

Abstract
We yoked anatomical brain Magnetic Resonance Imaging to a randomized, double-blind, placebo-controlled trial (RCT) of antidepressant medication for 10-week’s duration in patients with dysthymia. The RCT study design mitigated ascertainment bias by randomizing patients to receive either duloxetine or placebo, and it supported true causal inferences about treatment effects on the brain by controlling treatment assignment experimentally. We acquired 121 anatomical scans: at baseline and endpoint in 41 patients, and once in 39 healthy controls. At baseline, patients had diffusely thicker cortices than did healthy participants, and patients who had thicker cortices had proportionately less severe symptoms. During the trial, symptoms improved significantly more in medication- compared with placebo-treated patients; concurrently, thicknesses in medication-treated patients declined toward values in healthy controls, but they increased slightly, away from control values, in placebo-treated patients. Changes in symptom severity during the trial mediated the association of treatment assignment with the change in thickness, suggesting that the beneficial effects of medication on symptom severity were at least partially responsible for normalizing cortical thickness. Together our findings suggest that baseline cortical hypertrophy in medication-free patients likely represented a compensatory, neuroplastic response that attenuated symptom severity. Medication then reduced symptoms and lessened the need for compensation, thereby normalizing thickness. This is to the best of our knowledge the first study to report within an RCT a differential change in cortical morphology during medication treatment for depressive illness and the first to provide within an RCT in vivo evidence for the presence of neuroanatomical plasticity in humans.
INTRODUCTION
Dysthymic disorder (DD) is a chronic but comparatively less severe form of Persistent Depressive Disorder that affects approximately 5% of the US population(1). DD is characterized by the presence of depressed mood most of the time for 2 or more years accompanied by at least two additional depressive symptoms, including poor self-esteem, low energy, feelings of hopelessness, insomnia, poor appetite, or impaired concentration(2). Compared with patients who have episodic Major Depressive Disorder (MDD), DD has been associated with greater childhood adversity, more psychosocial impairment, greater suicidality(3), fewer disturbances in the hypothalamic-pituitary-adrenal axis, and elevated cytokines(4, 5). DD and MDD patients, however, respond similarly to medications(6), and more than 60% of DD patients have a lifetime history of MDD(7, 8). A comprehensive analysis of self-reported depressive symptoms suggests a latent continuum of symptoms across all forms of unipolar depression(9). Thus the preponderance of evidence suggests that DD and MDD lie along a phenotypic and pathophysiological continuum, although whether they also constitute a continuum at the level of brain architecture and function is unknown, as no imaging studies have yet compared brain measures across DD and MDD participants at the same time and using the same image processing techniques.
Prior magnetic resonance imaging (MRI) studies of MDD have reported abnormalities in brain regions involved in mood regulation, emotional processing, memory formation, and decision-making, particularly larger volumes and greater activity in the amygdala and reduced volumes in orbitofrontal, subgenual prefrontal(10), and prefrontal(11) cortices, and in the hippocampus(12). We previously showed that individuals at high compared with low familial risk for MDD had thinner cortices across the entire lateral aspect of the right and mesial wall of the left hemisphere; individuals who actually developed depression had additional thinning of the lateral aspect of the left hemisphere(13). Consistent with studies in MDD, the few neuroimaging studies in DD have also implicated abnormalities in brain regions that regulate mood, including enlarged amygdala(14), increased amygdala activation(15), and increased functional connectivity in the default-mode network(16). No studies of DD thus far have assessed measures of cortical thickness compared to values in healthy control participants.
A serious and pervasive limitation of nearly all prior brain imaging studies has been their use of a case-control design, which renders studies vulnerable to a wide range of ascertainment biases in the selection of participants. Most importantly, the absence of control over experimental variables renders case-control studies merely associational and incapable of supporting causal inferences concerning mechanisms of pathogenesis or treatment response(17). The most common independent variable studied in case-control designs is diagnosis (e.g. MDD vs control), which in humans ethically cannot be controlled experimentally. One variable that can be controlled in human studies, however, is treatment assignment. Experimental control of intervention through randomization and double blinding in treatment(17) or prevention studies(18) is why the randomized controlled trial (RCT) has become the gold standard for demonstrating that the experimentally controlled variable, such as treatment assignment, influences an outcome variable, such as symptom severity. A placebo arm in addition controls for non-specific treatment effects. We can understand the causal influences of treatment on the brain by using the change in brain imaging measures instead of the change in symptoms as the primary outcome measure in an RCT. This requires acquisition of images at the beginning and end of the RCT, then analyzing those images while blind to treatment assignment and time point of image acquisition (i.e., the beginning or end of the trial). Yoking imaging to an RCT study design in this way mitigates the problem of ascertainment bias that vitiates case-control studies, because patients are randomly assigned to the treatment arms, and most importantly it supports true causal inferences about the effects that the experimentally controlled treatment has on the brain.
We yoked anatomical MRI to a 10-week, prospective, double-blind, placebo-controlled RCT of the use of antidepressant duloxetine for treating DD(19), which permitted us to ascribe the effects of antidepressant medication on cortical thickness. We acquired MRI scans in dysthymic patients both at baseline and end of the trial. We also acquired scans in age- and sex-matched heathy participants at a single point in time to assess whether the medication in DD patients changed cortical thickness towards or away from healthy values. We previously showed in a larger clinical trial of 57 dysthymic patients that duloxetine treatment produced a greater reduction in symptoms than did placebo, and that a greater number of duloxetine- compared to placebo-treated patients experienced remission of symptoms by the end of the trial(19). We also reported in the same subsample as the present study that duloxetine treatment normalized resting-state BOLD fMRI activity in an otherwise hyperactive default-mode network, especially in the functional interactions between the posterior cingulate cortex and amygdala(16). We wanted to determine for the same participants as those in the fMRI study whether duloxetine treatment for 10 weeks changes brain structure and, if it does, how those changes relate to changes in symptom severity. Based on our prior resting state fMRI findings, we hypothesized that duloxetine treatment would normalize baseline alterations of cortical thickness in DD patients.
MATERIALS/SUBJECTS AND METHODS
See Supplemental Information for further details of participant recruitment and characterization, image processing, statistical modeling, and results. The study was approved by the New York State Institute/Columbia University Department of Psychiatry Institutional Review Board (IRB).
Participants
Patients were recruited for the study through advertisements, website postings, and the hospital’s telephone referral service; 350 individuals expressed interest in the study, 70 of these met inclusion/exclusion criteria, and 60 agreed to participate and were randomized to either active treatment or placebo. All provided informed written consent prior to participation. Three patients dropped out before the start of the trial, and therefore at baseline we treated and acquired clinical and behavioral data in 57 patients. Of these 57 patients, 53 were consented for MRI at baseline. However, 11 patients declined to have an MRI scan at the end of the trial, and the MRI data for 1 patient had motion artifact, leaving 41 patients who had MRI data at both baseline and end of the study. At the time of recruitment, 22 patients were medication-naïve; another 15 had discontinued medication use for more than a year. Four patients who were taking psychotropic medications underwent a 4-week medication wash-out prior to participation. We also acquired MRI data at one time point in 39 healthy participants who were group-matched to the patients on age and sex, and who were recruited through mailings and phone calls to households identified in telemarketing lists of the local community. DD patients were diagnosed by a board certified psychiatrist using clinical interviews and the Structured Clinical Interview for DSM-IV-TR (SCID).(19) Patients met DSM-IV-TR(2) criteria for either dysthymic disorder (DD) or Depressive Disorder not otherwise specified, but they did not meet criteria for Major Depressive Disorder (MDD). Symptom severity was assessed using both the 24-item Hamilton Depression Rating Scale (HDRS)(20) and Cornell Dysthymia Rating scale (CDRS)(21). At the time of recruitment, patients had their current episode of DD for at least 2 years before entering the clinical trial, and they scored 12 or higher on the Hamilton Depression Rating Scale (HDRS)(20). Healthy controls had no current illness and minimal lifetime history of Axis I illness on the SCID (Supplemental Information).
We randomized 21 patients to the active medication arm, where they were treated with up to 120 mg/day of duloxetine; 20 patients randomized to the placebo arm received gelatin shell capsules with sugar balls. Patients in the 2 arms did not differ on age, symptom severity at baseline, or lifetime history of MDD; they did differ significantly in their sex composition, however, with 14 females receiving duloxetine and 5 placebo (χ2=7.15,df=1,p=0.007)(Table 1). Comorbid illnesses either in the past or at the time of study entry in the patients are listed in Table 2; 10 patients in each treatment arm had any comorbid illness in their lifetimes. All participants agreed not to take other psychotropic medication or receive psychotherapy during the trial.
Table 1. Demographics and Symptom Severity of the Participants in this Study.
The 41 dysthymic patients did not differ significantly from the 39 healthy controls in age (p=0.97) or sex (χ2=0.508, df=1, p=0.47). At baseline, patients randomized to the duloxetine arm did not differ significantly from those randomized the placebo arm in age (p=0.44), symptom severity measured using either the HDRS (p=0.26) or the CDRS (p=0.76) scale, or lifetime history of Major Depressive Disorder (χ2=1.17, df=1, p=0.27). However, the sex composition differed significantly in the two treatment arms (χ2=7.152, df=1, p<0.01). By the end of the trial (i.e. Week 10), symptom severity had decreased significantly for both the duloxetine-treated patients (p<1.0*10−15 for HDRS scores and p<4.0*10−13 for CDRS scores) and placebo-treated patients (p<0.002 for HDRS scores and p<0.01 for CDRS scores). However, the duloxetine-treated compared with placebo-treated patients had significantly lower symptom severity (p<9.0*10−6 for HDRS scores and p<2.0*10−5 for CDRS scores). Furthermore, a significantly (χ2=15.14, df=1, p<0.0001) greater proportion of the duloxetine-treated patients had a remission of illness by the end of the trial. The average and standard deviation of age and symptom severity are shown in the table.
| Age (Years) |
Sex | Symptom Severity (At Baseline) |
Symptom Severity (At Week 10) |
Lifetime MDD |
Remitted at Week 10 |
|||
|---|---|---|---|---|---|---|---|---|
| HDRS | CDRS | HDRS | CDRS | |||||
| Healthy, N=39 | 39.55±10.26 | 22 Males | N/A | N/A | N/A | N/A | 0 | N/A |
| Duloxetine Arm, N=21 | 39.08±10.00 | 7 Males | 20.66±4.09 | 37.62±8.16 | 5.76±3.12* | 12.14±7.37* | 13 | 15 |
| Placebo Arm, N=20 | 40.82±10.81 | 15 Males | 22.23±4.79 | 38.38±8.22 | 15.76±7.71 | 29.04±13.09 | 9 | 2 |
HDRS = Hamilton Depression Rating Scale; CDRS = Cornell Dysthymia Rating Scale; N/A = not applicable; MDD = Major Depressive disorder
Table 2. Comorbid Illnesses in the Dysthymic Patients.
The table lists the number of dysthymic patients who had a comorbid illness either previously or at the time of study entry; 10 patients in the duloxetine arm and 10 patients in the placebo arm had a comorbid illness other than Major Depressive Disorder in their lifetimes.
| Comorbid Illnesses | Previous | At Study Entry | Duloxetine Arm | Placebo Arm |
|---|---|---|---|---|
| Major Depressive Disorder | 22 | 0 | 13 | 9 |
| Generalized Anxiety Disorder | 9 | 0 | 5 | 4 |
| Post Traumatic Stress Disorder | 1 | 2 | 1 | 2 |
| Panic Disorder | 2 | 2 | 2 | 2 |
| Social Phobia | 1 | 7 | 5 | 3 |
| Specific Phobia | 3 | 4 | 4 | 3 |
| Obsessive Compulsive Disorder | 2 | 2 | 3 | 1 |
MRI Scanning
We acquired a total of 121 anatomical MRI scans – one at baseline and one immediately after trial week 10 in the DD patients, and at one time point for the healthy participants. The scans were obtained on a 3Tesla GE Signa MRI scanner using a 3D fast spoiled gradient recall sequence: Resolution=0.98x0.98x1.0 mm; Repetition Time=4.7 ms; Echo Time=1.3 ms; Inversion Time=500 ms; Flip Angle=11°; Matrix=256x256; Field of View=25 cm; Phase Field of View=100%; Slice Thickness=1.0 mm.
MRI Processing
Scans received a clinical reading by a neuroradiologist to exclude the presence of any clinically significant findings in all study participants. All MRI data were processed blind to the order of scan acquisition (baseline or week 10), treatment assignment, and participant clinical characteristics.
Preprocessing
We applied automated tools to correct for large-scale variations in image intensities(22) and to remove extracerebral tissue(23). We then removed connecting dura manually on each slice in the sagittal, axial, and coronal views.
Segmentation of Cortical Gray Matter
We used a semi-automated method to segment brain tissue as gray or white matter, with an expert neuroanatomist sampling gray-scale values of both cortical gray matter and white matter at 4 standard locations throughout the brain. The sampled values were averaged to generate mean values for gray and white matter; these values were used to threshold and generate initial tissue segmentations, which were then edited to remove the subcortical gray matter. The test-retest intraclass correlation coefficient (ICC)(24) for the cortex was >0.98.
Mapping Cortical Thickness
We spatially normalized all brains to a template brain (see Supplementary Information) and established point-by-point correspondence across their surfaces. We then applied to each coregistered brain a 3D morphological operator to distance-transform the brain without its cortical mantle to the surface of the cerebrum(25) and calculated cortical thickness as the smallest distance of each point on the cerebral surface to the outermost surface of the white matter. Cortical thickness was calculated with brains scaled to the template, thereby accounting for overall scaling effects on thickness measures.
Statistical Analyses
We used voxel-wise linear regression and repeated measures analyses of variance, while covarying for age and sex, to assess respectively the correlations of cortical thickness with symptom severity and the effects of treatment on cortical thickness. We used a procedure for False Discovery Rate (FDR), with FDR = 0.05, to control for Type I error in the multiple statistical tests performed across the cerebral surface. We did not control for the effects of comorbid illnesses in our analyses because only a small number of patients in each treatment arm had those specific illnesses (Table 2). We color-coded P-values that survived FDR correction, using cool colors (purple and blue) for inverse associations and warm colors (yellow and red) for positive associations. Detailed statistical models are presented in the methods section of Supplementary Information.
Baseline Analyses
We used multiple linear regression to compare cortical thickness across patient and healthy control participants at baseline and to aid understanding of whether treatment-induced changes in thickness during the 10-week trial were in a direction toward or away from normal values. In addition, we correlated thickness with symptom severity in DD patients.
Longitudinal Analyses
We applied linear mixed models to the longitudinal data to assess whether treatment differentially altered cortical thickness across the two treatment arms (i.e., to assess the treatment-by-time interaction). We also assessed treatment effects on thickness separately in the duloxetine- and placebo-treated patients.
Repeated Measures Analyses of Thickness Associations with Symptom Severity
We used a repeated measures analysis of longitudinal data in the 21 duloxetine-treated patients to assess within individuals how thickness changed over time with changes in symptom severity, and to assess across individuals the correlation of thickness with severity.
Mediation Analysis
We conducted post hoc mediation analyses to test whether the changes in thickness mediated the effects of treatment assignment on changes in symptom severity, or whether the changes in symptom severity mediated the effects of treatment on cortical thickness.
Code Availability
Computer code as executables in Windows Operating System will be made available upon request.
RESULTS
Although symptom severity declined significantly over the 10-week trial for both the duloxetine- and placebo-treated patients (placebo p=5.76*10−3; duloxetine p=2.51*10−15), at the end of the trial severity was significantly lower for the duloxetine- than for the placebo-treated patients (p=8.54*10−6). In addition, a significantly greater proportion of duloxetine-(15 out of 21) compared to placebo-(2 out of 20) treated patients experienced remission of illness(χ2=15.14,df=1, p<0.0001) (Table 1). Age was not associated with symptom severity in patients at baseline, nor did it moderate the decline in symptom severity over the trial.
Baseline Analyses
At baseline, cortical thickness did not differ significantly between patients who were randomized to duloxetine compared with those randomized to placebo (not shown). The dysthymic patients collectively, compared with healthy participants, had thicker cortices bilaterally across large portions of the brain, especially in the superior and middle temporal gyrus, cingulate gyrus, inferior parietal lobule, supramarginal gyrus, and pre- and post-central gyrus(Fig.1). Thickening was present in patients randomized to either treatment arm (not shown). The associations of cortical thickness with DD did not change when controlling for prior MDD (not shown), suggesting that prior MDD did not influence the effects of depressive illness on cortical morphology. Symptom severity correlated inversely with thickness, indicating that a thicker cortex was associated with less severe symptoms; this was especially true in regions where the cortex was thicker in patients than in healthy controls.
Figure 1. Baseline Data Analyses.
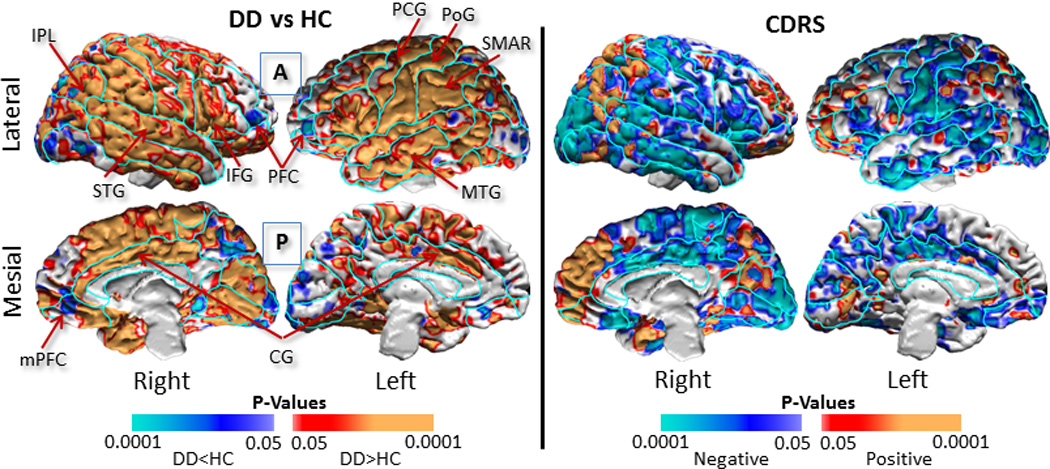
We compared cortical thickness in the 41 dysthymic patients with thickness in the 39 age- and sex-matched healthy participants, and we correlated cortical thickness with symptom severity within the dysthymic patients. We covaried for age and sex, and controlled for multiple hypothesis testing using a procedure for False Discovery Rate (FDR), with the FDR set at 0.05. We color-coded and displayed the P-values smaller than 0.05 across the surface of a template brain using the color bar shown at the bottom. The findings were unchanged when we covaried for a lifetime history of MDD in dysthymic patients.
Left Panel: Dysthymic patients compared to healthy controls had a thicker cortex bilaterally across large portions of the brain, especially in the superior and middle temporal gyrus, cingulate gyrus, inferior parietal lobule, supramarginal gyrus, and pre- and post-central gyrus. In addition, the patients had thinner lateral and mesial prefrontal cortices bilaterally. Warm colors (red and orange) represent thicker, cool colors (blue and purple) represent thinner, cortices in patients compared with healthy participants, and gray shows voxels where thickness did not differ between patients and controls.
Right Panel: Symptom severity correlated inversely with cortical thickness bilaterally across the parietal and temporal cortices, indicating that patients with thicker cortices had proportionately lower symptom severity scores. Warm colors (red and orange) show positive correlations, cool colors (blue and purple) show inverse correlations, and grey shows no significant correlations between thickness and symptom severity. Findings were similar for the correlations of cortical thickness with symptom severity measured using the Hamilton Depression Rating Scale (not shown).
DD=Dysthymic Disorder; HC=Healthy Controls; CDRS=Cornell Dysthymia Rating Scale; PFC=Prefrontal Cortex; mPFC=mesial Prefrontal Cortex; CG=Cingulate Gyrus; MTG=Middle Temporal Gyrus; STG=Superior Temporal Gyrus; IPL=Inferior Parietal Lobule; SMAR=Supramarginal Gyrus; PoG=Postcentral Gyrus; PCG=Precentral Gyrus; A=Anterior; P=Posterior
Longitudinal Analyses
These analyses revealed significant treatment-by-time effects on cortical thickness, deriving from cortical thinning over the course of the trial in the duloxetine-treated patients toward values in healthy participants, particularly in regions where the patients at baseline had thicker cortices than healthy participants(Fig.2 & Supplementary Fig.1). In contrast, the cortex thickened over the trial in placebo-treated patients, diverging further from control values than at baseline.
Figure 2. Longitudinal Data Analyses to Assess Change in Thickness with Time.
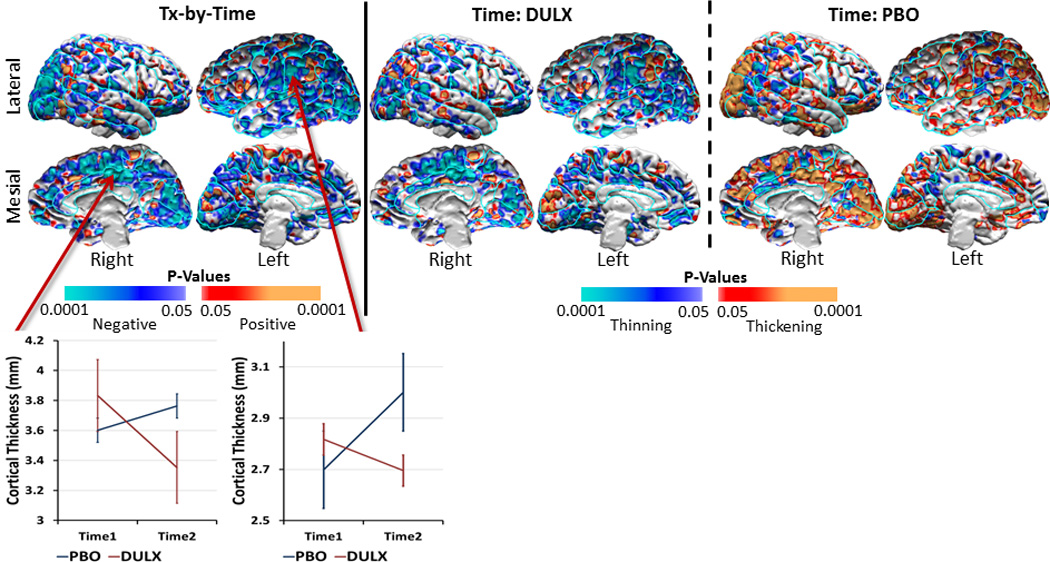
We used linear mixed models to assess the change in cortical thickness with time in the 41 dysthymic patients. Findings showed a significant treatment-by-time interaction on the cortical thickness (left panel), which was driven primarily by a decline in cortical thickness over the trial in the duloxetine-treated patients (middle panel) and a slight increase in thickness in the placebo-treated patients, especially in the occipital cortex bilaterally (right panel). Warm colors (orange and red) show thickening, cool colors (purple and blue) show thinning, and gray shows no significant change in thickness with time. We showed time effects only in voxels where we detected significant effects of treatment-by-time interaction on cortical thickness to understand better treatment effects over the 10-week period of the clinical trial. See Supplementary Figure 1 for maps of time effects across the entire cortical surface. Line graphs for representative locations in the right occipital cortex and right mesial parietal cortex illustrate the opposing change in thickness with time for patients in the two treatment arms. At baseline, the variance of the cortical thickness did not differ between duloxetine-treated patients and placebo-treated patients (scatterplot at mesial location: F-statistic=1.11; df1=19; df2=20; p=0.81; scatterplot at lateral location: F-statistic=0.65; df1=19; df2=20; p=0.36). At the end of the trial, the variance did differ between the two treatment arms for the lateral location (F-statistic=1.53; df1=19; df2=20; p=0.35) but was significantly different for the mesial location (F-statistic=0.31; df1=19; df2=20; p=0.016).
DULX=Duloxetine; PBO=Placebo
Repeated Measures Analyses
The between-individual analyses showed that severity correlated strongly and inversely with thickness across most of the brain, except for small regions of positive correlation in the mesial and lateral surfaces of the frontal pole and posterior parietal cortex(Fig.3). Within-individual changes in thickness generally correlated positively with changes in symptom severity–i.e., the cortex thinned as symptom severity declined over the 10-week clinical trial. Inverse associations, in which the cortex thickened within individuals as symptom severity declined, were evident in several brain regions, especially in the cingulate gyrus along the mesial wall of the left hemisphere and bilaterally in occipital cortex(Fig.3 & Supplementary Fig. 3). In contrast, within-individual changes in thickness for placebo-treated patients generally correlated inversely with changes in symptom severity(Supplementary Figure 4). In an exploratory analysis, we determined that cortical thinning varied with age in the duloxetine-treated patients, with thinning more prominent in the younger compared with older patients(Supplementary Fig.2).
Figure 3. Repeated Measures Analyses Associating Symptom Severity with Cortical Thickness.
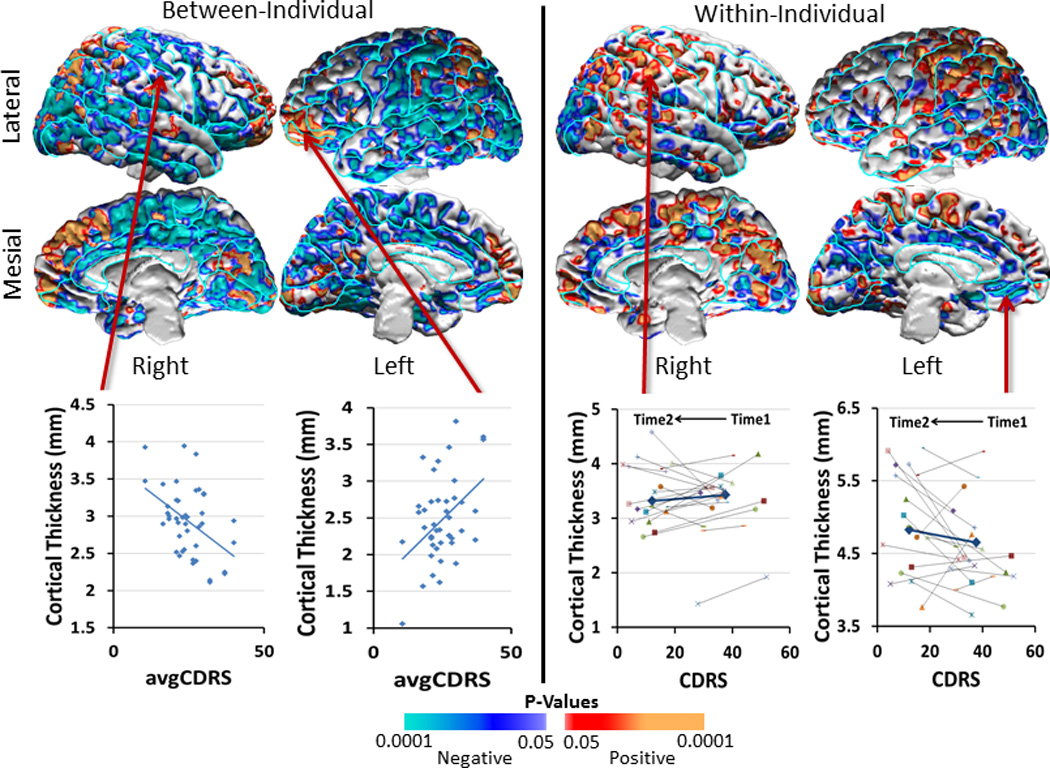
We assessed how cortical thickness in duloxetine-treated patients was associated with symptom severity across individuals, as well as how cortical thickness changed with the change in symptom severity within individuals over time. Symptom severity was measured with the Cornell Dysthymic Rating scale (CDRS). We covaried for age and sex effects and generated scatterplots using age- and sex-adjusted cortical thickness measures. We displayed findings only in voxels where the interaction of treatment-by-time on cortical thickness was statistically significant (Figure 2). Maps of findings across the entire brain are shown in Supplementary Figure 3 of the Supplementary Information.
Left Panel: The cross-sectional, across-individual correlation of cortical thickness with severity showed that thickness correlated inversely with CDRS values, averaged over pretreatment baseline (Time1) and end of trial (Time2), in large expanses of the lateral and mesial surfaces of both hemispheres, especially in regions where the dysthymic patients had thicker cortices than healthy participants at baseline (Figure 1). Scatterplots for representative voxels where inverse correlations were located illustrate that patients with thicker cortices generally had less severe symptoms. In contrast, cortical thickness correlated positively with symptom severity bilaterally in the anterior prefrontal and posterior parietal cortex.
Right Panel: The longitudinal, within-individual, correlation of change in thickness with the change in severity over the course of the trial showed positive associations (indicating that cortical thickness decreased as symptom severity decreased, as shown in orange and red) located bilaterally in the parietal and posterior temporal cortex, and inverse associations (indicating that cortical thickness increased as symptom severity decreased, shown in blue and violet) located bilaterally in the occipital, anterior cingulate, and anterior prefrontal cortex.
Note that the cross-sectional, across-individual maps generally show color codings for correlations that are opposite from those in the longitudinal, within-individual maps: In the left panel, individuals with fewer symptoms had thicker cortices in voxels shown in blue; in the right panel, cortical thickness decreased over the 10-week period of the trial within individuals for whom treatment with antidepressants reduced symptom severity. Thus, the findings in both panels, taken together, suggest that a thicker cortex at baseline is associated with fewer symptoms, likely representing a compensatory, neuroplastic hypertrophy of the cortex. Successful treatment and a decline in symptoms over the course of the trial seems to obviate the need for that compensatory hypertrophy and thereby produces a relative thinning and normalization of the cortical mantle.
Mediation Analyses
Mediation analyses showed that the change in thickness did not mediate treatment effects on the change in symptom severity. Instead, the change in symptom severity significantly mediated the effects of treatment on the change in cortical thickness across large portions of the brain(Fig.4 & Supplementary Fig.5).
Figure 4. Longitudinal Mediation Analyses.
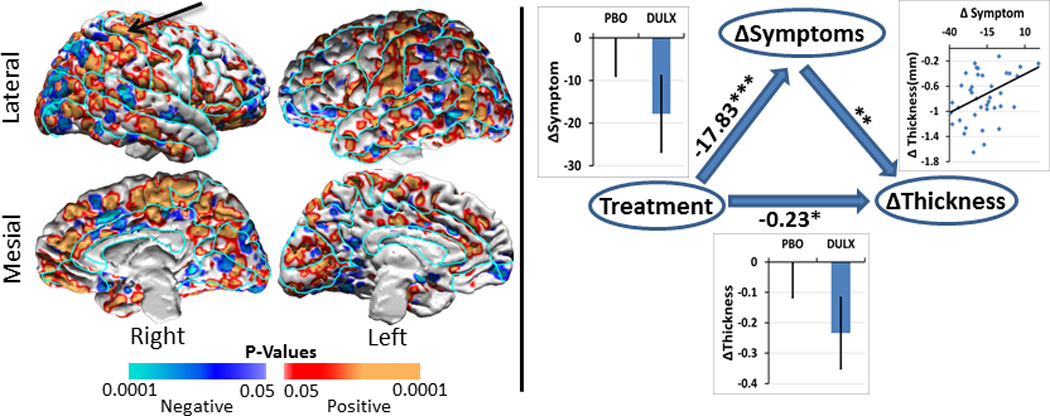
We applied a longitudinal mediation analysis to assess whether the change in symptom severity mediated the association between treatment arm and the change in cortical thickness in our 41 dysthymic patients. We covaried for age and sex and controlled for multiple hypothesis testing using a procedure for false discovery rate (FDR) with FDR=0.05. Here we display findings only for those voxels where we detected significant treatment-by-time interaction effects on cortical thickness (Figure 2). The findings across the entire brain are shown in Supplementary Figure 5.
Left Panel: At each voxel on the template surface we color-coded and plotted P-values in warm colors (red and orange) where the test statistic was positive and in cool colors (violet and blue) where the test statistic was negative for the mediation analyses. These plots showed that the decline in symptom severity over the trial significantly mediated the associations between treatment and change in cortical thickness across large portions of the brain where dysthymic patients at baseline had thicker cortices than healthy controls. We used cortical thickness from the brain region pointed to by the arrow for the scatterplot shown in the right panel.
Right Panel: We generated scatterplot for the associations among treatment, change in symptom severity, and change in cortical thickness variables for a representative voxel where the mediating effects of symptom severity were statistically significant. These plots showed that symptom severity declined significantly (p=4×10−6) in the duloxetine-treated patients, the decrease in symptom severity was significantly (p=0.01) associated with a decrease in cortical thickness, and treatment was significantly (p=0.045) associated with the decrease in cortical thickness. The overall P-value was < 0.008 for the mediating effect of change in symptom severity on the association of treatment with change in cortical thickness.
* = P-value<0.05; ** = P-value<0.01; *** = P-value<0.00001.
To understand better the mediation findings, we generated scatterplots representing the associations of the treatment (independent variable), change in symptom severity (mediator variable), and change in thickness (dependent variable) in a representative location in the superior parietal cortex. These plots showed that (1) duloxetine treatment significantly decreased symptom severity (p<4.0*10−6), (2) the decline in symptom severity correlated significantly with cortical thinning (p<0.01), and (3) duloxetine treatment also correlated significantly with cortical thinning (p<0.04)(Fig.4).
DISCUSSION
To the best of our knowledge, this study is the first to report a differential change in thickness of the cerebral cortex during medication compared with placebo treatment for a depressive illness within an RCT. Combining brain imaging with an RCT study design allowed us to infer that medication treatment not only was associated with changes in brain structure in DD patients, but it also allowed us to conclude that medication actually caused those changes. Inclusion of healthy controls allowed us to determine whether the treatment-related changes in brain structure were toward or away from normal values. We found that the cortex at the beginning of the trial was significantly and diffusely thicker in DD patients than in healthy participants. Moreover, the baseline thickening was associated with less severe depressive symptoms, suggesting that the thickening may have been a neuroplastic response to illness that served a compensatory function in this patient population. Although symptom severity declined in both treatment arms over the course of the trial, the duloxetine-treated patients improved to a significantly greater extent, and more frequently achieved clinical remission, than did placebo-treated patients. The cortex thinned significantly during the trial in duloxetine-treated patients in the direction of healthy control values, whereas the cortex thickened slightly in placebo-treated patients, diverging even further from healthy values at baseline. We also showed that the change in symptom severity mediated the association of treatment assignment with the change in cortical thickness during the trial, suggesting that medication exerted its normalizing effects on cortical thickness by reducing the severity of symptoms, and supporting further our interpretation that baseline thickening in DD patients was a neuroplastic compensation to chronic illness. When medication successfully reduced symptoms, this putative neuroplastic compensation was no longer needed, which presumably then permitted the return of cortical thickness toward normal values. Finally, we showed that younger compared with older patients had more prominent cortical thinning, and across larger expanses of the cortical surface over the course of the trial (Supplementary Fig. 2), suggesting that medication was more effective at normalizing cortical thickness in younger patients.
At baseline when they were medication-free, patients had cortices that were diffusely thicker than in the healthy participants, an effect that did not differ between patients randomized to the duloxetine or placebo arms. These findings are consistent with those in recent studies reporting thicker cortices across several brain regions in medication-naive adults with MDD(26). We also detected cortical thinning in the lateral and mesial prefrontal cortices, consistent with the majority of anatomical studies of MDD reporting thinner cortices in brain regions that support emotion regulation, including orbitofrontal, dorsal anterolateral, and ventrolateral prefrontal cortices(27). Our findings, however, could initially seem at odds with our previously reported findings that individuals at high, compared with low, familial risk for MDD have thinner cortices across the entire lateral aspect of the right hemisphere and mesial wall of the left hemisphere(13). It seems likely that the high-risk individuals in that prior study did not generate the compensatory cortical thickening that we have detected in DD patients because the high risk participants were largely free of illness at the time of scan. Their cortical thinning likely represented instead a trait vulnerability for developing MDD, particularly in relation to thickness in low-risk individuals, whose thicker cortices presumably protected against the development of MDD. Consistent with this interpretation, we also reported that high-risk individuals who endorsed more religious beliefs had thicker cortices relative to high-risk individuals who endorsed less religiosity, and they also had fewer lifetime MDD episodes, suggesting that cortical thickening associated with religiosity was protective against developing MDD(28). Our DD patients, in contrast to our prior high-risk sample, had been ill for more than 2 years at the time of study, giving them abundant time to develop neuroplastic cortical hypertrophy, as presumably manifested in those who had thicker cortices and proportionately fewer symptoms. Therefore we suspect that the baseline cortical thickening in DD patients was a consequence of their chronic illness and represented a neuroplastic compensation that helped to attenuate their depressive symptoms. It is possible that the presence of neuroplastic compensation is precisely what distinguished DD from MDD; without compensation, DD patients presumably would become more severe and would then meet criteria for MDD, rather than DD. If this is the case, then an important question for future research is why MDD patients do not engage fully these neuroplastic compensatory processes.
We detected a significant treatment-by-time interaction on cortical thickness over the 10-week trial. The interaction derived from treatment-related changes in cortical thickness occurring in both arms, but in opposite directions: in duloxetine-treated patients, the cortex thinned in a direction toward values in healthy participants; in placebo-treated patients, the cortex thickened slightly, and in a direction even more divergent from control values than at baseline. These longitudinal, within-individual analyses further showed that symptom severity declined as the cortex thinned during the trial in the duloxetine-treated patients. Because cortical thickening at baseline and its association with fewer symptoms suggested the presence of neuroplastic compensation, the exaggeration of baseline thickening in the placebo arm during the RCT suggests the presence of continuing neuroplastic compensation with continuing illness. It is tempting to speculate, in fact, that cortical thickening during placebo administration could have contributed to the reduction in symptoms in that arm of the trial. If the same compensatory processes were operating in the duloxetine-treated patients as speculated in the placebo-treated patients, then the duloxetine-induced normalization of cortical thickness would presumably represent the reduced need for compensatory hypertrophy, thanks to the duloxetine-induced reductions in symptom severity. Alternatively, duloxetine-induced normalization of cortical thickness could be a direct consequence of the medication, with the reductions in symptoms following it. We therefore conducted mediation analyses to provide statistical evidence for or against each of these two competing interpretations. We found no statistical evidence that cortical thickness mediated the association of treatment assignment (duloxetine or placebo) with the change in symptom severity during the trial. Instead, we found that the change in symptom severity partially but significantly mediated the association of treatment with change in cortical thickness, supporting the interpretation that medication-induced improvement in symptoms during the trial reduced the need for neuroplastic compensation, thereby reducing cortical thickness toward normal values. Placebo-treated patients, however, continued to require compensation for their continuing symptoms, thereby thickening their cortices during the trial.
Our findings provide imaging-based evidence for the presence of an adaptive, neuroplastic compensation in response to chronic symptoms of dysthymia. Neuroplastic changes comprise persistent changes in dendritic length and branching, synaptic density and strength, and neurogenesis, which together produce structural and functional alterations in brain circuits and how they process information in response to changing functional demands on the central nervous system(29). Activity-dependent neuroplasticity has been reported in several prior imaging studies in healthy humans, including experience-dependent changes in gray and white matter volumes in response to learning(30), mindfulness meditation(31), physical exercise(32), and cognitive training(33). Animal studies have reported cortical thickening in response to environmental enrichment, likely due to neurotrophin-driven increases in dendritic branching(34), spine density(35), and glial cell density(36). Studies of older animals in enriched environments(37) and older humans who suffered either limb amputations(38) or strokes(39) suggest that neural plasticity is retained across the lifespan. Our findings similarly suggest that neuroplastic compensation is present at all adult ages, although the age-specific effects in the reversion of thickening toward normal values during the trial, being more prominent in younger than in older patients when successfully treated with medication, suggests that neuroplastic hypertrophy may be more dynamic in younger persons.
Evidence is growing for the presence of neuroplasticity and its compensatory functions in neuropsychiatric illnesses(40). The voluntary suppression of tics in patients with Tourette Syndrome, for example, is thought to generate hypertrophy of frontal and parietal cortices, which in turn attenuates the severity of tic symptoms and normalizes excess functional activity during cognitive tasks that require activation of these cortical regions(41). Youth who, for reasons unknown, are unable to generate this cortical hypertrophy have more severe symptoms and may be more likely to have illness that persists into adulthood. Additional hypertrophy of the hippocampus is thought to modulate the severity of not only tic symptoms, but also of comorbid Attention Deficit/Hyperactivity Disorder (ADHD) and Obsessive-Compulsive Disorder symptoms(42). Hypertrophy of the hippocampus is thought to serve a similar symptom-attenuating function in youth who have ADHD without tic disorder(43). Successful treatment of ADHD youth with stimulant medications has been associated with normalization of highly localized morphological abnormalities in the surfaces of the basal ganglia nuclei(44) and thalamus(45), presumably because the stimulant-induced increases in dopaminergic transmission supported neuroplastic changes in these subcortical regions. Similarly, studies of veterans with Post-Traumatic Stress Disorder (PTSD) showed that the veterans who completely recovered from their illness had larger hippocampal volumes than those with current PTSD(46), suggesting either that those with larger hippocampal volumes recovered, or those with neuroplastic reversal of hippocampal volume loss had positive outcomes of treatment and higher rates of symptom remission. Antipsychotic medications have been reported to moderate the age-related decline in gray matter volumes in patients with Schizophrenia(47). Together these studies provide compelling evidence that activity-induced neuroplastic changes in the brain may simultaneously attenuate symptom severity, help maintain cognitive and behavioral performance on tasks that depend on those neural systems, and even contribute to the remission of illness in individuals with neuropsychiatric disorders.
The findings of this study should be interpreted in light of its limitations. First, we cannot entirely exclude the possibility that a thicker cortex at baseline derived from prior use of psychotropic medications in 19 of our patients. However, at the time of recruitment, 15 patients had not used medication more than a year prior to entering the study, and 4 patients on medications underwent a 4-week wash-out period; furthermore, our findings did not change when we excluded those 4 patients from our analyses. We therefore believe that prior use of psychotropic medications did not affect baseline findings of cortical thickening. Second, we acquired MRI data in healthy individuals at only one time point, which limited our ability to assess and interpret fully the brain changes in placebo-treated patients. Nevertheless, the data in healthy participants at even a single time point permitted us to assess whether the medication-induced changes in cortical thickness were in a direction toward or away from values in healthy persons. Third, even though the RCT study design mitigated ascertainment bias by randomly assigning patients to either duloxetine or placebo arm of the study, the participants in the trial may not be representative of all patients in the general population who have depressive illness. Fourth, despite random assignment, patients differed in sex composition across the 2 treatment arms (15 males in placebo, 7 males in duloxetine); symptom severity did not differ between men and women at baseline or at the end of the trial in either of the two treatment arms, however, indicating that treatment effects did not differ by sex. We also incorporated sex as a covariate in all our analyses. Thus we believe that differing sex compositions in the two treatment arms did not confound the effects of treatment on cortical thickness. Finally, our participants, similar to participants in any RCT, may not have been representative of all patients in the general population who have depressive illness.
Despite these potential limitations, our findings provide strong evidence for compensatory, activity-dependent, neuroplastic thickening of the cortex that attenuates the chronic symptoms of dysthymia. The baseline cortical thickening may have protected DD patients from developing a more severe form of illness, MDD, which has been associated with thinning across large portions of the cortical surface(13). In this view, the severity and clinical course of depressive illness may depend upon whether an individual is able to engender in the brain an adaptive neuroplastic response to the presence of their distress. Disruptions of neuroplasticity from any cause may interfere with compensatory thickening and lead either to a new onset of illness, more severe symptoms, or clinical relapse. Understanding the molecular and cellular underpinnings of neuroplastic changes in the cortex and why those mechanism fail in certain individuals, and identifying ways to enhance neuroplastic response, will be important in the future to prevent new onset of illness, reduce symptom severity in individuals who are already ill, and induce compensation as a form of resilience in individuals who are at high-risk for developing depressive illnesses.
Supplementary Material
Acknowledgments
This study was supported by NIH grant K02-74677 (PI, Dr. Bradley S. Peterson), an Investigator-Initiated Grant from Eli Lilly company to Drs. Hellerstein and Peterson, and funding from Children’s Hospital Los Angeles and the University of Southern California. We are grateful to Zachary Toth, MS, Giancarlo Nati, MA, and Ming Qian, MS, for their technical assistance in data processing, archiving, and maintenance. The research was made possible by the provision of data by New York State Psychiatric Institute and Columbia University.
Drs. Hellerstein and Peterson received an investigator-initiated grant from Eli Lilly to support in part the costs of this study. Neither received salary support from that grant.
Footnotes
Trial Registration: ClinicalTrials.gov identifier: NCT00360724
CONFLICT OF INTEREST
Dr. Bansal reports no financial relationships with commercial interests.
Supplementary information is available at Molecular Psychiatry’s website.
REFERENCES
- 1.Sansone RA, Sansone LA. Dysthymic disorder: forlorn and overlooked? Psychiatry (Edgmont) 2009;6(5):46–51. Epub 2009/09/03. [PMC free article] [PubMed] [Google Scholar]
- 2.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Fourth. DSM-IV-TR: American Psychiatric Pub; 2000. [Google Scholar]
- 3.Hellerstein DJ, Eipper JW. Dysthymia and chronic depression. In: Mann J, Roose SP, McGrath PJ, editors. The Clinical Handbook for the Management of Mood Disorders. New York: Cambridge University Press; 2013. pp. 20–36. [Google Scholar]
- 4.Anisman H, Ravindran AV, Griffiths J, Merali Z. Endocrine and cytokine correlates of major depression and dysthymia with typical or atypical features. Mol Psychiatry. 1999;4(2):182–188. doi: 10.1038/sj.mp.4000436. Epub 1999/04/20. [DOI] [PubMed] [Google Scholar]
- 5.Schlatter J, Ortuno F, Cervera-Enguix S. Differences in interleukins' patterns between dysthymia and major depression. Eur Psychiat. 2001;16(5):317–319. doi: 10.1016/s0924-9338(01)00585-5. [DOI] [PubMed] [Google Scholar]
- 6.Griffiths J, Ravindran AV, Merali Z, Anisman H. Dysthymia: a review of pharmacological and behavioral factors. Mol Psychiatr. 2000;5(3):242–261. doi: 10.1038/sj.mp.4000697. [DOI] [PubMed] [Google Scholar]
- 7.Weissman MM, Leaf PJ, Bruce ML, Florio L. The epidemiology of dysthymia in five communities: rates, risks, comorbidity, and treatment. Am J Psychiatry. 1988;145(7):815–819. doi: 10.1176/ajp.145.7.815. Epub 1988/07/01. [DOI] [PubMed] [Google Scholar]
- 8.Keller MB, Klein DN, Hirschfeld RMA, Kocsis JH, Mccullough JP, Miller I, et al. Results of the Dsm-Iv Mood Disorders Field Trial. American Journal of Psychiatry. 1995;152(6):843–849. doi: 10.1176/ajp.152.6.843. [DOI] [PubMed] [Google Scholar]
- 9.Solomon A, Haaga DAF, Arnow BA. Is clinical depression distinct from subthreshold depressive symptoms? A review of the continuity issue in depression research. Journal of Nervous and Mental Disease. 2001;189(8):498–506. doi: 10.1097/00005053-200108000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Malykhin NV, Carter R, Hegadoren KM, Seres P, Coupland NJ. Fronto-limbic volumetric changes in major depressive disorder. J Affect Disorders. 2012;136(3):1104–1113. doi: 10.1016/j.jad.2011.10.038. [DOI] [PubMed] [Google Scholar]
- 11.McEwen BS. Mood disorders and allostatic load. Biological Psychiatry. 2003;54(3):200–207. doi: 10.1016/s0006-3223(03)00177-x. [DOI] [PubMed] [Google Scholar]
- 12.Schmaal L, Veltman DJ, van Erp TGM, Penninx BWJH, Thompson PM, Hibar DP, et al. Subcortical Brain Alterations in Major Depressive Disorder: Findings from the ENIGMA Major Depressive Disorder Working Group. Biological Psychiatry. 2015;77(9) doi: 10.1038/mp.2015.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peterson BS, Warner V, Bansal R, Zhu H, Hao X, Liu J, et al. Cortical thinning in persons at increased familial risk for major depression. Proc Natl Acad Sci U S A. 2009;106(15):6273–6278. doi: 10.1073/pnas.0805311106. Epub 2009/03/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Elst LT, Woermann FG, Lemieux L, Trimble MR. Amygdala enlargement in dysthymia - A volumetric study of patients with temporal lobe epilepsy. Biological Psychiatry. 1999;46(12):1614–1623. doi: 10.1016/s0006-3223(99)00212-7. [DOI] [PubMed] [Google Scholar]
- 15.Ravindran AV, Smith A, Cameron C, Bhatla R, Cameron I, Georgescu TM, et al. Toward a functional neuroanatomy of dysthymia: A functional magnetic resonance imaging study. J Affect Disorders. 2009;119(1-3):9–15. doi: 10.1016/j.jad.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 16.Posner J, Hellerstein DJ, Gat I, Mechling A, Klahr K, Wang ZS, et al. Antidepressants Normalize the Default Mode Network in Patients With Dysthymia. Jama Psychiat. 2013;70(4):373–382. doi: 10.1001/jamapsychiatry.2013.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horga G, Kaur T, Peterson BS. Annual research review: Current limitations and future directions in MRI studies of child- and adult-onset developmental psychopathologies. Journal of child psychology and psychiatry, and allied disciplines. 2014;55(6):659–680. doi: 10.1111/jcpp.12185. Epub 2014/01/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peterson BS. From correlations to causation: the value of preventive interventions in studying pathogenic mechanisms in childhood psychiatric disorders. Journal of child psychology and psychiatry, and allied disciplines. 2013;54(8):813–815. doi: 10.1111/jcpp.12122. Epub 2013/07/20. [DOI] [PubMed] [Google Scholar]
- 19.Hellerstein DJ, Stewart JW, McGrath PJ, Deliyannides DA, Batchelder ST, Black SR, et al. A randomized controlled trial of duloxetine versus placebo in the treatment of nonmajor chronic depression. The Journal of clinical psychiatry. 2012;73(7):984–991. doi: 10.4088/JCP.11m07230. Epub 2012/08/21. [DOI] [PubMed] [Google Scholar]
- 20.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. Epub 1960/02/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hellerstein DJ, Batchelder ST, Lee A, Borisovskaya M. Rating dysthymia: an assessment of the construct and content validity of the Cornell Dysthymia Rating Scale. J Affect Disorders. 2002;71(1-3):85–96. doi: 10.1016/s0165-0327(01)00371-8. [DOI] [PubMed] [Google Scholar]
- 22.Sled GJ, Zijdenbos AP, Evans AC. A Nonparametric Method for Automatic Correction of Intensity Nonuniformity in MRI Data. IEEE Trans of Medical Imaging. 1998;17(1):87–97. doi: 10.1109/42.668698. [DOI] [PubMed] [Google Scholar]
- 23.Shattuck DW, Leahy RM. BrainSuite: An Automated Cortical Surface Identification Tool. Medical Image Analysis. 2002;8(2):129–142. doi: 10.1016/s1361-8415(02)00054-3. [DOI] [PubMed] [Google Scholar]
- 24.Shrout PE, Fleiss JL. Intraclass correlations: uses in assessing rater reliability. Psychol Bull. 1979;86:420–428. doi: 10.1037//0033-2909.86.2.420. [DOI] [PubMed] [Google Scholar]
- 25.Haralick R, Shapiro L. Computer and Robot Vision, volume 1: Addison-Wesley Publishing Company. 1992. [Google Scholar]
- 26.Qiu L, Lui S, Kuang W, Huang X, Li J, Li J, et al. Regional increases of cortical thickness in untreated, first-episode major depressive disorder. Translational Psychiatry. 2014;4 doi: 10.1038/tp.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kempton MJ, Salvador Z, Munafo MR, Geddes JR, Simmons A, Frangou S, et al. Structural Neuroimaging Studies in Major Depressive Disorder Meta-analysis and Comparison With Bipolar Disorder. Arch Gen Psychiat. 2011;68(7):675–690. doi: 10.1001/archgenpsychiatry.2011.60. [DOI] [PubMed] [Google Scholar]
- 28.Miller L, Bansal R, Wickramaratne P, Hao X, Tenke CE, Weissman MM, et al. Neuroanatomical correlates of religiosity and spirituality: a study in adults at high and low familial risk for depression. Jama Psychiat. 2014;71(2):128–135. doi: 10.1001/jamapsychiatry.2013.3067. Epub 2013/12/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pascual-Leone A, Amedi A, Fregni F, Merabet LB. The plastic human brain cortex. Annu Rev Neurosci. 2005;28:377–401. doi: 10.1146/annurev.neuro.27.070203.144216. Epub 2005/07/19. [DOI] [PubMed] [Google Scholar]
- 30.Zatorre RJ, Fields RD, Johansen-Berg H. Plasticity in gray and white: neuroimaging changes in brain structure during learning. Nature Neuroscience. 2012;15(4):528–536. doi: 10.1038/nn.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fox KCR, Nijeboer S, Dixon ML, Floman JL, Ellamil M, Rumak SP, et al. Is meditation associated with altered brain structure? A systematic review and meta-analysis of morphometric neuroimaging in meditation practitioners. Neurosci Biobehav R. 2014;43:48–73. doi: 10.1016/j.neubiorev.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 32.Hotting K, Roder B. Beneficial effects of physical exercise on neuroplasticity and cognition. Neurosci Biobehav R. 2013;37(9):2243–2257. doi: 10.1016/j.neubiorev.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 33.Stine-Morrow EAL, Basak C. Cognitive Interventions. In: Schaie KW, Willis SL, editors. Handbook of the Psychology of Aging. Seventh. Academic Press; 2011. pp. 153–171. [Google Scholar]
- 34.Greenoug Wt, Volkmar FR, Juraska JM. Effects of Rearing Complexity on Dendritic Branching in Frontolateral and Temporal Cortex of Rat. Experimental Neurology. 1973;41(2):371–378. doi: 10.1016/0014-4886(73)90278-1. [DOI] [PubMed] [Google Scholar]
- 35.Kozorovitskiy Y, Gross CG, Kopil C, Battaglia L, McBreen M, Stranahan AM, et al. Experience induces structural and biochemical changes in the adult primate brain. P Natl Acad Sci USA. 2005;102(48):17478–17482. doi: 10.1073/pnas.0508817102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blumenfeld-Katzir T, Pasternak O, Dagan M, Assaf Y. Diffusion MRI of Structural Brain Plasticity Induced by a Learning and Memory Task. PLoS One. 2011;6(6) doi: 10.1371/journal.pone.0020678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nilsson M, Perfilieva E, Johansson U, Orwar O, Eriksson PS. Enriched environment increases neurogenesis in the adult rat dentate gyrus and improves spatial memory. J Neurobiol. 1999;39(4):569–578. doi: 10.1002/(sici)1097-4695(19990615)39:4<569::aid-neu10>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 38.Ramachandran VS, Hirstein W. The perception of phantom limbs - The D.O. Hebb lecture. Brain. 1998;121:1603–1630. doi: 10.1093/brain/121.9.1603. [DOI] [PubMed] [Google Scholar]
- 39.Taub E, Uswatte G, Elbert T. New treatments in neurorehabilitation founded on basic research. Nature Reviews Neuroscience. 2002;3(3):228–236. doi: 10.1038/nrn754. [DOI] [PubMed] [Google Scholar]
- 40.Kays JL, Hurley RA, Taber KH. The dynamic brain: neuroplasticity and mental health. J Neuropsychiatry Clin Neurosci. 2012;24(2):118–124. doi: 10.1176/appi.neuropsych.24.1.118. Epub 2012/07/10. [DOI] [PubMed] [Google Scholar]
- 41.Plessen KJ, Royal JM, Peterson BS. Neuroimaging of tic disorders with co-existing attention-deficit/hyperactivity disorder. Eur Child Adolesc Psychiatry. 2007;16(Suppl 1):60–70. doi: 10.1007/s00787-007-1008-2. Epub 2007/09/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peterson BS, Choi HA, Hao XJ, Amat JA, Zhu H, Whiteman R, et al. Morphologic features of the amygdala and hippocampus in children and adults with Tourette syndrome. Arch Gen Psychiat. 2007;64(11):1281–1291. doi: 10.1001/archpsyc.64.11.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plessen KJ, Bansal R, Zhu HT, Whiteman R, Amat J, Quackenbush GA, et al. Hippocampus and amygdala morphology in attention-deficit/hyperactivity disorder. Arch Gen Psychiat. 2006;63(7):795–807. doi: 10.1001/archpsyc.63.7.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sobel LJ, Bansal R, Maia TV, Sanchez J, Mazzone L, Durkin K, et al. Basal ganglia surface morphology and the effects of stimulant medications in youth with attention deficit hyperactivity disorder. Am J Psychiatry. 2010;167(8):977–986. doi: 10.1176/appi.ajp.2010.09091259. Epub 2010/07/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller AM, Bansal R, Hao XJ, Sanchez-Pena JP, Sobel LJ, Liu J, et al. Enlargement of Thalamic Nuclei in Tourette Syndrome. Arch Gen Psychiat. 2010;67(9):955–964. doi: 10.1001/archgenpsychiatry.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Apfel BA, Ross J, Hlavin J, Meyerhoff DJ, Metzler TJ, Marmar CR, et al. Hippocampal volume differences in Gulf War veterans with current versus lifetime posttraumatic stress disorder symptoms. Biol Psychiatry. 2011;69(6):541–548. doi: 10.1016/j.biopsych.2010.09.044. Epub 2010/11/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vita A, De Peri L, Deste G, Barlati S, Sacchetti E. The Effect of Antipsychotic Treatment on Cortical Gray Matter Changes in Schizophrenia: Does the Class Matter? A Meta-analysis and Meta-regression of Longitudinal Magnetic Resonance Imaging Studies. Biological Psychiatry. 2015;78(6):403–412. doi: 10.1016/j.biopsych.2015.02.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.