

Abstract
Glioblastoma multiforme (GBM) is highly invasive and uniformly fatal, with median survival < 20 months after diagnosis even with the most aggressive treatment that includes surgery, radiation, and systemic chemotherapy. Cisplatin is a particularly potent chemotherapeutic agent, but its use to treat GBM is limited by severe systemic toxicity and inefficient penetration of brain tumor tissue even when it is placed directly in the brain within standard delivery systems. We describe the development of cisplatin-loaded nanoparticles that are small enough (70 nm in diameter) to move within the porous extracellular matrix between cells and that possess a dense polyethylene glycol (PEG) corona that prevents them from being trapped by adhesion as they move through the brain tumor parenchyma. As a result, these “brain penetrating nanoparticles” penetrate much deeper into brain tumor tissue compared to nanoparticles without a dense PEG corona following local administration by either manual injection or convection enhanced delivery. The nanoparticles also provide controlled release of cisplatin in effective concentrations to kill the tumor cells that they reach without causing toxicity-related deaths that were observed when cisplatin was infused into the brain without a delivery system. Median survival time of rats bearing orthotopic glioma was significantly enhanced when cisplatin was delivered in brain penetrating nano-particles (median survival not reached; 80% long-term survivors) compared to cisplatin in conventional un-PEGylated particles (median survival = 40 days), cisplatin alone (median survival = 12 days) or saline-treated controls (median survival = 28 days).
Keywords: Brain tumor, Convection enhanced delivery, Cisplatin, Therapeutic nanoparticle, Non-adhesive surface
1. Introduction
Glioblastoma multiforme (GBM) is the most aggressive primary brain tumor with high rates of recurrence and a median survival time of <20 months even following aggressive surgical resection and subsequent irradiation and chemotherapy [1]. Temozolomide (TMZ) is the frontline chemotherapy since it is able to cross the blood brain barrier (BBB) andit provides some improvement inpatient survival [1,2]. However, TMZ effectiveness is limited by its low potency as an alkylating agent, dose-limiting systemic toxicity due to myelosuppression and lymphopenia [3–5], rapid degradation in the body, and development of drug resistance [2,4,6]. As a result, new chemotherapeutic approaches for GBM are desperately needed [7].
cis-Diamminedichloroplatinum (Cisplatin, CDDP) is a highly potent agent used to treat numerous types of cancer, including testicular, ovarian, bladder and lung cancers [8]. Additionally, systemic CDDP has been widely adopted as anadjuvant therapy for several brain tumors in pediatric patients, such as neuroblastoma and medulloblastoma [9]. However, CDDP administered systemically to adults with GBM causes severe nephrotoxicity and neurotoxicity at sub-therapeutic drug levels [10–13]. Thus, clinical trials for GBM have investigated alternative platinum-based agents (i.e. carboplatin) that are less toxic, but also less effective [14].
FDA approval in 1996 of Gliadel, composed of bischloroethylnitrosourea (BCNU) impregnated into a biodegradable polyanhydride wafer roughly the size of a quarter, proved that local delivery of chemotherapy in the brain can be both safe and effective in the treatment of GBM [15]. Importantly, local delivery eliminates the severe systemic toxicity observed with most systemically-administered chemotherapeutic drugs, including BCNU. However, the effectiveness of Gliadel is limited primarily because BCNU released from the wafers only penetrates a few millimeters into brain tissue [16], but also because BCNU is not a particularly potent chemotherapeutic agent. Intracerebral infusion of CDDP at sub-therapeutic doses was safely conducted in humans with malignant glioma [17,18], but higher doses that might be effective were not tested due to safety concerns. Recent preclinical studies have revealed promising therapeutic outcomes following intratumoral administration of CDDP [19,20], but neurotoxicity of CDDP administered without a drug delivery system remains a significant limitation [20,21]. The encapsulation of chemotherapeutics into nanoparticles (NP) that slowly mete the drug out at therapeutic levels can greatly improve drug safety and efficacy [8,15]. Thus, it is logical to administer chemo-loaded NP locally to treat brain tumors. However, most NP do not penetrate tumors well since they are either too large to fit through the spaces between cells or their surfaces are adhesive to the extracellular matrix (ECM) [22]. Convection enhanced delivery (CED) has been widely explored to facilitate drug distribution in the brain [23,24]. However, most NP still remain at the infusion site even with pressure-driven flow provided by CED [25,26], likely due to the nanoporous and highly adhesive ECM [27,28]. We have previously demonstrated that a dense polyethylene glycol (PEG) corona on NP eliminates adhesive trapping in the brain parenchyma [29], and that such particles rapidly penetrate healthy brain tissue if they are <114 nm in diameter [29] and brain tumor tissue if they are <70 nm in diameter. We termed such particles “brain penetrating particles”, or BPN, whereas we call the same particles without a dense PEG corona “un-PEGylated particles”, or UPN [29]. Here, we describe the development of CDDP-loaded polymer nanoparticles, with and without a dense PEG corona, that release CDDP in a controlled manner. We then compare the ability of the BPN and UPN to penetrate healthy brain tissue and brain tumor tissue, both ex vivo and in vivo, and to treat rats with orthotopic GBM.
2. Materials and methods
2.1. PEGylation of polypeptide
Poly(aspartic acid) (PAA) with a molecular weight of 27 kDa (Alamanda Polymers, Huntsville, AL) was reacted with PEG with a molecular weight of 5 kDa (Creative PEGworks, Winston Salem, NC) at a 1:10 M ratio, as facilitated by the addition of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC, Invitrogen, Carlsbad, CA) at stoichiometric concentrations, in 50 mM 2-(N-morpholino)ethanesulfonic acid (MES Buffer, pH 5.0, Sigma Aldrich, St. Louis, MO). The reaction was carried out for 72 h at room temperature followed by dialysis against deionized water using a 20 kDa MWCO cassette (Spectrum Lab, Rancho Dominguez, CA) for 120 h. The solution was then lyophilized to obtain the PAA-PEG polypeptide which was then stored at −20 °C until use. The PAA:PEG ratio was confirmed through nuclear magnetic resonance (NMR) to be ∼1:10 (Fig. S1): 1H NMR (500 MHz, D2O): δ 2.70–2.80 (br, CHCH2COOH) 3.55–3.75 (br, CH2CH2O), 4.40–4.55 (br, NHCHCH2). Immediately prior to NP formulation, the lyophilized polymers were dissolved in ultrapure distilled water.
2.2. Fluorescent labeling
Fluorescent labeling dyes, Alexa Fluor® 555 and 647 (AF555 and AF647, Molecular Probes, Eugene, OR), were conjugated to PAA and PAA-PEG polypeptide, respectively, by dissolving in 200 mM borate buffer (pH 8.2) and reacting for 72 h at room temperature. The solution was dialyzed against deionized water using a 20 kDa MWCO cassette (Spectrum Lab) for 120 h, followed by lyophilization. The labeled poly-peptides were stored at −20 °C until use.
2.3. NP preparation and characterization
CDDP-loaded BPN (CDDP-BPN) and UPN (CDDP-UPN) were formulated using the following protocol. The CDDP-UPN were formulated by mixing 5 mM of CDDP (Sigma Aldrich) with 7.5 mM of aspartic acid (i.e. PAA only) in RNase-free water for 72 h at room temperature. For the CDDP-BPN, 5 mM of CDDP was mixed with 5 mM of aspartic acid (i.e. 9:1 aspartic acid ratio of PAA-PEG:PAA) in RNase-free water for 72 h atroom temperature. NP were then collected using ultracentrifuge filters (Amicon Ultra, 100 kDa MWCO; Millipore, Billerica, MA) by centrifuging at 1000 ×g for 10 min and stored at room temperature until further use. Physicochemical characteristics of NP were determined using a Zetasizer NanoZS (Malvern Instruments, Southborough, MA). All particles were diluted in 10 mM NaCl (diluted from phosphate buffered saline) and dynamic light scattering (DLS) was employed to determine the hydrodynamic diameter and polydispersity index (PDI) at a backscattering angle of 173°. The surface charge (ζ-potential) of the particles was determined using laser Doppler anemometry. Quantification of CDDP within the NPwas conducted through flameless atomic absorbance spectroscopy (AAS) (Perkin Elmer, Waltham, MA) and loading was calculated as the % mass of CDDP in the total sample. NP imaging was conducted using a Hitachi H7600 transmission electron microscope (TEM, Hitachi, Japan).
2.4. CDDP release kinetics
To determine the CDDP release over time, CDDP-UPN or CDDP-BPN were dispersed in 1 mL of artificial cerebrospinal fluid (ACSF) (Harvard Apparatus, Holliston, MA) within a 100 kDa MWCO dialysis membrane (Spectrum Laboratories, Rancho Dominguez, CA). The chamber was then placed in a 14 mL ACSF sink and shaken at 37 °C. At specific time points, the entire sink volume was removed and replaced with 14 mL of fresh ACSF. Samples were quantified using AAS.
2.5. In vitro cell viability
Rat brain tumor cells, including 9 L gliosarcoma and F98 glioma lines, were cultured and passaged in Dulbecco's Modified Eagles Medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin streptomycin (Invitrogen). Cells were seeded at a concentration of 2000 cells/well in 100 μL of media and allowed to attach overnight in 96 well plates. On the following day, the media was replaced with 100 μL of fresh media and 10 μL of either CDDP or CDDP-BPN (20 μM to 0.002 μM in 10-fold dilutions) was administered. Cells were incubated for 3 days at 37 °C and 5% CO2. To quantify the number of live cells, media was replaced with 100 μL of fresh medium and 10 μL of Dojindo Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc., Rockville, MD) solution was added. Cells were incubated for 2 h at 37 °C, followed by the measurement of absorbance at 450 nm using a Synergy Mx Multi-Mode Microplate Reader (Biotek, Instruments Inc. Wi-nooski, VT). The % cell viability was normalized to the untreated cell control.
2.6. Neocortical slice preparation and multiple particle tracking
Healthy or tumor-bearing rat brain tissue slices were prepared according to a slightly modified protocol of a previously publication [29]. F98 tumor-bearing Fischer 344 rats were sacrificed 12 days after the tumor inoculation (1 × 105 cells) to ensure a significantly large tumor bulk for investigation. Healthy or tumor-bearing brain tissue was sliced into 1.5 mm thick slices using a Zivic Mouse Brain slicer (Zivic instruments, Pittsburgh, PA) and placed in custom-made microscopy chambers. Half a microliter of fluorescently labeled particles was injected at a depth of 1 mm into the cerebral cortex using a 10 μL Hamilton Neuros Syringe (Hamilton, Reno, NV). The chambers were sealed using a coverslip to minimize convective bulk flow so that particle movement could be fully attributed to Brownian diffusion. The particle transport rate was calculated by analyzing the particle trajectories in brain tissue slices (N = 3 for each particle type) as described previously [29]. The particle trajectories were recorded as 20 s movies at an exposure of 66 ms, using an EMCCD camera (Evolve 512; Photometrics, Tuscon, AZ) mounted on an inverted epifluorescence microscope (Axio Observer D1, Carl Zeiss, Hertfordshire, UK) equipped with a 100× oil-immersion objective (NA1.3). Median mean squared displacement (MSD) of the various nanoparticle types was calculated and compared at a timescale of τ = 1 s. The median value as opposed to an ensemble average of the individual nanoparticle MSD was calculated due to the inherent heterogeneity and non-gaussian distributions of particle transport in the rat brain [30]. Theoretical MSD of NP in ACSF was calculated using theoretical Stokes-Einstein equation and the mean particle diameter calculated through DLS.
2.7. Fluorescence-based imaging of in vivo NP distribution
Fluorescently labeled CDDP-UPN and CDDP-BPN (12 μg of CDDP) at a ratio of 1:1 were loaded in a 50 μL Hamilton Neuros Syringe (Hamilton) and co-administered into the striatum of male Sprague Dawley rats (200–220 g). A burr hole was drilled 3 mm lateral and 1 mm posterior to the bregma. For manual injection, the catheter was lowered to a depth of 4.0 mm and raised up 0.5 mm. The NP mixture was manually administered at a rate of 2 μL/min for a total volume of 10 μL. Catheter was then withdrawn at a rate of 1 mm/min. For CED, the catheter was vertically mounted on a Chemyx Nanojet Injector Module (Chemyx, Stafford, TX), which was held on a small animal stereotactic frame (Stoelting, Wood Dale, IL). The catheter tip was lowered to a depth of 3.5 mm and NP were infused at a rate of 0.33 μL/min, followed by catheter withdrawal at a rate of 1 mm/min. Animals were sacrificed 1 h post-administration and the brains were removed and immediately frozen on dry ice. Tissues were cryosectioned (Leica CM 3050S, Leica Biosystems, Buffalo Grove, Il) into 100 μm coronal slices and imaged using a Zeiss confocal 710 laser scanning microscope through DS Red (AF555, CDDP-UPN), and Cy5 (AF647, CDDP-BPN) channels. As previously conducted [31], confocal images of brain slices were quantified for fluorescent distribution of NP within the striatum using a custom-made MATLAB script which thresholded the images at 10% of the maximum intensity. Fluorescent distribution of NP in the ventricles or white matter tracts (WMT) was avoided and not included in the volume of distribution quantification. The area of distribution calculated from each slice was multiplied by the slice thickness of 100 μm and summated across all images to obtain a total volume of distribution.
2.8. Toxicity of locally administered CDDP in healthy rats
A 10 μL solution of CDDP (12 μg and 24 μg) or CDDP-BPN (12 μg, 24 μg, and 48 μg) was administered intracranially using a manual injection (e.g., bolus injection) or CED. For the next 5 days following the CDDP administration, rats were evaluated 3 times a day for physical deficiencies and subsequently once every day. Weights were recorded every other day and rats were sacrificed as needed for clinical evaluation of symptoms. The study was terminated 30 days after CDDP administration.
2.9. In vivo tumor inoculation
Female Fischer F344 rats (100–140 g) were anesthetized for the inoculation of F98 glioma cells with a mixture of ketamine-xylazine, as previously described [32]. Briefly, a sagittal incision was made to expose the skull followed by the drilling of a burr hole 1 mm posterior and 3mm left of the bregma. A10 μLsolution of F98 glioma cells was administered into the striatumata depth of 3.5 mm. The head was then closed using biodegradable sutures (Polysorb™ Braided Absorbable Sutures 5-0, Covidien, Mundelein, Il) and Bacitracin was applied gently.
2.10. Efficacy of CDDP-BPN against an orthotopic brain tumor model following CED
For the first study, F98 glioma cells (1 × 103 cells in 10 μL) were inoculated on Day 0. We treated the animals on Day 7 according to a prior study that determined that the tumors were established at that time point [33]. Therefore, on Day 7, the burr holes were re-accessed and animals were treated via CED with 10 μL of the following treatment groups: normal saline (NS), CDDP, CDDP-UPN and CDDP-BPN. The concentration of CDDP in each CDDP treatment group was 1.2 mg/mL.
Further investigation of the therapeutic efficacy following CED of CDDP-BPN took place in a similar manner but with a higher tumor cell inoculum. F98 glioma cells (1 × 105 cells in 10 μL) were administered on Day 0, and animals were randomized into 3 separate groups: normal saline, radiotherapy only and CDDP-BPN. On Day 3, rats in the last group were anesthetized and received CDDP-BPN via CED. The concentration of CDDP was 1.2 mg/mL. On Day 4, rats undergoing radiotherapy only were anesthetized and a focused radiation beam (2 Gy/min for a total of 15 Gy) was applied on the ipsilateral hemisphere where the tumor was implanted.
Following the inoculation or treatment, rat brains were sealed using biodegradable sutures and placed on a heating pad until anesthesia effects wore off. Rats were monitored for deficiencies daily and weighed every other day. Animals were sacrificed when deemed humanely necessary in a blinded manner according to John's Hopkins Animal Care and Use policy. Rat brains were harvested and sent to Johns Hopkins Reference Histology for processing and H&E staining.
2.11. Statistical analysis
Statistical analysis between two groups was conducted using a two-tailed Student's t-test assuming unequal variances. If multiple comparisons were involved, one-way analysisofvariance (ANOVA), followed by post hoc test, was employed, using SPSS 18.0 software (SPSS Inc., Chicago, IL). Differences were determined to be statistically significant at p < 0.05.
3. Results
3.1. Physicochemical characterization of CDDP nanoparticles
We encapsulated CDDP into nanoparticles composed of either PAA alone (CDDP-UPN), or of a 9:1 mixture of PEGylated PAA with un-PEGylated PAA (CDDP-BPN), by mixing the polymers with CDDP in defined ratios in water followed by stirring for 72 h. PEGylated PAA contained an average of ten PEG chains (each 5 kDa) per 27 kDa PAA (Fig. S1). Table 1 shows that CDDP-BPN possessed a diameter of 74 ± 2 nm, a low PDI (0.11 ± 0.02), and near neutral surface charge as indicated by ζ-potential (−8.6 ± 1.1 mV), whereas CDDP-UPN were slightly smaller at 65 ± 3 nm in diameter, and possessed a higher PDI (0.2 ± 0.01) and a highly anionic surface charge (−35 ± 2.0 mV) since the charge of the carboxyl side groups of PAA were not shielded by a PEG corona. Both NP formulations were spherical in shape (Fig. 1A and B) and encapsulated a high concentration of CDDP, with a higher drug quantity loaded in a CDDP-BPN (19.5 ± 1.4% w/w) compared to the CDDP-UPN (9.5 ± 1.6% w/w). Sustained CDDP release kinetics were observed over a period of 24 h for both formulations incubated in ASCF (Fig. 1C). The overall percentages of encapsulated CDDP released from CDDP-BPN and CDDP-UPN over 72 h were ∼60% and 80%, respectively.
Table 1.
Physicochemical properties and diffusion behaviors of CDDP NP.
| Type of CDDP NP | Hydrodynamic Diameter ± SD (nm)a | ζ-potential ± SD (mV)b | Polydispersity index (PDI)a | CDDP loading% (w/w) | MSDACSF/MSDhealthyc | MSDACSF/MSDtumorc |
|---|---|---|---|---|---|---|
| CDDP-UPN | 65 ± 3 | − 35 ± 2 | 0.20 ± 0.01 | 9.5 ± 1.6% | 17,000 | 1000 |
| CDDP-BPN | 74 ± 2 | − 8.6 ±1.1 | 0.11 ± 0.02 | 19.4 ± 1.4% | 285 | 115 |
Hydrodynamic diameters and PDI were measured in 10 mM NaCl at pH 7.0. Data are presented as average of at least 3 measurements.
ζ-potentials were measured by laser Doppler anemometry in 10 mM NaCl at pH 7.0. Data are presented as average of at least 3 measurements.
These ratios indicate the extents to which diffusion of CDDP NP (median MSD) in healthy and tumor-bearing brain tissues is reduced compared to their diffusion in ACSF.
Fig. 1.
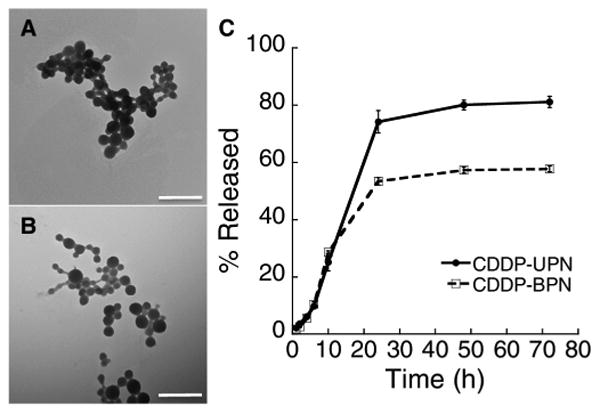
Characterization of CDDP NP. Transmission micrographs of (A) CDDP-UPN and (B) CDDP-BPN formulations. Scale bar = 500 nm. (C) Quantified in vitro release kinetics in ACSF (pH 7.0) demonstrating the release of CDDP from both CDDP-UPN and CDDP-BPN formulations.
3.2. In vitro cytotoxicity of CDDP-BPN against rat glioma cells
In vitro tests in F98 glioma and 9 L gliosarcoma lines showed that a 20 μM dose of unencapsulated CDDP in aqueous solution or CDDP-BPN imparted significant cytotoxicity (Fig. S2). In general, CDDP and CDDP-BPN treatments did not yield significant differences in cytotoxicity in either cell line, suggesting that delivering CDDP in BPN did not reduce the intrinsic potency of CDDP. Both CDDP and CDDP-BPN treatments exhibited higher levels of efficacy against F98 cells compared to 9 L cells at dosages of 2 and 20 μM.
3.3. Ex vivo diffusion of CDDP nanoparticles in healthy and tumor brain tissue
To test the rate of NP diffusion within the brain tissue parenchyma, we tracked the movements of fluorescently labeled CDDP-UPN and CDDP-BPN in freshly excised healthy rat brain tissue and in F98 tumor tissue that had been freshly excised from rat brains. Individual particle trajectories were tracked and their MSD were quantified using multiple particle tracking (MPT). The MSD represents a square of distance traveled by a particle within a given time interval (i.e., timescale or τ), and thus the greater the MSD, the faster the particle diffusion [34]. As shown by representative trajectories, CDDP-BPN traversed greater distances over 20 s in healthy (Fig. 2A) and tumor-bearing (Fig. 2B) rat brain tissues compared to the highly confined motions of CDDP-UPN. The diffusion rates of CDDP-BPN and CDDP-UPN in brain tissues were 285-fold and 17,000-fold lower, respectively, than their theoretical diffusion rates in ACSF at τ = 1 s (Table 1). Overall, CDDP-BPN diffused significantly faster than CDDP-UPN in both healthy and tumor-bearing brain tissues (Fig. 2C).
Fig. 2.
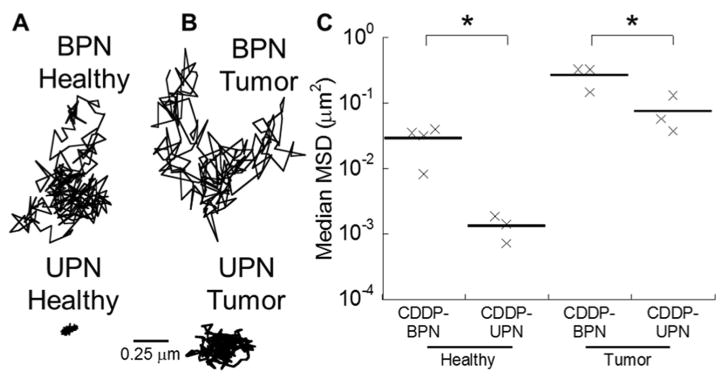
Diffusion of CDDP NP in healthy and tumor-bearing rat brain tissues. Representative trajectories of CDDP-BPN (upper panel) and CDDP-UPN (lower panel) in (A) healthy and (B) tumor-bearing brain tissues. (C) Median MSD of CDDP-BPN and CDDP-UPN at a time scale of τ= 1 sin least N = 3 healthy or tumor-bearing rat brain tissue samples. Greater than 100 NP were tracked per sample. *p < 0.05 denotes statistically significant differences between comparing groups.
3.4. In vivo distribution of CDDP nanoparticles following intracranial administration
To verify our ex vivo findings, we co-infused fluorescently labeled CDDP-UPN and CDDP-BPN into the striatum of live rats and measured the volume of distribution (Vd) of each. When administered as a manual co-injection, the Vd of CDDP-BPN in the striatum was 14-fold higher than that of CDDP-UPN (Fig. 3A, C). Of note, however, over 50% of CDDP-BPN and 70% of CDDP-UPN, respectively, were found in WMT following manual co-injection (Fig. 3A, D). As previous studies have described [35], backflow of the infused solution leads to preferential flow through the WMT, thus reducing therapeutic distribution in the striatum. We further co-administered CDDP-UPN and CDDP-BPN using CED to determine whether the pressure-driven flow could improve the Vd. CDDP-BPN administered by CED homogeneously distributed throughout the striatum (Fig. 3B), with a 6.5-fold enhancement in Vd compared to administration of the same particles by manual injection (Fig. 3C). The Vd of CDDP-UPN was also enhanced by CED, albeit only by a factor of 3. Overall, the Vd of CDDP-BPN was 29-fold higher that of CDDP-UPN when both formulations were administered by CED (Fig. 3C). Furthermore, <20%of CDDP-BPN and CDDP-UPN were found within WMT after CED (Fig. 3D).
Fig. 3.
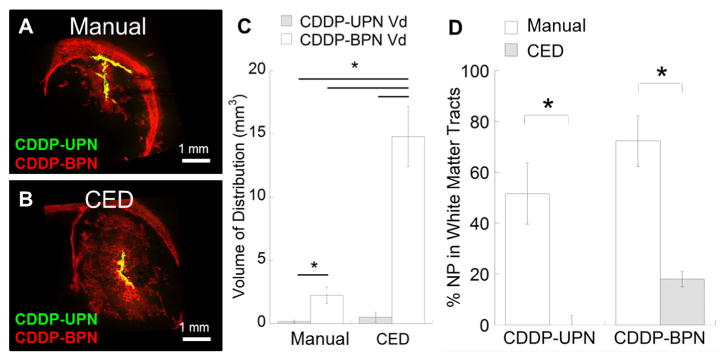
In vivo distribution of CDDP-UPN and CDDP-BPN following local administration. Representative confocal images of CDDP-UPN (green) and CDDP-BPN (red) following administration via (A) manual injection or (B) CED (N ≥ 3 for each administration). NP overlay is depicted as yellow. Scale bar = 1 mm. (C) Volume of distribution (Vd) of CDDP-BPN and CDDP-UPN following manual injection or CED. Calculations were conducted using image-based MATLAB quantification. *p < 0.05 denotes statistically significant differences. (D) Distribution of CDDP-BPN and CDDP-UPN present in WMT as a percentage of total distribution. Higher % of both NP formulations are found in WMT when administered using a manual injection as opposed to CED (*p < 0.05). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.5. Maximum tolerated dose following local administration
Given that CDDP is highly toxic when administered directly into the brain [21], we next evaluated whether the controlled release of CDDP from BPN provided animproved safety profile following intracranial administration. Healthy rats were treated with various intracranial doses of either CDDP in aqueous solution or CDDP-BPN in 10 μL solution by manual injection. Rats treated with CDDP-BPN at doses of 12 μg (100% survival) and 24 μg (80% survival) fared better than those treated with CDDP at the same doses (47% survival and 23% survival, respectively) (Fig. 4). Furthermore, 40% of rats treated with 48 μg CDDP-BPN were long-term survivors, representing an improvement in CDDP tolerability compared to animals treated with CDDP at half that dose. A dose of 48 μg of CDDP could not be tested due to the limited solubility of CDDP in water. Similarly, we administered CDDP-BPN and CDDP using CED to determine whether the method of administration had an effect on CDDP tolerability. CED-administered CDDP-BPN at 12 and 24 μg were both better tolerated than CDDP administered at 12 μg. Thus, the CDDP-BPN formulation enables the administration of higher CDDP doses compared to CDDP without encapsulation. Prior studies have safely administered 6 μg of CDDP intracranially [21]. Here, we found that the maximum tolerable dose (MTD) of CDDP delivered in CDDP-BPN is 2-fold higher at 12 μg (Fig. 4A, B) with both manual injection and CED administration methods.
Fig. 4.
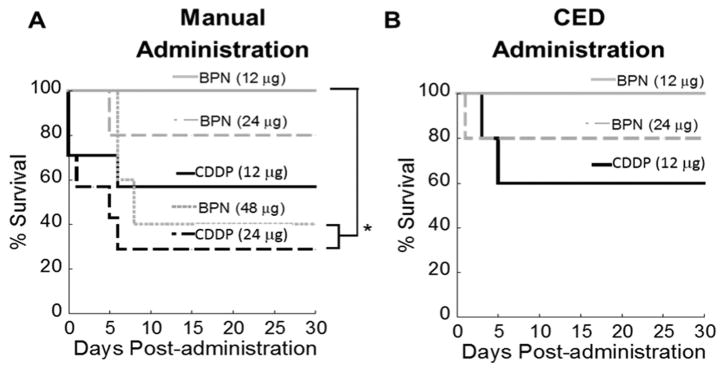
In vivo toxicity of CDDP and CDDP-BPN following local administration. (A) Kaplan-Meier survival curve of CDDP and CDDP-BPN (N ≥ 5 for each group) administered into healthy rat brain using (A) manual injection or (B) CED. *p < 0.05 denotes the statistically significant difference.
3.6. Efficacy in an aggressive rat brain tumor model
We next evaluated the ability of CDDP-BPN, CDDP-UPN, and CDDP (each delivered by CED at a total dose of 12 μg CDDP) to enhance survival of F98 tumor-bearing rats, with one group receiving NS alone as control (Fig. 5A). F98 forms highly aggressive tumors that mimic many of the hallmarks of human GBM, including a highly invasive pattern of growth and overexpression of protein-based tumor markers such as PDGFβ and EGFR [36]. Moreover, F98 glioma is lethal following intracranial inoculation of only 500 cells in Fischer rats [37] due to its low im-munogenicity; we have extensive experience with implanting the F98 glioma and have had 100% tumor inoculation efficiency.
Fig. 5.
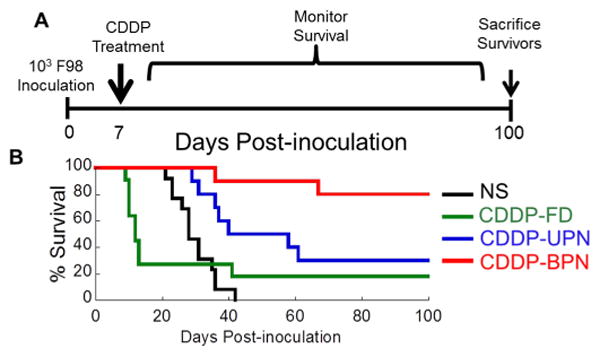
In vivo survival of tumor-bearing rats following CED of CDDP in varying formulation. (A) Tumor inoculation and CDDP treatment scheme for investigating long-term survival of a rat model of orthotopic brain tumor. (B) Rats inoculated with 1 × 103 F98 rat glioma cells were treated on Day 7 following the inoculation with CED of normal saline (NS), CDDP, CDDP-UPN or CDDP-BPN. Kaplan-Meier survival curve depicts significantly improved (p < 0.05) long-term survival of animals treated with CDDP-BPN as compared to all other treatment groups.
In the first study, animals were inoculated with 1 × 103 F98 cells and subsequently treated after tumor establishment (on day 7). Control animals that did not receive CDDP (infusion of NS alone) had a median survival of 28 days with 0% long-term survivors (Figs. 5B, S3A and Table 2). Animals treated with CDDP had a median survival of only 12 days, asthe majority of rats (> 50%) demonstrated seizure-like symptoms and required humane euthanization (Fig. 5B and Table 2). Treatment with CDDP-UPN significantly enhanced median survival (40 days) compared to CDDP treatment. However, the majority of rats treated with CDDP-UPN eventually died (70% mortality; Fig. 5B and Table 2) due to tumor growth and significant brain herniation (Fig. S3B). On the other hand, 80% of animals treated with CDDP-BPN were long-term survivors, which was statistically significant (p < 0.05) compared to all other treatment groups (Fig.5B and Table 2). Long-term survivors displayed no obvious neuropathological symptoms 100 days after tumor inoculation (S. Video 1). Histopathological analysis of brains harvested from survivors revealed that there was no tumor remnant (Fig. S3C, S3F). Brain tissues from surviving rats exhibited various levels of ventricular distension, which is often observed and generally well managed by ventriculoperitoneal shunts in brain tumor patients [38].
Table 2.
Summary of in vivo anti-tumor efficacy in anorthotopic rat F98 brain tumor model following CED of varying formulations of CDDP.
| Treatment | Rats per group (N) | Survival time (days) | p-Value compared to CDDP-BPN | |
|---|---|---|---|---|
|
|
||||
| Range | Median | |||
| NS | 13 | 21–42 | 28 | p < 0.001 |
| CDDP | 11 | 9–100<a | 12 | p < 0.005 |
| CDDP-UPN | 10 | 29–100<a | 40 | p < 0.05 |
| CDDP-BPN | 10 | 36–100<a | N/Ab | N/A |
Presence of long-term survivals (i.e. >100 days).
Median survival could not be determined for the CDDP-BPN group, as 80% of the treated animals survived the study period of 100 days.
We further tested the therapeutic efficacy of CDDP-BPN administered by CED against orthotopic F98 rat brain tumors established by inoculating a higher number of cells (1 × 105) (Fig. S4A). Even in these more advanced tumors, treatment with CDDP-BPN cured a majority of animals (60% long-term survivors). The brains of surviving rats, processed 120 days after initial tumor inoculation, demonstrated no signs of remaining tumor. Of note, one animal in this group required early euthanization, which was likely due to CDDP-related toxicity. In contrast, non-treated control animals and animals treated with focused radiotherapy alone had median survival times of 20 and 28 days, respectively (Fig. S4B, S4C).
4. Discussion
Improving the distribution of a chemotherapeutic within the tumor parenchyma following intracranial administration is essential for achieving improved therapeutic outcome [23,39], but current drug delivery strategies have been unable to effectively address this requirement. Small molecule drugs administered directly into the brain fail to distribute as they are rapidly metabolized or eliminated [39]. On the other hand, while conventionally designed drug delivery NP avoid rapid clearance, they remain largely immobilized within the tumor extracellular space at the immediate vicinity of the point of administration [25,26]. We carefully engineered CDDP-BPN by tailoring its physico-chemical characteristics in order to achieve highly uniform and widespread distribution throughout the brain tumor tissues [22,29,39]. These densely PEGylated CDDP-BPN provide sustained delivery of high CDDP concentrations to a large population of tumor cells in the tumor cores and likely at the infiltrating ends, thereby decreasing the tumor recurrence rates and improving therapeutic outcomes.
In order to formulate CDDPNP possessing highly PEGylated surfaces, we have conjugated PEG polymers to PAA at a high PEG:PAA ratio of 10. However, effective CDDP complexation into a PAA-based NP is dependent on the number of available carboxyl groups for the formation of coordination bonds [40]. Hence, we anticipated that the conjugation of a large number of PEG chains to the carboxyl groups of PAA would compromise CDDP loading. Employing a strategy previously described by our group to improve NP condensation [31,41,42], a small weight % (10%) of un-PEGylated PAA was incorporated into the CDDP-BPN formulation. This provided additional carboxyl groups that bind with CDDP, thereby improving the CDDP loading (Table S1). A dense PEG layeronthe resulting CDDP-BPN was confirmed by the near neutral surface ζ-potential and the ability of the NP to rapidly diffuse ex vivo and distribute in vivo within rat brain tissues. This is in good agreement with our previous findings with various BPN formulations specifically designed for the delivery of different types of chemotherapeutics [22, 29] and nucleic acids [31,42].
CDDP is known for its high anti-tumor potency, but its applicability for treating human GBM has been limited due to the significant toxicities that arise when locally or systemically administered [8]. Two clinical studies to date have investigated local administration of CDDP for treating malignant glioma and managed CDDP-associated toxicity, but it was achieved only at low CDDP dosages where therapeutic benefit is negligible [15,43]. Controlling the release kinetics of CDDP has been shown to alleviate its toxicities in a variety of tumor models by preventing the exposure of cytotoxic concentrations to the surrounding healthy tissues [44–47]. Here, both CDDP-UPN and CDDP-BPN formulations exhibited sustained release of CDDP over 24 h in ACSF, likely triggered by the presence of chloride, calcium, and magnesium ions that compete with CDDP for carboxylate coordinate bonds [48]. However, less overall percentage of encapsulated CDDP was released from CDDP-BPN (∼60%) compared to CDDP-CP (∼80%). We speculate that the discrepancy may be attributed to the dense surface PEG coatings of CDDP-BPN that interfere with an access of ions into the core of the particles. Overall, we confirmed that while locally administered CDDP-BPN were well tolerated by rats, dose-matched free CDDP led to significant acute toxicity. We also note that CDDP delivered in a NP with sustained release enables the administration of significantly higher overall doses of CDDP that can further lead to improved therapeutic efficacy. Given that CDDP-associated toxicities at therapeutically relevant doses have led to the early termination of several clinical trials, the improved safety profile of CDDP provided by a BPN formulation opens a path to reevaluate this highly potent chemotherapeutic for treating GBM [12,49].
The expected benefit of localized chemotherapeutic delivery in providing therapeutically effective concentrations within the tumor has resulted in the clinical development of the Gliadel® wafer [50]; however, limited diffusion of the encapsulated drugs away from the wafer has minimized its therapeutic efficacy [16]. Here, we characterized the distribution of CDDP NP using two, well-detailed strategies, including manual injection [24] and CED [23,24]. When administered using a manual injection, CDDP-BPN achieved a higher Vd within the rat striatum as compared to that of the conventionally designed CDDP-UPN. However, a significant fraction of injected CDDP-BPN was found in the WMT, likely due to fluid backflow resulted from the pressure buildup mediated by rapid injection. When administered using CED, CDDP-BPN was able to harness the advantages of the pressure-driven fluid flowprovidedbyCED andachieved more homogeneous and significantly greater Vd within the striatum while minimizing its presence in WMT.
CED has been widely employed in clinical trials for intracranial administration of therapeutics following tumor resection of patients diagnosed with high grade gliomas [51–53]. These studies were conducted under the notion that CED can achieve therapeutic distribution up to centimeters away from the administration site [54]; however, investigational studies following the failure of these clinical trials have revealed that the distribution remains poor and nonhomogeneous [55]. Similarly, we demonstrated that conventional, un-PEGylated nanoparticles (i.e. CDDP-UPN) administered via CED distributed poorly in the rat brain, and thus provided only marginal therapeutic benefit over free CDDP. In contrast, CED of CDDP-BPN, due to its unique ability to efficiently percolate brain tissue, resulted in widespread therapeutic distribution within the rat striatum, thereby leading to a markedly greater survival compared to all other conditions tested. The translational applicability of this CDDP NP platform for treatment of tumors has been established by a similar, PEG coated, peptide-based CDDP NP which has demonstrated promising Phase I/II results and is currently under investigation in a Phase III clinical trial for pancreatic cancer [56]. We note that the PEGylated CDDP NP tested in this clinical study possess diameters of ∼30 nm, which is significantly smaller than the CDDP-BPN detailed in this study. However, it is yet to be determined whether the surface PEG density on these particles is high enough to provide efficient penetration through the brain tissue, as achieved by CDDP-BPN. Importantly, prior studies have demonstrated that the extent of distribution is far more dependent upon the surface chemistry of the nanoparticle rather than their sizes [57], as long as particle diameters remain small enough to fit through the brain extracellular space [29]. Overall, we anticipate that local, intratumoral administration of CDDP-BPN via CED may provide a promising adjuvant therapy for patients diagnosed with high grade gliomas.
5. Conclusion
The development of a CDDP-BPN addresses two major concerns pertaining to the use of CDDP as a chemotherapeutic agent and enables us to harness the potency of CDDP for the treatment of GBM. Namely, administering CDDP-BPN using CED (1) reduces the inherent toxicities associated with CDDP and (2) delivers high overall concentrations of CDDP throughout the tumor bulk, yielding improved therapeutic efficacy. As a therapeutic platform, we anticipate that CDDP-BPN delivered using CED can be combined with additional adjuvant therapies such as radiotherapy that may further improve the treatment and outcome of patients with GBM.
Supplementary Material
Acknowledgments
The funding for this work was provided by the National Institutes of Health (R01CA164789, R01EB020147, R01CA197111, R01CA204968 and P30EY001765) and Focused Ultrasound Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also thank B. Schuster for his help in high-throughput MPT analysis.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jconrel.2017.03.007.
References
- 1.Stupp R, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glio-blastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Zhang J, Stevens MF, Bradshaw TD. Temozolomide: mechanisms of action, repair and resistance. Curr Mol Pharmacol. 2012;5(1):102–114. doi: 10.2174/1874467211205010102. [DOI] [PubMed] [Google Scholar]
- 3.Trinh VA, Patel SP, Hwu WJ. The safety of temozolomide in the treatment of malignancies. Expert Opin Drug Saf. 2009;8(4):493–499. doi: 10.1517/14740330902918281. [DOI] [PubMed] [Google Scholar]
- 4.Chamberlain MC. Temozolomide: therapeutic limitations in the treatment of adult high-grade gliomas. Expert Rev Neurother. 2010;10(10):1537–1544. doi: 10.1586/ern.10.32. [DOI] [PubMed] [Google Scholar]
- 5.Ostermann S, et al. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin Cancer Res. 2004;10(11):3728–3736. doi: 10.1158/1078-0432.CCR-03-0807. [DOI] [PubMed] [Google Scholar]
- 6.Hegi ME, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 7.Palanichamy K, Chakravarti A. Combining drugs and radiotherapy: from the bench to the bedside. Curr Opin Neurol. 2009;22(6):625–632. doi: 10.1097/WCO.0b013e3283327d33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oberoi HS, et al. Nanocarriers for delivery of platinum anticancer drugs. Adv Drug Deliv Rev. 2013;65(13–14):1667–1685. doi: 10.1016/j.addr.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hargrave DR, Zacharoulis S. Pediatric CNS tumors: current treatment and future directions. Expert Rev Neurother. 2007;7(8):1029–1042. doi: 10.1586/14737175.7.8.1029. [DOI] [PubMed] [Google Scholar]
- 10.Kim IH, et al. Radiotherapy followed by adjuvant temozolomide with or without neoadjuvant ACNU-CDDP chemotherapy in newly diagnosed glioblastomas: a prospective randomized controlled multicenter phase III trial. J Neuro-Oncol. 2011;103(3):595–602. doi: 10.1007/s11060-010-0427-y. [DOI] [PubMed] [Google Scholar]
- 11.Silvani A, et al. Cisplatinum and BCNU chemotherapy in primary glioblastoma patients. J Neuro-Oncol. 2009;94(1):57–62. doi: 10.1007/s11060-009-9800-0. [DOI] [PubMed] [Google Scholar]
- 12.Grossman SA, et al. Phase III study comparing three cycles of infusional carmustine and cisplatin followed by radiation therapy with radiation therapy and concurrent carmustine in patients with newly diagnosed supratentorial glioblastoma multiforme: Eastern Cooperative Oncology Group Trial 2394. J Clin Oncol. 2003;21(8):1485–1491. doi: 10.1200/JCO.2003.10.035. [DOI] [PubMed] [Google Scholar]
- 13.Cepeda V, et al. Biochemical mechanisms of cisplatin cytotoxicity. Anti Cancer Agents Med Chem. 2007;7(1):3–18. doi: 10.2174/187152007779314044. [DOI] [PubMed] [Google Scholar]
- 14.de Groot JF, et al. Phase II study of carboplatin and erlotinib (Tarceva, OSI-774) in patients with recurrent glioblastoma. J Neuro-Oncol. 2008;90(1):89–97. doi: 10.1007/s11060-008-9637-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheleg SV, et al. Local chemotherapy with cisplatin-depot for glioblastoma multiforme. J Neuro-Oncol. 2002;60(1):53–59. doi: 10.1023/a:1020288015457. [DOI] [PubMed] [Google Scholar]
- 16.Fung LK, et al. Pharmacokinetics of interstitial delivery of carmustine, 4-hydroperoxycyclophosphamide, and paclitaxel from a biodegradable polymer implant in the monkey brain. Cancer Res. 1998;58(4):672–684. [PubMed] [Google Scholar]
- 17.Bouvier G, et al. Intratumoral chemotherapy with multiple sources. Ann N Y Acad Sci. 1988;531:213–214. doi: 10.1111/j.1749-6632.1988.tb31830.x. [DOI] [PubMed] [Google Scholar]
- 18.Bouvier G, et al. Stereotactic administration of intratumoral chronic chemotherapy of recurrent malignant gliomas. Appl Neuropsychol. 1987;50(1–6):223–226. doi: 10.1159/000100713. [DOI] [PubMed] [Google Scholar]
- 19.Rousseau J, et al. Efficacy of intracerebral delivery of cisplatin in combination with photon irradiation for treatment of brain tumors. J Neuro-Oncol. 2010;98(3):287–295. doi: 10.1007/s11060-009-0074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huo T, et al. Preparation, biodistribution and neurotoxicity of liposomal cisplatin following convection enhanced delivery in normal and F98 glioma bearing rats. PLoS One. 2012;7(11):e48752. doi: 10.1371/journal.pone.0048752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olivi A, et al. Direct delivery of platinum-based antineoplastics to the central nervous system: a toxicity and ultrastructural study. Cancer Chemother Pharmacol. 1993;31(6):449–454. doi: 10.1007/BF00685034. [DOI] [PubMed] [Google Scholar]
- 22.Nance E, et al. Brain-penetrating nanoparticles improve paclitaxel efficacy in malignant glioma following local administration. ACS Nano. 2014;8(10):10655–10664. doi: 10.1021/nn504210g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou J, et al. Highly penetrative, drug-loaded nanocarriers improve treatment of glioblastoma. Proc Natl Acad Sci U S A. 2013;110(29):11751–11756. doi: 10.1073/pnas.1304504110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin D, et al. Convection-enhanced delivery improves distribution and efficacy of tumor-selective retroviral replicating vectors in a rodent brain tumor model. Cancer Gene Ther. 2013;20(6):336–341. doi: 10.1038/cgt.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Voges J, et al. Imaging-guided convection-enhanced delivery and gene therapy of glioblastoma. Ann Neurol. 2003;54(4):479–487. doi: 10.1002/ana.10688. [DOI] [PubMed] [Google Scholar]
- 26.MacKay JA, Deen DF, Szoka FC., Jr Distribution in brain of liposomes after convection enhanced delivery; modulation by particle charge, particle diameter, and presence of steric coating. Brain Res. 2005;1035(2):139–153. doi: 10.1016/j.brainres.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 27.Zamecnik J. The extracellular space and matrix of gliomas. Acta Neuropathol. 2005;110(5):435–442. doi: 10.1007/s00401-005-1078-5. [DOI] [PubMed] [Google Scholar]
- 28.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nance EA, et al. A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Sci Transl Med. 2012;4(149) doi: 10.1126/scitranslmed.3003594. 149ra119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feinstein AR. Principles of Medical Statistics. Chapman and Hall/CRC. 2001 [Google Scholar]
- 31.Mastorakos P, et al. Highly PEGylated DNA nanoparticles provide uniform and widespread gene transfer in the brain. Adv Healthc Mater. 2015;4(7):1023–1033. doi: 10.1002/adhm.201400800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Recinos VR, et al. Combination of intracranial temozolomide with intracranial carmustine improves survival when compared with either treatment alone in a rodent glioma model. Neurosurgery. 2010;66(3):530–537. doi: 10.1227/01.NEU.0000365263.14725.39. discussion 537. [DOI] [PubMed] [Google Scholar]
- 33.Fournier E, et al. Therapeutic effectiveness of novel 5-fluorouracil-loaded poly(methylidene malonate 2.1.2)-based microspheres on F98 glioma-bearing rats. Cancer. 2003;97(11):2822–2829. doi: 10.1002/cncr.11388. [DOI] [PubMed] [Google Scholar]
- 34.Schuster BS, et al. Particle tracking in drug and gene delivery research: state-of-the-art applications and methods. Adv Drug Deliv Rev. 2015;30(91):70–91. doi: 10.1016/j.addr.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raghavan R, et al. Convection-enhanced delivery of therapeutics for brain disease, and its optimization. Neurosurg Focus. 2006;20(4):E12. doi: 10.3171/foc.2006.20.4.7. [DOI] [PubMed] [Google Scholar]
- 36.Barth RF, Kaur B. Rat brain tumor models in experimental neuro-oncology: the C6, 9L, T9, RG2, F98, BT4C, RT-2 and CNS-1 gliomas. J Neuro-Oncol. 2009;94(3):299–312. doi: 10.1007/s11060-009-9875-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Volovitz I, et al. Split immunity: immune inhibition of rat gliomas by subcutaneous exposure to unmodified live tumor cells. J Immunol. 2011;187(10):5452–5462. doi: 10.4049/jimmunol.1003946. [DOI] [PubMed] [Google Scholar]
- 38.Pinto FC, et al. Role of endoscopic third ventriculostomy and ventriculoperitoneal shunt in idiopathic normal pressure hydrocephalus: preliminary results of a randomized clinical trial. Neurosurgery. 2013;72(5):845–853. doi: 10.1227/NEU.0b013e318285b37c. discussion 853-4. [DOI] [PubMed] [Google Scholar]
- 39.Allard E, Passirani C, Benoit JP. Convection-enhanced delivery of nanocarriers for the treatment of brain tumors. Biomaterials. 2009;30(12):2302–2318. doi: 10.1016/j.biomaterials.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 40.Nishiyama N, et al. Preparation and characterization of self-assembled polymer-metal complex micelle from cis-dichlorodiammineplatinum(II) and poly(ethylene glycol)-poly(alpha,beta-aspartic acid) block copolymer in an aqueous medium. Langmuir. 1999;15(2):377–383. [Google Scholar]
- 41.Suk JS, et al. Lung gene therapy with highly compacted DNA nanoparticles that overcome the mucus barrier. J Control Release. 2014;178C:8–17. doi: 10.1016/j.jconrel.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mastorakos P, et al. Biodegradable DNA nanoparticles that provide widespread gene delivery in the brain. Small. 2015;12(5):678–685. doi: 10.1002/smll.201502554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bouvier G, et al. Direct delivery of medication into a brain tumor through multiple chronically implanted catheters. Neurosurgery. 1987;20(2):286–291. doi: 10.1227/00006123-198702000-00015. [DOI] [PubMed] [Google Scholar]
- 44.Uchino H, et al. Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br J Cancer. 2005;93(6):678–687. doi: 10.1038/sj.bjc.6602772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dhar S, et al. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc Natl Acad Sci U S A. 2011;108(5):1850–1855. doi: 10.1073/pnas.1011379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paraskar AS, et al. Harnessing structure-activity relationship to engineer a cisplatin nanoparticle for enhanced antitumor efficacy. Proc Natl Acad Sci U S A. 2010;107(28):12435–12440. doi: 10.1073/pnas.1007026107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Plummer R, et al. A phase I clinical study of cisplatin-incorporated polymeric micelles (NC-6004) in patients with solid tumours. Br J Cancer. 2011;104(4):593–598. doi: 10.1038/bjc.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dudev T, Lim C. Effect of carboxylate-binding mode on metal binding/selectivity and function in proteins. Acc Chem Res. 2007;40(1):85–93. doi: 10.1021/ar068181i. [DOI] [PubMed] [Google Scholar]
- 49.Buckner JC, et al. Phase III trial of carmustine and cisplatin compared with carmustine alone and standard radiation therapy or accelerated radiation therapy in patients with glioblastoma multiforme: North Central Cancer Treatment Group 93-72-52 and Southwest Oncology Group 9503 Trials. J Clin Oncol. 2006;24(24):3871–3879. doi: 10.1200/JCO.2005.04.6979. [DOI] [PubMed] [Google Scholar]
- 50.Fung LK, et al. Chemotherapeutic drugs released from polymers: distribution of 1,3-bis(2-chloroethyl)-1-nitrosourea in the rat brain. Pharm Res. 1996;13(5):671–682. doi: 10.1023/a:1016083113123. [DOI] [PubMed] [Google Scholar]
- 51.Vogelbaum M, Inc IT. Topotecan Using Convection-Enhanced Delivery (CED) in High Grade Glioma. [2015 April 14];2014 Available from: https://clinicaltrials.gov/ct2/show/NCT02278510.
- 52.Elder J. Carboplatin in Treating Patients with Recurrent High-grade Gliomas. [2015 April 14];2012 Available from: https://clinicaltrials.gov/ct2/show/NCT01644955.
- 53.Inc IT. The PRECISE Trial: Study of IL13-PE38QQR Compared to GLIADEL Wafer in Patients With Recurrent Glioblastoma Multiforme. [2015 April 14];2004 Available from: https://clinicaltrials.gov/ct2/show/NCT00076986.
- 54.Chaichana KL, Pinheiro L, Brem H. Delivery of local therapeutics to the brain: working toward advancing treatment for malignant gliomas. Ther Deliv. 2015;6(3):353–369. doi: 10.4155/tde.14.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sampson JH, et al. Poor drug distribution as a possible explanation for the results of the PRECISE trial. J Neurosurg. 2010;113(2):301–309. doi: 10.3171/2009.11.JNS091052. [DOI] [PubMed] [Google Scholar]
- 56.Su W, et al. Phase I/ii study of Nc-6004, a novel micellar formulation of cisplatin, in combination with gemcitabine in patients with pancreatic cancer in Asia - results of phase I. Ann Oncol. 2012;23:247. [Google Scholar]
- 57.Chen MY, et al. Surface properties, more than size, limiting convective distribution of virus-sized particles and viruses in the central nervous system. J Neurosurg. 2005;103(2):311–319. doi: 10.3171/jns.2005.103.2.0311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.