 h of TG and TM treatment. The number of cells was clearly decreased as shown by the nuclei (Hoechst) of the same fields. The insets show examples of fragmented (arrow) and condensed (arrowheads) nuclei seen in apoptotic cells under TG and TM conditions. F) Western blot of cell lysates clearly indicates the presence of cleaved caspase-3 (cCasp3) as early as 48 h and more so in TM treated cells. N= 3 for the 72 h time point. G) Cells treated with TM for 24 h show reduced mitochondrial uptake of TMRM (red) in confocal microscope images, indicating a reduction in mitochondrial membrane potential. DAPI (blue) stains nuclei. H) Spectrofluorometric measurement of TMRM normalized to DAPI fluorescence, to account for cell numbers, shows a progressive reduction in mitochondrial membrane potential. Data are percent of control ± SEM from three independent experiments. *p<0.05, ** p<0.01, ***p<0.001 compared to control, # p<0.05 TG vs TM. I) DAPI measurements from the same cell lysates confirm the cell death most notably at 72 h. J) ER stress for 24 h causes bioenergetic failure as measured by oxygen consumption rate (OCR) and shown in a representative Seahorse XF24 trace. H) Quantification of basal respiration (first three values), ATP production (difference after adding oligomycin), maximum reserve capacity (difference after adding FCCP), and maximum respiratory capacity (difference after adding antimycin). N = 3 independent experiments. * p<0.05, *** p<0.001 compared to control, #p<0.05, ## p<0.01, ### p<0.001, TG vs TM
3.2. Excessive ER stress causes mitochondrial dysfunction and damage in endothelial cells
Mitochondrial membrane pore formation is a marker of mitochondrial damage, resulting in reduced membrane potential and is key to ER stress-induced apoptosis [13]. Reduced membrane potential was observed at 72 h with TG and 24 h with TM and more so at 72 h, as documented by TMRM fluorescence (Fig. 1G, H). Cell death in this experiment was confirmed by the DAPI measurements (Fig. 1I), and were consistent with the cell counts (Fig. 1B) and LDH release (Fig. 1D). ER stress caused a marked reduction in mitochondrial bioenergetic function as early as 24 h and more extensively with TM than TG (Fig. 1J, K). The great reduction in reserve and respiratory capacity is indicative of loss of mitochondrial membrane potential and mitochondrial damage.
Severe mitochondrial damage induces mitophagy, which involves mitochondrial-lysosomal co-localization, and this was also seen after TG and TM induced ER stress in the bEnd5 cells. Thus, lysosomal content per cell was increased and the number of contacts with mitochondria were increased, as shown by co-localization in confocal images (Fig. 2A-D). Quantification of co-localized pixels in confocal Z-stacks suggests that mitophagy was observed at 48 h and 72 h with both TG and TM (Fig. 2E). Increased mitophagy also was confirmed by the increase in the initiating proteins LC3II and parkin that translocate to mitochondria, in mitochondrial cell fractions (Fig. 2F). General autophagy was also increased as shown in the cytoplasmic fractions.
Figure 2. ER stress induces mitophagy in bEnd5 cells.
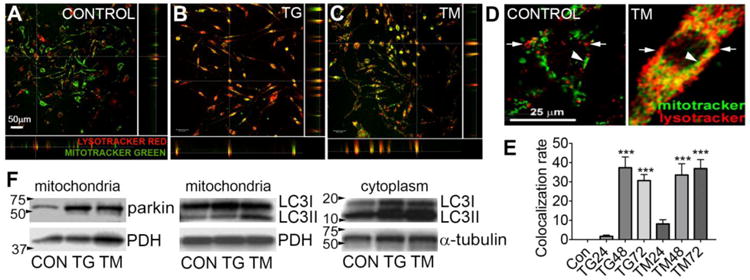
Confocal images show that compared to controls (A), TG (B) and TM (C) increase lysosomes (identified by lysotracker green) and increase their association with mitochondria (identified by mitotracker red), at 48 h of ER stress. D) Higher magnification images showing individual lysosomes (arrowheads) and mitochondria (arrows) in controls with essentially no co-localization, but a high degree of overlap (yellow) after ER stress, shown here with TM. E) Analyses of co-localization in 3-D Z-stacks showed significantly greater co-localization of green and red pixels with TG and TM at 48 and 72 h compared to controls (CON). N = 3 independent experiments. ***p<0.001 compared to control. F) TG and TM induced mitophagy also shown by Western blots of mitochondrial cell fractions probed for parkin, which translocates to the mitochondria as part of the mitophagy process. PDH = mitochondrial loading control. The autophagy and mitophagy marker, LC3II was increased in both mitochondrial and cytoplasmic fractions. Blots are representative of n=2.
3.3. ER stress causes loss of mitochondrial pS727-STAT3 in concert with reduced pFAK
PS727-STAT3 is known to translocate to the mitochondria and promote their function and integrity. Here, ER stress caused a progressive reduction in mitochondrial pS727-STAT3, most notably at 72 h, and more so with TM than with TG (Fig. 3A). The reduction at 72 h, was to a similar level as seen with the STAT3 inhibitor stattic. Levels of total STAT3 in the mitochondria appeared similar between controls and TG or TM, and at all time points (Fig. 3A), suggesting that the reduction was not due to altered translocation. In whole cell lysates, a similar time-dependent reduction in pS727-STAT3 without changes in total STAT3 was also observed (Fig.3B), suggesting that it is caused by signaling events rather than STAT3 synthesis or changes in translocation.
Figure 3. ER stress reduces mitochondrial pS727-STAT3 and pFAK.
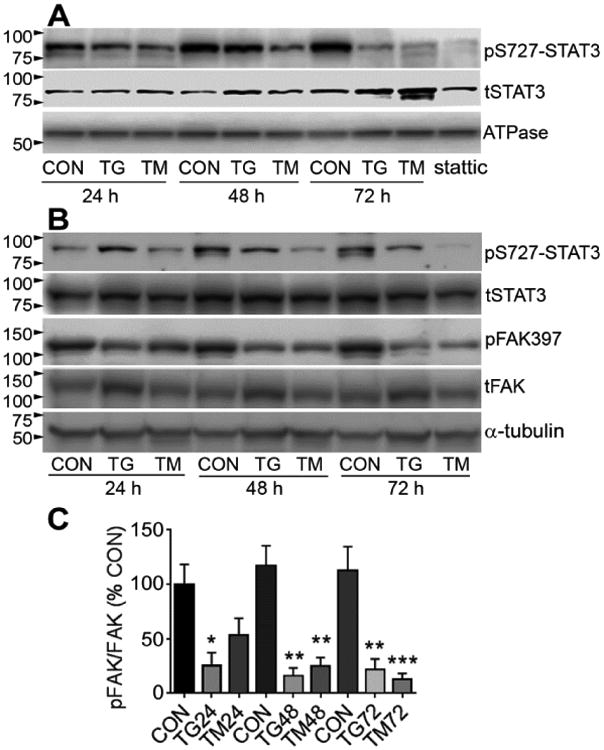
A) Treatment with TG and TM caused a progressive and pronounced decrease in pS727-STAT3 in the mitochondrial cell fraction, as shown by Western blot (representative of n=3).Treatment for 24 h with the STAT3 inhibitor stattic was used as a positive control. tSTAT3 =total STAT3, which remained stable under ER stress over time. Mitochondrial ATPase was used as a loading control. B) pS727-STAT3 was also reduced in whole cell lysates. A decrease in phosphorylation of FAK on Y397, which is critical for its activation, was seen in largely the same time-dependent manner. tFAK = total FAK, α-tubulin was used as loading control. Blots are representative of n=3. C) Densitometry of blots probed for pFAK/total FAK shows significant decreases in pFAK397 with TG and TM treatment. N=4-5 independent experiments.
Our previous study showed that integrin-FAK signaling promotes STAT3 phosphorylation specifically on the S727 residue [21]. Here, ER stress induced by TG and TM caused a reduction in phosphorylation of the key Y397 residue of FAK, as early as 24 h (Fig. 3B), as confirmed by densitometry (Fig. 3C). Total FAK densitometry values, normalized to α-tubulin, did not change (data not shown).
3.4. Calcium and phosphatases reduce pFAK during ER stress, contributing to reduced pS727
A reduction of pFAK could be due to reduced enzymatic cleavage but we did not detect the expected 90 kD cleavage product in blots for total FAK [42], suggesting that dephosphorylation played the main role. In fact, treatment with the general tyrosine phosphatase inhibitor, bpV (bisperoxovanadium), preserved pFAK at 48 and 72 h of ER stress, most notably after TM and to levels similar as seen under control conditions without ER stress (Fig. 4A). FAK phosphorylation can also be affected by increased intracellular calcium [34], which is also a hall-mark of ER stress, especially after TG treatment. Indeed, APB, a calcium channel blocker, had a remarkably protective effect preserving pFAK at normal levels under ER stress conditions caused by 72 h of TG (Fig. 4B, upper rows). The APB effects were less striking with TM (Fig. 4B). APB treatment also resulted in a complete preservation of pS727-STAT3 in the same cells under TG, but not TM, conditions (Fig. 4B, middle rows). The reduced levels of pS727-STAT3 were not affected by bpV treatment under the ER stress conditions, perhaps because bpV affects many different signaling pathways, thus interfering with intermediaries between FAK and STAT3.
Figure 4. ER stress reduces pFAK through tyrosine phosphatases and calcium.
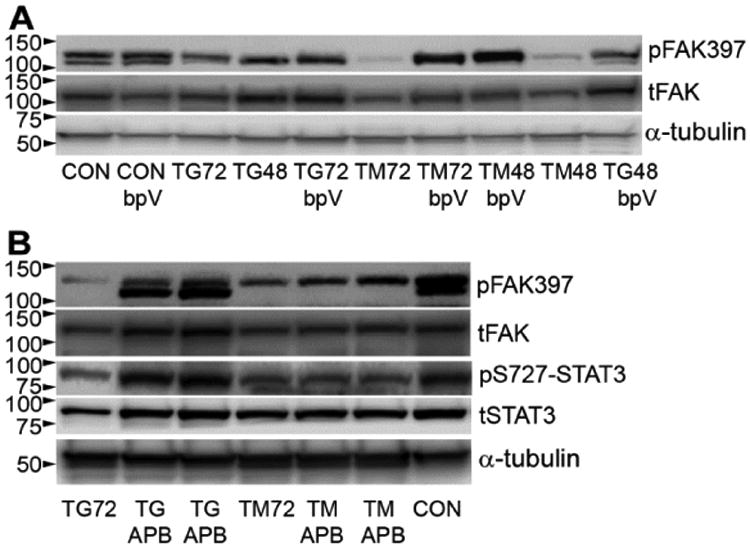
bEnd5 cells were treated with TG or TM for 48 or 72 h with or without the tyrosine phosphatase inhibitor bpV or calcium channel blocker APB.A) bpV prevented the loss of pFAK caused by ER stress, as shown in a Western blot. tFAK = total FAK, α-tubulin was used as a loading control. B) APB completely rescued pFAK against TG, but not against TM. This was accompanied by similar effects on pS727-STAT3. tSTAT3 = total STAT3. Blots in A and B are representative of n=3.
3.5. Calcium channel inhibition promotes endothelial cell survival damage during ER stress
The calcium channel blocker APB did not noticeably affect the changes in bEnd5 cell morphology caused by TG or TM (Fig. 5A) but improved their viability to ∼70% in the presence of TG for 72 h, compared to 50% survival seen with TG alone (Fig. 5B). APB had no significant survival-promoting effect under TM conditions. The bpV treatment did not noticeably affect changes in cell morphology after TG, but preserved more cells with an adherent morphology under the TM condition (Fig. 5A). The bpV treatment improved cell survival under both TG and TM conditions (Fig. 5B). In a different set of experiments, LDH release into the media, another marker for cell death, affirmed the cell counts (Fig. 5C).
Figure 5. APB and bpV rescue endothelial cells from ER stress.
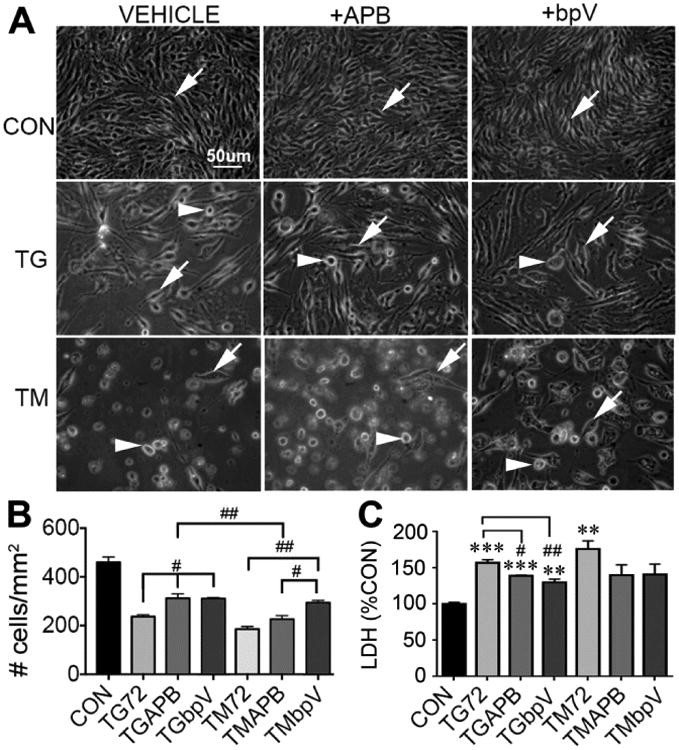
bEnd5 cells were treated with TG or TM for 72 h followed by cell counts or mitochondrial membrane potential (TMRM) measurements. A) Representative images show the expected loss of cells in with TG and TM with vehicle treatment (left column) compared to controls (CON). APB (middle column) appeared more protective against TG than TM. The bpV (right column) treatment appeared to protect the cells equally against TG and TM, but had different effects on the cell morphology. Arrows indicate adhered cells and arrowheads rounded dying cells. P<0..001 compared to control (CON) for all conditions. # p<0.05, ## p<0.01, ### p<0.001 B) Quantification showed that APB protected∼20% of the cells against TG, but not TM, and that bpV protected against TG and TM. Data are mean ± SEM from three independent experiments. C) In separate experiments, LDH release by cells, a marker for cell death, confirmed the cell counts. * p<0.05, ** p<0.01, *** p<0.001 compared to control, ## p<0.01, ### p<0.001 for treatment vs. TG alone. N = 3 independent experiments.
3.6. STAT3 protects endothelial cells against ER stress via residue S727
To determine the protective effects of STAT3 against ER stress, we overexpressed WT, S727A or S727D mutants in STAT3 KO cells. Counts of GFP+ cells at 72 h were normalized to those in the same wells seen at 24 h. STAT3 KO cells transfected with GFP alone (Fig. 6A) often showed an aberrant morphology indicative of dying cells compared to WT STAT3 (Fig. 6B) in response to 72 hours of TM treatment. Most cells transfected with S727A mutants were rounded and condensed (Fig. 6C), indicative of cell death. Cells transfected with the constitutively active S727D mutants, which mimic pS727-STAT3, looked healthy (Fig. 6D). More cells were lost when transfected with GFP only (∼60%) after 72 hours of TM treatment in the controls than those transfected with WT STAT3 (∼40%), whereas more S727A cells were lost than with WT (∼75%, Fig. 6E). This suggests that S727-STAT3 is protective against TM. S727D mutants were significantly more protected compared to 727A but not different from WT, suggesting that there is either a maximum level of potential protection by phosphorylated S727-STAT3 or that constitutively active S727-STAT3 also has detrimental effect that counterbalance the beneficial effects.
Figure 6. S727-STAT3 protects endothelial cells against ER stress.
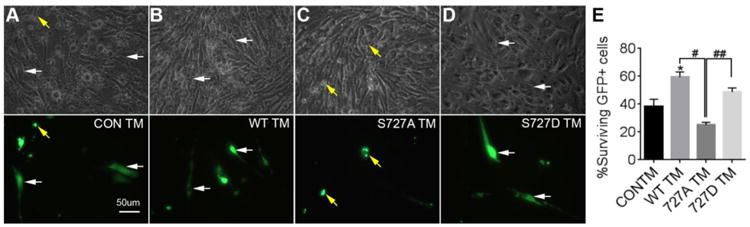
STAT3 KO bEnd5 endothelial cells were transfected with GFP-containing constructs and treated with TM for 72 h. Top panels are bright-field images. Bottom images are fluorescent images. A) With control transfections (GFP alone, CON TM), many transfected cells show aberrant small rounded morphology (yellow arrows). B) WT-STAT3-GFP cells looked for the most part healthy, i.e., long-flattened attached cells (white arrows). C) S727A mutants were mostly rounded, whereas D) S727D mutants looked healthy. E) Significantly more GFP+ cells survived TM treatment when overexpressing GFP-WT STAT3 relative to control plasmids. The number of cells transfected with S727A mutants was significantly lower than the WT and S727D mutant transfected cells. Cell counts performed at 72 h are representative of 4 independent experiments, normalized to transfection efficiency obtained at 24 h. * p<0.05 compared to control. # p<0.05, ## p<0.01 compared to S727A.
4. Discussion
One of the main findings of this study is that ER stress causes reductions in pS727-STAT3 at the same time as mitochondrial dysfunction, and when substantial loss of pS727-STAT3 occurs, endothelial cells die. Our mutant data suggests that this very functionally important serine residue both protects cells against and is a target of ER stress. This phosphorylated form of STAT3 is known to be important for mitochondrial function in a non-transcriptional manner [22, 26, 43]. The role of ER stress-induced loss of pS727-STAT3 in mitochondrial failure is also suggested by our findings that it is more severe with TM than with TG treatments, and that TM causes more bioenergetic failure, mitophagy and cell death. Moreover, only the APB treatment preserved pS727-STAT3, and only against TG induced ER stress. The tyrosine phosphatase inhibitor, bpV, which rescued the cells, did not rescue pS727-STAT3. A protective role for pS727-STAT3 under ER stress is consistent with what is known about its non-transcriptional, mitochondrial, role. Mitochondrial STAT3 reduces mitochondrial damage markers such as calcium overload and ROS production, as well as mPTP formation [23, 24, 44], one of the critical and last steps in mitochondrial failure [45]. Mitochondria play a role in apoptosis [46], among other factors, by releasing cytochrome-c through the mPTP, which is inhibited by STAT3 [43]. ER stress causes mitochondrial damage, resulting in the terminal step of mPTP formation [47, 48]. Lengthy bioenergetic dysfunction can induce apoptosis [46]. Our data suggests that ER stress also causes early mitochondrial dysfunction by reducing bioenergetic function at 24 h when reductions in pS727-STAT3 were seen, though less pronounced than at later times. Others have shown that STAT3 knockdown or S727A mutations cause a reduction in mitochondrial bioenergetic function [22], which we also observed and confirmed with a pharmacological inhibitor [26]. The association of a mitophagy initiator protein, parkin with mitochondria has also been found to promote bioenergetic function under ER stress conditions [49, 50]. However, here, mitophagy markers were not increased at 24 h when ATP production was severely compromised.
Thus, it seems that the reduction in pS727-STAT3 plays a key role in driving the sequence of events that leads to ER stress-induced cell loss in endothelial cells. Our data are consistent with an evolving pathological mechanism that includes reduced bioenergetics, failure of mitochondrial membrane potential, damage to mitochondria as indicated by mitophagy, and finally cell death. Earlier on, mitophagy of damaged mitochondria might play a protective role [51, 52], as also suggested by our finding of substantial early mitophagy. Thus, at 48 h of ER stress cell loss was still minimal. On the other hand, excessive autophagy can lead to cell death [53-55], which we observed at 72 h. The reduced protective role of STAT3 in mitochondria is most likely not the only contributor to ER stress-induced cell loss. This is suggested by our finding that APB provided complete protection of pS727-STAT3 at 72 h of TG treatment, while rescuing only ∼20% of the endothelial cells. Also, CHOP, a major effector of ER stress, was increased to similar levels at 24 h as at later times, while the loss of pS727-STAT was more protracted. Other contributors to ER stress pathology involve other ER stress signaling pathways, mitochondrial calcium toxicity, increased mitochondrial fission and apoptotic signaling [56-58].
Despite similar levels of ER stress, as indicated by CHOP protein expression, TM was overall more devastating to the endothelial cells, with greater reductions in bioenergetic function, pSTAT3, and cell survival. The mechanism(s) underlying these differences remain to be determined. TM-mediated ER stress is effected through the UPR, which results in toxic calcium influx from the ER into mitochondria [50, 59], whereas TG leads to increased cytosol calcium, thus affecting mitochondria [60]. With both TG and TM, reduced mitochondrial STAT3 might be involved, as it is thought to maintain mitochondrial calcium homeostasis [25].
The total mitochondrial STAT3 protein levels appeared to remain at normal levels under ER stress conditions at all time points, suggesting that the reduced mitochondrial pS727-STAT3 was not caused by inhibition of translocation. Total STAT3 was also not markedly changed in whole cell lysates, indicating that STAT3 synthesis was not affected and that the loss of pS727-STAT3 resulted from dysfunctional upstream signaling.
We recently identified an FAK-STAT3 pathway that activates the S727 residue of STAT3 [21], and determined that FAK inhibition caused dramatic changes in mitochondrial bioenergetic function [26]. Here, the ER stress-induced loss of pS727-STAT3 coincided with reductions in pFAK. Moreover, APB treatment led to protection of pFAK and pS727-STAT3 under TG conditions and improved cell survival. APB did not affect pFAK under TM conditions and also did not protect pS727-STAT3. The mechanisms by which APB maintains pFAK under ER stress remain to be determined. APB can block calcium release from the ER [61] which under physiological conditions is involved in signaling and might have direct access to FAK in focal adhesions [62]. Our finding is consistent with the observation that calcium spikes are associated with reduced pFAK in neurons [34]. In apparent contrast, increased intracellular calcium levels [63, 64], induced even by a brief TG treatment [65], or virally-induced release of calcium from the ER can stimulate FAK phosphorylation [66]. A short exposure to hydrogen peroxide, which induces ROS, can also stimulate pFAK in endothelial cells [67] but without inhibiting tyrosine phosphatases [68]. This suggests that calcium levels and tyrosine phosphatases regulate pFAK separately. We propose that excessive and prolonged exposure to calcium causes dephosphorylation of FAK through a yet to be determined mechanism.
The bpV treatment provided substantial protection of pFAK while not affecting pS727-STAT3, most likely because it interferes with many tyrosine phosphatases [69, 70], some of which might be inhibitors of tyrosine kinase pathways that lead to inhibition of pS727-STAT3. It will be important to identify these because they might be good therapeutic targets. BpV also protects human endothelial cells in vitro and in rats after spinal cord injury [38], which is characterized by ischemia and mitochondrial dysfunction, resulting in microvascular endothelial cell death [71, 72]. The finding that bpV protects cells against both TG and TM induced ER stress at the same time as increased pFAK is notable and suggests that additional pFAK signaling pathways are key to cell survival. The phosphatases responsible for dephosphorylating FAK during ER stress remain to be identified, but could include SHP2 [28, 29], PTP-PEST/PTPN12 [30-32] and PTP 1B [29]. PTP1B is a good candidate because it is increased during ER stress in the liver and its deletion improves outcomes after high fat diet or TM-induced ER stress in mice [73]. ER-associated PTP1B seems to potentiate ER stress-induced mechanisms and apoptosis [74], additionally its localization would put it close to ER-associated FAK. On the other hand, a PTP1B inhibitor did not have effects on pFAK in endothelial cells [75]. The effects of ER stress on SHP2 or PTPN12 expression are not known.
Finally, our data with STAT3 KO cells confirm the importance of S727-STAT3 in promoting the survival of endothelial cells under pathological ER stress conditions. More STAT3 KO endothelial cells survived under ER stress when transfected with WT STAT3. Together with the finding that preservation of pS727 improves outcomes, this suggests that stimulation of pS727 would be protective against cellular insults. The S727D mutant showed more protection than the S727A mutant, again confirming the important protective role of pSTAT3-S727 under ER stress. Others had shown that S727D mutants increased bioenergetic function more than WT in STAT3 KO cells [22]. Therefore it was surprising that the S727D mutant did not provide more protection against TM-induced ER stress than the WT. This suggests that there is a limit to being able to rescue cells from chronic ER stress. Alternatively, this might indicate the necessity for a proper balance of phosphorylation and dephosphorylation of mitochondrial STAT3 or a balance between p705-STAT3 and pS727-STAT3. Interestingly, the S727A mutant seemed more detrimental than the absence of STAT3. It is possible that this mutant enables phosphorylation of predominantly the p705 site, which is known to mediate some of the mechanisms related to ER stress damage [76].
5. Conclusion
In summary, this study identifies that ER stress causes a reduction in pFAK and mitochondrial STAT3 signaling, leading to mitochondrial dysfunction and damage and ultimately, endothelial cell loss. Altogether, this suggests that mitochondria and their STAT3-regulated function represent a node that can be modulated to promote cell survival. Our data also suggest that FAK and its downstream signaling pathways are important regulators of cell survival which can be therapeutically targeted in disease processes characterized by excessive ER stress.
Highlights.
ER stress causes mitochondrial dysfunction and cell loss by reducing pS727-STAT3
ER stress reduces pFAK via PTPs and calcium, causing reduced pS727-STAT3
S727-STAT3 confers protection against ER stress, as shown by mutant studies
FAK-STAT3 stimulation may be a therapeutic approach against pathological ER stress
Acknowledgments
We are grateful for the technical support by Richard Sante, Chiharu Lovins and Aruna Visavadiya, Dr. Gary Wright is thanked for the use of his Seahorse equipment. bEnd5 cells were a kind gift from Dr. Britta Engelhardt of the Theodor Kocher Institute. Supported by NIH Grants NS45734 and AG029493, and in part by NIH grant C06RR0306551, and by funds from the Quillen College of Medicine at ETSU.
Abbreviations
- APB
2-Aminoethoxydiphenylborane
- ANOVA
One way analysis of variance
- ATPase
Adenosine triphosphate synthase
- bEnd5
Mouse brain endothelioma cell line
- bpV
Bisperoxovanadium
- BSA
Bovine serum albumin
- Cas9
CRISPR-associated protein 9
- CHOP
CCAAT-enhancer-binding protein homologous protein
- CO2
Carbon-di-oxide
- CRISPR
Clustered Regularly Interspersed Short Palindromic Repeats
- DAPI
4′,6-diamidino-2-phenylindole
- DMEM
Dulbecco's Modified Eagle Medium
- ECL
Enhanced chemiluminescence
- ECM
Extracellular matrix
- EGTA
Ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- ER
Endoplasmic reticulum
- FAK
Focal adhesion kinase
- FAK14
1,2,4,5-Benzenetetramine tetrahydrochloride
- FBS
Fetal bovine serum
- FCCP
Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone
- GFP
Green Fluorescent Protein
- HBSS
Hank's balanced salt solution
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- KO
Knockout
- LC3
Microtubule-associated protein 1A/1B-light chain 3
- LDH
Lactate dehydrogenase
- mPTP
Mitochondrial permeability transition pore
- OCR
Oxygen consumption rate
- PBS
Phosphate buffer saline
- PDH
Pyruvate dehydrogenase
- PI
Propidium iodide
- PVDF
Polyvinylidene difluoride
- ROS
Reactive oxygen species
- SDS-PAGE
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- STAT3
Signal transducer and activator of transcription 3
- TBST
Tris-buffered saline Tween 20
- TG
Thapsigargin
- TM
Tunicamycin
- TMRM
Tetramethylrhodamine, methyl ester
- WT
Wild type
Footnotes
Author Contributions: KB, MPK, VR, NPV, CJ and TH performed experiments and analyzed and interpreted data. KB and TH wrote the paper.
Competing Interests: The authors declare no competing interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ron D, Habener JF. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992;6(3):439–53. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- 2.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 3.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 4.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A. 1990;87(7):2466–70. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larsson O, Carlberg M, Zetterberg A. Selective killing induced by an inhibitor of N-linked glycosylation. J Cell Sci. 1993;106(Pt 1):299–307. doi: 10.1242/jcs.106.1.299. [DOI] [PubMed] [Google Scholar]
- 6.Dricu A, Carlberg M, Wang M, Larsson O. Inhibition of N-linked glycosylation using tunicamycin causes cell death in malignant cells: role of down-regulation of the insulin-like growth factor 1 receptor in induction of apoptosis. Cancer Res. 1997;57(3):543–8. [PubMed] [Google Scholar]
- 7.Xu D, Perez RE, Rezaiekhaligh MH, Bourdi M, Truog WE. Knockdown of ERp57 increases BiP/GRP78 induction and protects against hyperoxia and tunicamycin-induced apoptosis. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L44–51. doi: 10.1152/ajplung.90626.2008. [DOI] [PubMed] [Google Scholar]
- 8.Placido AI, Oliveira CR, Moreira PI, Pereira CM. Enhanced amyloidogenic processing of amyloid precursor protein and cell death under prolonged endoplasmic reticulum stress in brain endothelial cells. Mol Neurobiol. 2015;51(2):571–90. doi: 10.1007/s12035-014-8819-1. [DOI] [PubMed] [Google Scholar]
- 9.Fassbender JM, Saraswat-Ohri S, Myers SA, Gruenthal MJ, Benton RL, Whittemore SR. Deletion of endoplasmic reticulum stress-induced CHOP protects microvasculature post-spinal cord injury. Curr Neurovasc Res. 2012;9(4):274–81. doi: 10.2174/156720212803530627. [DOI] [PubMed] [Google Scholar]
- 10.Myers SA, Andres KR, Hagg T, Whittemore SR. CD36 deletion improves recovery from spinal cord injury. Exp Neurol. 2014;256:25–38. doi: 10.1016/j.expneurol.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han S, Arnold SA, Sithu SD, Mahoney ET, Geralds JT, Tran P, Benton RL, Maddie MA, D'Souza SE, Whittemore SR, Hagg T. Rescuing vasculature with intravenous angiopoietin-1 and alpha v beta 3 integrin peptide is protective after spinal cord injury. Brain. 2010;133(Pt 4):1026–42. doi: 10.1093/brain/awq034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fassbender JM, Whittemore SR, Hagg T. Targeting microvasculature for neuroprotection after SCI. Neurotherapeutics. 2011;8(2):240–51. doi: 10.1007/s13311-011-0029-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boya P, Cohen I, Zamzami N, Vieira HL, Kroemer G. Endoplasmic reticulum stress-induced cell death requires mitochondrial membrane permeabilization. Cell Death Differ. 2002;9(4):465–7. doi: 10.1038/sj.cdd.4401006. [DOI] [PubMed] [Google Scholar]
- 14.Kaufman RJ, Malhotra JD. Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim Biophys Acta. 2014;1843(10):2233–9. doi: 10.1016/j.bbamcr.2014.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta. 2014;1837(4):461–9. doi: 10.1016/j.bbabio.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 16.Flis VV, Daum G. Lipid transport between the endoplasmic reticulum and mitochondria. Cold Spring Harb Perspect Biol. 2013;5(6) doi: 10.1101/cshperspect.a013235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruoslahti E, Reed JC. Anchorage dependence, integrins, and apoptosis. Cell. 1994;77(4):477–8. doi: 10.1016/0092-8674(94)90209-7. [DOI] [PubMed] [Google Scholar]
- 18.Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134(3):793–9. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu Q, Rounds S. Focal adhesion kinase and endothelial cell apoptosis. Microvasc Res. 2012;83(1):56–63. doi: 10.1016/j.mvr.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jan Y, Matter M, Pai JT, Chen YL, Pilch J, Komatsu M, Ong E, Fukuda M, Ruoslahti E. A mitochondrial protein, Bit1, mediates apoptosis regulated by integrins and Groucho/TLE corepressors. Cell. 2004;116(5):751–62. doi: 10.1016/s0092-8674(04)00204-1. [DOI] [PubMed] [Google Scholar]
- 21.Keasey MP, Kang SS, Lovins C, Hagg T. Inhibition of a novel specific neuroglial integrin signaling pathway increases STAT3-mediated CNTF expression. Cell Commun Signal. 2013;11:35. doi: 10.1186/1478-811X-11-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323(5915):793–7. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol. 2010;105(6):771–85. doi: 10.1007/s00395-010-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boengler K, Ungefug E, Heusch G, Schulz R. The STAT3 inhibitor stattic impairs cardiomyocyte mitochondrial function through increased reactive oxygen species formation. Curr Pharm Des. 2013;19(39):6890–5. doi: 10.2174/138161281939131127115940. [DOI] [PubMed] [Google Scholar]
- 25.Yang R, Rincon M. Mitochondrial Stat3, the Need for Design Thinking. Int J Biol Sci. 2016;12(5):532–44. doi: 10.7150/ijbs.15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Visavadiya NP, Keasey MP, Razskazovskiy V, Banerjee K, Jia C, Lovins C, Wright GL, Hagg T. Integrin-FAK signaling rapidly and potently promotes mitochondrial function through STAT3. Cell Commun Signal. 2016;14(1):32. doi: 10.1186/s12964-016-0157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116(Pt 8):1409–16. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 28.Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol. 2000;2(2):62–9. doi: 10.1038/35000008. [DOI] [PubMed] [Google Scholar]
- 29.Sorenson CM, Sheibani N. Altered regulation of SHP-2 and PTP 1B tyrosine phosphatases in cystic kidneys from bcl-2 -/- mice. Am J Physiol Renal Physiol. 2002;282(3):F442–50. doi: 10.1152/ajprenal.00184.2001. [DOI] [PubMed] [Google Scholar]
- 30.Davidson D, Veillette A. PTP-PEST, a scaffold protein tyrosine phosphatase, negatively regulates lymphocyte activation by targeting a unique set of substrates. EMBO J. 2001;20(13):3414–26. doi: 10.1093/emboj/20.13.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng Y, Xia Y, Hawke D, Halle M, Tremblay ML, Gao X, Zhou XZ, Aldape K, Cobb MH, Xie K, He J, Lu Z. FAK phosphorylation by ERK primes ras-induced tyrosine dephosphorylation of FAK mediated by PIN1 and PTP-PEST. Mol Cell. 2009;35(1):11–25. doi: 10.1016/j.molcel.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng Y, Yang W, Xia Y, Hawke D, Liu DX, Lu Z. Ras-induced and extracellular signal-regulated kinase 1 and 2 phosphorylation-dependent isomerization of protein tyrosine phosphatase (PTP)-PEST by PIN1 promotes FAK dephosphorylation by PTP-PEST. Mol Cell Biol. 2011;31(21):4258–69. doi: 10.1128/MCB.05547-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rafiq K, Kolpakov MA, Abdelfettah M, Streblow DN, Hassid A, Dell'Italia LJ, Sabri A. Role of protein-tyrosine phosphatase SHP2 in focal adhesion kinase down-regulation during neutrophil cathepsin G-induced cardiomyocytes anoikis. J Biol Chem. 2006;281(28):19781–92. doi: 10.1074/jbc.M513040200. [DOI] [PubMed] [Google Scholar]
- 34.Conklin MW, Lin MS, Spitzer NC. Local calcium transients contribute to disappearance of pFAK, focal complex removal and deadhesion of neuronal growth cones and fibroblasts. Dev Biol. 2005;287(1):201–12. doi: 10.1016/j.ydbio.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 35.Wagner EF, Risau W. Oncogenes in the study of endothelial cell growth and differentiation. Semin Cancer Biol. 1994;5(2):137–45. [PubMed] [Google Scholar]
- 36.Yang T, Roder KE, Abbruscato TJ. Evaluation of bEnd5 cell line as an in vitro model for the blood-brain barrier under normal and hypoxic/aglycemic conditions. J Pharm Sci. 2007;96(12):3196–213. doi: 10.1002/jps.21002. [DOI] [PubMed] [Google Scholar]
- 37.Steiner O, Coisne C, Engelhardt B, Lyck R. Comparison of immortalized bEnd5 and primary mouse brain microvascular endothelial cells as in vitro blood-brain barrier models for the study of T cell extravasation. J Cereb Blood Flow Metab. 2011;31(1):315–27. doi: 10.1038/jcbfm.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakashima S, Arnold SA, Mahoney ET, Sithu SD, Zhang YP, D'Souza SE, Shields CB, Hagg T. Small-molecule protein tyrosine phosphatase inhibition as a neuroprotective treatment after spinal cord injury in adult rats. J Neurosci. 2008;28(29):7293–303. doi: 10.1523/JNEUROSCI.1826-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Decker T, Lohmann-Matthes ML. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J Immunol Methods. 1988;115(1):61–9. doi: 10.1016/0022-1759(88)90310-9. [DOI] [PubMed] [Google Scholar]
- 40.Dranka BP, Benavides GA, Diers AR, Giordano S, Zelickson BR, Reily C, Zou L, Chatham JC, Hill BG, Zhang J, Landar A, Darley-Usmar VM. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med. 2011;51(9):1621–35. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banerjee K, Munshi S, Xu H, Frank DE, Chen HL, Chu CT, Yang J, Cho S, Kagan VE, Denton TT, Tyurina YY, Jiang JF, Gibson GE. Mild mitochondrial metabolic deficits by alpha-ketoglutarate dehydrogenase inhibition cause prominent changes in intracellular autophagic signaling: Potential role in the pathobiology of Alzheimer's disease. Neurochem Int. 2016;96:32–45. doi: 10.1016/j.neuint.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wen LP, Fahrni JA, Troie S, Guan JL, Orth K, Rosen GD. Cleavage of focal adhesion kinase by caspases during apoptosis. J Biol Chem. 1997;272(41):26056–61. doi: 10.1074/jbc.272.41.26056. [DOI] [PubMed] [Google Scholar]
- 43.Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, Cichy J, Kukreja RC, Dulak J, Lesnefsky EJ, Larner AC. Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J Biol Chem. 2011;286(34):29610–20. doi: 10.1074/jbc.M111.226209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meier JA, Larner AC. Toward a new STATe: the role of STATs in mitochondrial function. Semin Immunol. 2014;26(1):20–8. doi: 10.1016/j.smim.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwong JQ, Molkentin JD. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015;21(2):206–14. doi: 10.1016/j.cmet.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mattson MP, Kroemer G. Mitochondria in cell death: novel targets for neuroprotection and cardioprotection. Trends Mol Med. 2003;9(5):196–205. doi: 10.1016/s1471-4914(03)00046-7. [DOI] [PubMed] [Google Scholar]
- 47.Koo HJ, Piao Y, Pak YK. Endoplasmic reticulum stress impairs insulin signaling through mitochondrial damage in SH-SY5Y cells. Neurosignals. 2012;20(4):265–80. doi: 10.1159/000333069. [DOI] [PubMed] [Google Scholar]
- 48.Bronner DN, Abuaita BH, Chen X, Fitzgerald KA, Nunez G, He Y, Yin XM, O'Riordan MX. Endoplasmic Reticulum Stress Activates the Inflammasome via NLRP3- and Caspase-2-Driven Mitochondrial Damage. Immunity. 2015;43(3):451–62. doi: 10.1016/j.immuni.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cali T, Ottolini D, Negro A, Brini M. Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim Biophys Acta. 2013;1832(4):495–508. doi: 10.1016/j.bbadis.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 50.Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015;40(3):141–8. doi: 10.1016/j.tibs.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282(7):4702–10. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- 52.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan Y, Zhang X, Zheng Y, Chen Z. Regulation of mitophagy in ischemic brain injury. Neurosci Bull. 2015;31(4):395–406. doi: 10.1007/s12264-015-1544-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015;22(3):367–76. doi: 10.1038/cdd.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yonekawa T, Thorburn A. Autophagy and cell death. Essays Biochem. 2013;55:105–17. doi: 10.1042/bse0550105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Joshi AU, Kornfeld OS, Mochly-Rosen D. The entangled ER-mitochondrial axis as a potential therapeutic strategy in neurodegeneration: A tangled duo unchained. Cell Calcium. 2016 doi: 10.1016/j.ceca.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.La Rovere RM, Roest G, Bultynck G, Parys JB. Intracellular Ca2+ signaling and Ca2+ microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium. 2016 doi: 10.1016/j.ceca.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 58.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833(12):3460–70. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tadic V, Prell T, Lautenschlaeger J, Grosskreutz J. The ER mitochondria calcium cycle and ER stress response as therapeutic targets in amyotrophic lateral sclerosis. Front Cell Neurosci. 2014;8:147. doi: 10.3389/fncel.2014.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hom JR, Gewandter JS, Michael L, Sheu SS, Yoon Y. Thapsigargin induces biphasic fragmentation of mitochondria through calcium-mediated mitochondrial fission and apoptosis. J Cell Physiol. 2007;212(2):498–508. doi: 10.1002/jcp.21051. [DOI] [PubMed] [Google Scholar]
- 61.Yoon MJ, Lee AR, Jeong SA, Kim YS, Kim JY, Kwon YJ, Choi KS. Release of Ca2+ from the endoplasmic reticulum and its subsequent influx into mitochondria trigger celastrol-induced paraptosis in cancer cells. Oncotarget. 2014;5(16):6816–31. doi: 10.18632/oncotarget.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Q, Wang Y, Downey GP, Plotnikov S, McCulloch CA. A ternary complex comprising FAK, PTPalpha and IP3 receptor 1 functionally engages focal adhesions and the endoplasmic reticulum to mediate IL-1-induced Ca2+ signalling in fibroblasts. Biochem J. 2016;473(4):397–410. doi: 10.1042/BJ20150907. [DOI] [PubMed] [Google Scholar]
- 63.Giannone G, Ronde P, Gaire M, Beaudouin J, Haiech J, Ellenberg J, Takeda K. Calcium rises locally trigger focal adhesion disassembly and enhance residency of focal adhesion kinase at focal adhesions. J Biol Chem. 2004;279(27):28715–23. doi: 10.1074/jbc.M404054200. [DOI] [PubMed] [Google Scholar]
- 64.Alessandro R, Masiero L, Lapidos K, Spoonster J, Kohn EC. Endothelial cell spreading on type IV collagen and spreading-induced FAK phosphorylation is regulated by Ca2+ influx. Biochem Biophys Res Commun. 1998;248(3):635–40. doi: 10.1006/bbrc.1998.8705. [DOI] [PubMed] [Google Scholar]
- 65.Giannone G, Ronde P, Gaire M, Haiech J, Takeda K. Calcium oscillations trigger focal adhesion disassembly in human U87 astrocytoma cells. J Biol Chem. 2002;277(29):26364–71. doi: 10.1074/jbc.M203952200. [DOI] [PubMed] [Google Scholar]
- 66.Cheshenko N, Del Rosario B, Woda C, Marcellino D, Satlin LM, Herold BC. Herpes simplex virus triggers activation of calcium-signaling pathways. J Cell Biol. 2003;163(2):283–93. doi: 10.1083/jcb.200301084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vepa S, Scribner WM, Parinandi NL, English D, Garcia JG, Natarajan V. Hydrogen peroxide stimulates tyrosine phosphorylation of focal adhesion kinase in vascular endothelial cells. Am J Physiol. 1999;277(1 Pt 1):L150–8. doi: 10.1152/ajplung.1999.277.1.L150. [DOI] [PubMed] [Google Scholar]
- 68.Natarajan V, Scribner WM, al-Hassani M, Vepa S. Reactive oxygen species signaling through regulation of protein tyrosine phosphorylation in endothelial cells. Environ Health Perspect. 1998;106(5):1205–12. doi: 10.1289/ehp.98106s51205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Posner BI, Faure R, Burgess JW, Bevan AP, Lachance D, Zhang-Sun G, Fantus IG, Ng JB, Hall DA, Lum BS, et al. Peroxovanadium compounds. A new class of potent phosphotyrosine phosphatase inhibitors which are insulin mimetics. J Biol Chem. 1994;269(6):4596–604. [PubMed] [Google Scholar]
- 70.Ruff SJ, Chen K, Cohen S. Peroxovanadate induces tyrosine phosphorylation of multiple signaling proteins in mouse liver and kidney. J Biol Chem. 1997;272(2):1263–7. doi: 10.1074/jbc.272.2.1263. [DOI] [PubMed] [Google Scholar]
- 71.Muradov JM, Ewan EE, Hagg T. Dorsal column sensory axons degenerate due to impaired microvascular perfusion after spinal cord injury in rats. Exp Neurol. 2013;249:59–73. doi: 10.1016/j.expneurol.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ewan EE, Hagg T. Intrathecal Acetyl-L-Carnitine Protects Tissue and Improves Function after a Mild Contusive Spinal Cord Injury in Rats. J Neurotrauma. 2016;33(3):269–77. doi: 10.1089/neu.2015.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Agouni A, Mody N, Owen C, Czopek A, Zimmer D, Bentires-Alj M, Bence KK, Delibegovic M. Liver-specific deletion of protein tyrosine phosphatase (PTP) 1B improves obesity- and pharmacologically induced endoplasmic reticulum stress. Biochem J. 2011;438(2):369–78. doi: 10.1042/BJ20110373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gu F, Nguyen DT, Stuible M, Dube N, Tremblay ML, Chevet E. Protein-tyrosine phosphatase 1B potentiates IRE1 signaling during endoplasmic reticulum stress. J Biol Chem. 2004;279(48):49689–93. doi: 10.1074/jbc.C400261200. [DOI] [PubMed] [Google Scholar]
- 75.Wang Y, Yan F, Ye Q, Wu X, Jiang F. PTP1B inhibitor promotes endothelial cell motility by activating the DOCK180/Rac1 pathway. Sci Rep. 2016;6:24111. doi: 10.1038/srep24111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meares GP, Liu Y, Rajbhandari R, Qin H, Nozell SE, Mobley JA, Corbett JA, Benveniste EN. PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol Cell Biol. 2014;34(20):3911–25. doi: 10.1128/MCB.00980-14. [DOI] [PMC free article] [PubMed] [Google Scholar]