

Abstract
Mechanical forces between cells and their extracellular matrix (ECM) are mediated by hundreds of different receptors. These biophysical interactions play fundamental roles in processes ranging from cellular development to tumor progression. However, mapping the spatial and temporal dynamics of tension among various receptor-ligand pairs remains a significant challenge. To address this issue, we have developed a synthetic strategy to generate modular tension probes combining the native chemical ligation (NCL) reaction with solid phase peptide synthesis (SPPS). In principle, this approach accommodates virtually any peptide or expressed protein amenable to NCL. We generated a small library of tension probes displaying different ligands, flexible linkers, and fluorescent reporters, thus mapping integrin and cadherin tension, and demonstrating the first example of long-term (∼3 days) molecular tension imaging. This approach provides a toolset to better understand mechanotransduction events fundamental to cell biology.
Graphical Abstract
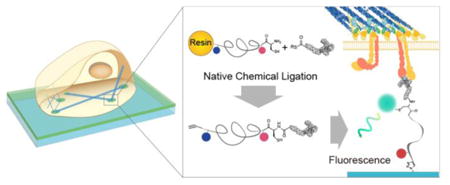
Cells sense and transduce physical signals from their external environment using hundreds of different cell surface receptors. Transmembrane adhesion receptors, such as integrins and cadherins, mediate cell-cell and cell-extracellular matrix (ECM) adhesions, and transmit the mechanical forces responsible for giving tissue their intrinsic architecture.1 Because integrin2–4and cadherin5 receptor function is highly dependent on mechanics, there is a pressing need to develop specific molecular probes to map receptor forces throughout the chemo-mechanical coupling cycles controlling cell fate.
We recently developed molecular tension-based fluorescence microscopy (MTFM) to visualize receptor forces.6–10 In MTFM, an extendable polymer (eg. polyethylene glycol (PEG) or DNA) is flanked by a fluorescent donor at one terminus and a quencher or acceptor at the second terminus and the construct is immobilized on a surface. Receptor-mediated forces extend the polymer “spring”, which de-quenches the fluorophore allowing the transduction of a mechanical signal into an optical readout. Recently, we found that biotin-streptavidin-immobilized MTFM probes were dissociated due to integrin receptor forces.8 This was unexpected because this association is described as the strongest non-covalent bond in nature. Given that integrins exert sufficient tension to dissociate the biotin-streptavidin bond, we next developed nanoparticle-based MTFM probes that employ thiol-gold binding for immobilization.7,10 Nonetheless, thiolated ligands are known to dissociate from the Au surface within 24 hrs.11 Ligand exchange is further exasperated in biological media containing ∼100 μM thiol bearing molecules.12 Alternatively, Blakely and colleagues used amine-thiol heterobifunctional linkers to immobilize DNA tension probes.13 However, this crosslinking chemistry limits the choice of ligands to molecules lacking lysine and cysteine. Therefore, new bio-orthogonal approaches for covalent immobilization of molecular tension probes are needed.
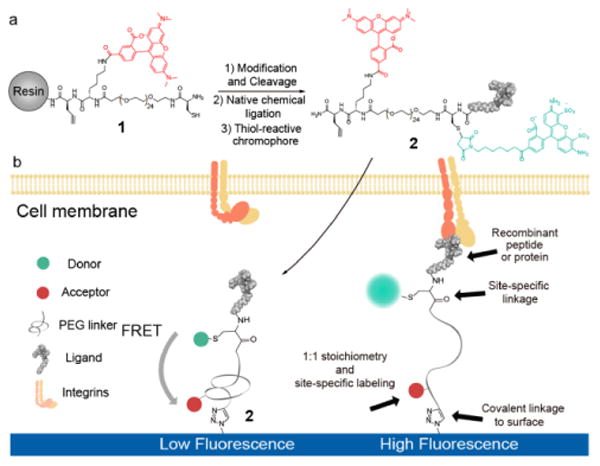
We overcome this challenge by developing a new class of stable and modular MTFM probes that is generated using solid phase peptide synthesis (SPPS) along with the NCL reaction (Scheme 1). This approach allows one to site-specifically incorporate a ligand and a pair of chromophores, and is compatible with virtually any peptide of interest. SPPS was used to generate a “spring” composed of a C-terminal alkyne-modified amino acid followed by lysine, discrete PEG24/PEG48, and terminated with an N-terminal cysteine, 1 (Scheme 1). The lysine was coupled to an NHS-ester modified tetramethylrhodamine (TAMRA) fluorophore. The peptide conjugate was then cleaved and purified using reverse phase HPLC (RP-HPLC) and verified using MALDI-TOF (Figure S1). We also synthesized a small library of α-thioester modified peptide ligands selected based on their specificity toward the integrin and cadherin receptors (Figure S2-S3, SI Material and Methods). These peptide ligands recapitulate many of the cell responses in cell-cell and cell-ECM adhesions.14,15 These ligands included: 1) cyclic RGDfK (cRGDfK) and linear GRGDS peptides derived from the fibronectin-III repeat 10 (FN-III10), which primarily bind the α5β1 and αvβ3 integrins,16 2) the synergy site PHSRN derived from FN-III9, which is reported to increase cell adhesion when combined with the RGD sequence, 3) the PHSRN(SG)4RGDS peptide, which includes a spacer between the PHSRN and GRGDS peptides and better mimics the distance between the two binding domains in FN17 and 4) the SHAVSS and LRAHAVDING peptides, which bind the E-cadherin and N-cadherin receptors, respectively.15,18 Standard SPPS protocols were used to generate these α-thioester linear peptides, except for the cRGDfK sequence for which we used a protocol adapted from Xiao et al.19 Standard NCL conditions were used to couple the peptide to 1 (Figure S4). We subsequently took advantage of the free thiol group inherent to the NCL reaction to site-specifically couple maleimide-modified dyes that take part in FRET with the TAMRA fluorophore, 2. Typically, we employed Alexa 488 due to its appropriate Forster radius R0 = 5.9 nm with TAMRA (Supplementary Note 1). The molecular probes were then covalently immobilized onto an azide-modified glass slide using standard click reaction conditions. Fluorescence microscopy and FRET analysis showed uniform surface coverage of the tension probes and high quenching efficiency (Figure S5-7). Withholding Cu(II) or using surfaces that lacked the azide did not lead to significant binding of the probe, thus confirming the specificity of the click reaction. The surface density of tension probes ranged from 9000 to 11000 probes/μm2, as determined by quantitative fluorescence imaging (Figure S8).20
Scheme 1. Synthetic scheme for generating ligand-general molecular tension fluorescence microscopy (MTFM) probes.
To maximize the interaction of the cell receptors with tension probes, we passivated the surface against non-specific binding of cell-generated ECM and serum proteins. Typically, PEG polymers are used for passivation21. However, we found that employing PEG polymers for passivation increased background signal and diminished sensitivity (Figure 1). To solve this problem, we tested the zwitterionic silane (3-(dimethyl-(3-(trimethoxysilyl)propyl)ammonio) propane-1-sulfonate), (SBS) (Figure S5),22 and compared its efficiency of passivation against that of PEG polymers (Mw = 2000, 5000 Da).23 SBS provided enhanced passivation against cell adhesion in comparison to PEG polymers (Figure 1a-1b). Given the importance of the molecular extension of the tension probes, we next aimed to estimate the mean inter-fluorophore distance when the surface was PEG passivated rather than SBS passivated. This was achieved by measuring the FRET efficiency using a TAMRA-PEG24-fluorescein probe and reporting the acceptor-normalized donor intensity (Figure S9). The donor-acceptor distance was greater for the PEG2000 and PEG5000 surfaces in comparison to the SBS passivated surface (Figure 1c). The mean inter-fluorophore distance for SBS surfaces was approximately 2.6 nm, which is in agreement with the predicted 2.4 nm distance based on the Flory model. The TAMRA-fluorescein distance increases by ∼1 nm when the surface is passivated using PEG5000, which leads to a ∼15% decrease in quenching efficiency. These results indicate that the PEG polymer passivation (∼2000 Da and ∼5000 Da) leads to molecular crowding of the tension probe, thus placing it in a more extended conformation compared to the samples prepared with SBS passivation. The extension of the tension probe upon passivation with PEG polymers is consistent with the literature showing that polymers with fixed grafting density and increasing molecular weight tend to increase crowding and the transition of polymers to more brush-like conformations.24,25 In contrast, the 1.3 nm length of the SBS was insufficient to crowd the tension probes (Figure 1d). Therefore, SBS provides superior passivation against biofouling and concurrently improves probe sensitivity by reducing background signal. SBS passivation was used in all subsequent cell studies. This result represents the first demonstration of using SBS for cell culture, outperforming PEG polymers, the most widely used reagent for passivation against biofouling.
Figure 1.
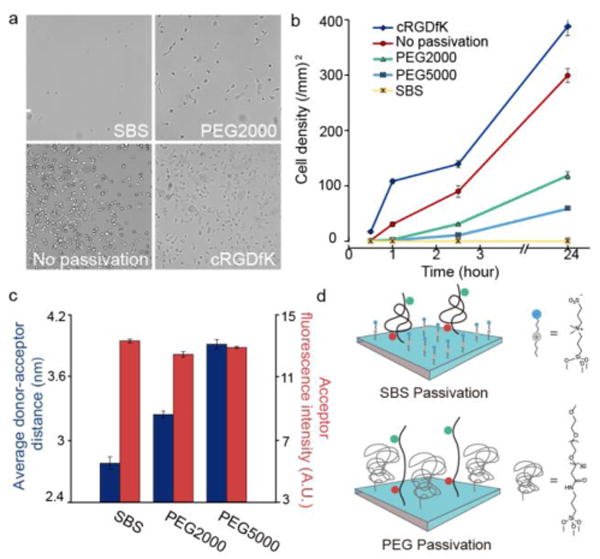
Role of passivation in MTFM probe conformation. (a) Representative brightfield images of NIH 3T3 cells plated for 2.5 hrs on SBS, PEG2000, bare glass, and cRGDfK substrates. Scale bar = 100 μm. (b) Plot showing average cell density on cRGDfK, bare glass, PEG2000, PEG5000, and SBS substrates as a function of time (24 hrs). Error bars represent the standard error of the mean (SEM) of n = 3 substrates where a total of 10 regions of interest were averaged from these samples. (c) Bar graph showing the average extension (left y-axis) and acceptor intensity (right y-axis) of MTFM probes as a function of passivation. Error bars represent the SEM of n=3 substrates, where a total of 10 regions were imaged. (d) Model showing the influence of passivation molecule on crowding and extension of tension probes.
To test the cRGDfK tension probe, NIH 3T3 cells were plated onto TAMRA-QSY9 sensor modified surfaces for 1 hr to allow the cells to adhere. We used epifluorescence microscopy to image the live cell tension response and reflection interference contrast microscopy (RICM) to monitor cell-substrate binding. A dynamic increase in fluorescence in the tension channel (TAMRA) was observed in regions associated with the cell-binding pattern in RICM (Figure 2a and Movie S1). Tension signal colocalized with GFP-tagged β3 integrin, confirming that tension signal is integrin mediated (Figure S10, 11). The spatial distribution of tension was relatively dynamic, changing on a time scale of tens of minutes (Figure S12a). We used the worm-like chain (WLC) model and the measured quenching efficiency to estimate the average tension per integrin ligand (Figure 2a and Supplementary Note 2-3). Tension signal dissipated upon treating cells with latruculin B (latB), an actin polymerization inhibitor, indicating that the signal can be reduced by inhibiting the cellular cytoskeleton (Figure S12b and Movie S2). Taken together, the dynamic fluctuations in tension signal, live cell dual channel integrin/tension imaging, along with the latB experiment show that the reversible fluorescence response is due to mechanical tension exerted by integrin receptors engaged to the MTFM sensor. Given the predicted force-fluorescence curve for the PEG24 probes, we expect that the dynamic range of the probes is limited to ∼15 pN; the stiffness of the probes increases drastically as the polymer approaches its contour length (Figure S13). The PEG48-based tension probes performed similarly to the PEG24 probes, confirming the modularity of the synthetic approach. The calculated dynamic range of the PEG48 probes is limited to ∼5 pN, which suggests that these are more appropriate for signaling pathways that employ a lower magnitude of tension such as the Notch pathway.4,26
Figure 2.
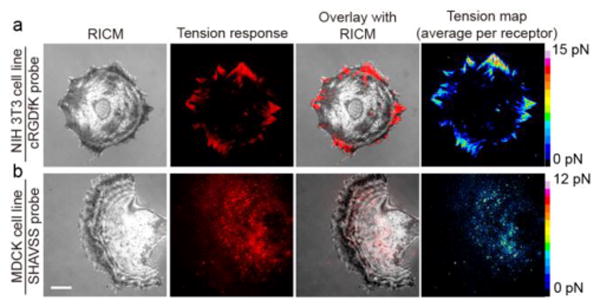
Representative RICM, fluorescence, overlay of fluorescence and RICM, and quantified heat map of tension for cells cultured on the cRGDfK (TAMRA-QSY9) and SHAVSS (TAMRA-Alexa 488) tension probe surfaces. Top row shows an NIH 3T3 cell cultured on the cRGDfK tension probe for 1 hr, while the bottom row shows an MDCK cell cultured on the SHAVSS peptide tension probe for 3 hrs. Raw fluorescence data were converted to a force map. Scale bar = 10 μm.
To study the tension generated by E-cadherin receptors, we plated endothelial cells (MDCK) on the SHAVSS peptide tension probes. In contrast to the FA tension patterns observed for the cRGDfK peptide sensor, we observed punctate tension across the cell-substrate junction. The intensity of tension signal was significantly lower for the SHAVSS peptide compared to the cRGDfK peptide. The SHAVSS tension signal was abolished upon treating cells with latB, showing that the signal is generated by the cellular cytoskeleton (Figure S14). Immunostaining for the E-cadherin extracellular domain EC4 displayed puncta at the basal cell surface resembling the signal associated with E-cadherin tension in our assays (Figure S14). We also found that 3T3 fibroblasts did not adhere to the SHAVSS surface, confirming that E-cadherin expression is necessary for cell adhesion. Importantly, tension sensors specific to the N-cadherin ligand, LRAHAVDING, did not yield detectable signal when rat dorsal root ganglion (DRG) neurons were cultured onto substrates (data not shown). Taken together these results indicate that E-cadherin-binding ligands experience lower values of tension than that of integrin ligands, which may reflect the mechanics of cadherin signaling or binding affinity and receptor density differences among these cell types.
To test the capability of the tension probes in long-term imaging, we incubated NIH 3T3 fibroblasts on cRGDfK-Alexa488-TAMRA tension probes. Cells displayed tension patterns at the cell periphery similar to that of the FA markers (Figure 3). After 20 min of incubation, the tension signal became more elongated as cells polarized (Figure 3a). We followed the cRGDfK tension pattern for a period of 64 hrs. During this time, the average tension signal within FAs increased initially and then decreased as cells spread (Figure 3b). Note that the magnitude of integrin tension was correlated with the average size of FAs. As cells fully spread, FAs became slightly smaller in area, and this was accompanied by a decrease in integrin tension. This represents the longest imaging window for mapping receptor-ligand tension using a molecular probe. Minimal change was observed in the fluorescence background of both the donor and the acceptor during the 64-hour imaging window (Figure S15).
Figure 3.
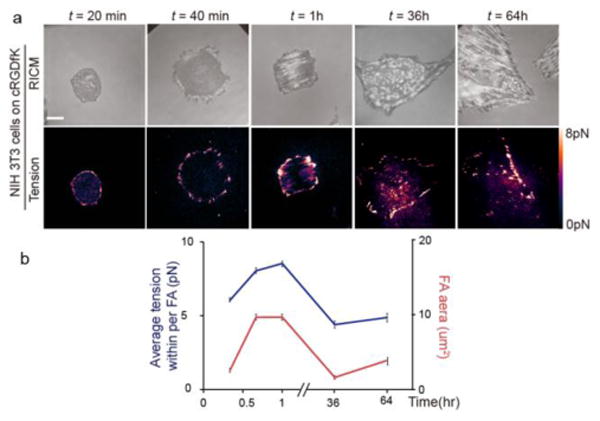
Long-term live cell mechano-imaging using TAMRA-Alexa 488 sensor. (a) RICM and fluorescence images showing the cell-substrate contact zone along with a map of integrin tension at the indicated time points that spanned from 20 min to 64 hrs. Note the changes in cell morphology and force pattern. Scale bar = 10 μm. (b) Plot showing the average tension within FAs (left y-axis), and average FA area (right y-axis) as a function of time over a period of 64 hrs. The error bars represent the SEM from n=3-4 cells, where 10-30 FAs were analyzed from each cell.
The peptide PHSRN is found in the FN-III9, adjacent to the 10th domain that contains the RGD peptide.27 PHSRN has been identified as a synergy ligand that enhances the spreading of cells on the RGD peptide.27–30 We asked whether PHSRN carries a mechanical load much like the RGD peptide that supports adhesion. Tension probes with the PHSRN, cRGDfK, PHSRN(SG)4RGDS and linear GRGDS peptides were immobilized on glass slides. Cells attached and spread inefficiently on PHSRN substrates (Figure 4a). In contrast, cells plated onto surfaces comprised a binary mixture of PHSRN and GRGDS/cRGDfK probes (1:1) spread efficiently. This is in agreement with recent reports showing that PHSRN enhances cell spreading with RGD but is inactive when presented exclusively.31,32 The tension signals for cells cultured on PHSRN(SG)4RGDS, GRGDS and cRGDfK were similar and greater than that of substrates with the binary mixture of GRGDS/cRGDfK and PHSRN (Figure 4b). Note that the samples modified with the binary mixture of RGD ligands and PHSRN display the same total density of MTFM probes but 50% of the RGD ligand density compared to the single component surfaces. Although the affinity of integrins for cRGDfK is greater than that of linear GRGDS, the signals were similar for both ligands, which is in agreement with results obtained using DNA-based tension probes.9 We were not able to detect tension signals on the substrates presenting PHSRN exclusively. These data indicate that mechanical tension is not transmitted through the PHSRN synergy ligand, rather its role is most likely in enhancing integrin-ligand affinity. This conclusion clarifies a long-standing question regarding the mechanical role of the PHSRN ligand in cell adhesion. We expect that MTFM probes generated using this modular approach will help elucidate the role of various ECM components in mediating mechanotransduction processes. A general caveat of this approach is that the dynamic range of the sensor is limited to ∼15 pN; thus, while we are able to detect differences in the ensemble average tension signal, receptor forces that are ≫15 pN are not distinguishable from lower magnitude signals. Therefore, the lack of statistical difference in tension signal between the GRGDS, cRGDfK, and PHSRN(SG)4RGDS probes may be due to probe sensitivity rather than the lack of biophysical difference.
Figure 4.
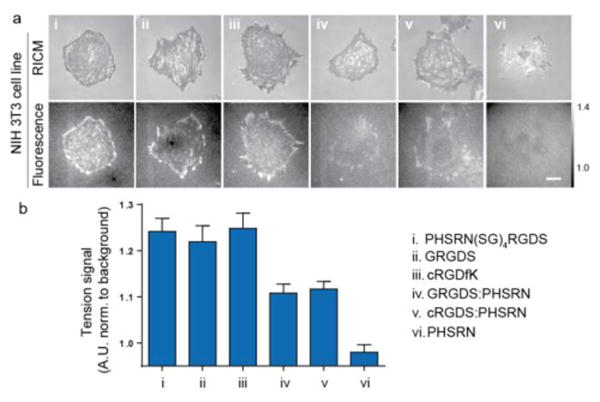
The role of RGD and PHSRN peptides in mediating integrin tension. a) Representative RICM and fluorescence tension images of 3T3 fibroblasts cultured onto PHSRN(SG)4RGDS, GRGDS, cRGDfK, 1:1 GRGDS: PHSRN, 1:1 cRGDfK: PHSRN, and PHSRN MTFM probes (TAMRA-Alexa 488). Scale bar = 10 μm and contrasts are set identically. b) Bar graph showing the average tension normalized to the background for cells cultured onto the above substrates (a). Data obtained in triplicate from n=8 cells in each category for a total of 40 cells, where 10-30 FAs were analyzed from each cell. Note that the average tension for the PHSRN probe was ∼2% below the background signal likely due to optical effects from cell adhesion.
In summary, we demonstrated that the integration of SPPS and NCL for MTFM synthesis provides a general and modular approach overcoming stability issues and providing improved sensitivity over previous strategies for tension probe design. This probe can be used to image receptor-ligand forces for peptides amendable to the NCL reaction, which are generally smaller peptides and proteins. There are also some limitations in the choice of dyes that emit in the near-infrared, as these are not generally compatible with SPPS.
Supplementary Material
Acknowledgments
K.S. is grateful for support from the NIH (R01-GM097399), the Alfred P. Sloan Research Fellowship. We also thank Dr. Benjamin Geiger, Department of Molecular Cell Biology at Weizmann Institute, for rat embryonic fibroblast cells transfected with the β3-integrin-GFP.
Footnotes
The authors declare no competing financial interests.
Supporting Information: Supporting Information and movies. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hynes RO. Cell. 2002;110(6):673. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 2.Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS. Proc Natl Acad Sci U S A. 2003;100(4):1484. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Legant WR, Choi CK, Miller JS, Shao L, Gao L, Betzig E, Chen CS. Proc Natl Acad Sci U S A. 2013;110(3):881. doi: 10.1073/pnas.1207997110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Ha T. Science. 2013;340(6135):991. doi: 10.1126/science.1231041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borghi N, Sorokina M, Shcherbakova OG, Weis WI, Pruitt BL, Nelson WJ, Dunn AR. Proc Natl Acad Sci U S A. 2012;109(31):12568. doi: 10.1073/pnas.1204390109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stabley DR, Jurchenko C, Marshall SS, Salaita KS. Nat Methods. 2012;9(1):64. doi: 10.1038/nmeth.1747. [DOI] [PubMed] [Google Scholar]
- 7.Liu Y, Yehl K, Narui Y, Salaita K. J Am Chem Soc. 2013;135(14):5320. doi: 10.1021/ja401494e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jurchenko C, Chang Y, Narui Y, Zhang Y, Salaita KS. Biophys J. 2014;106(7):1436. doi: 10.1016/j.bpj.2014.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Ge C, Zhu C, Salaita K. Nat Commun. 2014;5:5167. doi: 10.1038/ncomms6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Medda R, Liu Z, Galior K, Yehl K, Spatz JP, Cavalcanti-Adam EA, Salaita K. Nano Lett. 2014;14(10):5539. doi: 10.1021/nl501912g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhatt N, Huang PJJ, Dave N, Liu J. Langmuir. 2011;27:6132. doi: 10.1021/la200241d. [DOI] [PubMed] [Google Scholar]
- 12.Mansoor MA, Svardal AM, Ueland PM. Anal Biochem. 1992;200(2):218. doi: 10.1016/0003-2697(92)90456-h. [DOI] [PubMed] [Google Scholar]
- 13.Blakely BL, Dumelin CE, Trappmann B, McGregor LM, Choi CK, Anthony PC, Duesterberg VK, Baker BM, Block SM, Liu DR, Chen CS. Nat Methods. 2014;11(12):1229. doi: 10.1038/nmeth.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajagopalan P, Marganski Wa, Brown XQ, Wong JY. Biophys J. 2004;87(4):2818. doi: 10.1529/biophysj.103.037218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ung P, Winkler, Winkler DA. J Med Chem. 2011;54(5):1. doi: 10.1021/jm1012984. [DOI] [PubMed] [Google Scholar]
- 16.Pfaff M, Tangemann K, Müller B, Gurrath M, Müller G, Kessler H, Timpl R, Engel J. J Biol Chem. 1994;269(32):20233. [PubMed] [Google Scholar]
- 17.Craig Ja, Rexeisen EL, Mardilovich A, Shroff K, Kokkoli E. Langmuir. 2008;24(18):10282. doi: 10.1021/la702434p. [DOI] [PubMed] [Google Scholar]
- 18.Noë V, Willems J, Vandekerckhove J, Van Roy F, Bruyneel E, Mareel M. J Cell Sci. 1999;112:127. doi: 10.1242/jcs.112.1.127. [DOI] [PubMed] [Google Scholar]
- 19.Xiao J, Chen R, Pawlicki MA, Tolbert TJ. J Am Chem Soc. 2009;131(38):13616. doi: 10.1021/ja9045179. [DOI] [PubMed] [Google Scholar]
- 20.Galush WJ, Nye Ja, Groves JT. Biophys J. 2008;95(5):2512. doi: 10.1529/biophysj.108.131540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elbert DL, Hubbell JA. Annu Rev Mater Sci. 1996;26(1):365. [Google Scholar]
- 22.Estephan ZG, Jaber Ja, Schlenoff JB. Langmuir. 2010;26(22):16884. doi: 10.1021/la103095d. [DOI] [PubMed] [Google Scholar]
- 23.Jain a, Liu R, Xiang YK, Ha T. Nat Protoc. 2012;7(3):445. doi: 10.1038/nprot.2011.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Backmann N, Kappeler N, Braun T, Huber F, Lang HP, Gerber C, Lim RYH. Beilstein J Nanotechnol. 2010;1:3. doi: 10.3762/bjnano.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Gennes PG. Macromolecules. 1980;13(19):1069. [Google Scholar]
- 26.Kopan R, Ilagan MXG. Cell. 2009;137(2):216. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aota S, Nomizu M, Yamada KM. J Biol Chem. 1994;269(40):24756. [PubMed] [Google Scholar]
- 28.Redick SD. J Cell Biol. 2000;149(2):521. doi: 10.1083/jcb.149.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petrie Ta, Capadona JR, Reyes CD, García AJ. Biomaterials. 2006;27(31):5459. doi: 10.1016/j.biomaterials.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 30.Li F, Redick SD, Erickson HP, Moy VT. Biophys J. 2003;84(2):1252. doi: 10.1016/S0006-3495(03)74940-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng Y, Mrksich M. Biochemistry. 2004;43(50):15811. doi: 10.1021/bi049174+. [DOI] [PubMed] [Google Scholar]
- 32.Eisenberg JL, Piper JL, Mrksich M. Langmuir. 2009;25(24):13942. doi: 10.1021/la901528c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.