

Abstract
The characterization of integral membrane proteins presents numerous analytical challenges on account of their poor activity under non-native conditions, limited solubility in aqueous solutions, and low expression in most cell culture systems. Nanodiscs are synthetic model membrane constructs that offer many advantages for studying membrane protein function by offering a native-like phospholipid bilayer environment. The successful incorporation of membrane proteins within Nanodiscs requires experimental optimization of conditions. Standard protocols for Nanodisc formation can require large amounts of time and input material, limiting the facile screening of formation conditions. Capitalizing on the miniaturization and efficient mass transport inherent to microfluidics, we have developed a microfluidic platform for efficient Nanodisc assembly and purification, and demonstrated the ability to incorporate functional membrane proteins into the resulting Nanodiscs. In addition to working with reduced sample volumes, this platform simplifies membrane protein incorporation from a multi-stage protocol requiring several hours or days into a single platform that outputs purified Nanodiscs in less than one hour. To demonstrate the utility of this platform, we incorporated Cytochrome P450 into Nanodiscs of variable size and lipid composition, and present spectroscopic evidence for the functional active site of the membrane protein. This platform is a promising new tool for membrane protein biology and biochemistry that enables tremendous versatility for optimizing the incorporation of membrane proteins using microfluidic gradients to screen across diverse formation conditions.
Introduction
Membrane proteins play pivotal roles in cellular processes as the primary units of biomolecular transport and cellular communication. Because of their importance, membrane proteins are the most common targets for pharmaceutical agents.1,2 Key to the study of membrane proteins is maintaining protein function in vitro. Purified membrane proteins exhibit substantially reduced activity outside of a native lipid bilayer environment, primarily because of protein misfolding in aqueous solutions.3–7 Soluble lipid bilayer systems, such as protein-lipid micelles and liposomes, act as water-soluble and semi-native environments that have facilitated the characterization of many membrane proteins.4,8,9 However, limitations of these systems have inspired exploration into alternative lipid bilayer mimetics for structural and functional studies of membrane proteins. For example, liposome preparations often have high viscosities and/or turbidity that present major challenges for cell-free expression systems and many biophysical methods to interrogate functional protein activity.10,11 Proteins solubilized in detergent micelles often demonstrate structural changes caused by the non-native environment.5
Nanodiscs are soluble, protein stabilized discoidal lipid bilayers that offer enabling advantages over liposomes and micelles for membrane protein studies.12 In comparison to liposomes, micelles, and other soluble lipid bilayer systems, such as those made with styrene-maleic acid copolymers,13–15 Nanodiscs are remarkably homogenous and stable in aqueous solutions. Beyond bilayer and protein stability, Nanodiscs have added advantages of access to both sides of the bilayer and precise control of bilayer composition, stoichiometry, and size.16–18 The variable sizes of Nanodiscs allow for the incorporation of mono- or dimeric membrane proteins and can even support incorporation of multiprotein complexes essential for maintaining protein function.19–21 The enhanced functionalities of Nanodiscs have resulted in their wide adoption as the preferred lipid bilayer mimetic system across diverse facets of membrane protein biology.12,22
For balance, it is worth noting that micellar and liposomal systems do have advantages in certain applications--particularly those that require compartmentalization or asymmetry across the bilayer. Furthermore, though commercially available, Nanodiscs do require a membrane scaffold protein (MSP), which adds a potential level of complexity, and the spectroscopic overlap between the MSP and incorporated membranes might complicate some assays, such as protein content determination via simple UV absorbance. Nevertheless, Nanodiscs have emerged as a powerful technology that continues to enable many studies that require model membrane interfaces.
Conventional Nanodisc assembly is achieved by solubilizing phospholipids and membrane proteins with detergents in the presence of a MSP. Upon removal of detergent, Nanodiscs self-assemble with MSP wrapping around a discoidal phospholipid bilayer with an integrated membrane protein.16,23 A variety of membrane proteins have been incorporated into Nanodiscs, demonstrating the generality of the platform.12,22,24 Despite this versatility, the incorporation of new membrane proteins within Nanodiscs does require empirical optimization, which typically involves serial screening of different detergents and phospholipids, as well as determining ideal ratios of reagents (e.g., phospholipid to MSP ratio). This laborious task can consume unacceptably large amounts of starting material—an impediment that is particularly limiting for many membrane proteins, which are natively expressed at low levels and are notoriously challenging to recombinantly express.7,25,26
Microfluidic technologies have emerged as robust alternatives to many bulk-scale molecular biology protocols featuring intrinsic miniaturization and low reagent consumption.27–29 The ability to precisely manipulate small fluid volumes facilitates precise timing of fluid handling steps, and the short distances involved in microfluidics leads to efficient diffusion and expedient reaction processing. The additional benefits of parallelization and modular device design have further positioned microfluidics as powerful tools for improved protein processing and characterization.30–34
This study describes an integrated platform for Nanodisc assembly and purification using a microfluidic device that supports rapid Nanodisc assembly and reduced membrane protein consumption. Moreover, we demonstrate that these membrane proteins retain functional activity, which is indicative of incorporation into a fully reconstituted model membrane system. In series with Nanodisc assembly, a purification module can be incorporated into the device and tuned to specific sample processing applications, adding additional functional capabilities to the platform. Importantly, the device architecture incorporates multiple inlets for on-chip reagent mixing at user-defined ratios and times. This multi-port design allows for avoidance of conditions where prolonged reagent mixing results in deleterious effects, such as reduced protein activity after exposure to detergents. This design also allows for the generation of temporal reagent gradients (e.g. lipids or surfactants), facilitating screening of variable membrane protein incorporation conditions within a single experiment. The capabilities of this new platform are demonstrated by the formation and characterization of Nanodiscs without incorporated protein (termed empty Nanodiscs) having variable lipid composition, and through the on-chip incorporation of Cytochrome P450 into Nanodiscs and subsequent confirmation of functional enzyme activity.11,12 Though the focus in this study is on well-characterized proteins that have been previously shown to incorporate into Nanodiscs, we anticipate that this platform will offer broad utility for determining incorporation conditions for new proteins in which minimal reagent consumption and high throughput, gradient-based screening approaches are advantageous.
Experimental Methods
Materials
Amberlite XAD-2 hydrophobic beads, ethylenediaminetetraacetic acid (EDTA), dimethyl sulfoxide, 3-[(3-Cholamidopropyl)dimethylammonio]-1- propanesulfonate (CHAPS), sodium cholate, and other chemicals were purchased from Sigma Aldrich (St. Louis, MO, USA) unless otherwise indicated. Pierce Detergent Removal Resin was purchased from ThermoFisher Scientific (Waltham, MA, USA). The phospholipids 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1-palmitoyl-2-oleoyl- sn-glycero-3-phosphocholine (POPC), and 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine- N-(lissamine rhodamine B sulfonyl) (Liss Rhod PE) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). The membrane scaffold proteins (MSP) used were MSP1D1 and MSP1E3D1, which were expressed and purified as previously described.16,17 Both MSPs have an N-terminal His-tag that can be used for affinity purification. The His-tag can be removed using a Tobacco Etch Virus protease to completely and specifically cleave the tag. Cytochrome P450 3A4 (CYP3A4) was expressed from the NF-14 construct in the pCWOri+ vector with a histidine affinity tag. CYP3A4 was purified and incorporated into Nanodiscs as previously described.35–38 All buffers were prepared with deionized water and filtered prior to use.
Microfluidic Design & Fabrication
Microfluidic device masters were designed using AutoCad (Autodesk Inc., San Rafael, CA), and photomasks were printed by CAD/Art Services,Inc. (Bandon, OR, USA). SU-8 2100, an epoxy-based negative photoresist, was purchased from Microchem (Westborough, MA, USA) and used to fabricate masters according to standard photolithography methods.39 Device features were designed to be 200 μm in height and confirmed using profilometry. Polydimethylsiloxane (PDMS) was purchased from Momentive (Waterford, NY, USA) under the name RTV615 silicone rubber kit. The two-part mixture was combined 10:1 monomer:initiator, thoroughly mixed, and degassed under vacuum prior to pouring onto the negative master mold. PDMS was cured at 70°C for a minimum of 1 h. Device stamps were cut out of the PDMS mold, and access ports were added using Integra Miltex biopsy punches. Stamps were cleaned with Scotch Magic Tape to remove dust and other particulates prior to bonding to glass slides.
Silastic tubing with an inner diameter of 0.040″ (Dow Corning, Midland, MI, USA) was used for the bead filling port, and 0.022″ inner diameter Teflon tubing (Cole-Parmer, Vernon Hills, IL, USA) was used for all other ports. Filling of the bead bed was performed using either a custom-built pressure system or manually with a disposable syringe attached to the Silastic tubing. For manual filling, a density-balanced bead slurry was prepared from Pierce Detergent Removal Resin, Optiprep density gradient medium, and water. To prevent the loss of beads, the bead inlet tubing was clamped with a hemostat. The detergent removal capacity for this resin was previously demonstrated to be 1-10 mg of detergent per 1 mL of detergent removal resin across a wide variety of detergents, including sodium cholate and CHAPS.40
Microfluidic Nanodisc Assembly
Prior to Nanodisc assembly, all devices were washed with methanol for at least 20 min at 30 μL/min followed by a water rinse for at least 10 min at the same flow rate. Detergent bead beds could be regenerated by first rinsing device with water for 10 min and then following the same washing procedure for new devices. The lower limits of rinsing times were not determined, but could likely be shortened. Phospholipids used for Nanodisc assembly were stored in chloroform at -20°C. Prior to use, the phospholipids were dried to a lipid film and stored under vacuum for a minimum of 4 h. Nanodisc reagents were prepared in Standard Disc Buffer (SDB; 20 mM Tris HCl pH 7.4, 0.1 M NaCl, 0.5 mM EDTA, and 0.01% NaN3). In cases where temperature control was needed, the microfluidic device was placed directly into a temperature controlled environment. For example, Nanodisc assembly with POPC lipids, which is optimum at 4°C, was performed with the devices on ice.
Nanodisc Assembly with Single Port Devices
Reagents for Nanodisc assembly were prepared according to desired ratios for lipid:MSP and MSP:CYP3A4. Supplementary Table 1 provides an example reagent sheet for Nanodisc assembly with a single port device. Reagents were mixed immediately prior to Nanodisc assembly, loaded into a syringe, and flowed through the device. The typical flow rate used for single port devices was 30 μL/min controlled by a Pump 11 Pico Plus Elite Dual Syringe Pump from Harvard Apparatus (Holliston, MA, USA). The eluent was collected in fractions from 5-100 μL and analyzed with SEC and/or AFM. Nanodisc self-assembly is initiated upon removal of detergent via adsorption onto the detergent removal resin beads. Some sample loss is likely to occur caused by adsorption on the resin beads and onto the walls of the PDMS device. The resin has been previously shown to preserve more than 90% of protein samples.40 Because the surface area of the detergent removal resin is much higher than that of the PDMS device, adsorption onto the PDMS walls is likely a minor contributor to sample loss as opposed to the much larger surface area of the resin beads in the packed bed.
Nanodisc Assembly with Multi-Port Devices
Multi-port devices were prepared following the same sample protocol as single port devices. The reagents were divided into three syringes: (1) lipid with detergent, (2) MSP with detergent, (3) either buffer or membrane protein of interest. An example reagent sheet for a multi-port device is provided in Supplementary Table 2. Reagent concentrations were determined such that optimal reagent ratios were achieved when all syringes flowed at the same rate, usually 10 μL/min for each syringe. Gradient experiments were performed by changing the flow rate of one of the syringes. The Pico Plus Elite pumps can be programmed with linear gradients for automated gradient experiments. See Supplementary Figure 7 for additional details on the microfluidic gradients.
Colorimetric Quantitation of Detergent Removal
The amount of either sodium cholate or CHAPS in a solution can be determined colorimetrically by oxidation of the detergents with concentrated sulfuric acid. To quantitate the detergent removal capacity of the devices, detergent-containing solutions were flowed across the packed beds of the devices and fractions were collected from the eluent. The concentration of detergent in each fraction was quantified according to a previously described method (Supplemental Figure 3).41
Nanodisc Characterization by Size Exclusion Chromatography
Eluent fractions collected from the microfluidic devices were characterized by size exclusion chromatography (SEC) to assess the quality of Nanodisc assembly. Fractions were injected onto a Superdex 200 Increase 3.2/300 or 10/300 column (GE Healthcare, Pittsburgh, PA, USA). The 10/300 column was operated at a flow rate of 0.75 mL/min, and the 3.2/300 column was operated at a flow rate of 50 μL/min. Absorbances were measured at 280 nm to monitor Nanodisc formation and 417 nm to follow CYP3A4 incorporation into Nanodiscs. The following proteins (with known hydrodynamic radii) were used as chromatographic standards: Thyroglobulin (17 nm), Ferritin (12.2 nm), Bovine Liver Catalase (10.4 nm), and Bovine Serum Albumin (7.1 nm).
Nanodisc Characterization by AFM
Characterization of Nanodiscs with atomic force microscopy (AFM) was performed with a Cypher ES Environmental AFM (Asylum Research, Santa Barbara, CA, USA) equipped with a fluid cell. To prepare the surface for Nanodisc analysis, mica was glued to a stainless-steel disc and cleaned with cellophane tape. Nanodiscs were diluted between 10- and 100-fold, and 10 μL of diluted sample was applied to the mica surface. The surface was then rinsed with 10-20 μL of imaging buffer (10 mM Tris-HCL pH 7.4, 0.15 M NaCl, 10 mM MgCl2). A PAP pen (Ted Pella Inc., Redding, CA, USA) was used to circumscribe an area of mica with a hydrophobic border, which was used to prevent flow of solution off of the mica. After 10 min, 5-10 mL of imaging buffer was passed through the cell to remove any unadsorbed material. The sample was then mounted onto the imaging stage. Contact imaging was performed under imaging buffer with a thin-legged 310 μm cantilever with a nominal spring constant of 0.01 N/m.
Nanodisc Characterization by Dynamic Light Scattering
Characterization of Nanodiscs with dynamic light scattering (DLS) was performed using the Litesizer 500 Particle Analyzer (Anton Parr, Ashland, VA, USA). DLS was performed on Nanodiscs with DMPC phospholipids and MSP1D1 with Nanodiscs eluted from the assembly device. No sample purification was performed prior to analysis, and samples were analyzed at a concentration of 25 μM following the manufacturer's recommended protocol.
SDS-PAGE Analysis of Nanodiscs
Nanodisc assembly and purification was assessed using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on a 4-12% gradient gel (Bio-Rad, Hercules, CA, USA) under reducing conditions. The gel was stained with Coomassie Blue (Bio-Rad) after rinsing with deionized water. Both electrophoresis and staining followed the manufacturer's recommended protocol. SDS-PAGE gels were imaged with a ChemiDoc MP Imaging System (Bio-Rad), and further analysis was performed using Fiji.42
Nanodisc Purification with Affinity Chromatography
A slurry of Ni-NTA agarose resin was prepared in water. Using a syringe, methanol and water of sufficient volume to completely fill the device (approximately 100 μL each) were pushed through the device by hand. A syringe was then filled with Ni-NTA slurry and an 18-gauge needle was attached to the syringe with silastic tubing securely nested over needle. The silastic tubing was inserted into the filling port, and the resin bed was filled by applying steady pressure to the syringe plunger. Once the device was filled, the silastic tubing was clamped immediately above the filling port with either a hemostat or cable tie. To prepare the filled device for purification, the device was washed with 4 bed volumes of water followed by 8 bed volumes of Purification Buffer (250 mM NaH2PO4, pH 8.0 50 mM NaCl) at a flow rate of 30 μL/min. The Nanodisc solution was then flowed across the packed bed at a flow rate of 10 μL/min. Then, the device was washed with the Wash Buffer (250 mM NaH2PO4, pH 8.0 50 mM NaCl, and 10 mM imidazole) at 30 μL/min for 6 bed volumes. After washing, 5 bed volumes of Elution Buffer (250 mM NaH2PO4, pH 8.0 50 mM NaCl, and 250 mM imidazole) were flowed through the device at 10 μL/min. Elution fractions were collected typically at volumes between 5 and 60 μL. The protein content of each fraction was determined using a Qubit 3.0 Fluorometer (ThermoFisher) following the manufacturer's recommended protocol. The purification modules could be reused after washing with 0.5M NaOH flowing at 30 μL/min for 30 min.
Per the manufacturer (Sigma), Ni-NTA resin can bind 5-10 mg of protein per mL of resin. The standard device design has 60 μL of resin. At a Nanodisc concentration of 30 μM (60 μM MSP), there is ∼0.135 mg of MSP per 90 μL fraction collected from a single device. This equates to more than 3 fractions (90 μL fraction from a standard Nanodisc assembly device) per 60 μL Ni-NTA device. Fractions containing 0.5 mg/mL total protein content or greater were combined into a single fraction. For subsequent spectroscopic analysis, imidazole was removed from the fractions using 3.5 kDa MWCO filters (ThermoFisher) per manufacturer-recommended protocols. SDB was used as dialysis buffer.
Spin-Shift Assays with CYP3A4 in Nanodiscs
Prior to performing the spin shift assay, Nanodiscs were formed using the microfluidic assembly and purification modules according to the specified protocol. Purified Nanodiscs containing CYP3A4 were added to a quartz cuvette. Absorption spectra were acquired using a StellarNet Black Comet UV-visible spectrometer optically connected to a StellarNet cuvette holder and Halogen lamp light source using two 400 μm fiber-optic cables with 0.22 numerical aperture (Thorlabs). A baseline spectrum was collected for the CYP3A4 Nanodisc solution. Bromocriptine was dissolved in DMSO at 5 mg/mL and stored at -20° for short term storage. Prior to use for the spin-shift assay, the solution was allowed to equilibrate to ambient temperature and diluted to 0.1 mg/mL in a 1:9 DMSO:SDB solution. The bromocriptine solution was then added to the cuvette with pipette mixing before collecting absorption spectra. For the imidazole spin-shift assay, the absorption spectrum was collected after purification but prior to dialysis to remove imidazole. This spectrum was then compared to the baseline absorption spectrum for the bromocriptine spin shift assay.
Results and Discussion
This microfluidic platform is modular and can be divided into two primary functions: (1) Nanodisc assembly and (2) purification of assembled Nanodiscs. The Nanodisc assembly consists of reagent inlets, a larger inlet for loading resin material, a packed bed of detergent removal resin, and an outlet for the collection of Nanodiscs (Figure 1a). Mixing of Nanodisc reagents can be performed either on- or off-chip. For on-chip mixing, the devices include multiple reagent inlets and a serpentine mixing chamber with alternative jutting structures to ensure efficient mixing (Supplementary Figure 1a). The bead bed consists of capture structures at the inlet and outlet of the bed along with posts for structural support throughout the bed. The design uses a three-port reagent inlet for on-chip mixing and a bed volume of either 20 or 60 μL, though the modularity of the platform allows for individual devices to be tailored for specific experiments (Supplementary Figure 1). Flow through the various device designs was visualized with dye, demonstrating rapid and efficient mixing for multiport devices (Supplementary Figure 2). Components were flowed through the device and Nanodiscs self-assemble as the detergent was removed by the on-chip packed resin bed.40 To demonstrate this assembly, Nanodiscs were formed on the device through the removal of two types of bile-salt detergents: sodium cholate and 3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate (CHAPS) (Supplementary Figure 3). Detergent choice for Nanodisc assembly is typically dictated by the stability of the membrane protein target,4,12 and the ability of the resin to remove many types of detergents ensures the generality of this microfluidic device for many model membrane mimetic systems.40
Figure 1. Platform Device Designs.
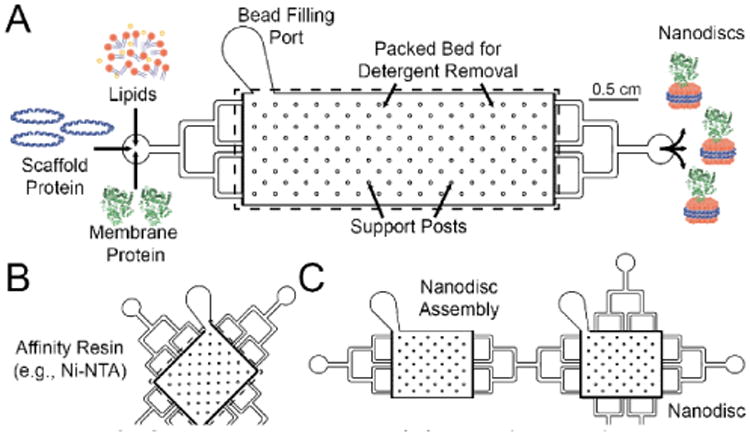
(A) A single-port device consists of reagent and detergent removal bead inlets, a bead bed with integrated posts for structural support, and an outlet for Nanodisc elution. The device has a bead bed volume of 60 μL and yields 0.1-2 nmol of Nanodiscs. (B) The purification devices feature multi directional flow for loading of Nanodiscs formed using devices from A. (C) Interfacing Nanodisc self-assembly and purification can be achieved as a single, integrated platform.
The purification module (Figure 1b) is conceptually similar to the Nanodisc assembly device in that they both rely on microstructures to capture a bead-based bed of resin that achieves the end function. However, the resin used in this module can be tuned to enable affinity-based purification, and subsequent elution, of Nanodisc-incorporated membrane proteins. For proof-of-principle, Ni-nitrilotriacetic acid (Ni-NTA) agarose resin was used as the affinity resin for purification. For the present study, we only used Ni-NTA as a purification resin, though other bead-based purification systems are compatible with the current design (e.g., immunoaffinity purification). The standalone design consists of a (1) single inlet, (2) a packed bed of affinity purification resin, and (3) an outlet for the collection of purified Nanodisc material. The purification module can also be integrated directly downstream of the Nanodisc assembly module through the simple addition of inlet and outlet ports with a flow direction perpendicular to the flow from the assembly module (Figure 1c).
For both modules, microfluidic devices were fabricated from polydimethylsiloxane (PDMS) stamps bonded to glass using standard soft lithography. Briefly, a master mold was fabricated using 2-D photolithography with silicon wafers and an epoxy-based negative photoresist. PDMS stamps were made from the master molds and plasma bonded to glass.
To demonstrate this approach for microfluidic Nanodisc assembly, initial experiments focused on the creation of Nanodiscs without incorporation of a membrane protein. Beyond providing a simple system for assessing Nanodisc assembly, “empty” Nanodiscs of precise lipid composition have found broad utility in probing protein-lipid interactions of fundamental importance to several biological processes, including the blood coagulation cascade.43–45 Nanodiscs were formed using either 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) or 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), and two different MSPs (MSP1D1 or MSP1E3D1). MSP1D1 results in Nanodiscs 9.7 nm in diameter with 120 to 160 lipids per Nanodisc and a lipid:MSP ratio of 60:1 to 80:1 (there are two MSPs per Nanodisc), depending on the packing density of the lipids. MSP1E3D1 gives larger 12.7 nm diameter Nanodiscs with lipid:MSP ratios ranging from 120:1 to 150:1.16
The Nanodisc reagents were initially mixed off-chip and then flowed through the single-port inlet device and across the packed bed of detergent removal resin. Nanodisc assembly occurs immediately upon removal of detergent as the solution flows across the packed bed. Size exclusion chromatography (SEC) was used to assess Nanodisc purity, size, and dispersity. Chromatograms showed a single, narrow peak at the appropriate elution times relative to a mixture of protein standards (Figure 2a-b). Successful microfluidic assembly of Nanodiscs was also orthogonally confirmed by atomic force microscopy (Figure 2c) and dynamic light scattering (Figure 2d). Nanodisc assembly was found to be independent of device flow rates after testing from 1 μL/min to 100 μL/min (Supplementary Figure 4). Since Nanodisc assembly is entirely based upon detergent removal, the removal capacity of the basic assembly module (Figure 1a) was experimentally determined to be ∼1 mg of detergent, for both sodium cholate and CHAPS (Supplementary Figure 3). Once the detergent capacity is reached for a given bed volume, the detergent removal resin can be regenerated by rinsing with methanol. Nanodisc assembly can be performed repeatedly on a single device with no observable degradation in Nanodisc quality.
Figure 2. Microfluidic Self-Assembly with Empty Nanodiscs.
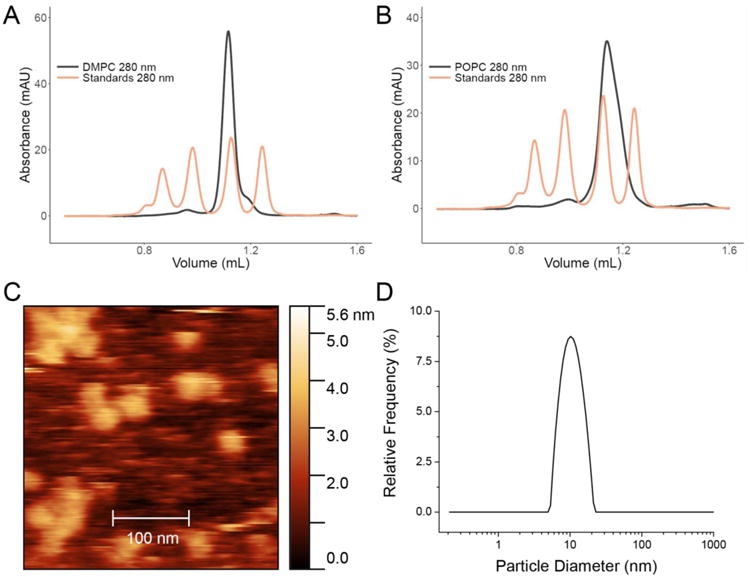
SEC analysis of Nanodiscs formed from a single-port device with DMPC and a DMPC:MSP ratio of 80:1 (A) and POPC and a POPC:MSP ratio of 60:1 (B) with MSP1D1. Approximate Nanodisc concentrations for each are 25 μM. (C) Atomic force microscopy (AFM) of DMPC Nanodiscs formed with the Nanodisc assembly module without prior purification show Nanodiscs of appropriate dimension with no evidence of large lipid aggregates. (D) Dynamic light scattering (DLS) analysis of DMPC Nanodiscs indicating a single, monodisperse peak corresponding to Nanodiscs.
In contrast to the single port module that requires all reagents to be combined off-chip, the multi-port detergent removal device allows on-chip reagent mixing so that the Nanodisc components are only combined immediately before detergent removal and Nanodisc assembly. A device with three inlet ports (SupplementaryFigure 1a) was used for Nanodisc assembly with both DMPC and POPC. The three inlets were used to flow: (1) detergent solubilized phospholipids, (2) MSP, and (3) SDB. There were no observed differences in Nanodisc quality as compared to premixing with single port devices (Supplementary Figure 5).
Beyond on-chip mixing, the multi-port design also offers the ability to tune reagent composition as a function of time. For example, flow rates at different inlets can be tuned over time to generate temporal microfluidic gradients that offer dynamically varying conditions over which Nanodisc assembly can be screened. To demonstrate this capability, we used a programmable syringe pump to dynamically change the lipid composition of Nanodiscs over time. Nanodisc lipid composition was determined using a fluorescent phospholipid, 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (LR-PE), in addition to DMPC (Supplementary Figure 6). For the 3-port device, the flow rate for the MSP containing inlet was held constant while the rate of the DMPC inlet was decreased and the rate of the fluorescent lipid was increased over the course of the Nanodisc assembly. The fluorescence intensity associated with the LR-PE lipid steadily increased with elution volume indicating an increase in Nanodisc formed with fluorescent lipid. The composition of the Nanodiscs with the addition of the fluorescent lipid was assessed with SEC (Supplementary Figure 6c). It is worth noting that screening Nanodisc assembly conditions will result in poor Nanodisc formation when Nanodisc reagent stoichiometries are suboptimal. Key to gradient analysis, however, is delivery of Nanodisc reagents across the packed bed of detergent removal resin in the proportion to their delivery into the mixing device. Longitudinal mixing would blur the microfluidic gradient, and Supplementary Figure 6b suggests that this form of mixing is minimal for the Nanodisc assembly module.
To demonstrate the platform's utility for the incorporation of membrane proteins into Nanodiscs, cytochrome P450 3A4 (CYP3A4) was used as a model system. Cytochromes P450 are ubiquitous membrane proteins that predominantly serve as oxidase enzymes in electron transfer chains.46 CYP3A4 is the most abundant cytochrome P450 expressed in the human liver and small intestines, and approximately half of small molecule pharmaceuticals are thought to be metabolized by CYP3A4.47 Alone, CYP3A4 forms aggregates in solution after isolation and purification; however, the incorporation of CYP3A4 into Nanodiscs prevents aggregation and also allows precise control over the protein's oligomeric state.48 The role of CYP3A4 in drug metabolism has motivated a wide variety of studies incorporating CYP3A4 into Nanodiscs.11,38,48–52 As such, CYP3A4 was chosen as an important proof-of-principle membrane protein with which to demonstrate the utility of this microfluidic Nanodisc assembly platform.
The strong optical absorbance of the CYP3A4 heme cofactor (λmax = 417 nm) provides a useful spectroscopic handle for monitoring protein incorporation into Nanodiscs. Using the single port inlet device with off-chip reagent mixing, the CYP3A4 protein was found to readily incorporate into Nanodiscs composed of DMPC as a model lipid. Nanodisc assembly was characterized by SEC with absorbance detection at both 280 nm, for general protein (including MSP) absorbance, and at 417 nm for the heme cofactor of CYP3A4, and compared against the same SEC analysis for empty Nanodiscs (Figure 3a). The elution peaks for filled Nanodiscs shows clear absorption at both 280 and 417 nm, which is consistent with well-formed Nanodiscs incorporating CYP3A4. Empty Nanodiscs have only an absorbance at 280 nm. Notably, the 417 nm absorbance for filled Nanodiscs is shifted to a slightly earlier elution compared to 280 nm, which is in agreement with the CYP3A4-containing Nanodiscs having a slightly larger hydrodynamic radius compared with empty Nanodiscs. We also demonstrated that CYP3A4 can be incorporated into Nanodiscs using on-chip reagent mixing (Supplementary Figure 7). The ability to mix on-chip is important given that some classes of membrane proteins can denature or deactivate with prolonged detergent exposure.4 It is also important to point out that at this CYP3A4:lipid:MSP incorporation stoichiometry filled Nanodiscs are assembled in a large background of empty Nanodiscs, a point that will be discussed later. SDS-PAGE and SEC analysis of Nanodiscs filled with CYP3A4 after purification indicate that 5-20% of Nanodiscs contain incorporated CYP3A4 (Supplementary Figure 8).
Figure 3. Incorporation of CYP3A4 into Nanodiscs.
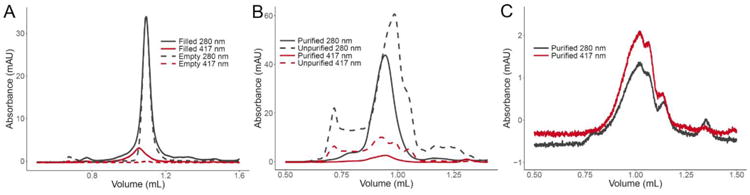
Size exclusion chromatograms recorded at 280 nm and 417 nm (A) demonstrate the successful incorporation of CYP3A4 into Nanodiscs using the microfluidic assembly module. Equivalently sized Nanodiscs were formed either with (red) or without (black) CYP3A4 in combination with DMPC lipids, MSP1D1, and CHAPS detergent. The filled Nanodiscs, which had a MSP:CYP3A4 ratio of 20:1, were confirmed by the strong absorbance at 417 nm with minimal signal for the empty Nanodiscs. (B) Purification with His-tag with POPC, MSP1E3D1, and CYP3A4 (both CYP3A4 and MSP have His-tag) at a ratio of 10:1 MSP:CYP3A4 (C) Purification of CYP3A4 Nanodiscs made with DMPC MSP1D1(-) (that indicates His-tag is removed) at a ratio of 20:1 MSP:CYP3A4
As demonstrated above, the Nanodisc assembly module produces well-formed Nanodiscs of homogenous distribution. However, the incorporation of membrane proteins into Nanodiscs, as with all other lipid bilayer systems, does not result in the incorporation of all solubilized membrane proteins into Nanodiscs. As such, membrane protein aggregates that can interfere with downstream assays of membrane protein structure or function will remain in the sample after Nanodisc assembly. Often the most time-consuming process when using Nanodiscs for membrane protein studies is purification.23 Two approaches are typically combined for this process: affinity purification and fractionation with SEC. Each step can result in loss of Nanodisc product or dilution of Nanodiscs, requiring additional re-concentrating processing steps for many applications. To improve the purification process, we designed an affinity purification module for the platform, as described above (Figure 1b-c). Ni-NTA was used for affinity-based purification. MSP1D1 and MSP1E3D1 scaffold proteins have N-terminal His-tags to allow for efficient Nanodisc purification (Scheme S1).
As demonstration of the utility of the affinity purification module, CYP3A4 was incorporated into Nanodiscs using POPC as the lipid, cholate as the detergent, and the larger MSP1E3D1. POPC has a single point of unsaturation in the lipid tail that, while helping create a more native-like environment for membrane proteins, also forms more loosely packed bilayers and a wider dispersity of resulting Nanodiscs. Chromatograms of CYP3A4-containing Nanodiscs before and after purification with the Ni-NTA module (dotted traces in Figure 3b) clearly demonstrate the improvement in collected Nanodiscs. Specifically, faster eluting contaminants such as cytochrome and lipid aggregates are removed, as are smaller lipid aggregates at longer elution times.
Purification using engineered affinity tags on MSP offers a generalizable approach for Nanodisc purification, though this results in a mixture of Nanodiscs containing the protein of interest with a background of empty Nanodiscs. For applications requiring only Nanodiscs with incorporated protein, affinity purification using features of the incorporated protein is needed. To demonstrate this capability, Nanodiscs filled with His-tagged CYP3A4 were made using MSP from which the N-terminal His-tag was cleaved prior to Nanodisc assembly. Purification via the Ni-NTA resin module described above showed only filled Nanodiscs, as evidenced by the overlap of the absorbance signals measured at both 280 nm and 417 nm (Figure 3c). Importantly, this approach is generalizable to other affinity purification approaches, such as antibody-based affinity chromatography.
It is clearly essential that membrane proteins retain their function upon Nanodisc incorporation, and CYP3A4 provides an opportunity to spectroscopically verify substrate binding and protein activity.11 Specifically, binding induced changes to the spin state of the 3d electrons of the Fe3+ in the heme cofactor cause the optical absorption to shift. Type I binders induce a change in the coordination of Fe3+ from six- to five-coordinate with a corresponding decrease in the Soret absorption band (417 nm) and increase in absorbance at 390 nm.53 To demonstrate type I binding with CYP3A4-containing Nanodiscs assembled with the platform, bromocriptine, a type I binder of CYP3A4, was titrated into the Nanodisc solution resulting in a reduction in the absorbance at 417 nm and an increase at 390 nm (shown as a difference spectra in Figure 4a). Minimal low to high spin shift is observed for CYP3A4 not incorporated into Nanodiscs, which is only detergent-stabilized. Type II binders of CYP3A4 have an unobstructed nitrogen atom that coordinates with Fe3+ resulting in a six-coordinate geometry at the activation site and a shift in maximal absorbance from 417 nm to 422 nm. Imidazole, a type II binder of CYP3A4, is a component of the elution buffer and so the as-eluted Nanodiscs show an absorbance maximum at 422 nm; however, after removal of imidazole via buffer exchange, the absorbance shifts to 417 nm (Figure 4b). Taken together, these substrate binding spectroscopic shifts demonstrate the viability of proteins incorporated into Nanodiscs assembled with the microfluidic platform.
Figure 4. Spin Shift Assays for Nanodiscs Filled with CYP3A4.
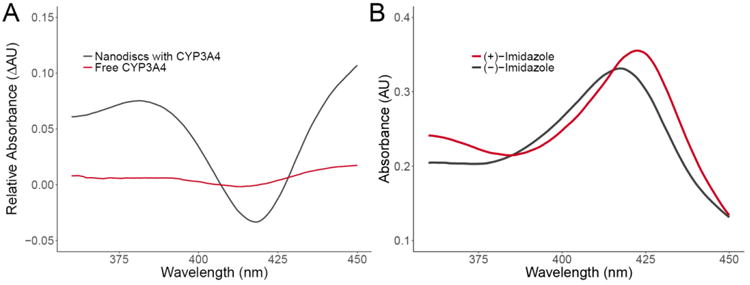
(A) UV/Vis absorption difference spectrum demonstrates the low to high spin shift for CYP3A4 in Nanodiscs (black) and free CYP3A4 (red) induced by the binding of bromocriptine (BCT), a type I CYP3A4 binder. Binding of BCT results in a decrease in the absorbance maximum of 417 nm and an increase at 390 nm. (B) Imidazole, a type II CYP3A4 binder, induces a shift in the absorbance maximum for CYP3A4 from 417 nm to 422 nm.
Conclusions
In conclusion, Nanodiscs have emerged as a powerful construct that are enabling for numerous biochemical and biophysical studies of model membrane environments; however, the determination of optimal assembly and membrane protein incorporation conditions can require time- and material-consuming iterations. We have developed a microfluidic Nanodisc assembly platform that is capable of rapidly assembling Nanodiscs with generality for different lipid and detergent compositions. We also demonstrated the successful incorporation of a membrane protein and show that its activity towards substrate binding was preserved through microfluidic preparation. This platform will serve as a powerful tool for the facile assembly of Nanodiscs and screening for incorporation conditions while minimizing reagent consumption and time. Furthermore, continued miniaturization and automation of the technology will further increase the accessibility of the Nanodisc platform across the broad biochemical research community.
Supplementary Material
Acknowledgments
The authors acknowledge support from the National Institutes of Health (NIH) for funding under awards GM110432 and CA177462. JHW acknowledges support from the National Science Foundation Graduate Research Fellowship Program and American Chemical Society Division of Analytical Chemistry Graduate Fellowship Program.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Yildirim MA, Goh KI, Cusick ME, Barabási AL, Vidal M. Nat Biotechnol. 2007;25:1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- 2.Engelman DM. Nature. 2005;438:578–580. doi: 10.1038/nature04394. [DOI] [PubMed] [Google Scholar]
- 3.Silvius JR. Annu Rev Biophys Biomol Struct. 1992;21:323–348. doi: 10.1146/annurev.bb.21.060192.001543. [DOI] [PubMed] [Google Scholar]
- 4.Seddon AM, Curnow P, Booth PJ. Biochim Biophys Acta. 2004;1666:105–117. doi: 10.1016/j.bbamem.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 5.Cross TA, Sharma M, Yi M, Zhou HX. Trends Biochem Sci. 2011;36:117–125. doi: 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernaudat F, Frelet-Barrand A, Pochon N, Dementin S, Hivin P, Boutigny S, Rioux JB, Salvi D, Seigneurin-Berny D, Richaud P, Joyard J, Pignol D, Sabaty M, Desnos T, Pebay-Peyroula E, Darrouzet E, Vernet T, Rolland N. PLoS One. 2011;6:e29191. doi: 10.1371/journal.pone.0029191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goehring A, Lee CH, Wang KH, Michel JC, Claxton DP, Baconguis I, Althoff T, Fischer S, Garcia KC, Gouaux E. Nat Protoc. 2014;9:2574–2585. doi: 10.1038/nprot.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garavito RM, Ferguson-Miller S. J Biol Chem. 2001;276:32403–32406. doi: 10.1074/jbc.R100031200. [DOI] [PubMed] [Google Scholar]
- 9.Akbarzadeh A, Rezaei-Sadabady R, Davaran S, Joo SW, Zarghami N, Hanifehpour Y, Samiei M, Kouhi M, Nejati-Koshki K. Nanoscale Res Lett. 2013;8:102. doi: 10.1186/1556-276X-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Proverbio D, Henrich E, Orbán E, Dötsch V, Bernhard F. In: Membrane Proteins Production for Structural Analysis. Mus-Veteau I, editor. Springer New York; New York, NY: 2014. pp. 45–70. [Google Scholar]
- 11.Denisov IG, Sligar SG. Biochim Biophys Acta. 2011;1814:223–229. doi: 10.1016/j.bbapap.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denisov IG, Sligar SG. Chem Rev. 2017;117:4669–4713. doi: 10.1021/acs.chemrev.6b00690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan CR, Hebling CM, Rand KD, Stafford DW, Jorgenson JW, Engen JR. Mol Cell Proteomics. 2011;10:M111.010876. doi: 10.1074/mcp.M111.010876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka M, Hosotani A, Tachibana Y, Nakano M, Iwasaki K, Kawakami T, Mukai T. Langmuir. 2015;31:12719–12726. doi: 10.1021/acs.langmuir.5b03438. [DOI] [PubMed] [Google Scholar]
- 15.Jamshad M, Grimard V, Idini I, Knowles TJ, Dowle MR, Schofield N, Sridhar P, Lin Y, Finka R, Wheatley M, Thomas ORT, Palmer RE, Overduin M, Govaerts C, Ruysschaert JM, Edler KJ, Dafforn TR. Nano Res. 2015;8:774–789. doi: 10.1007/s12274-014-0560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. J Am Chem Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- 17.Bayburt TH, Grinkova YV, Sligar SG. Nano Lett. 2002;2:853–856. [Google Scholar]
- 18.Bayburt TH, Sligar SG. FEBS Lett. 2010;584:1721–1727. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mineev KS, Goncharuk SA, Kuzmichev PK, Vilar M, Arseniev AS. Biophys J. 2015;109:772–782. doi: 10.1016/j.bpj.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gregersen JL, Fedosova NU, Nissen P, Boesen T. In: P-Type ATPases. Bublitz M, editor. Springer New York; pp. 403–409. [Google Scholar]
- 21.Duan H, Civjan NR, Sligar SG, Schuler MA. Arch Biochem Biophys. 2004;424:141–153. doi: 10.1016/j.abb.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 22.Denisov IG, Sligar SG. Nat Struct Mol Biol. 2016;23:481–486. doi: 10.1038/nsmb.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi L, Howan K, Shen QT, Wang YJ, Rothman JE, Pincet F. Nat Protoc. 2013;8:935–948. doi: 10.1038/nprot.2013.048. [DOI] [PubMed] [Google Scholar]
- 24.Ritchie TK, Grinkova YV, Bayburt TH, Denisov IG, Zolnerciks JK, Atkins WM, Sligar SG. Methods Enzymol. 2009;464:211–231. doi: 10.1016/S0076-6879(09)64011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grisshammer R. Curr Opin Biotechnol. 2006;17:337–340. doi: 10.1016/j.copbio.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Duong-Ly KC, Gabelli SB. Laboratory Methods in Enzymology: Protein Part C. Vol. 541. Elsevier; 2014. pp. 209–229. [DOI] [PubMed] [Google Scholar]
- 27.Hansen C, Quake SR. Curr Opin Struct Biol. 2003;13:538–544. doi: 10.1016/j.sbi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 28.Whitesides GM. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 29.Duncombe TA, Tentori AM, Herr AE. Nat Rev Mol Cell Biol. 2015;16:554–567. doi: 10.1038/nrm4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Funakoshi K, Suzuki H, Takeuchi S. Anal Chem. 2006;78:8169–8174. doi: 10.1021/ac0613479. [DOI] [PubMed] [Google Scholar]
- 31.He M, Herr AE. Nat Protoc. 2010;5:1844–1856. doi: 10.1038/nprot.2010.142. [DOI] [PubMed] [Google Scholar]
- 32.Hu R, Feng X, Chen P, Fu M, Chen H, Guo L, Liu BF. J Chromatogr A. 2011;1218:171–177. doi: 10.1016/j.chroma.2010.10.090. [DOI] [PubMed] [Google Scholar]
- 33.Hughes AJ, Herr AE. Proc Natl Acad Sci U S A. 2012;109:21450–21455. doi: 10.1073/pnas.1207754110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Millet LJ, Lucheon JD, Standaert RF, Retterer ST, Doktycz MJ. Lab Chip. 2015;15:1799–1811. doi: 10.1039/c5lc00094g. [DOI] [PubMed] [Google Scholar]
- 35.Gillam EM, Baba T, Kim BR, Ohmori S, Guengerich FP. Arch Biochem Biophys. 1993;305:123–131. doi: 10.1006/abbi.1993.1401. [DOI] [PubMed] [Google Scholar]
- 36.Hosea NA, Miller GP, Guengerich FP. Biochemistry. 2000;39:5929–5939. doi: 10.1021/bi992765t. [DOI] [PubMed] [Google Scholar]
- 37.Domanski TL, He YA, Khan KK, Roussel F, Wang Q, Halpert JR. Biochemistry. 2001;40:10150–10160. doi: 10.1021/bi010758a. [DOI] [PubMed] [Google Scholar]
- 38.Denisov IG, Baas BJ, Grinkova YV, Sligar SG. J Biol Chem. 2007;282:7066–7076. doi: 10.1074/jbc.M609589200. [DOI] [PubMed] [Google Scholar]
- 39.Levario TJ, Zhan M, Lim B, Shvartsman SY, Lu H. Nat Protoc. 2013;8:721–736. doi: 10.1038/nprot.2013.034. [DOI] [PubMed] [Google Scholar]
- 40.Antharavally BS, Mallia KA, Rosenblatt MM, Salunkhe AM, Rogers JC, Haney P, Haghdoost N. Anal Biochem. 2011;416:39–44. doi: 10.1016/j.ab.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 41.Urbani A, Warne T. Anal Biochem. 2005;336:117–124. doi: 10.1016/j.ab.2004.09.040. [DOI] [PubMed] [Google Scholar]
- 42.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morrissey JH, Tajkhorshid E, Sligar SG, Rienstra CM. Thromb Res. 2012;129 Suppl 2:S8–10. doi: 10.1016/j.thromres.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sloan CDK, Marty MT, Sligar SG, Bailey RC. Anal Chem. 2013;85:2970–2976. doi: 10.1021/ac3037359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gajsiewicz JM, Morrissey JH. Seminars in thrombosis and hemostasis. Vol. 41. Thieme Medical Publishers; 2015. pp. 682–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Danielson PB. Curr Drug Metab. 2002;3:561–597. doi: 10.2174/1389200023337054. [DOI] [PubMed] [Google Scholar]
- 47.Guengerich FP. Annual Review of Pharmacology and Toxicology. 1999;39:1–17. doi: 10.1146/annurev.pharmtox.39.1.1. [DOI] [PubMed] [Google Scholar]
- 48.Baas BJ, Denisov IG, Sligar SG. Arch Biochem Biophys. 2004;430:218–228. doi: 10.1016/j.abb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 49.Nath A, Grinkova YV, Sligar SG, Atkins WM. J Biol Chem. 2007;282:28309–28320. doi: 10.1074/jbc.M703568200. [DOI] [PubMed] [Google Scholar]
- 50.Kijac AZ, Li Y, Sligar SG, Rienstra CM. Biochemistry. 2007;46:13696–13703. doi: 10.1021/bi701411g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Das A, Zhao J, Schatz GC, Sligar SG, Van Duyne RP. Anal Chem. 2009;81:3754–3759. doi: 10.1021/ac802612z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baylon JL, Lenov IL, Sligar SG, Tajkhorshid E. J Am Chem Soc. 2013;135:8542–8551. doi: 10.1021/ja4003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.M. D. Segall, Cambridge, 1997.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.