

Graphical abstract
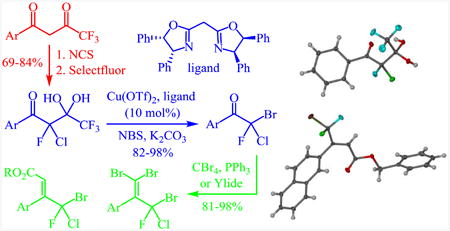
Sequential chlorination/fluorination of aromatic trifluoroacetylated ketones gives 1-aryl 2-chloro-2,4,4,4-tetrafluoro-butan-1,3-dione hydrates that are used for the synthesis of ketones and alkenes exhibiting a terminal bromochlorofluoromethyl group. The hydrates undergo detrifluoroacetylative cleavage and subsequent bromination in the presence of a copper(II)bisoxazoline catalyst, K2CO3 and NBS at room temperature. The corresponding bromochlorofluoromethyl ketones can be applied in Wittig and Horner-Wadsworth-Emmons reactions and dibromoalkenylations.
Introduction
The development of synthetic methods that produce halogenated compounds continues to receive increasing attention. The general interest in organohalide chemistry originates at least in part from the widespread use of organofluorines and organochlorines in the pharmaceutical and agrochemical industries as well as the outstanding synthetic utility of carbon-halide bonds.1 The presence of fluorine and chlorine substituents can improve bioavailability, lipophilicity, metabolic stability and other important pharmacological properties of drugs and the incorporation of halides into synthetic intermediates provides unique opportunities for subsequent modifications aimed at the synthesis of increasingly complex target structures. While chlorinated and fluorinated pharmaceuticals play central roles in the pharmaceutical market,2 drug discovery efforts are gradually extended to the screening of candidates containing less frequently used bromides and iodides. Despite a continuing interest in the synthesis and use of compounds exhibiting a bromochlorofluoromethyl group3 and some success based on ring opening of fluorinated cyclopropanes with bromine or trapping of Julia-Kocienski intermediates with a brominating agent,4 the preparation of this moiety has remained challenging (Scheme 1).5
Scheme 1.

Synthesis of bromochlorofluoromethyl-derived alcohols, ketones and alkenes.
Traditional synthetic routes and established methods developed with nonhalogenated substrates and reagents can often not be applied to halogenated analogues because the presence of halogen atoms in close proximity to the reaction center can alter the stability of intermediates and ultimately change reactivity and reaction pathways. These complications have limited the versatility and general usefulness of halogenated enolates in aldol type reactions. In the search for practical alternatives to reactions with halogenated silyl enolates, several groups have focused on an orthogonal strategy that is based on mild in situ generation of halogenated nucleophiles from readily available trifluoroacetyl derived precursors.6 Prager and Ogden demonstrated almost 50 years ago that the cleavage of hexafluoroacetone and its ester derivatives with excess of base, metal salts and other additives produces fluorinated carbanions suitable for C-C bond formation.7 The full potential of this concept in aldol reactions with difluoroenolates generated in situ from a bench-stable trifluoroacetyl-derived precursor was first demonstrated by Colby et al.8 This seminal work has led to a variety of interesting applications and developments reported by Prakash,9 Colby,10 Zhu,11 Soloshonok and Han,12 Hong and Wang,13 Fang and Wu,14 and our group.15 Inspired by the successful halogenation of difluoroenolates prepared by detrifluoroacetylation in the presence of excess of lithium salts and triethylamine,16 we initiated the synthesis of a small series of 1-aryl 2-chloro-2,4,4,4-tetrafluoro-butan-1,3-dione hydrates. We now wish to describe the mild catalytic cleavage of these gem-diols and subsequent bromination of intermediate chlorofluoroenolates to chiral bromochlorofluoromethyl ketones, and the use of these products in alkene synthesis.
Results and Discussion
The difluorination of 1-aryl 4,4,4-trifluorobutane-1,3-diones with Selectfluor is a well established reaction that typically proceeds with high yields. For example, the fluorination of the trifluoroacetylated acetophenone derivative 1 gives the geminal diol of 1-phenyl-2,2,4,4,4-pentafluorobutan-1,3-dione, 2, in 95% yield (Scheme 2).15a The adjacent trifluoromethyl and difluoromethylene groups significantly increase the electrophilicity of the central ketone unit which readily attracts water to afford the hydrate 2. We expected that the diol form would also predominate over the free diketone in the corresponding 1-aryl 2-chloro-2,4,4,4-tetrafluorobutane-1,3-diones. The development of a consecutive fluorination/chlorination procedure for the synthesis of 3, however, proved difficult. Our first attempts to prepare the mixed dihalide via monofluorination of 1 with Selectfluor failed because the second fluorination step is fast and thus favors formation of 2. We therefore decided to reverse the halogenation sequence and started investigating the possibility of monochlorination of 1. Stavber et al. reported successful monohalogenation of trifluoromethyl substituted 1,3-diketones with N-halosuccinimides but they isolated the hydrates which cannot be further fluorinated.17 We therefore resorted to a two-step protocol in which 1 is first chlorinated by grinding with N-chlorosuccinimide, NCS, under inert atmosphere to avoid the water addition. The crude reaction mixture is then dissolved in anhydrous acetonitrile and subjected to fluorination with Selectfluor. This gave 2-chloro-2,4,4,4-tetrafluoro-3,3-dihydroxy-1-phenylbutan-1-one, 3, in 76% yield. With this procedure in hand, we were able to synthesize the 2-naphthyl, 4-chlorophenyl and 2-furanyl analogues 3-6 in 69 to 84% yield (Scheme 2). As expected, all pentahalogenated compounds spontaneously add water to form the gem-diols upon exposure to air.
Scheme 2.

Synthesis of 1-aryl 2-chloro-2,4,4,4-tetrafluorobutane-1,3-diones 3-6 by stepwise chlorination/fluorination of trifluoromethylated 1,3-diones and X-ray structure of 3.
In analogy to the copper(II) bisoxazoline catalyzed C-C bond formation of difluoroenolates prepared in situ via base promoted cleavage of 2 or derivatives thereof,15b we anticipated that the diol 3 can be converted to α-bromo-α-chloro-α-fluoroacetophenone, 8, in the presence of triethylamine and a brominating agent (Scheme 3). We rationalized that the synthesis of haloform 8 and other bromochlorofluoromethyl ketones would provide new access toward a variety of perhalogenated compounds, for example via modification of the carbonyl or carbon-bromide bond.
Scheme 3.

In situ cleavage of 3 and formation of 8 and 9.
Initial studies showed that bromination of the intermediate enolate 7 in the presence of 10 mol% of Cu(II) triflate, bromine and triethylamine is relatively slow compared to the formation of the α-chloro-α-fluoroacetophenone, 9. To minimize the effect of the competing protonation path, we employed inorganic bases (potassium carbonate or phosphate) and NBS in the reaction, and we obtained 8 and 9 in 60% and 40% yield, respectively. We found that the cleavage of 3 requires 2 equivalents of base but predominantly produces 9 unless Cu(OTf)2 is present. In fact, a mixture containing only 19% of 8 and 81% of the chlorofluoromethyl ketone 9 was formed when the reaction was conducted with equimolar amounts of NBS and 2.5 equivalents of K2CO3 in THF in the absence of a copper complex. The effect of several solvents and ligands on the reaction outcome was then investigated and the results of the optimization study are summarized in Table 1.
Table 1.
Optimization of the cleavage/bromination sequence using hydrate 3.

| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Ligand | Solvent | Time (h) | % Yield 8 (9)a |
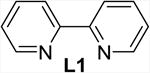
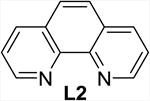
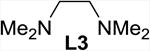
| 1 | none | THF | 2 | 60 (40) |
| 2 |
|
THF | 2 | 57 (43) |
| 3 |
|
THF | 2 | 57 (43) |
| 4 |
|
THF | 2 | 38 (62) |
| 5 |
|
THF | 2 | 79 (21) |
| 6 |
|
THF | 2 | 89 (11) |
| 7 |
|
THF | 2 | 75 (25) |
| 8 |
|
THF | 1 | 94 (6) |
| 9 | L7 | CH2Cl2 | 18 | 96 (4) |
| 10 | L7 | CHCl3 | 46 | 88 (n.d.) |
| 11 | L7 | (CH2Cl)2 | 46 | 97 (3) |
| 12 | L7 | ACN | 18 | <70 (n.d.) |
Based on NMR analysis. n.d. not determined.
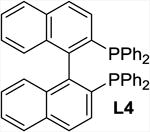
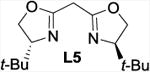
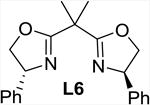
The addition of bipyridine or phenanthroline gave no improvement and TMEDA proved detrimental compared to the ligand-free procedure (entries 1-4). The introduction of BINAP and bisoxazolines L5 and L6, however, showed promise and favored conversion to 8 (entries 5-7). Finally, we found that the cleavage and subsequent bromination of 3 gives 8 almost exclusively within 1 hour at room temperature when 10 mol% of copper triflate and bisoxazoline L7, NBS and 2 equivalents of potassium carbonate are used in THF (entry 8). The reaction is fairly slow in chlorinated solvents and unidentified by-products were observed in chloroform and acetonitrile (entries 9-12). With an optimized procedure in hand, we were able to prepare the bromochlorofluoromethyl ketones 8 and 10-12 in 82-98% yield (Scheme 4). The bromination reaction of the in situ enolate can also be accomplished under noncatalytic conditions when large excess of lithium bromide is used to suppress the competing protonation.16 For example, we obtained 11 from diol 5 in 95% yield in the presence of 5 equivalents of LiBr under otherwise similar conditions.18
Scheme 4.

Copper catalyzed synthesis of bromochlorofluoromethyl ketones 8 and 10-12.
The high-yielding synthesis of these mixed trihalo ketones via mild cleavage of the diol precursors 3-6 effectively complements previously reported routes aimed at generating a bromochlorofluoromethyl group.4,5,19 At this point, we decided to further explore the general synthetic versatility of bromochlorofluoromethyl ketones including the possibility of Wittig and Horner-Wadsworth-Emmons reactions. We were pleased to find that treatment of α-bromo-α-chloro-α-fluoroacetophenone, 8, with carbon tetrabromide and triphenylphosphine gives the dibromoalkene 13 in almost quantitative amounts (Scheme 5).20 The same reaction furnished the dibromoalkenes 14 and 15 in 86-88% yield. Similarly, both Wittig and HWE reactions were successful. Treatment of 8 with benzyl (triphenylphosphoranylidene)acetate at room temperature gave the (E)-alkene 16 with high diastereoselectivity in 81% yield.21 When we employed the triaholmethyl ketones 10 and 11 in the same protocol we obtained (E)-17 and (E)-18 in 91-92% yield. The formation of the (E)-isomer was confirmed by crystallographic analysis and is consistent with literature procedures using α-halogenated acetophenone derivatives.22 Finally, the Horner-Wadsworth-Emmons reaction using 8 and triethyl phosphonoacetate produced ethyl (E)-4-bromo-4-chloro-4-fluoro-3-phenylbut-2-enoate, 19, in almost quantitative amounts.
Scheme 5.

Results of dibromoalkenylations, Wittig and Horner-Wadsworth-Emmons reactions of bromochlorofluoromethyl ketones. Crystallographic analysis of 17 shows E-configuration.
Conclusion
In summary, we have developed a mild catalytic procedure that exploits detrifluoroacetylative in situ generation of dihalogenated enolates from readily available geminal diols for the synthesis of bromochlorofluoromethyl ketones which were obtained in 82-98% yield. The copper(bisoxazoline) catalyzed reaction successfully favors bromination of intermediate chlorofluoroenolates over the competing protonation pathway. We like to point out that the base promoted cleavage of the chlorofluoro-derived diols 3-6 and analogues thereof provides simple access to chlorofluoromethyl ketones such as 9 and other dihalomethyl ketones if desired. The synthetic utility of bromochlorofluoromethyl ketones was demonstrated with high-yielding dibromoalkenylations, Wittig and Horner-Wadsworth-Emmons reactions.
Experimental Section
General Information
Commercially available bisoxazoline ligands, trifluoromethyl diketones, reagents and solvents were used as purchased without further purification. NMR spectra were obtained at 400 MHz (1H NMR), 376 MHz (19F NMR) and 100 MHz (13C NMR) in deuterated chloroform or DMSO. Chemical shifts are reported in ppm relative to TMS or relative to the DMSO-d6 solvent peak. Reaction products were purified by column chromatography on silica gel (particle size 40-63 μm) as described below.
General procedure for the synthesis of 1-aryl-2-chloro-2,4,4,4-tetrafluoro-3,3-dihydroxybutanones
The corresponding 1-aryl-4,4,4-trifluoro-butane-1,3-dione (1.0 equiv.) and N-chlorosuccinimide (1.2 equiv.) were triturated together in a mortar under inert atmosphere. The reaction was monitored by 19F NMR and trituration was continued until full conversion was achieved. The crude reaction mixture was then dissolved in anhydrous acetonitrile and stirred together with Selectfluor (1.5 equiv.) at room temperature. After full conversion was achieved based on 19F NMR analysis, the solvent was removed and replaced with dichloromethane. The insoluble Selectfluor was filtered from the reaction mixture and the filtrate was extracted with water. The combined organic layers were dried over sodium sulfate and the solvent was removed in vacuo. The crude product was purified by flash chromatography on silica gel using with hexanes-ethyl acetate (90:10) as mobile phase.
2-Chloro-2,4,4,4-tetrafluoro-3,3-dihydroxy-1-phenylbutan-1-one (3)
Compound 3 was obtained as a colorless solid in 76% yield (4.34 g, 15.2 mmol) from 4,4,4-trifluoro-1-phenylbutane-1,3-dione (4.32 g, 20.0 mmol) by following the general procedure described above. mp: 53-55 °C; Rf = 0.4 (hexanes / EtOAc, 8:2); 1H NMR (400 MHz, Chloroform-d): δ = 8.09 (d, J = 7.4 Hz, 2H), 7.70 (dd, J = 7.5, 7.4 Hz, 1H), 7.53 (dd, J = 7.5, 7.4 Hz, 2H), 5.14 (s, 1H), 4.99 (s, 1H) ppm; 13C NMR (100 MHz, Chloroform-d): δ = 193.8 (d, JC-F = 29.2 Hz), 135.3, 131.4 (d, JC-F = 3.0 Hz), 130.6 (d, JC-F = 6.6 Hz), 128.8, 121.5 (qd, JC-F = 289.6, 2.5 Hz), 103.9 (d, JC-F = 275.9 Hz), 94.9 (qd, JC-F = 32.5, 23.9 Hz) ppm; 19F NMR (376 MHz, Chloroform-d) δ = -78.8 (d, J = 14.8 Hz), -127.9 (q, J = 14.7 Hz) ppm; Anal. Calcd. for C10H7ClF4O3: C, 41.91; H, 2.46. Found: C, 41.84; H, 2.44.
2-Chloro-2,4,4,4-tetrafluoro-3,3-dihydroxy-1-(naphthalen-2-yl)butan-1-one (4)
Compound 4 was obtained as a pale yellow solid in 84% yield (2.82 g, 8.4 mmol) from 4,4,4-trifluoro-1-(naphthalen-2-yl)butane-1,3-dione (2.66 g, 10.0 mmol) by following the general procedure described above. mp: 82-84 °C; Rf = 0.3 (hexanes / EtOAc, 8:2); 1H NMR (400 MHz, DMSO-d6) δ 8.78 (s, 1H), 8.59 (m, 1H), 8.12 – 8.05 (m, 4H), 7.73 – 7.58 (m, 2H) ppm; 13C NMR (100 MHz, Chloroform-d) δ 193.5 (d, JC-F = 28.7 Hz), 171.4, 136.4, 133.8 (d, JC-F = 9.3 Hz), 132.3, 130.3, 130.1, 128.7, 127.8 (d, JC-F = 1.6 Hz), 127.3, 124.9 (d, JC-F = 4.4 Hz), 121.6 (qd, JC-F = 289.7, 2.4 Hz), 104.2 (d, JC-F = 276.1 Hz), 95.0 (qd, JC-F = 32.3, 23.9 Hz) ppm; 19F NMR (376 MHz, DMSO-d6) δ = -77.6 (d, J = 13.4 Hz), -118.3 (d, J = 13.4 Hz); Anal. Calcd. for C14H9ClF4O3: C, 49.95; H, 2.69. Found: C, 49.91; H, 2.64.
2-Chloro-1-(4-chlorophenyl)-2,4,4,4-tetrafluoro-3,3-dihydroxybutan-1-one (5)
Compound 5 was obtained as a colorless solid in 81% yield (2.08 g, 6.48 mmol) from 1-(4-chlorophenyl)-4,4,4-trifluorobutane-1,3-dione (2.0 g, 8.0 mmol) by following the general procedure described above. mp: 68-70 °C; Rf = 0.4 (hexanes / EtOAc, 8:2); 1H NMR (400 MHz, DMSO-d6) δ = 8.83 (s, 1H), 8.61 (s, 1H), 8.09 (d, J = 8.2 Hz, 2H), 7.63 (d, J = 8.6 Hz, 2H) ppm; 13C NMR (100 MHz, Chloroform-d) δ = 192.4 (d, JC-F = 29.3 Hz), 142.3, 132.0 (d, JC-F = 6.8 Hz), 129.7 (d, JC-F = 3.7 Hz), 129.3, 121.4 (qd, JC-F = 289.7, 2.4 Hz), 103.9 (d, JC-F = 275.2 Hz), 94.8 (qd, JC-F = 32.5, 23.6 Hz) ppm; 19F NMR (376 MHz, DMSO-d6) δ = -77.7 (d, J = 13.5 Hz), -118.6 (q, J = 13.4 Hz) ppm; Anal. Calcd. for C10H6Cl2F4O3: C, 37.41; H, 1.88. Found: C, 37.53; H, 1.92.
2-Chloro-2,4,4,4-tetrafluoro-1-(furan-2-yl)-3,3-dihydroxybutan-1-one (6)
Compound 6 was obtained as a colorless solid in 69% yield (1.90 g, 6.9 mmol) from 4,4,4-trifluoro-1-(furan-2-yl)butane-1,3-dione (2.06 g, 10.0 mmol) by following the general procedure described above. mp: 48-50 °C; Rf = 0.5 (hexanes / EtOAc, 8:2); 1H NMR (400 MHz, Chloroform-d) δ = 7.87 (bs, 1H), 7.69 (m, 1H), 6.71 (m, 1H), 4.92 (s, 2H) ppm; 13C NMR (100 MHz, Chloroform-d) δ = 179.8 (d, JC-F = 28.4 Hz), 150.8, 146.7 (d, JC-F = 4.4 Hz), 126.4 (dd, JC-F = 14.8, 2.0 Hz), 121.5 (qd, JC-F = 289.4, 2.4 Hz), 113.6 (d, JC-F = 2.9 Hz), 103.8 (d, JC-F = 272.9 Hz), 94.6 (qd, JC-F = 32.6, 23.3 Hz) ppm; 19F NMR (376 MHz, Chloroform-d) δ = -79.1 (d, J = 14.3 Hz), -131.0 (q, J = 14.3 Hz) ppm; Anal. Calcd. for C8H5ClF4O4: C, 34.74; H, 1.82. Found: C, 34.82; H, 1.85.
2.2 General procedure for the synthesis of 2-bromo-2-chloro-2-fluoromethyl ketones
Copper(II) triflate (0.10 equiv.) and (4R,5S)-bis(4,5-diphenyl-4,5-dihydrooxazol-2-yl)methane (0.12 equiv.) in 0.5 mL of anhydrous tetrahydrofuran were stirred together under inert atmosphere for one hour. The complex solution was added to a solution of the corresponding 1-aryl-2-chloro-2,4,4,4-tetrafluoro-3,3-dihydroxybutanone (1.0 equiv.), N-bromosuccinimide (1.5 equiv.) and potassium carbonate (2.5 equiv.) in 0.5 mL of anhydrous tetrahydrofuran under inert atmosphere. The mixture was stirred vigorously until conversion of the starting material was complete based on 19F NMR analysis. The crude product was loaded onto a silica gel column and purified by flash chromatography using hexanes as mobile phase.
2-Bromo-2-chloro-2-fluoro-1-phenylethan-1-one (8)
Compound 8 was obtained as a colorless liquid in 90% yield (45 mg, 0.18 mmol) from 2-chloro-2,4,4,4-tetrafluoro-3,3-dihydroxy-1-phenylbutan-1-one (57 mg, 0.2 mmol) by following the general procedure described above. Rf = 0.5 (hexanes); 1H NMR (400 MHz, Chloroform-d): δ = 8.19 (d, J = 7.9 Hz, 2H), 7.66 (dd, J = 7.9, 7.5 Hz, 1H), 7.52 (dd, J = 7.5, 7.5 Hz, 2H) ppm; 13C NMR (100 MHz, Chloroform-d): δ = 182.3 (d, JC-F = 24.7 Hz), 134.7, 131.0 (d, JC-F = 3.5 Hz), 129.0 (d, JC-F = 2.2 Hz), 128.7, 103.6 (d, JC-F = 319.3 Hz) ppm; 19F NMR (376 MHz, Chloroform-d) δ = -60.6 ppm; Anal. Calcd. for C8H5BrClF: C, 38.21; H, 2.00. Found: C, 37.91; H, 1.88.
2-Bromo-2-chloro-2-fluoro-1-(naphthalen-2-yl)ethan-1-one (10)
Compound 10 was obtained as a colorless liquid in 98% yield (59 mg, 0.19 mmol) from 2-chloro-2,4,4,4-tetrafluoro-3,3-dihydroxy-1-(naphthalen-2-yl)butan-1-one (67 mg, 0.2 mmol) by following the general procedure described above. Rf = 0.4 (hexanes); 1H NMR (400 MHz, Chloroform-d): δ = 8.80 (s, 1H), 8.16 (d, J = 8.9 Hz, 1H), 8.00 (d, J = 8.1 Hz, 1H), 7.92 (dd, J = 10.1, 10.7 Hz, 2H), 7.67 (dd, J = 8.2, 8.1 Hz, 1H), 7.60 (dd, J = 7.1, 7.9 Hz, 1H) ppm; 13C NMR (100 MHz, Chloroform-d): δ = 182.3 (d, JC-F = 24.6 Hz), 136.0, 133.6 (d, JC-F = 4.5 Hz), 132.1, 130.1, 129.7, 128.5, 127.8, 127.2, 126.1 (d, JC-F = 2.1 Hz), 125.6 (d, JC-F = 2.6 Hz), 103.9 (d, JC-F = 319.5 Hz) ppm; 19F NMR (376 MHz, Chloroform-d) δ = -59.9 ppm; Anal. Calcd. for C12H7BrClFO: C, 47.80; H, 2.34. Found: C, 47.78; H, 2.36.
2-Bromo-2-chloro-1-(4-chlorophenyl)-2-fluoroethan-1-one (11)
Compound 11 was obtained as a colorless liquid in 82% yield (47 mg, 0.16 mmol) from 2-chloro-1-(4-chlorophenyl)-2,4,4,4-tetrafluoro-3,3-dihydroxybutan-1-one (64 mg, 0.2 mmol) by following the general procedure described above. Rf = 0.5 (hexanes); 1H NMR (400 MHz, Chloroform-d): δ = 8.14 (d, J = 8.4 Hz, 2H), 7.49 (d, J = 8.5 Hz, 2H) ppm; 13C NMR (100 MHz, Chloroform-d): δ = 181.3 (d, JC-F = 25.1 Hz), 141.5, 132.4 (d, JC-F = 3.7 Hz), 129.1, 127.5, 103.5 (d, JC-F = 318.8 Hz) ppm; 19F NMR (376 MHz, Chloroform-d) δ = -61.0 ppm; Anal. Calcd. for C8H4BrCl2FO: C, 33.61; H, 1.41. Found: C, 33.85; H, 1.39.
2-Bromo-2-chloro-2-fluoro-1-(furan-2-yl)ethan-1-one (12)
Compound 12 was obtained as a colorless liquid in 85% yield (41 mg, 0.17 mmol) from 2-chloro-2,4,4,4-tetrafluoro-1-(furan-2-yl)-3,3-dihydroxybutan-1-one (55 mg, 0.2 mmol) by following the general procedure described above. Rf = 0.6 (hexanes); 1H NMR (400 MHz, Chloroform-d): δ = 7.78 (d, J = 1.8 Hz, 1H), 7.55 (m, 1H), 6.66 (dd, J = 3.7, 1.7 Hz, 1H) ppm; 13C NMR (100 MHz, Chloroform-d): δ = 171.5 (d, JC-F = 26.5 Hz), 149.3, 144.9, 123.9 (d, JC-F = 6.6 Hz), 112.9, 102.4 (d, JC-F = 317.8 Hz) ppm; 19F NMR (376 MHz, Chloroform-d) δ = -64.1 ppm; Anal. Calcd. for C6H3BrClFO2: C, 29.85; H, 1.25. Found: C, 29.97; H, 1.29.
2.3 General procedure for the dibromo olefination of 2-bromo-2-chloro-2-fluoroketones
A solution of triphenylphosphine (4 equiv.) in 2 mL of anhydrous dichloromethane was added dropwise to a solution of carbon tetrabromide (2 equiv.) in 2 mL of anhydrous dichloromethane at 0 °C over 30 minutes. The solution was stirred for 10 minutes and 2-bromo-2-chloro-2-fluoromethyl ketone (1 equiv.) in 2 mL of anhydrous dichloromethane was then added over 10 minutes. The resulting solution was stirred at 0 °C until conversion was complete based on TLC and 19F NMR analysis and 5 mL of water was added. The organic phase was washed with brine, dried over anhydrous sodium sulfate and the solvent was removed in vacuo. The crude product was purified by flash chromatography on silica gel using hexanes as mobile phase.
1,1,3-Tribromo-3-chloro-3-fluoro-2-phenylprop-1-ene (13)
Compound 13 was obtained after 4 hours as a colorless liquid in 98% yield (47 mg, 0.117 mmol) from 2-bromo-2-chloro-2-fluoro-1-phenylethan-1-one (30 mg, 0.119 mmol) by following the general procedure described above. Rf = 0.6 (hexanes); Rotation about the aryl-vinyl bond appears to be slow on the NMR time scale and more than one conformer is observed. 1H NMR (400 MHz, Chloroform-d): δ = 7.44 – 7.41 (m, 3H), 7.33 – 7.28 (m, 2H) ppm; 13C NMR (100 MHz, Chloroform-d): δ = 146.3 (d, JC-F = 24.5 Hz), 137.7 (d, JC-F = 2.4 Hz), 129.2, 129.1, 128.8, 128.7, 128.6, 103.0 (d, JC-F = 308.6 Hz), 99.1 ppm; 19F NMR (376 MHz, Chloroform-d) δ = -40.5 ppm; Anal. Calcd. for C9H5Br3ClF: C, 26.54; H, 1.24. Found: C, 26.77; H, 1.49.
2-(1,1,3-Tribromo-3-chloro-3-fluoroprop-1-en-2-yl)naphthalene (14)
Compound 14 was obtained after 4 hours as a colorless solid in 88% yield (67 mg, 0.146 mmol) from 2-bromo-2-chloro-2-fluoro-1-(naphthalen-2-yl)ethan-1-one (50 mg, 0.166 mmol) by following the general procedure described above. mp: 66-67 °C; Rf = 0.5 (hexanes); Rotation about the aryl-vinyl bond appears to be slow on the NMR time scale and more than one conformer is observed. 1H NMR (400 MHz, Chloroform-d): δ = 7.93 – 7.88 (m, 2H), 7.88 (d, J = 6.5 Hz, 1H), 7.83 (d, J = 8.9 Hz, 1H), 7.60 – 7.50 (m, 2H), 7.39 (d, J = 7.8 Hz, 1H) ppm; 13C NMR (100 MHz, Chloroform-d): δ = 146.3 (d, JC-F = 13.5 Hz), 146.1 (d, JC-F = 12.5 Hz), 135.0 (d, JC-F = 2.3 Hz), 134.9 (d, JC-F = 2.5 Hz), 133.2, 132.9, 128.7, 128.4, 128.4, 127.8, 127.1, 126.6, 126.3, 126.1, 103.1 (d, JC-F = 308.3 Hz), 103.0 (d, JC-F = 307.0 Hz), 99.4 (d, JC-F = 6.8 Hz) ppm; 19F NMR (376 MHz, Chloroform-d) δ = -40.3, -40.6 ppm; Anal. Calcd. for C13H7Br3ClF: C, 34.14; H, 1.54. Found: C, 34.26; H, 1.56.
1-Chloro-4-(1,1,3-tribromo-3-chloro-3-fluoroprop-1-en-2-yl)benzene (15)
Compound 15 was obtained after 6 hours as a colorless solid in 86% yield (52 mg, 0.12 mmol) from 2-bromo-2-chloro-1-(4-chlorophenyl)-2-fluoroethan-1-one (40 mg, 0.14 mmol) by following the general procedure described above. mp: 70-72 °C; Rf = 0.6 (hexanes); Rotation about the aryl-vinyl bond appears to be slow on the NMR time scale and more than one conformer is observed. 1H NMR (400 MHz, Chloroform-d): δ = 7.41 (d, J = 8.6 Hz, 2H), 7.25 (d, J = 8.5 Hz, 2H) ppm; 13C NMR (100 MHz, Chloroform-d): δ = 145.2 (d, JC-F = 24.5 Hz), 136.0 (d, JC-F = 2.5 Hz), 135.5, 130.5, 130.3 (d, JC-F = 1.5 Hz), 129.0 (d, JC-F = 1.4 Hz), 102.6 (d, JC-F = 308.8 Hz), 99.7 ppm; 19F NMR (376 MHz, Chloroform-d) δ = -41.1 ppm; Anal. Calcd. for C9H4Br3Cl2F: C, 24.47; H, 0.91. Found: C, 24.72; H, 1.18.
2.4 General procedure for the Wittig olefination of 2-bromo-2-chloro-2-fluoromethyl ketones
The 2-bromo-2-chloro-2-fluoromethyl ketone (1.0 equiv.) and benzyl 2-(triphenyl-phosphanylidene)acetate (1.2 equiv.) were dissolved in 1 mL of anhydrous tetrahydrofuran under inert atmosphere at room temperature. The reaction was monitored by TLC using hexanes-ethyl acetate (96:4) as mobile phase. The crude product was purified by flash chromatography on silica gel using hexanes-ethyl acetate (96:4).
Benzyl (E)-4-bromo-4-chloro-4-fluoro-3-phenylbut-2-enoate (16)
Compound 16 was obtained as a colorless solid in 81% yield (62 mg, 0.16 mmol) from 2-bromo-2-chloro-2-fluoro-1-phenylethan-1-one (50.3 mg, 0.2 mmol) by following the general procedure described above. Rf = 0.4 (hexanes / EtOAc, 9:1); 1H NMR (400 MHz, Chloroform-d) δ = 7.45 – 7.35 (m, 5H), 7.29 (m, 3H), 7.16 – 7.05 (m, 2H), 6.57 (s, 1H), 4.97 (s, 2H) ppm; 13C NMR (100 MHz, Chloroform-d) δ = 164.1 (d, JC-F = 1.2 Hz), 152.9 (d, JC-F = 18.4 Hz), 135.0, 132.4 (d, JC-F = 1.5 Hz), 130.0, 129.1, 128.5, 128.4, 128.3, 127.8, 120.1 (d, JC-F = 9.8 Hz), 105.4 (d, JC-F = 310.7 Hz), 66.9 ppm; 19F NMR (376 MHz, Chloroform-d) δ = -54.2 ppm. Anal. Calcd. for C17H13O2BrClF: C, 53.22; H, 3.42. Found: C, 53.30; H, 3.04.
Benzyl (E)-4-bromo-4-chloro-4-fluoro-3-(naphthalen-2-yl)but-2-enoate (17)
Compound 17 was obtained as a colorless solid in 92% yield (43 mg, 0.11 mmol) from 2-chloro-2,4,4,4-tetrafluoro-3,3-dihydroxy-1-(naphthalen-2-yl)butan-1-one (36 mg, 0.12 mmol) by following the general procedure described above. mp: 102-105 °C; Rf = 0.2 (hexanes / EtOAc, 98:2); 1H NMR (400 MHz, Chloroform-d): δ = 7.88 – 7.81 (m, 4H), 7.57 – 7.51 (m, 2H), 7.47 (d, J = 8.3 Hz, 1H), 7.20 (dd, J = 7.5, 7.4 Hz, 1H), 7.10 (dd, J = 7.6, 7.5 Hz, 2H), 6.91 (d, J = 7.4 Hz, 2H), 6.67 (s, 1H), 4.93 (s, 2H) ppm; 13C NMR (100 MHz, Chloroform-d): δ = 164.2, 152.6 (d, JC-F = 18.5 Hz), 134.7, 133.3, 132.5, 129.9, 129.4, 128.4, 128.3, 128.2, 128.2, 127.8, 127.6, 127.3, 126.9, 126.4, 120.5 (d, JC-F = 9.8 Hz), 105.5 (d, JC-F = 310.8 Hz), 66.9 ppm; 19F NMR (376 MHz, Chloroform-d) δ = -53.8 ppm; Anal. Calcd. for C21H15BrClFO2: C, 58.16; H, 3.49. Found: C, 58.22; H, 3.55.
Benzyl (E)-4-bromo-4-chloro-3-(4-chlorophenyl)-4-fluorobut-2-enoate (18)
Compound 18 was obtained as a colorless solid in 91% yield (62 mg, 0.15 mmol) from 2-bromo-2-chloro-1-(4-chlorophenyl)-2-fluoroethan-1-one (46 mg, 0.16 mmol) by following the general procedure described above. mp: 51-52 °C; Rf = 0.3 (hexanes / EtOAc, 98:2); 1H NMR (400 MHz, Chloroform-d) δ = 7.36 – 7.28 (m, 7H), 7.15 – 7.10 (m, 2H), 6.58 (s, 1H), 5.00 (s, 2H) ppm; 13C NMR (100 MHz, Chloroform-d) δ = 163.8 (d, JC-F = 1.6 Hz), 151.6 (d, JC-F = 18.8 Hz), 135.4, 134.7, 131.4, 130.8 (d, JC-F = 1.5 Hz), 128.6, 128.5, 128.4, 128.1, 120.6 (d, JC-F = 9.8 Hz), 105.0 (d, JC-F = 310.7 Hz), 67.1 ppm; 19F NMR (376 MHz, Chloroform-d) δ = -54.71 ppm. Anal. Calcd. for C17H12BrCl2FO2: C, 48.84; H, 2.89. Found: C, 48.83; H, 2.89.
Ethyl (E)-4-bromo-4-chloro-4-fluoro-3-phenylbut-2-enoate (19)
To a solution of 2-bromo-2-chloro-2-fluoro-1-phenylethan-1-one (50 mg, 0.2 mmol) in anhydrous tetrahydrofuran was added sodium hydride (10 mg, 0.24 mmol) at 0 °C under inert atmosphere. After stirring for 30 minutes, triethyl phosphonoacetate (0.047 mL, 0.24 mmol) was added. The resulting solution was stirred for 2 h at 0 oC and quenched with water. The organic phase was washed with brine, dried over anhydrous sodium sulfate and the solvent was removed in vacuo. The crude product was purified by flash chromatography on silica gel using hexanes-ethyl acetate (95:5) as mobile phase. Compound 19 was obtained in 95% yield as a colorless liquid (61 mg, 0.19 mmol); Rf = 0.4 (hexanes / EtOAc, 19: 1); 1H NMR (400 MHz, Chloroform-d): δ = 7.51 – 7.33 (m, 5H), 6.53 (s, 1H), 3.99 (q, J = 7.1 Hz, 2H), 1.02 (t, J = 7.2 Hz, 3H) ppm; 13C NMR (100 MHz, Chloroform-d): δ = 164.3, 152.4 (d, JC-F = 18.5 Hz), 132.6 (d, JC-F = 1.5 Hz), 130.1, 129.0, 127.7, 120.4 (d, JC-F = 9.6 Hz), 105.4 (d, JC-F = 310.5 Hz), 60.9, 13.7 ppm; 19F NMR (376 MHz, Chloroform-d) δ = -54.3 ppm; Anal. Calcd. for C12H11BrClFO2: C, 44.82; H, 3.45. Found: C, 44.35; H, 3.61.
Crystallographic Analysis
A single crystal of 3 was obtained by slow evaporation of a solution of the compound in a mixture of ethyl acetate and hexanes (5% EtOAc in hexanes). Single crystal X-ray analysis was performed at 296 K using a Siemens platform diffractometer with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). Data were integrated and corrected using the APEX II program. The structures were solved by SHELXT and refined with full-matrix least-square analysis using SHELX-97-2 software. Non-hydrogen atoms were refined with anisotropic displacement parameter. Crystal data: C10H7ClF4O3, M = 286.61, colorless prism, 0.3 × 0.2 × 0.1 mm3, monoclnic, space group P21/c, a = 8.0622(11), b = 18.116(2), c = 7.6291(10) Å, V = 1113.3(3) Å3, Z = 4. A single crystal of 17 was obtained by slow evaporation of a solution of the compound in a mixture of ethyl acetate and hexanes (5% EtOAc in hexanes). Single crystal X-ray analysis was performed as described above. Crystal data: C21H15BrClFO2, M = 433.69, colorless needle, 0.12 × 0.07 × 0.05 mm3, orthorombic, space group P212121, a = 5.9238(9), b = 8.0989(12), c = 37.622(6) Å, V =1805.0(5) Å3, Z = 4.
Supplementary Material
Acknowledgments
We greatfully acknowledge financial support from the National Institutes of Health (GM106260).
Footnotes
Supplementary Information. Full characterization and NMR spectra for all compounds.
Notes and References
- 1.Smith BR, Eastman CM, Njardarson JTJ. Med Chem. 2014;57:9764773. doi: 10.1021/jm501105n. [DOI] [PubMed] [Google Scholar]; Wang J, Sanchez-Rosello M, Acena JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem Rev. 2014;114:2432506. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Y, Wang J, Gu Z, Wang S, Zhu W, Acena JL, Soloshonok VA, Izawa K, Liu H. Chem Rev. 2016;116:42218. doi: 10.1021/acs.chemrev.5b00392. [DOI] [PubMed] [Google Scholar]
- 3.a) Hargreaves MK, Barrett GC, Hall DM, Modarai BJ. Chem Soc C. 1971:279–282. [Google Scholar]; b) Hargreaves MK, Modarai B. J Chem Soc C. 1971:1013–1015. [Google Scholar]; c) Doyle TR, Vogl O. J Am Chem Soc. 1989;111:8510511. [Google Scholar]; d Doyle TR, Vogl O. Monatsh Chem. 1990;121:313. [Google Scholar]
- 4.a) Yang ZYJ. Org Chem. 2003;68:4410416. [Google Scholar]; b) Zhao Y, Gao B, Hu J. J Am Chem Soc. 2012;134:5790793. doi: 10.1021/ja301601b. [DOI] [PubMed] [Google Scholar]
- 5.a) Kvicala J, Stambasky J, Skalicky M, Paleta OJ. Fluorine Chem. 2005;126:1390395. [Google Scholar]; b) Petko KI, Kot SY, Yagupolskii LM. J Fluorine Chem. 2008;129:1119123. [Google Scholar]; c) Matsnev AV, Qing SY, Stanton MA, Berger KA, Haufe G, Thrasher JS. Org Lett. 2014;16:2402405. doi: 10.1021/ol500766v. [DOI] [PubMed] [Google Scholar]; d) Shiosaki M, Inoue M. J Fluorine Chem. 2015;175:16068. [Google Scholar]
- 6.Mei H, Nie C, Acena JL, Soloshonok VA, Roeschenthaler GV, Han J. Eur J Org Chem. 2015:6401–6412. [Google Scholar]
- 7.Prager JH, Ogden PHJ. Org Chem. 1968;33:2100102. [Google Scholar]
- 8.Han C, Kim EH, Colby DAJ. Am Chem Soc. 2011;133:5802805. doi: 10.1021/ja202213f. [DOI] [PubMed] [Google Scholar]
- 9.Prakash GKS, Zhang Z, Wang F, Munoz S, Olah GAJ. Org Chem. 2013;78:3300305. doi: 10.1021/jo400202w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Han C, Kim EH, Colby DA. Synlett. 2012:1559–1563. [Google Scholar]; b) Riofski MV, Hart AD, Colby DA. Org Lett. 2013;15:20811. doi: 10.1021/ol303291x. [DOI] [PubMed] [Google Scholar]
- 11.Li W, Zhu X, Mao H, Tang Z, Cheng Y, Zhu C. Chem Commun. 2014;50:7521523. doi: 10.1039/c4cc02768j. [DOI] [PubMed] [Google Scholar]
- 12.a) Xie C, Wu L, Mei H, Soloshonok VA, Han J, Pan Y. Tetrahedron Lett. 2014;55:5908910. [Google Scholar]; b) Xie C, Wu L, Zhou J, Mei H, Soloshonok VA, Han J, Pan Y. J Fluorine Chem. 2015;172:131. [Google Scholar]; c) Xie C, Wu L, Mei H, Soloshonok VA, Han J, Pan Y. Org Biomol Chem. 2014;12:7836843. doi: 10.1039/c4ob00489b. [DOI] [PubMed] [Google Scholar]; d) Xie C, Wu L, Han J, Soloshonok VA, Pan Y. Angew Chem Int Ed. 2015;54:6019–6023. doi: 10.1002/anie.201500908. [DOI] [PubMed] [Google Scholar]; e) Xie C, Dai Y, Mei H, Han J, Soloshonok VA, Pan Y. Chem Commun. 2015;51:9149152. doi: 10.1039/c5cc02256h. [DOI] [PubMed] [Google Scholar]
- 13.Wu C, Li G, Sun W, Zhang M, Hong L, Wang R. Org Lett. 2014;16:1960963. doi: 10.1021/ol500517d. [DOI] [PubMed] [Google Scholar]
- 14.a) Saidalimu I, Fang X, He XP, Liang J, Yang XY, Wu FH. Angew Chem Int Ed. 2013;52:5566–5570. doi: 10.1002/anie.201301443. [DOI] [PubMed] [Google Scholar]; b) Saidalimu I, Fang X, Lv W, Yang X, He X, Zhang J, Wu FH. Adv Synth Catal. 2013;355:85763. [Google Scholar]
- 15.a) Zhang P, Wolf WJ. Org Chem. 2012;77:8840844. [Google Scholar]; b) Zhang P, Wolf W. Angew Chem Int Ed. 2013;52:7869873. doi: 10.1002/anie.201303551. [DOI] [PubMed] [Google Scholar]
- 16.John JP, Colby DAJ. Org Chem. 2011;76:9163168. [Google Scholar]
- 17.Pravst I, Zupan M, Stavber S. Tetrahedron. 2008;64:5191199. [Google Scholar]
- 18.The reaction was carried out with 5 equivalents of LiBr, 2.5 equivalents of K2CO3 and 2 equivalents of NBS using THF as solvent at rt. Complete conversion of 5 was observed within 1 hour.
- 19.The trapping of chlorofluoro-substituted enolates such as 7 with NIS or iodine is also possible but the corresponding chlorofluoroiodomethyl ketones are considerable less stable than 8 and its analogues.
- 20.Ramirez F, Desai NB, McKelvie NJ. Am Chem Soc. 1962;84:1745747. [Google Scholar]
- 21.The (Z)-isomer was not detected.
- 22.a) Burton DJ, Koppes WMJ. Org Chem. 1975;40:3026032. [Google Scholar]; b) Eguchi T, Aoyama T, Kakinuma K. Tetrahedron Lett. 1992;33:5545546. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.