

Abstract
BRD4 has emerged as an important factor in tumorigenesis by promoting the transcription of genes involved in cancer development. However, how BRD4 is regulated in cancer cells remains largely unknown. Here, we report that the stability and functions of BRD4 are positively regulated by prolyl-isomerase PIN1 in gastric cancer cells. PIN1 directly binds to phosphorylated threonine (T) 204 of BRD4 as revealed by peptide binding and crystallographic studies and enhances BRD4’s stability by inhibiting its ubiquitination. PIN1 also catalyses the isomerization of proline 205 of BRD4 and induces its conformational change, which promotes its interaction with CDK9 and increases BRD4’s transcriptional activity. Substitution of BRD4 with PIN1 binding-defective BRD4-T204A mutant in gastric cancer cells reduces BRD4’s stability, attenuates BRD4-mediated gene expression by impairing its interaction with CDK9, and suppresses gastric cancer cell proliferation, migration and invasion, and tumor formation. Our results identify BRD4 as a new target of PIN1 and suggest that interfering with their interaction could be a potential therapeutic approach for cancer treatment.
Keywords: BRD4, PIN1, isomerization, transcription, tumorigenesis
Introduction
The multi-functional BRD4 belongs to the two tandem bromodomains and an extra terminal (ET) domain-containing family proteins (BETs), which have emerged as important epigenetic modulators for gene transcription and cancer development11, 23, 45. BRD4 binds to acetylated histone or non-histone proteins via its two bromodomains and recruits different transcription components, such as Mediator and P-TEFb (positive transcriptional elongation factor b), to selective target gene promoters, leading to the phosphorylation of RNA polymerase II (RNAPII) and the activation of gene expression11, 45. BRD4 has been shown to modulate the expression and activity of cancer promoting factors, such as c-Myc, ERα and NF-κB, and regulates a variety of properties of cancer cells, including cell proliferation, transformation, invasion, and drug-resistance11, 14, 21, 26, 33.
BRD4 has been implicated in the development of hematological malignancies, including mixed-lineage leukemia (MLL), multiple myeloma, and adult T cell leukemia8, 25, 33, 48. For example, BRD4 was identified as an essential protein for the development and maintenance of acute myeloid leukemia57. BRD4 is also implicated in a number of solid tumors, including lung, breast, prostate cancer and NUT midline carcinoma (NMC), and BRD4 is critical for the proliferation and survival of these solid tumor cells4, 14, 17, 28, 56. Furthermore, BRD4 is overexpressed in melanoma, glioblastoma and colorectal cancer and inhibition of BRD4 suppresses cell proliferation and invasion in these cancers18, 20, 39,36. Due to its essential role in cancer, BRD4 has become a promising therapeutic target in cancer treatment9, 25, 33. Small molecules targeting bromodomains of BRD4 exert strong anti-tumor activities, suppressing the proliferative and transformation potential of cancer cells13, 15, 33, 57. These BET inhibitors (BETi) bind to the acetylated lysine recognition pocket and competitively block BET bromodomains from binding to histones or non-histone proteins12, 15, 34, 56. While accumulating evidence indicates that BRD4 has a pivotal role in the tumorigenesis by activating the genes responsible for cell proliferation and survival, how BRD4 itself is regulated in cancer remains elusive.
PIN1 is a peptidyl-prolyl cis-trans isomerase (PPIase) and specifically recognizes phosphorylated Ser/Thr-Pro motif and induces protein conformational changes by isomerization27, 30, 31. PIN1 contains an N-terminal WW domain for protein interaction, and a catalytic C-terminal PPIase domain for isomerization30. PIN1 has been shown to be an important signaling molecule in cancer. It regulates many cancer related proteins via isomerization-mediated conformational change, leading to altered protein complex formation or ubiquitin-mediated proteasomal degradation30, 31. The importance of PIN1 in cancer development is supported by its overexpression in many human cancers, including gastric, prostate and breast cancer6, 7, 40, 50. Overexpression of PIN1 correlates with poor prognosis in many types of cancer31, 40. Overexpression of Pin1 in mouse mammary glands results in mammary hyperplasia and malignant mammary tumors42. Conversely, ablation of Pin1 in Her2 or Has-Ras transgenic mice or p53-knockout mice suppresses tumorigenesis and prevents cancer development38, 43, 49. The oncogenic activity of PIN1 is largely attributed to its ability to stabilize or activate oncoproteins and to destabilize or inactivate tumor suppressors27, 31, 35.
Many transcription factors or transcription regulators important for tumor development are regulated by PIN130, 31 and BRD4 has recently emerged as a key epigenetic regulator in cancer development10, 33. In addition, BRD4 is a phosphorylated protein with multiple Ser/Thr-Pro motifs, raising an intriguing question whether BRD4 might be a target of PIN1. Therefore, we explored the possibility that BRD4 might be regulated by PIN1 in cancer cells. Our studies reveal that phosphorylated BRD4 at threonine (T) 204 is specifically recognized by the WW domain of PIN1. Binding of PIN1 to phosphorylated T204 prevents the ubiquitination and degradation of BRD4 and facilitates its interaction with CDK9 for the transcription of genes involved in cancer development. Our results uncover a mechanism by which BRD4 is regulated by PIN1 in cancer cells and suggest that targeting the interaction between PIN1 and BRD4 could have therapeutic potential.
Results
BRD4 abundance is positively correlated with PIN1 expression in human gastric cancer tissues and cells
To investigate the possibility that PIN1 might regulate BRD4 in cancer cells, we first employed immunohistochemistry to examine the possible pathological correlation of the expression of PIN1 and BRD4 in human gastric cancer since PIN1 is overexpressed and correlates with poor prognosis in gastric cancer40. While BRD4 was predominantly in the nucleus, PIN1 could be found in both the nucleus and the cytoplasm (Fig. 1A). In a tissue array with a cohort of 58 human gastric cancer samples, more than half of the samples showed high expression levels of BRD4 (30 out of 58) (Figs. 1A&1B) and about 90% of samples (27 out of 30) with high levels of BRD4 displayed high levels of PIN1 (Fig. 1B). Approximately 82% of samples (23 out of 28) with low levels of BRD4 had lower expression levels of PIN1. Statistic analysis reveals a positive correlation between the expression of PIN1 and BRD4 in these cancer samples with a Spearman coefficient for correlation (PIN1 and BRD4) of 0.90 (p<0.01) (Fig. 1B). Furthermore, in a panel of gastric epithelial cells, the overall protein levels of PIN1 seemed to positively correlate with the levels of BRD4 in most gastric cancer cell lines (Fig. 1C), although the mRNA levels of PIN1 and BRD4 in these cells varied and did not precisely correspond to their protein levels (Fig. S1).
Fig. 1. BRD4 abundance is positively correlated with PIN1 expression in human gastric cancer tissues.
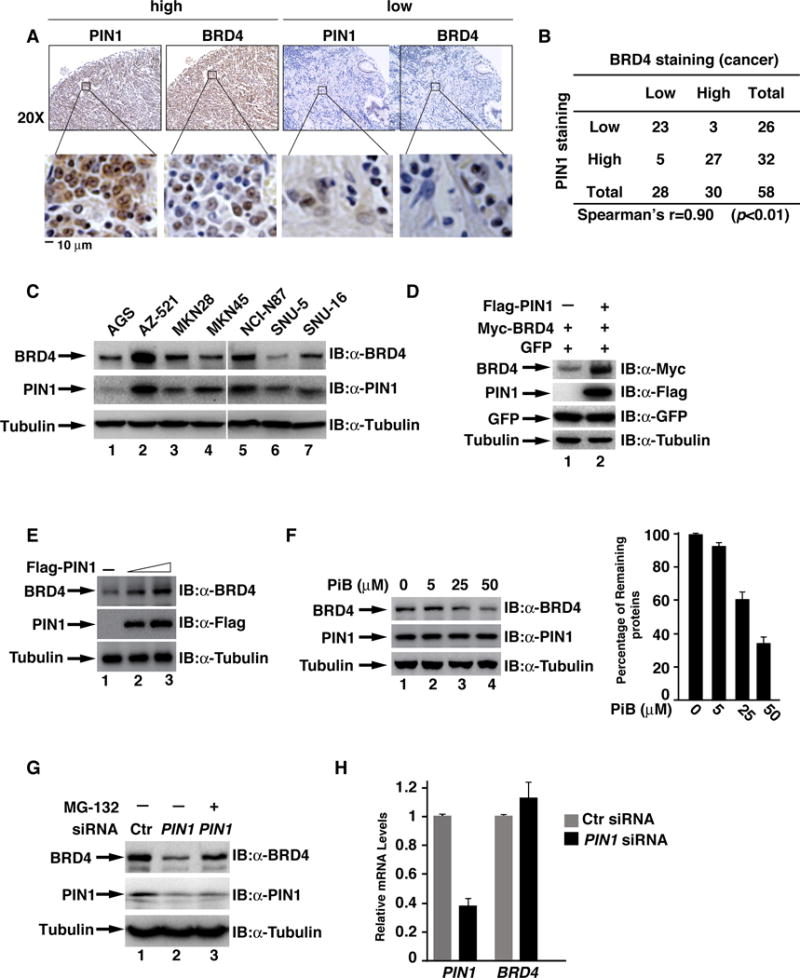
(A) Representative of immunohistochemical staining of PIN1 and BRD4 in human gastric cancer tissues. Boxed regions are enlarged to the bottom of each image. (B) Tissue sections of 58 gastric cancer samples were immunostained with anti-PIN1 or anti-BRD4 antibodies and their correlation was analyzed by Spearman rank correlation test (P<0.01). (C) Protein expression levels of BRD4 and PIN1 in gastric cancer cells. (D) Expression levels of BRD4, PIN1, GFP or Tubulin in HEK293T cells transfected with indicated expression plasmids (plasmid ratio of BRD4 vs PIN1: 1:3). (E) Expression levels of PIN1, endogenous BRD4 or Tubulin in MKN28 cells transfected with increasing amount of PIN1 plasmids. (F) Expression levels of BRD4 and Tubulin in MKN28 cells treated with PiB with indicated concentration for 24 hr. Quantification of the relative BRD4 levels normalized to Tubulin is shown in the right panel. (G) Expression levels of BRD4, PIN1 and Tubulin in MKN28 cells transfected with control or PIN1 siRNA for 48 hr, followed by the treatment with MG-132 for 8 hr. (H) PIN1 or BRD4 mRNA levels in MKN28 cells transfected with control or PIN1 siRNA.
Co-expression of PIN1 enhanced the cellular levels of exogenous BRD4 in HEK293 cells and endogenous BRD4 in MKN28 cells (Figs. 1D&1E). In contrast, PiB, a PIN1 inhibitor with the least non-specific toxicity44, reduced the cellular levels of endogenous BRD4 (Fig. 1F). Depletion of PIN1 in MKN28 cells also reduced the levels of endogenous BRD4 (Fig. 1G), but not the mRNA of BRD4 (Fig. 1H). The reduced levels of BRD4 were reversed by proteasome inhibitor MG-132 (Fig. 1G), suggesting that PIN1 might regulate the stability of BRD4.
PIN1 interacts with BRD4 via its WW domain in a phosphorylation-dependent manner
Since PIN1 regulates protein functions by selectively binding to its substrates that contain a pSer/Thr-Pro motif30, 31, we next determined the interaction between PIN1 and BRD4. Endogenous PIN1 co-immunoprecipitated with endogenous BRD4 in MKN28 cells (Figs. 2A&2B). Endogenous BRD4 also colocalized with endogenous PIN1 in the nucleus of MKN28 as revealed by the indirect immunofluorescence staining (Fig. S2). In an in vitro binding assay, GST-PIN1 but not GST pulled down BRD4 from cell lysates of HEK293T transfected with Myc-BRD4 (Fig. 2C). Treatment of cell lysates with the calf intestinal alkaline phosphatase (CIP) greatly reduced GST-PIN1-associated BRD4 (Fig. 2D), indicating that PIN1 binds to phosphorylated BRD4. The phosphorylation of BRD4 appears to occur on the Ser/Thr-Pro motif since PIN1-associated phosphorylated BRD4, recognized by the pSer/Thr-Pro (MPM-2) antibodies, was significantly decreased with the CIP treatment (Fig. 2D).
Fig. 2. PIN1 interacts with BRD4 in a phosphorylation-dependent manner in vitro and in vivo.
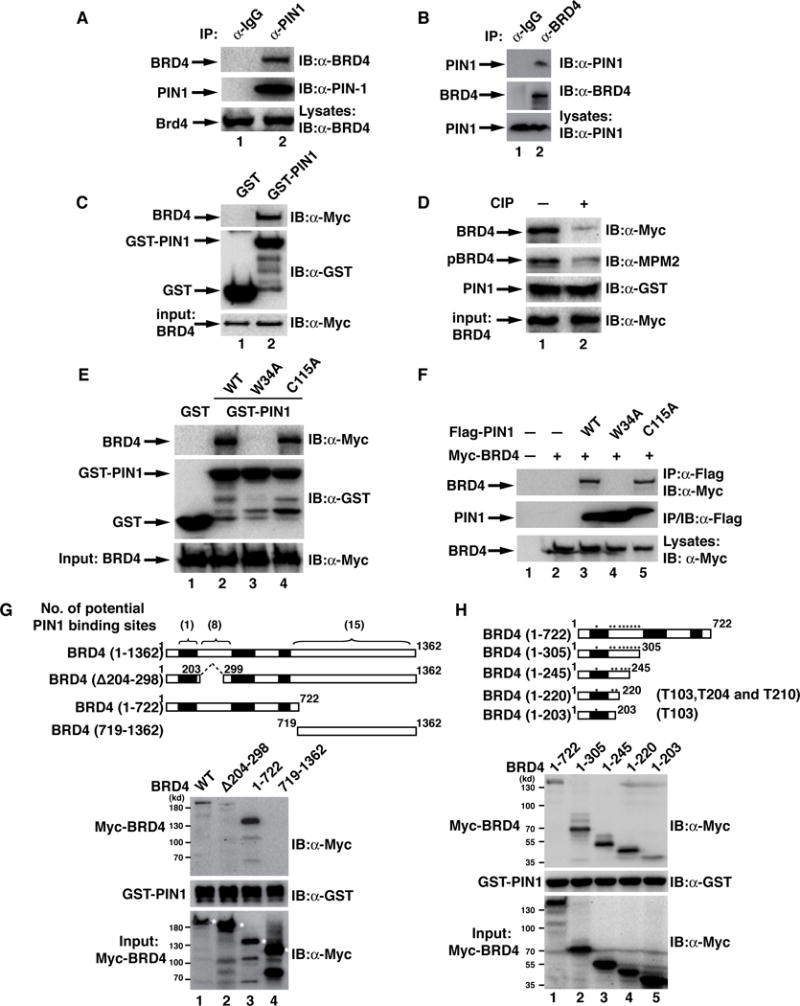
(A&B) Endogenous PIN1 (A) or BRD4 (B) was immunoprecipitated from MKN28 cells and immunoblotted for associated BRD4 (A) or PIN1 (B), respectively. IgG was used as a control. (C) GST or GST-PIN1 conjugated agarose beads were incubated with HEK293T cell lysates transfected with Myc-BRD4. GST-PIN1 associated BRD4 was detected with the indicated antibodies. (D) HEK293T cell lysates containing Myc-BRD4 were pretreated with CIP for 1 hr, followed by the incubation with GST-PIN1 conjugated agarose. GST-PIN1-associated BRD4 was detected as in (A). (E) HEK293T cell lysates containing transfected Myc-BRD4 were subjected to GST pull-down assay with GST-PIN1, GST-PIN1-W34A or GST-PIN1-C115A. (F) HEK293T cells were co-transfected with Myc-BRD4 and Flag-PIN1, or various PIN1 mutants as indicated (plasmid ratio of PIN1 vs BRD4: 1:2). Flag-PIN1 immunoprecipitates were immunoblotted for the associated Myc-BRD4. (G) Upper: Schematic diagram showing the domain structure of BRD4 and various deletion mutants of BRD4. Below: HEK293T cell lysates containing transfected Myc-BRD4 or its deletion mutants were subjected to GST pull-down assay with GST-PIN1. Positions of various deletion mutants are marked with asterisks. (H) Upper: Schematic diagram showing BRD4 (1–722) and its various deletion mutants. Positions of potential PIN1-binding motifs are marked with asterisks. Below: HEK293T cell lysates containing transfected Myc-BRD4 (1–722) or its deletion mutants were subjected to GST pull-down assay with GST-PIN1.
PIN1 contains an N-terminal WW domain which is responsible for substrate recognition and a C-terminal catalytic PPIase domain which is responsible for isomerization30. The substrate binding-deficient mutant of PIN1, PIN1-W34A, failed to pull-down BRD4 or co-immunoprecipitate BRD4 (Figs. 2E&2F). In contrast, the catalytically inactive mutant, PIN1-C115A, had no effect on PIN1’s binding to BRD4 (Figs. 2E&2F), indicating that WW domain but not the isomerization domain of PIN1 is essential for the interaction with BRD4.
To identify the potential pSer/Thr-Pro motif in BRD4, we generated three deletion mutants of BRD4 (Fig. 2G) and determined which region of BRD4 was involved in its association with PIN1. In GST-PIN1 pull-down assay, full-length BRD4 as well as its C-terminal deletion mutant (deleted up to amino acid 722) retained the ability to associate with PIN1 (Fig. 2G). However, N-terminal deletion mutant of BRD4 (deleted up to amino acid 718) completely lost its interaction with PIN1 and deletion of amino acids 204 to 298 dramatically reduced the interaction with PIN1 (Fig. 2G). These data suggest that the pSer/Thr-Pro motif resides in the N-terminal region of BRD4 and most likely from amino acids 204 to 298. Further deletion of the C-terminal regional of BRD4 revealed that a major PIN1 binding site might reside from amino acids 203 to 220 and an additional PIN1 binding site might exist from amino acids 1 to 203 since deletion up to 203 significantly reduced the interaction with PIN1 (Fig. 2H).
PIN1 binds to phosphorylated threonine 204 of BRD4
There are three potential PIN1 binding motifs from amino acids 1 to 220, including motifs containing threonines (T) 103, 204, and 210 (Fig. 3A). We mutated each threonine to alanine alone or in combination and examined their abilities to bind to PIN1. Mutation of T103 or T210 alone did not affect BRD4’s binding to GST-PIN1 while mutation of T204 alone significantly reduced BRD4’s binding to GST-PIN1 (Fig. 3A). Mutation of T204 together with T210 or mutation of three threonines completely destroyed the interaction (Fig. 3A). In addition, BRD4-T103A or BRD4-T210A but not BRD4-T204A co-immunoprecipitated PIN1 at the same levels as WT BRD4 (Figs. 3B&S3). Mutation of T204 in combination with T210 or T103 also disrupted BRD4’s interaction with PIN1 (Fig. 3B). These data suggest that T204 of BRD4 is the major PIN1 binding site and T204 is likely phosphorylated since PIN1 only binds to phosphorylated serine or threonine30, 31. Supporting this notion, we found that a site-specific polyclonal antibody detected the phosphorylated T204 of the endogenous BRD4 in MKN28 cells and exogenous WT BRD4 but not the BRD4-T204A (Figs. 3C&3D). Only the phosphorylated T204 was found to bind to PIN1 when peptides containing the nonphosphorylated or phosphorylated T204 were overlaid with recombinant PIN1 (Fig. 3E).
Fig. 3. PIN1 interacts with phosphorylated T204 of BRD4.
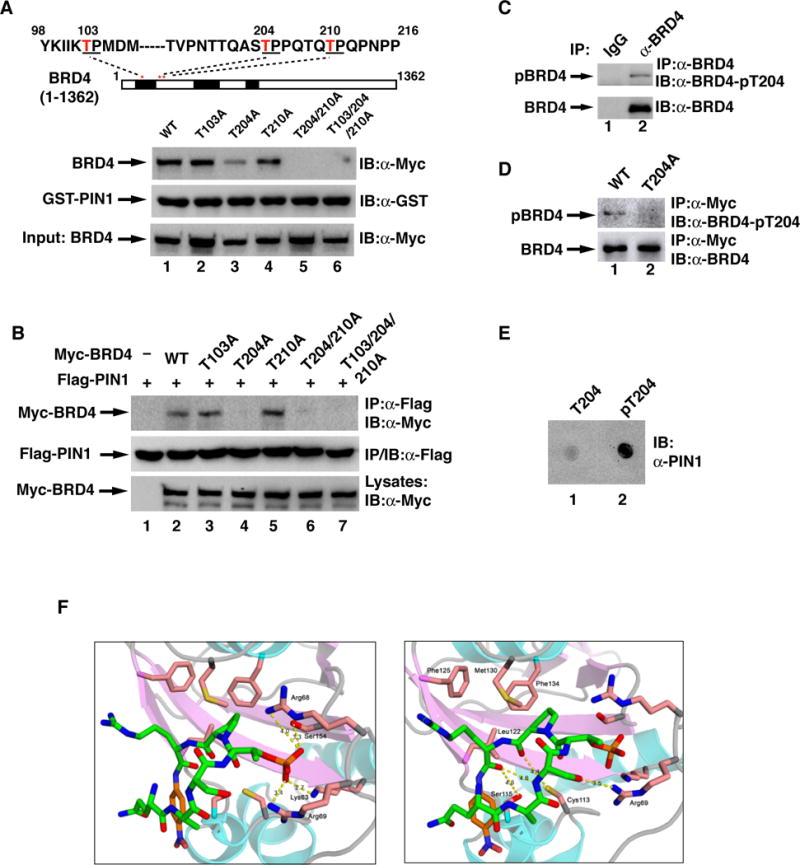
(A) HEK293T cell lysates containing transfected Myc-BRD4 or its various mutants were subjected to GST pull-down assay with GST-PIN1. Sequence from amino acids 98 to 216 containing three potential PIN1-binding motifs is shown above. (B) HEK293T cells were co-transfected with Flag-PIN1 and Myc-BRD4 or its mutants as indicated (plasmid ratio of PIN1 vs BRD4: 1:2). Flag-PIN1 immunoprecipitates were immunoblotted for the associated BRD4. (C) MKN28 cell lysates were immunoprecipitated with IgG or anti-BRD4 antibodies followed by immunoblotting with anti-pT204 BRD4 antibodies. (D) HEK293T cells transfected with WT or T204A of Myc-BRD4 were immunoprecipitated with anti-Myc antibodies, followed by immunoblotting with anti-pT204 BRD4 antibodies. (E) BRD4 peptides containing nonphosphorylated or phosphorylated T204 (25 μM) spotted on nitrocellulose membranes were incubated with recombinant GST-PIN1, followed by immunoblotted with anti-PIN1 antibodies. (F) Structures of PIN1 in complex with the pT204-BRD4 peptide at 1.85 Å resolution, showing the substrate-binding pocket of PIN1, with interactions of the pT204 (left panel) and with other peptidic residues (right panel).
To further characterize the interaction between PIN1 and BRD4, we determined the 1.85 Å resolution structure of PIN1 in complex with a pT204-peptide (6 amino acids) (Fig. 3F). As expected, the pT204-peptide was bound to PIN1 in a same pocket with previously reported PIN1 structures55. The phosphate group of the pT204-peptide pointed into a positively charged triad that consists of 3 basic residues, Lys63, Arg68, and Arg69 (Fig. 3F, left panel). However, the interaction distance with Arg68 is much shorter than in other structures (4 Å) and additional interactions are also observed with Ser154 (3.3 Å) (Fig. 3F, left panel). Potential hydrogen bond interactions were also found between PIN1 and pT204-peptide, which includes the interactions between the side chains of Arg69 and Ser203 of BRD4, the side chain of Cys113 and the backbone of Pro205 of BRD4 (Fig. 3F, right panel). Collectively, these data demonstrate that phosphorylated T204 directly interacts with PIN1.
PIN1 regulates the stability of BRD4
One of the major functions of PIN1 is to regulate the stability of its substrates27. We next investigated the possibility that PIN1 might regulate the stability of BRD4. While co-expression of WT PIN1 increased the levels of BRD4 in a dose-dependent manner, PIN1-W34A or PIN1-C115A failed to increase the levels of BRD4 (Fig. 4A), suggesting that binding of PIN1 to BRD4 and the isomerase activity of PIN1 are both essential for the enhanced cellular levels of BRD4. Furthermore, the cellular levels of BRD4-T204A remained unchanged even with the co-expression of PIN1 (Fig. 4B).
Fig. 4. PIN1 increases the stability of BRD4 by inhibiting its ubiquitination.
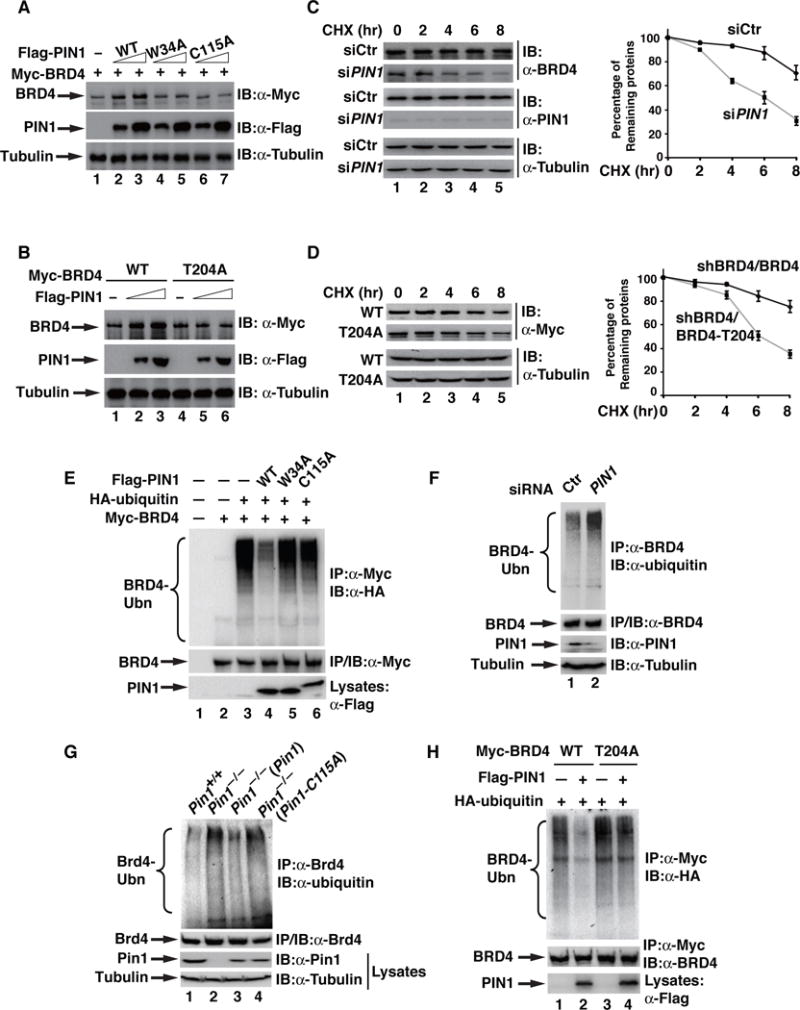
(A) Immunoblotting of BRD4 and PIN1 in HEK293T cells transfected with indicated plasmids. Transfected plasmid ratio (PIN1 vs BRD4): 1:3 (lanes: 2, 4&6); 1:5 (lanes: 3, 5&7). (B) Immunoblotting of BRD4 and PIN1 in HEK293T cells transfected with indicated plasmids. Transfected plasmid ratio (PIN1 vs BRD4): 1:3 (lanes: 2 &5); 1:5 (lanes: 3&6). (C) MKN28 cells transfected with control or PIN1 siRNAs were treated with cycloheximide for the indicated time points and immunoblotted for the expression of BRD4, PIN1, and Tubulin. A representative result from three independent experiments is shown in the left panels. Quantification of the results is shown in the right panels. Data represent the average of three independent experiments ± SD. (D) shBRD4/BRD4 or shBRD4/BRD4-T204A MKN28 cells were treated with CHX for indicated time points and immunoblotted for the expression of BRD4 and Tubulin, and levels were quantified as described in (C). (E) HEK293T cells were transfected with indicated plasmids expressing Myc-BRD4, HA-ubiquitin, Flag-PIN1, Flag-PIN1-W34A or Flag-PIN1-C115A. BRD4 immunoprecipitates were immunoblotted for ubiquitination with anti-HA antibody. (F) Endogenous BRD4 immunoprecipitates from MKN28 cells transfected with control or PIN1 siRNA were immunoblotted for ubiquitination with anti-ubiquitin antibody. (G) Endogenous BRD4 immunoprecipitates from MEFs of Pin1+/+, Pin1−/−, Pin1−/− (Pin1) or Pin1−/− (Pin1-C115A) were immunoblotted for ubiquitination with anti-ubiquitin antibody. (H) BRD4 immunoprecipitates from HEK293T cells transfected with indicated plasmids were immunoblotted for ubiquitination with anti-HA antibody.
We next determined the effect of PIN1 knockdown on the half-life of BRD4. BRD4 was relatively stable with a half-life more than 8 h in MKN28 cells transfected with control siRNA (Fig. 4C). Depletion of PIN1 shortened the half-life of BRD4 to less than 6 h (Fig. 4C). Consistently, Brd4 was relatively stable with a longer half-life in WT mouse embryonic fibroblasts (MEFs) compared to Pin1-deficient MEFs (Fig. S4). Importantly, reconstitution of Pin1-deficient MEFs with WT Pin1 but not Pin1-C115A extended the half-life of Brd4 (Fig. S4).
We then compared the half-life of WT BRD4 and BRD4-T204A mutant in BRD4 knockdown MKN28 cells stably expressing shBRD4-resistant WT BRD4 or BRD4-T204A (designated as shBRD4/BRD4 and shBRD4/BRD4-T204A MKN28 cells, respectively). WT BRD4 was relatively stable compared to BRD4-T204A in MKN28 cells and in HEK293T cells (Figs. 4D&S5). Together, these data suggest that binding of PIN1 to BRD4 plays an essential role in regulating the stability of BRD4.
PIN1 inhibits the ubiquitination of BRD4
Given that PIN1 prevents the degradation of BRD4, we sought to determine whether PIN1 inhibits BRD4 ubiquitination. Co-expression of PIN1 but not PIN1-C115A or PIN1-W34A significantly reduced the ubiquitination of BRD4 in HEK293T cells (Fig. 4E). Consistently, enhanced ubiquitination of endogenous BRD4 was found in PIN1 knockdown MKN28 cells or Pin1-deficient MEFs (Figs. 4F&4G). Reconstitution of Pin1-deficient MEFs with WT Pin1 but not Pin1-C115A reduced the ubiquitination of Brd4 (Fig. 4G). When we compared the ubiquitination of BRD4 and BRD4-T204A, we found that, the ubiquitination of BRD4-T204A was slightly stronger than WT BRD4 in the absence of co-transfected PIN1 (Fig. 4H, lanes 1&3). However, the ubiquitination of WT BRD4 but not BRD4-T204A was significantly reduced by PIN1 (Fig. 4H, lanes 2&4). All together, these data demonstrate that binding of PIN1 to pT204 of BRD4 stabilizes BRD4 by reducing its ubiquitination in an isomerase activity-dependent manner.
PIN1 mediates the conformational change of BRD4
Binding of PIN1 to its substrates often results in the cis-trans proline conformational change of the substrates and the associated functional changes27, 30. To determine whether PIN1 mediated the cis-trans conformational change in the identified PIN1 binding motif (Fig. 4), we designed BRD4 peptides containing the phosphorylated T204 and proline 205 followed by a phenyalanine (F) and paranitroanaline (pNA) group and measured the ability of recombinant PIN1 to catalyze the cis-trans isomerization of the synthesized peptides (Fig. 5A). Chymotrypsin exclusively cleaves the peptides in the trans-proline conformation and the cis-trans conversion rate can be measured as chymotrypsin-dependent release of pNA16. Incubation of the peptides with GST-PIN1 but not with GST-PIN1-C115A or GST was associated with an increased release of pNA (Fig. 5A), suggesting that PIN1 stimulates the cis-trans isomerization of BRD4 at proline 205. Further supporting this, we found that trypsin diggestion of BRD4 isolated from PIN1 knockdown MKN28 cells pre-incubated with WT PIN1 but not with PIN1-C115A or GST yielded an additional band (Fig. 5B, lane 4), suggesting that PIN1 binding changes the conformation of BRD4 via isomerization, likely result from cis to trans of proline of 205 of BRD4 (Fig. 5A).
Fig. 5. PIN1 changes the conformation of BRD4 and regulates BRD4’s interaction with CDK9.
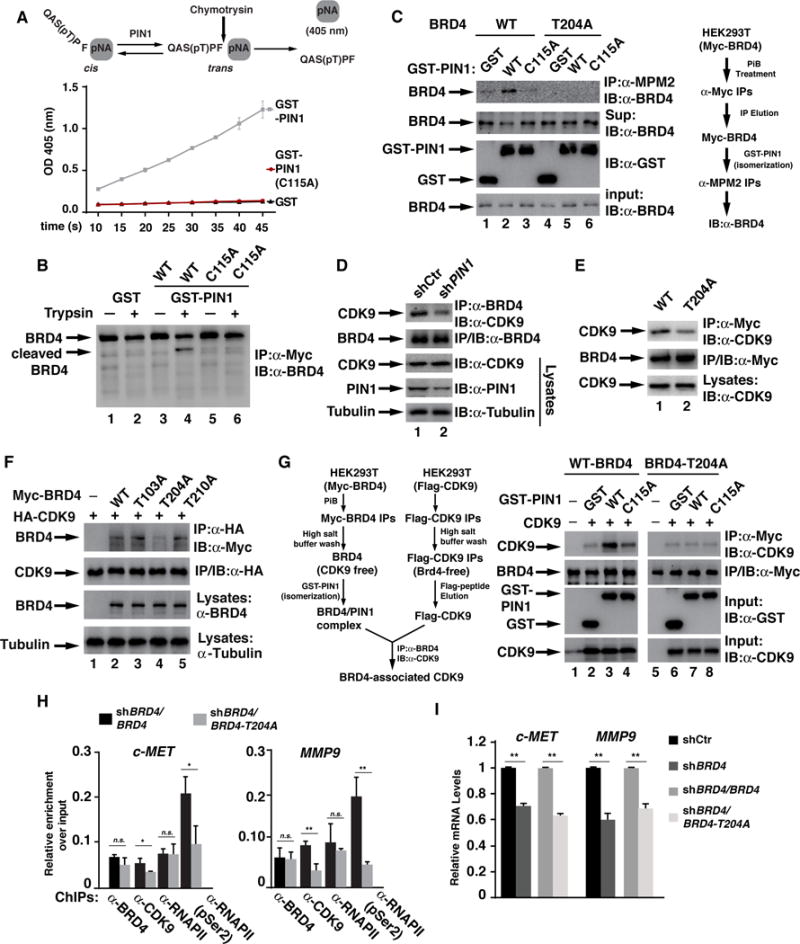
(A) Synthetic BRD4-pNA peptides were incubated with GST, GST-PIN1-WT or catalytically inactive GST-PIN1-C115A and isomerase activity was measured as absorbance of free pNA at 405 nm. (B) Myc-BRD4 immunoprecipitates from PIN1 knockdown shBRD4/BRD4 MKN28 cells were incubated with 0.5 μg/mL of recombinant GST, GST-PIN1, or GST-PIN1-C115A, followed by digestion with trypsin (10 ng/ml). Myc-BRD4 immunoprecipitates were analyzed by immunoblotting. (C) BRD4 was immunoprecipitated from PiB treated HEK293T cells expressing Myc-BRD4 or Myc-BRD4-T204A, and washed with high salt buffer, eluted with Myc-peptides, then incubated with GST, GST-PIN1 or GST-PIN1-C115A, followed by immunoprecipitation with anti-MPM-2 antibody. BRD4 was detected with anti-Brd4 antibodies. A diagram for the experiment procedure is shown on the right. (D) BRD4 immunoprecipitates from PIN1 knockdown MKN28 cells were immunoblotted with co-purified CDK9. (E) BRD4 immunoprecipitates from shBRD4/BRD4 or shBRD4/BRD4-T204A MKN28 cells were immunoblotted with co-purified CDK9. (F) HEK293T cells transfected with indicated plasmids were immunoprecipitated with anti-HA antibodies and immunoblotted with anti-Myc antibody for co-purified BRD4 or its various mutants. (G) Myc-BRD4 immunoprecipitates from PiB-treated HEK293T cells washed with high salt buffer were incubated with 0.5 μg/mL GST, GST-PIN1 or GST-PIN1-C115A. After wash, BRD4 immunoprecipitates were used to pull-down BRD4-free Flag-CDK9 from transfected HEK293T cells and the associated CDK9 was detected with anti-CDK9 antibodies. A diagram for the experiment is shown on the left. (H) ChIPs for the recruitment of BRD4, CDK9, RNAPII and pRNAPII on the promoter of c-MET or MMP9 in indicated cells. (I) mRNA levels of c-MET or MMP9 from indicated MKN28 cells were analyzed by RT-PCR.
We next assessed whether binding of PIN1 to pT204 is essential for PIN1-mediated conformational change of BRD4. We measured the reactivity of BRD4 and BRD4-T204A to the MPM-2 antibody with or without PIN1 since differential reactivity to MPM-2 antibody, which preferentially recognizes trans form of phosphorylated substrates, has been shown to be an effective index for PIN1-mediated protein conformational change41. The eluted BRD4 immunoprecipitates from PiB-treated HEK293T cells were incubated with or without recombinant PIN1 or PIN1-C115A before subject to MPM-2 immunoprecipitation. BRD4 was barely immunoprecipitated by MPM-2 antibody in the absence of PIN1 (Fig. 5C). Incubation of BRD4 with WT PIN1 but not PIN1-C115A significantly increased the amount of BRD4 immunoprecipitated by MPM-2 antibody (Fig. 5C), confirming that PIN1 facilitates the cis-trans conformational change of BRD4. In contrast, MPM-2 did not immunoprecipitate BRD4-T204A even in the presence of PIN1 (Fig. 5C). Collectively, these data suggest that binding of PIN1 to pT204 of BRD4 changes its conformation.
PIN1 facilitates BRD4’s interaction with CDK9
PIN1-mediated conformational change of its substrates plays a key role in regulating protein functions via protein-protein interaction27, 30. Therefore, we examined the effect of PIN1 on the interaction of BRD4 with CDK9, one of the major BRD4-interacting proteins45. A significantly reduced interaction of BRD4 and CDK9 was found in PIN1 knockdown MKN28, AGS or Pin1-deficient MEFs (Figs. 5D, S8&S9). Furthermore, BRD4-T204A co-immunoprecipitated less CDK9 than WT BRD4 (Fig. 5E). The impaired interaction between BRD4 and CDK9 was also found with exogenously expressed BRD4-204A but not BRD4-T103A or BRD4-T210A (Fig. 5F). These data suggest that PIN1 regulates BRD4’s interaction with CDK9.
We then employed an in vitro pull-down assay in combination with the isomerization assay to determine whether PIN1-mediated isomerization was involved in the altered interaction between BRD4 and CDK9. Purified CDK9-free BRD4 from PiB-treated cells was pre-incubated with or without GST-PIN1 and then used to pull-down BRD4-free CDK9 purified from transfected HEK293T cells (Fig. 5G). The interaction between BRD4 and CDK9 was dramatically enhanced when BRD4 was pre-incubated with PIN1 but not with PIN1-C115A or PIN1-W34A (Figs. 5G&S10). These data suggest that PIN1-mediated conformational change of BRD4, likely through isomerization, facilitates BRD4’s interaction with CDK9.
The expression of c-MET and MMP9 has been shown to be regulated by BRD419, 37 and c-MET and MMP9 are overexpressed in gastric cancer and play important roles in the development of gastric cancer32, 52. When we examined the recruitment of BRD4, CDK9 and RNAPII on the promoters of these two genes by chromatin immunoprecipitation in shBRD4/BRD4 or shBRD4/BRD4-T204A MKN28 cells, we observed a reduced binding of CDK9 and serine-2 phosphorylated RNAPII to the promoters of c-MET and MMP9 in shBRD4/BRD4-T204A cells (Fig. 5H). No significant change of recruitment of BRD4 and RNAPII was observed on these promoters (Fig. 5H). Consistent with the role of CDK9-mediated serine-2 phosphorylation of RNAPII in transcription elongation, the transcription of c-MET and MMP9 was down-regulated in shBRD4/BRD4-T204A cells, to a degree same as the BRD4 knockdown MKN28 cells (Fig. 5I). Collectively, these data suggest that PIN1-mediated conformational change of BRD4 enhances BRD4’s interaction with CDK9 and the subsequent activation of RNAPII for the transcription of tumor-promoting genes.
Binding of PIN1 to BRD4 regulates the tumor-promoting activity of BRD4
We next determined whether PIN1 played a regulatory role in BRD4-dependent cancer cell proliferation and tumorigenicity. Depletion of BRD4 by shRNA reduced the growth of MKN28 cells compared to MKN28 transfected with control shRNA (Fig. 6A). We then rescued the BRD4 knockdown cells with WT BRD4 or BRD4-204A. Although the levels of BRD4 in rescued cells were higher than the endogenous BRD4 (Fig. 6A), the similar proliferation patterns of the parental cells and the reconstituted shBRD4/BRD4 MKN28 cells indicate that the reconstituted BRD4 recapitulates the activity of endogenous BRD4. In contrast, shBRD4/BRD4-T204A cells displayed reduced cell proliferation, similar to that of the BRD4 knockdown MKN28 cells (Fig. 6A). Depletion of BRD4 significantly reduced the anchorage-independent growth of MKN28 cells (Figs. 6B&6C), indicating that BRD4 is important for the transformation potential of MKN28 cells. Notably, shBRD4/BRD4 but not the shBRD4/BRD4-T204A MKN28 cells restored the transformation potential of MKN28 cells (Figs. 6B&6C). These data indicate that binding of PIN1 to BRD4 regulates the growth and colony formation potential of MKN28 cells.
Fig. 6. The T204A mutant of BRD4 suppresses the tumorigenicity of gastric cancer cells.
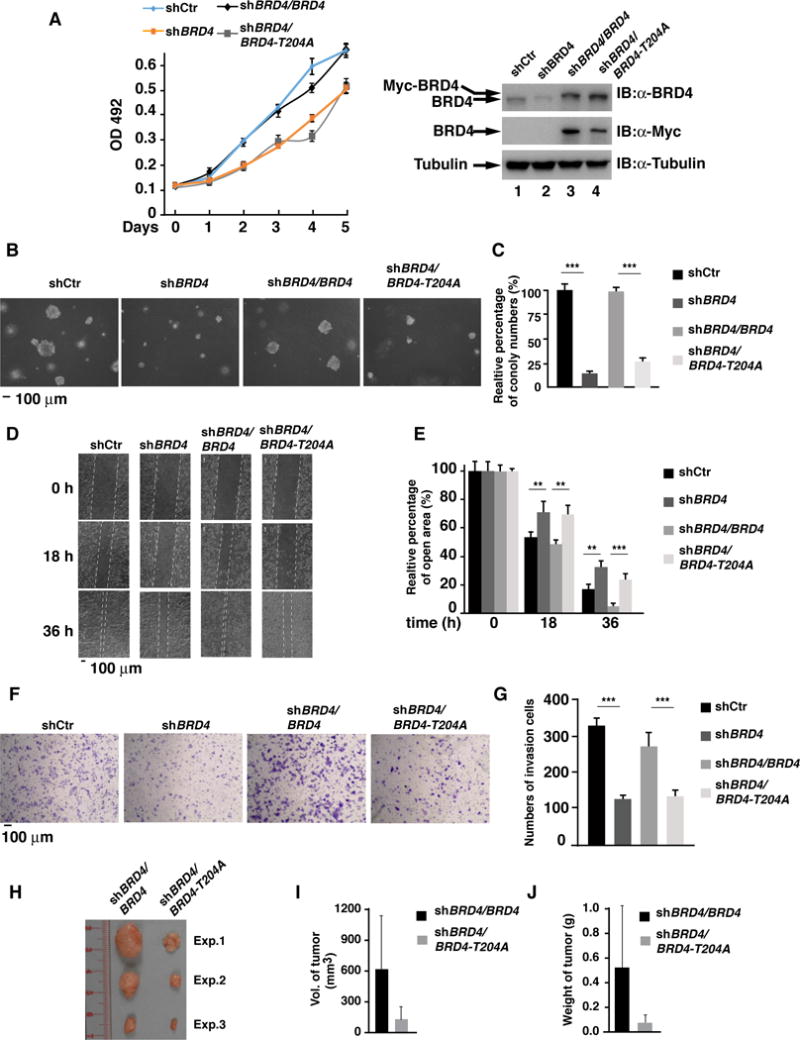
(A) Cell proliferation for MKN28 cells stably expressing control shRNA, BRD4 shRNA or shBRD4/BRD4 or shBRD4/BRD4-T204A. Data represent the average of three independent experiments ± SD (left). The cellular levels of endogenous or exogenous BRD4 in the indicated MKN28 cells (right). (B&C) Various MKN28 cells as in (A) were seeded in soft-agar and cultured for 15 days. Representative photographs were taken at day 15 (B). The relative percentage of colony numbers compared to shCtr cells (set as 100%) from three independent experiments ± SD is shown in (C). (D&E) The wound-healing migration assays for various MKN28 cells as indicated. Representative photographs were taken at 0, 18 and 36 hr (D). The percentage of average speed of wound closure from three independent experiments ± SD is shown in (E). (F&G) Invasion assays for various MKN28 cells as indicated. Representative photographs of invading cells stained with crystal violet are shown in (F). Quantification of cell invasion is shown in (G) and data represent the average of three independent experiments ± SD. (H) Tumorigenicity of shBRD4/BRD4 or shBRD4/BRD4-T204A MKN28 cells (1 × 107) after subcutaneously injection for 7 months in nude mice (n =3). (I&J) Summary of the average volume (I) and weight (J) of tumors from (H).
We next performed a wound-healing assay to determine whether PIN1 regulated BRD4 to affect cell migration. In BRD4 knockdown MKN28 cells, the scratch wound closed more slowly compared to MKN28 cells with control shRNA at 18 or 36 h (Fig. 6D). Importantly, shBRD4/BRD4 cells restored its wound closing activity, whereas shBRD4/BRD4-T204A cells retained the slow wound healing activity as the BRD4 knockdown cells (Figs. 6D&6E). In addition, depletion of BRD4 impaired the invasion of MKN28 cells (Figs. 6F&6G). However, rescue of BRD4 knockdown cells with WT BRD4 but not the BRD4-204A restored the ability of the BRD4 knockdown cell to pass through the gel (Fig. 6F).
Finally, we determined the effect of binding of PIN1 to pT204 on cancer formation and growth in the mouse xenograft model. While the tumor sizes varied in each mouse, tumors from shBRD4/BRD4-T204A-MKN28 cells were consistently smaller than tumors from shBRD4/BRD4-MKN28 cells (Figs. 6H, 6I&6J). These results indicate that abolishing the binding of PIN1 to BRD4 inhibits the ability of MKN28 cancer cells to grow as tumors. Together, these data reveal that binding of PIN1 to phosphorylated BRD4 at T204 is critical for the tumor-promoting activity of BRD4.
Discussion
While increasing numbers of studies support the cancer promoting function of BRD4 in various cancer signaling pathways, how BRD4 is regulated in cancer cells is relatively unclear. In this study, we have identified that peptidyl-prolyl isomerase PIN1 is a key regulatory protein of BRD4 and contributes to the tumor-promoting activity of BRD4 in human gastric cancer cells via isomerization-mediated protein conformation change (Fig. 7).
Fig. 7. Schematic model for the regulation of BRD4 by PIN1 in cancer cells.
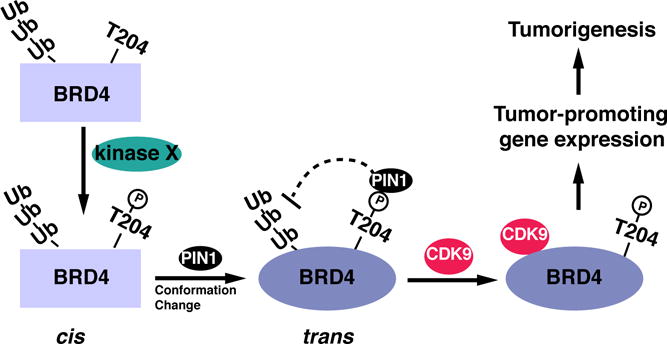
PIN1 regulates the stability, the transcriptional activity and oncogenic potential of BRD4. PIN1 directly binds to the phosphorylated T204 of BRD4, mediated by an unidentified kinase. PIN1-mediated cis-trans isomerization and conformational change of BRD4 increases the stability of BRD4 by suppressing its ubiquitination and degradation. At the same time, this conformational change enhances BRD4’s interaction with CDK9 to stimulate the transcription of cancer-promoting genes and tumorigenesis.
The expression of PIN1 positively correlated with the expression of BRD4 in human gastric cancer samples and cancer cells (Fig. 1). Depletion of PIN1 reduced cellular levels of BRD4 in multiple gastric cancer cell lines, including MKN28, AGS and AZ-521 cells (Figs. 1G&S6). While PIN1 binds to phosphorylated BRD4 and regulates its stability in gastric cancer cells, it appears that PIN1 could also regulate BRD4’s stability in normal cells since the phosphorylation of BRD4 and the interaction of BRD4 and PIN1 could be found in a normal gastric epithelial cell line GES-1 (Figs. S7&S15) and the half-life of Brd4 was reduced in Pin1-deficent MEFs (Fig. S4). It is likely that in gastric cancer cells the overexpressed PIN1 contributes to a more stable or more active BRD4, leading to enhanced interaction with CDK9 and the transcription of cancer promoting genes in gastric cancer cells (Fig. 7). Interestingly, overexpression of PIN1 is observed in a large number of human cancers, including breast, prostate and gastric cancer7, 40, whether BRD4 expression levels are similarly elevated in these cancer samples remains to be determined. Notably, overexpressed BRD4 has been found in some tumors, including melanoma and glioblastoma18, 39,36, where elevated levels of PIN1 have also been identified5, 24. It is possible that PIN1-mediated stabilization might contribute to the overexpressed BRD4 in these tumors. Additionally, expression levels of PIN1 have been suggested to be a prognostic marker in gastric and prostate cancer6, 40. Due to the positive correlation of PIN1 and BRD4 levels, it remains to be evaluated whether the levels of BRD4 could also have prognostic value in these cancers.
In addition to BRD4, BET family proteins include BRD2, BRD3 and testis specific BRDT. BRD2 and BRD3 have been shown to have certain redundant activities of BRD49. However, T204 is present only in BRD4 but not in other BET proteins, suggesting that PIN1 might uniquely regulate the activity of BRD4 through pT204. Consistently, we found that PIN1 only associated with BRD4 but not with BRD2 and BRD3 (Fig. S11). While T204 of BRD4 is not conserved in BET proteins, it is highly conserved among BRD4 of various species (Fig. S12), indicating that BRD4 might be similarly regulated by the corresponding PIN1 in these different species and this regulatory mechanism might be evolutionally conserved.
BRD4 is normally expressed as a long isoform (BRD4-L, amino acid 1–1362) and a short isoform (BRD4-S, amino acid 1–722)45. BRD4-L and BRD4-S share the same PIN1 binding motif of T204 and it is quite possible that BRD4-S can also be phosphorylated and recognized by PIN1. However, BRD4-S doesn’t seem to have tumor-promoting activity in gastric cancer cells since only BRD4-L but not the BRD4-S was able to rescue the halted cell proliferation of BRD4 knockdown MKN28 cells (Fig. S13) and the mRNA levels of BRD4-S were relatively low in gastric cancer cells compared to BRD4-L (Fig. S14). The BRD4-S has been shown to be involved in other cancer properties, including breast cancer metastasis2, 3. It is unclear and remains an interesting question whether the pro-metastatic activity of BRD4-S could be regulated by PIN1 in breast cancer cells.
Phosphorylation has been shown to be associated with the regulation and function of BRD446, 47. For example, BRD4 is phosphorylated at serines 484 and 488 by casein kinase II (CK2) and this phosphorylation dictates BRD4’s binding to chromatin and the recruitment of p53 to regulated promoters46. Different from CK2-mediated phosphorylation of Ser484 and S488, phosphorylation of BRD4 at T204 regulates BRD4’s interaction with PIN1 and the subsequent PIN1-mediated protein conformational change and protein function. While the identity of the kinase remains to be determined, the phosphorylated BRD4 was similarly detected by the site-specific antibody in MKN28, AGS cells and normal gastric epithelial GES-1 cells (Figs. 3C & S15) and could be further enhanced with cytokine stimulation (data not shown), indicating that the T204 kinase is constitutively activated in cells and the kinase activity could be further increased in response to certain stimulations. Additionally, we also noticed that cells had elevated levels of pT204 in G1 than in G0 (Fig. S16), suggesting that the activity of T204 kinase could also be regulated by cell cycle, consistent with a role of BRD4 in cell cycle regulation45.
PIN1 specifically recognizes the pSer/Thr-Pro motif within the substrates via its WW domain and isomerizes the substrates via its PPIase domain30. Similar to its binding to other substrates, PIN1 utilizes its WW domain to bind to phosphorylated BRD4 at pT204 (Fig. 3). Binding of PIN1 to BRD4 peptides was significantly enhanced when T204 was phosphorylated (Fig. 3E). The peptide binding and crystallographic studies further demonstrate a direct interaction between PIN1 and phosphorylated T204 (Fig. 3). Phosphorylated T204 binds to a three-basic-residue cluster (Lys63, Arg68 and Arg69) (Fig. 3) that provides the three magnitude higher preference of PIN1 for phosphorylated over unphosphorylated substrates29. Several additional amino acids of BRD4, including Cys113, Ser115, and Leu122, also make weak interaction with PIN1 (Fig. 3F). However, different from the positively charged triad of Lys63, Arg68 and Arg69, these additional interactions have been suggested to facilitate the PIN1-catalyzed isomerization reaction54. In consistent, we found that mutation of T204 to alanine abolished the interaction between PIN1 and BRD4 in cultured cells (Fig. 3B), indicating that phosphorylated T204 is the key residue responsible for BRD4’s interaction with PIN1. PIN1 binding defective BRD4-T204 reduced the transcriptional and tumor-promoting activity of BRD4 in both MKN28 and AGS gastric cancer cells (Figs. 5, 6 and S17), reflecting a critical role of PIN1 in regulating the functions of BRD4. Supporting this notion, we found that inhibiting the activity of PIN1 with PiB also suppressed the proliferation, anchorage-independent growth, migration and invasion of MKN28 cells (Fig. S18).
How does PIN1 regulate the activity of BRD4? It is well recognized that phosphorylation of Ser/Thr-Pro motifs plays a key role in controlling various cellular responses in which PIN1 serves as the post-phosphorylation regulatory factor by changing the conformation of its substrates30. In line with this, we found that binding of PIN1 to phosphorylated T204 changed the conformation of BRD4 through a cis-trans isomerization of BRD4 (Fig. 5). One of the consequences of this conformational change is the enhanced CDK9 binding to BRD4 and the enhanced recruitment of CDK9 to a subset of promoters of BRD4-mediated tumor-promoting genes, including c-MET and MMP9 (Figs. 5G&5H). It is likely that CDK9 has a higher affinity for the trans form of BRD4 than its cis form. BRD4 plays a key role in recruiting active CDK9 to the promoters for gene transcription22, 53. The chromatin-bound BRD4 needs to be released from chromatin before its association with CDK9 and recruitment of CDK9 to promoters1. While PIN1 interacted with pBRD4 and colocalized with BRD4 in the nucleus (Figs. 2&S2), it is not clear whether PIN1 binds to chromatin-bound BRD4 or chromatin-free BRD4. However, we could only detect weak Pin1 signals on the promoters of c-MET and MMP9 by ChIP (data not shown), suggesting that Pin1-mediated conformational change of BRD4 might not occur on chromatin. It is possible that PIN1 binds to the chromatin-free BRD4 and induces its conformational change before it interacts with CDK9. However, the detailed mechanism needs to be defined in the future experiments.
PIN1-mediated cis-to-trans conformation change of BRD4 doesn’t seem to affect BRD4’s binding to chromatin since BRD4’s recruitment to the promoters was not affected by the mutation of T204A (Fig. 5H). The impaired CDK9 recruitment led to decreased phosphorylation and the activation of RNAPII (Fig. 5H), which seems to be independent of PIN1’s ability to modulate the function of RNAPII51 since mutation of T204 to alanine without changing PIN1’s activity altered the activity of RNAPII and RNAPII-mediated gene transcription (Figs. 5H &5I). While our data clearly demonstrate that phosphorylation of T204 of BRD4 is recognized by PIN1, leading to the conformational change and increased CDK9 binding, whether phosphorylation of T204 might have some additional functions remains an interesting question and needs to be further explored.
Inhibiting the activity of PIN1 by siRNA or PIN1 inhibitors reduced the cellular levels of BRD4 (Figs. 1& S1), whereas inhibition of BRD4 has no effect on the expression of PIN1 (Fig. S19). It is clear that PIN1 regulates the stability of BRD4 by inhibiting its ubiquitination (Fig. 4). PIN1-mediated conformational change of BRD4 might also affect the stability and ubiquitination of BRD4 since WT PIN1 but not PIN1-C115A increased the cellular levels of BRD4 and inhibited the ubiquitination of BRD4 (Figs. 4A&4E). PIN1-mediated conformational change might decrease the accessibility of a BRD4 E3 ligase or increases the accessibility of a BRD4 deubiquitinating enzyme. Therefore, the overall tumor-promoting activity of BRD4 in cancer cells might result from the PIN1-mediated conformational change of BRD4, leading to more stabilized BRD4 and conformational change-associated increased transcriptional potential of BRD4.
Overall, our studies have explored the functional consequence of the positive correlation of PIN1 and BRD4 expression in gastric cancer cells and the molecular and structural mechanism of the PIN1 BRD4 interaction. Identification of PIN1 as a novel regulator of BRD4 not only provides new insights into the regulation of BRD4, but also provides potential alternative approaches for the treatment of gastric cancer by targeting the interaction between BRD4 and PIN1 or with combination therapy by targeting both PIN1 and BRD4.
Materials and methods
Detailed materials and methods can be found in supporting information.
Cell lines, recombinant proteins and plasmids
Human gastric cancer cell lines MKN28, AGS and human embryonic kidney HEK293T were purchased from ATCC (Manassas, VA, USA) and were cultured according to ATCC’s instructions. Wild-type or PIN1 knockout MEFs, shBRD4/BRD4 and shBRD4/BRD4-T204A MKN28 cells were maintained in DMEM supplemented with 10% FBS. Expression vectors for GST-PIN1, GST-PIN1-C115A, GST-PIN1-W34A, Flag-PIN1, Flag-PIN1-C115A, PIN1-W34A and Flag-BRD4 have been previous described21, 35. BRD4 deletion mutants were generated by PCR cloning and point mutation mutants were generated using Quickchange site-mutagenesis (Stratagene, San Diego, CA, USA) and all the mutants were confirmed by sequencing.
Statistical analysis
Statistical significance was determined using a two-tailed homoscedastic Student’s t-test. All data are presented as mean ± SD and a p value ≤ 0.05 was considered statistically significant. The data generated were representative of at least three experiments.
Supplementary Material
Acknowledgments
We thank members in the Chen lab for discussion. This work is supported in part by fund provided by UIUC (to L.F.C.) and NIH grants DK085158 and CA179511 (to L.F.C.) and Natural Science Foundation of China Grants 81361120386 (to R.C.).
Footnotes
Conflict of interest
The authors declare no competing financial interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Ai N, Hu X, Ding F, Yu B, Wang H, Lu X, et al. Signal-induced Brd4 release from chromatin is essential for its role transition from chromatin targeting to transcriptional regulation. Nucleic Acids Res. 2011;39:9592–9604. doi: 10.1093/nar/gkr698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alsarraj J, Walker RC, Webster JD, Geiger TR, Crawford NP, Simpson RM, et al. Deletion of the proline-rich region of the murine metastasis susceptibility gene Brd4 promotes epithelial-to-mesenchymal transition- and stem cell-like conversion. Cancer Res. 2011;71:3121–3131. doi: 10.1158/0008-5472.CAN-10-4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alsarraj J, Faraji F, Geiger TR, Mattaini KR, Williams M, Wu J, et al. BRD4 short isoform interacts with RRP1B, SIPA1 and components of the LINC complex at the inner face of the nuclear membrane. PLoS One. 2013;8:e80746. doi: 10.1371/journal.pone.0080746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–282. doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atkinson GP, Nozell SE, Harrison DK, Stonecypher MS, Chen D, Benveniste EN. The prolyl isomerase Pin1 regulates the NF-kappaB signaling pathway and interleukin-8 expression in glioblastoma. Oncogene. 2009;28:3735–3745. doi: 10.1038/onc.2009.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ayala G, Wang D, Wulf G, Frolov A, Li R, Sowadski J, et al. The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res. 2003;63:6244–6251. [PubMed] [Google Scholar]
- 7.Bao L, Kimzey A, Sauter G, Sowadski JM, Lu KP, Wang DG. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am J Pathol. 2004;164:1727–1737. doi: 10.1016/S0002-9440(10)63731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Basheer F, Huntly BJ. BET Bromodomain Inhibitors in leukemia. Exp Hematol. 2015 doi: 10.1016/j.exphem.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12:465–477. doi: 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen R, Yik JH, Lew QJ, Chao SH. Brd4 and HEXIM1: multiple roles in P-TEFb regulation and cancer. BioMed research international. 2014;2014:232870. doi: 10.1155/2014/232870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiang CM. Brd4 engagement from chromatin targeting to transcriptional regulation: selective contact with acetylated histone H3 and H4. F1000 Biol Rep. 2009;1:98. doi: 10.3410/B1-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung CW. Small molecule bromodomain inhibitors: extending the druggable genome. Progress in Medicinal Chemistry. 2012;51:1–55. doi: 10.1016/B978-0-12-396493-9.00001-7. [DOI] [PubMed] [Google Scholar]
- 13.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng Q, Zhang Z, Shea MJ, Creighton CJ, Coarfa C, Hilsenbeck SG, et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell research. 2014;24:809–819. doi: 10.1038/cr.2014.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2011;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer G, Bang H, Berger E, Schellenberger A. Conformational specificity of chymotrypsin toward proline-containing substrates. Biochim Biophys Acta. 1984;791:87–97. doi: 10.1016/0167-4838(84)90285-1. [DOI] [PubMed] [Google Scholar]
- 17.French CA. Pathogenesis of NUT midline carcinoma. Annu Rev Pathol. 2012;7:247–265. doi: 10.1146/annurev-pathol-011811-132438. [DOI] [PubMed] [Google Scholar]
- 18.Gallagher SJ, Mijatov B, Gunatilake D, Gowrishankar K, Tiffen J, James W, et al. Control of NF-kB activity in human melanoma by bromodomain and extra-terminal protein inhibitor I-BET151. Pigment Cell Melanoma Res. 2014;27:1126–1137. doi: 10.1111/pcmr.12282. [DOI] [PubMed] [Google Scholar]
- 19.Hsu TI, Lin SC, Lu PS, Chang WC, Hung CY, Yeh YM, et al. MMP7-mediated cleavage of nucleolin at Asp255 induces MMP9 expression to promote tumor malignancy. Oncogene. 2015;34:826–837. doi: 10.1038/onc.2014.22. [DOI] [PubMed] [Google Scholar]
- 20.Hu Y, Zhou J, Ye F, Xiong H, Peng L, Zheng Z, et al. BRD4 inhibitor inhibits colorectal cancer growth and metastasis. Int J Mol Sci. 2015;16:1928–1948. doi: 10.3390/ijms16011928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang B, Yang XD, Zhou MM, Ozato K, Chen LF. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol Cell Biol. 2009;29:1375–1387. doi: 10.1128/MCB.01365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Molecular cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 23.Jeanmougin F, Wurtz JM, Le Douarin B, Chambon P, Losson R. The bromodomain revisited. Trends Biochem Sci. 1997;22:151–153. doi: 10.1016/s0968-0004(97)01042-6. [DOI] [PubMed] [Google Scholar]
- 24.Jin J, Zhang Y, Li Y, Zhang H, Li H, Yuan X, et al. RNA-interference-mediated downregulation of Pin1 suppresses tumorigenicity of malignant melanoma A375 cells. Neoplasma. 2013;60:92–100. doi: 10.4149/neo_2013_013. [DOI] [PubMed] [Google Scholar]
- 25.Jung M, Gelato KA, Fernandez-Montalvan A, Siegel S, Haendler B. Targeting BET bromodomains for cancer treatment. Epigenomics. 2015;7:487–501. doi: 10.2217/epi.14.91. [DOI] [PubMed] [Google Scholar]
- 26.Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nature genetics. 2014;46:364–370. doi: 10.1038/ng.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liou YC, Zhou XZ, Lu KP. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem Sci. 2011;36:501–514. doi: 10.1016/j.tibs.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lockwood WW, Zejnullahu K, Bradner JE, Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:19408–19413. doi: 10.1073/pnas.1216363109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380:544–547. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- 30.Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8:904–916. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- 31.Lu Z, Hunter T. Prolyl isomerase Pin1 in cancer. Cell research. 2014;24:1033–1049. doi: 10.1038/cr.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morishita A, Gong J, Masaki T. Targeting receptor tyrosine kinases in gastric cancer. World journal of gastroenterology: WJG. 2014;20:4536–4545. doi: 10.3748/wjg.v20.i16.4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muller S, Filippakopoulos P, Knapp S. Bromodomains as therapeutic targets. Expert reviews in molecular medicine. 2011;13:e29. doi: 10.1017/S1462399411001992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nicole Tsang YH, Wu XW, Lim JS, Wee Ong C, Salto-Tellez M, Ito K, et al. Prolyl isomerase Pin1 downregulates tumor suppressor RUNX3 in breast cancer. Oncogene. 2013;32:1488–1496. doi: 10.1038/onc.2012.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pastori C, Daniel M, Penas C, Volmar CH, Johnstone AL, Brothers SP, et al. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics. 2014;9:611–620. doi: 10.4161/epi.27906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Proserpio V, Fittipaldi R, Ryall JG, Sartorelli V, Caretti G. The methyltransferase SMYD3 mediates the recruitment of transcriptional cofactors at the myostatin and c-Met genes and regulates skeletal muscle atrophy. Genes Dev. 2013;27:1299–1312. doi: 10.1101/gad.217240.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ryo A, Liou YC, Wulf G, Nakamura M, Lee SW, Lu KP. PIN1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Mol Cell Biol. 2002;22:5281–5295. doi: 10.1128/MCB.22.15.5281-5295.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Segura MF, Fontanals-Cirera B, Gaziel-Sovran A, Guijarro MV, Hanniford D, Zhang G, et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013;73:6264–6276. doi: 10.1158/0008-5472.CAN-13-0122-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi M, Chen L, Ji J, Cai Q, Yu Y, Liu B, et al. Pin1 is Overexpressed and Correlates with Poor Prognosis in Gastric Cancer. Cell Biochem Biophys. 2014 doi: 10.1007/s12013-014-0274-0. [DOI] [PubMed] [Google Scholar]
- 41.Stukenberg PT, Kirschner MW. Pin1 acts catalytically to promote a conformational change in Cdc25. Molecular cell. 2001;7:1071–1083. doi: 10.1016/s1097-2765(01)00245-3. [DOI] [PubMed] [Google Scholar]
- 42.Suizu F, Ryo A, Wulf G, Lim J, Lu KP. Pin1 regulates centrosome duplication, and its overexpression induces centrosome amplification, chromosome instability, and oncogenesis. Mol Cell Biol. 2006;26:1463–1479. doi: 10.1128/MCB.26.4.1463-1479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takahashi K, Akiyama H, Shimazaki K, Uchida C, Akiyama-Okunuki H, Tomita M, et al. Ablation of a peptidyl prolyl isomerase Pin1 from p53-null mice accelerated thymic hyperplasia by increasing the level of the intracellular form of Notch1. Oncogene. 2007;26:3835–3845. doi: 10.1038/sj.onc.1210153. [DOI] [PubMed] [Google Scholar]
- 44.Uchida T, Takamiya M, Takahashi M, Miyashita H, Ikeda H, Terada T, et al. Pin1 and Par14 peptidyl prolyl isomerase inhibitors block cell proliferation. Chem Biol. 2003;10:15–24. doi: 10.1016/s1074-5521(02)00310-1. [DOI] [PubMed] [Google Scholar]
- 45.Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. The Journal of biological chemistry. 2007;282:13141–13145. doi: 10.1074/jbc.R700001200. [DOI] [PubMed] [Google Scholar]
- 46.Wu SY, Lee AY, Lai HT, Zhang H, Chiang CM. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Molecular cell. 2013;49:843–857. doi: 10.1016/j.molcel.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu SY, Nin DS, Lee AY, Simanski S, Kodadek T, Chiang CM. BRD4 Phosphorylation Regulates HPV E2-Mediated Viral Transcription, Origin Replication, and Cellular MMP-9 Expression. Cell Rep. 2016;16:1733–1748. doi: 10.1016/j.celrep.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu X, Qi J, Bradner JE, Xiao G, Chen LF. Bromodomain and extraterminal (BET) protein inhibition suppresses human T cell leukemia virus 1 (HTLV-1) Tax protein-mediated tumorigenesis by inhibiting nuclear factor kappaB (NF-kappaB) signaling. The Journal of biological chemistry. 2013;288:36094–36105. doi: 10.1074/jbc.M113.485029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wulf G, Garg P, Liou YC, Iglehart D, Lu KP. Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis. EMBO J. 2004;23:3397–3407. doi: 10.1038/sj.emboj.7600323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V, et al. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 2001;20:3459–3472. doi: 10.1093/emboj/20.13.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu YX, Hirose Y, Zhou XZ, Lu KP, Manley JL. Pin1 modulates the structure and function of human RNA polymerase II. Genes Dev. 2003;17:2765–2776. doi: 10.1101/gad.1135503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang S, Zhao Z, Wu R, Lu H, Zhang X, Huan C, et al. Expression and biological relationship of vascular endothelial growth factor-A and matrix metalloproteinase-9 in gastric carcinoma. J Int Med Res. 2011;39:2076–2085. doi: 10.1177/147323001103900603. [DOI] [PubMed] [Google Scholar]
- 53.Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Molecular cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 54.Yogesha SD, Mayfield JE, Zhang Y. Cross-talk of phosphorylation and prolyl isomerization of the C-terminal domain of RNA Polymerase II. Molecules. 2014;19:1481–1511. doi: 10.3390/molecules19021481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, Daum S, Wildemann D, Zhou XZ, Verdecia MA, Bowman ME, et al. Structural basis for high-affinity peptide inhibition of human Pin1. ACS Chem Biol. 2007;2:320–328. doi: 10.1021/cb7000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zou Z, Huang B, Wu X, Zhang H, Qi J, Bradner J, et al. Brd4 maintains constitutively active NF-kappaB in cancer cells by binding to acetylated RelA. Oncogene. 2014;33:2395–2404. doi: 10.1038/onc.2013.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.