


Abstract
The cell type-specific, mutually-exclusive alternative splicing of the fibroblast growth factor receptor 2 (FGFR2) pre-mRNA is tightly regulated. A sequence termed ISAR (intronic splicing activator and repressor) has been implicated as an important cis regulatory element in both activation of exon IIIb and repression of exon IIIc splicing in epithelial cells. In order to better understand how this single sequence could have dual roles, we transfected minigenes containing a series of 2-bp mutations in the 18 3′-most nucleotides of ISAR that we refer to as the ISAR core. Transfection of cells with dual-exon (IIIb and IIIc) minigenes revealed that mutation of terminal sequences of the core led to decreased exon IIIb inclusion and increased exon IIIc inclusion. Transfection of cells with single-exon IIIb minigenes and single-exon IIIc minigenes revealed that mutation of terminal sequences of the ISAR core led to decreased exon IIIb inclusion and increased exon IIIc inclusion, respectively. Nucleotides of the ISAR core responsible for exon IIIb activation appear to overlap very closely with those required for exon IIIc repression. We describe a model in which ISAR and a 5′ intronic sequence known as IAS2 form a stem structure required for simultaneous exon IIIb activation and exon IIIc repression.
INTRODUCTION
Alternative splicing of pre-mRNAs is a method commonly used by metazoans to produce several similar but distinct proteins from a single gene transcript. This mechanism introduces a post-transcriptional level of gene regulation. In both developing and adult tissues that exhibit both epithelial and stromal compartments, alternative splicing of the fibroblast growth factor receptor 2 (FGFR2) pre-mRNA results in partitioning one protein isoform in epithelial cells (FGFR2IIIb) and a different isoform in stromal (mesenchymal) cells (FGFR2IIIc) (1–7). By regulating inclusion of exons 8 (IIIb) and 9 (IIIc) in a mutually exclusive manner, the splicing machinery can produce an mRNA encoding FGFR2IIIb, which binds to stromal-derived FGF-7 (also called keratinocyte growth factor) and FGF-10, or FGFR2IIIc that rejects FGF-7 and binds to other isoforms of FGF (8,9). This splicing decision is very important in cell-to-cell communication during early development as well as for maintenance of homeostasis among cellular compartments in the adult (for review see 10). Alterations in splicing of exons 8 and 9 in FGFR2 pre-mRNA are a hallmark in the progression of prostate cancer (8,11,12).
The rat Dunning prostate tumor is composed of well-defined epithelial and stromal compartments and is a model slow-growing, relatively benign and androgen-responsive tumor. Cells derived from the epithelial compartment of the tumor, DTE cells, express only FGFR2IIIb. When injected subcutaneously into a rat host in the absence of stromal cells, or when injected with stromal cells into castrated hosts, the DTE tumor progresses to an androgen-independent, fast-growing tumor known as the AT tumor. This tumor is composed of one cell type (AT3) that initially expresses only the IIIc isoform of FGFR2 and in later stages loses FGFR2 gene expression completely (8). Lack of FGFR2IIIb expression is thought to sever one point of normal communication between the stromal and epithelial compartments. Restoration of the FGFR2 signal by transfection actually slows the growth and leads to differentiation of the AT tumor (11). Therefore, defining the mechanisms that regulate FGFR2 splicing may have important implications for preventing prostate cancer progression.
Splicing of pre-mRNAs is a multi-step process involving several small ribonuclear proteins and other protein factors (13). While tissue- and cell-specific splicing factors in lower metazoans, such as Drosophila, have been described that can activate or repress exon inclusion in a cell-specific manner (14), few tissue-specific splicing factors have been identified in mammalian systems. Rather, regulated inclusion and exclusion of mammalian exons generally results from subtle changes in the levels of several factors that bind to either exonic or intronic regulatory sequences (15–17). Binding of members of the SR protein family to both exonic and intronic sequences has been shown to regulate splice site selection (15–17). SF2/ASF and hnRNPA1 are two such factors that have been shown to function antagonistically both in vitro and in vivo to influence the use of alternative splice sites. Regulators of splice site recognition necessarily must either promote or inhibit the ability of the splicing machinery to recognize either the 5′ splice site, the polypyrimidine tract, the branch point or the 3′ splice site.
A 57-nt element located upstream of exon IIIc in the FGFR2 gene was previously characterized (6) and shown to be necessary for both activation of exon IIIb and repression of exon IIIc splicing in an epithelial cell type. For this reason, the sequence was named ISAR for intronic splicing activator and repressor (Fig. 1). The 18 3′-most nucleotides within this element were previously found to be critical for its activity. In this study, we sequentially mutated dinucleotide components of ISAR and tested the effects of these mutations on splicing of transfected minigenes. We sought to determine whether mutations that decrease splicing activation have a coordinate effect on efficiency of splicing repression. We discovered that mutation of sequences participating in a previously proposed stem structure between IAS2 and ISAR (3) not only decreased exon IIIb inclusion, but also increased exon IIIc inclusion in DTE cells.
Figure 1.

Regulation of FGFR2 splicing in DTE and AT3 cells. In DTE cells, a model epithelial cell type which expresses exclusively exon IIIb from the native FGFR2 gene, the ISAR sequence (shaded box) upstream of exon IIIc functions to activate exon IIIb inclusion (+) and repress exon IIIc inclusion (–). Exon 7 (IIIa or C1) is spliced to exons 8 (IIIb) and 10 (C2) in DTE cells. AT3 cells splice exclusively exon 7 to exons 9 (IIIc) and 10.
MATERIALS AND METHODS
Plasmid constructions
Plasmid DNA constructions used in this study were made by standard cloning methods as previously described (6,7,18). Minigenes PI11-FS-Not/Cla, PI11-IIIb-plus and PI11-IIIc-plus have been previously described (6) and comprised the backbone of all minigenes used in this study. Minigenes NC-ISAR 1–9 resulted from ligation of 38-bp ISAR oligos (Fig. 2A) into PI11-FS-Not/Cla-ISAR digested by NotI and ClaI. PI11-IIIb-plus-Not/Cla and PI11-IIIc-plus-Not/Cla resulted from PCR of pI11-IIIb-plus and PI11-IIIc-plus respectively with primers NdeI/NotI-F (5′-CCGGCATATGGCGGCCGCCAAACAAATTCAAAGAGAAC-3′) and NsiI/ClaI-R (5′-CCGGATGCATATCGATGCGATTGAACACATGGAAAA-3′), digestion of these products with NdeI and NsiI, and cloning into the NdeI and NsiI sites in pBlue-FS. The minigene sequences were reNSmoved with SpeI and XhoI and cloned into the XbaI and XhoI sites of pI11-IIIb-plus and pI11-IIIc-plus to generate pI11-IIIb-plus-Not/Cla and pI11-IIIc-plus-Not/Cla respectively (Fig. 3). Minigenes BFS1-9 and CFS1-9 were constructed by annealing oligos ISAR 1–9 into pI-11-IIIb-plus-Not/Cla and pI-11-IIIc-plus-Not/Cla respectively. Minigene C-IAS-PacAsc resulted from PCR of PI11-IIIc-plus-Not/Cla with the PI11BCFor (5′-GGCAGCTTCTAGAATAACTCTTGTGGTCTT-3′), IASRev (5′-ACAGGCGCGCCTGCATTAATTAACCACTGGCATGACGCCAGCTGCACCACCAA-3′) and IASFor (5′-GTGGTTAATTAATGCAGGCGCGCCTGTTCAAAGTGCTTGAAGATTATCTTCCAC-3′), PI11BCRev (5′-ATGCATATCGATGCGATTGAACACATGGAA-3′) primer pairs followed by ligation into PI11-IIIc-plus-Not-Cla digested by XbaI and ClaI. Minigenes IIIcPA-WT and -MUT7 were constructed by ligation of oligos IASWT, and IAS7 into C-IAS-PA respectively digested by PacI and AscI (see Fig. 8A). Compensatory mutation-containing minigene IIIcPA-MUT7ISAR7 was constructed by ligation of oligo ISAR-7 into minigene IIIcPA-MUT7 that had been digested by NotI and ClaI.
Figure 2.



Terminal sequences in the ISAR core are critical for regulation of splicing. (A) pI11 is a previously described adenovirus splicing construct adapted for eukaryotic expression in which the adenoviral PIP11 exons can be spliced to exons cloned into the XbaI and XhoI sites (6). pI11-FS-Not/Cla+ is a previously described pI11 splicing construct containing FGFR2 genomic sequence cloned into the XbaI and XhoI sites and NotI and ClaI sites flanking the ISAR sequence upstream of exon IIIc (6). Sequences cloned into the NotI and ClaI sites are listed below the pI11-FS-Not/Cla+ schematic. SAR5′ is a previously described sequence and utilized as a negative control of splicing regulation (6). SAR-20 is a previously described sequence utilized as a positive control for splicing regulation (6). Numbers 1–9 indicate constructs identical to SAR-20 with the exception of the 2-bp mutations indicated. CMV indicates a cytomegalovirus promoter. T7 and SP6 indicate primer sites utilized for subsequent RT–PCR analysis. (B) Mutants numbered 1 and 5–9 display significant loss in splicing regulation. RT–PCR of RNA derived from DTE cells stably transfected by indicated FGFR2 minigenes. From our minigene constructions, four possible spliced products are predicted. When both exon IIIb and IIIc are spliced to the flanking PIP11 exons, a 525-nt U-3B-3C-D product will result. When either exon IIIb or IIIc is spliced to the flanking PIP11 exons, a 377-nt U-3B-D or 380-nt U-3C-D product will result respectively. When neither exon IIIb nor IIIc is spliced to the flanking PIP11 exons (i.e. both PIP11 exons spliced together), a 232-nt product results. Products were either loaded onto non-denaturing polyacrylamide without digestion (U = undigested) or following digestion with AvaI (A) or HincII (H). Products containing exon IIIb (U-3B-3C-D and U-3B-D) are digested by AvaI while products containing exon IIIc (U-3B-3C-D and U-3C-D) are digested by HincII. Percentages shown [UBD/(UBD+UCD)] were calculated by quantitating the intensity of the band at 377-bp (U-3B-D) following HincII digestion and dividing by the intensity of the U-3B-D band and the band at 380-bp (U-3C-D) following digestion by AvaI. Sizes of bands in bp are indicated at the left of the phosphorimage. Data presented is the mean of three independent experiments and ‘Std. dev’ represents the standard deviation from this mean. (C) Graphic representation of data from (B).
Figure 3.

Single-exon minigene controls and 2-bp ISAR mutants. The indicated pI11-IIIb and pI11-IIIc minigenes (Materials and Methods) were utilized to determine sequences involved in exon IIIb activation and exon IIIc repression. CMV indicates the cytomegalovirus promoter. T7 and SP6 indicate primer sites utilized for subsequent RT–PCR analysis. PIP11 boxes indicate adenoviral exons described in Figure 2 that can be spliced to exons cloned into the XbaI and XhoI sites. pI11-IIIb-Not/Cla+ represents the parent exon IIIb-containing minigene while pI11-IIIc-Not/Cla+ represents the parent exon IIIc-containing minigene. BLUE and SAR5′ represent sequences previously shown to confer no splicing regulation (6) and were utilized as negative controls. SAR-20 represents a sequence previously shown to provide full splicing regulation and was the positive control. Sequences 1–9 are identical to SAR-20 except for the mutations indicated (in italics) and are identical to the sequences cloned into the dual-exon minigenes described in Figure 2.
Figure 8.

Effect of mutations in rIAS2 that disrupt and restore putative stem structure between ISAR and rIAS2. (A) Schematic of minigene constructs. CMV indicates the cytomegalovirus promoter. T7 and SP6 indicate primer sites utilized for subsequent RT–PCR analysis. pI11 represents the previously described 2-exon splicing construct (Figs 2 and 6). IIIC-PacAsc-WT represents the exon IIIc-containing minigene with PacI and AscI sites flanking rIAS2. IIIC-PacAsc-MUT7 contains the indicated 2-bp mutation in rIAS2 that is compensatory to the ISAR7 mutation but contains a wild-type ISAR sequence. (B) Left, transfection of single-exon IIIc minigenes into DTE cells revealed that a 2-nt substitution in rIAS2 position-7 caused an increase in exon IIIc usage similar to placing the 2-nt substitution in position-7 of ISAR. Placing the rIAS2 mutation together with the compensatory ISAR mutation in minigene IIIC-PacAsc-MUT7-ISAR-7 leads to restoration of splicing regulation. Quantitation of inclusion ratios and standard deviations shown were derived from phosphorimager analysis of at least three independent experiments. The phosphorimage shown is representative of one experiment. Phosphorimage of electrophoresed RT–PCR of RNA from DTE cells following stable transfection with indicated single-exon IIIc minigenes. Exon IIIc inclusion and standard deviations are shown below the lanes. Right, normalized graphical representation of increase in exon IIIc inclusion from data on left after arbitrarily setting the 23% inclusion of the IIIC-Pac-Asc-WT minigene to 0% and normalizing the increases in inclusion data from the other minigenes proportionately.
RT–PCR and restriction enzyme analysis of minigene expression product
Total RNA was isolated from transfected DTE and AT3 cells using the Ultraspec RNA isolation reagent (Biotecx, Houston, TX) as per the manufacturer’s instructions. Total RNA (5 µg) was reversed transcribed with a random 6mer as primer in a total reaction volume of 25 µl. A 5-µl portion was used for subsequent PCR reactions. The PCR was performed at 94°C for 1 min, 60°C for 1 min and 72°C for 1 min for 40 cycles in a total volume of 50 µl and supplementation of the reaction with 10 µCi [α-32P]dCTP. Each stable transfection, RT–PCR and restriction enzyme analysis was carried out at least three times, and the figures displayed are a single representative of the three. In all amplification reactions, a water control and a mock RT control were included, and resulted in no amplified product in all experiments. All products from single exon minigenes were then loaded directly onto 5% non-denaturing polyacrylamide gels and electrophoresed at 100 V for 3–4 h and exposed to Molecular Dynamics phosphorimager screens. All products from dual exon minigenes were either loaded directly onto 5% non-denaturing gels or were incubated with AvaI or HincII restriction enzymes for 1 h prior to loading onto gels. Gels were subjected to autoradiography and the relative intensities of products quantitated by phosphorimager analysis using the ImageQuant program.
Cell culture
Stock cultures of cloned cell lines from the Dunning R3327PAP and R3327AT3 tumors were prepared and maintained by described methods (8).
Mammalian cell transfections
Liposomes were prepared according to manufacturer protocols (Life Technologies, Inc., Gaithersburg, MD). Transfection conditions were optimized for cell density, liposome reagent, liposome volume, DNA amount, time of liposome incubation and time after transfection by varying the conditions and assaying for luminescence following transfection of cells with the pGL-3 Control vector (Promega). Cells (2.5 × 105) were seeded in 6-well plates prior to transfection. DNA vector (2.5 µg) along with 7.5 µl of CellFectin (Life Technologies, Inc.) was used for transfecting DTE cells. DNA (2.5 µg) was mixed with 7.5 µl of liposomes in a total volume of 100 µl serum-free RD for a total of 45 min. Three days post-transfection, cells were selected with geneticin (Gibco) at an active concentration of 400 µg/ml. Stable cell lines were thereafter maintained in the presence of 100 µg/ml geneticin.
RESULTS
Terminal sequences in the ISAR core are required for either exon IIIb activation or exon IIIc repression
We stably transfected cells with minigenes derived from rat FGFR2 genomic sequences to further characterize cis elements required for the regulation of FGFR2 splicing in DTE and AT3 cells. Minigenes were constructed by placing rat FGFR2 genomic sequences into an adenovirus-derived splicing construct pI-11 (Fig. 2A) (6). Splicing of exons IIIb and IIIc was assayed by RT–PCR using primers T7 and SP6. Because exon IIIb contains an AvaI site not present in exon IIIc and because exon IIIc contains two HincII restriction sites not present in exon IIIb, these restriction enzymes were used to distinguish between exon IIIb- and IIIc-containing products following RT–PCR. We constructed both dual-exon (containing both exons IIIb and IIIc) and single-exon minigenes (containing either exon IIIb or exon IIIc). Four possible products are expected from the dual-exon minigenes: products containing the upstream exon (U) spliced to exons IIIb and IIIc and to the downstream adenovirus exon (D) designated U-IIIb-IIIc-D as well as products designated U-IIIb-D, U-IIIc-C2 and U-D. U-IIIb-IIIc-D will hereafter be referred to as the double-inclusion product. U-D will hereafter be referred to as the double-exclusion or skipped product. From single-exon IIIb or IIIc minigenes only two products are possible: the exon-included and exon-excluded products.
Previous studies have shown that stable transfection of DTE cells with minigene pI11-FS-Not/Cla+SAR-20 (that contains 37 nt of ISAR) is able to recapitulate the splicing pattern of the endogenous FGFR2 gene. When the ISAR sequence was deleted or replaced by pBluescript sequences, exon IIIb inclusion decreased while exon IIIc inclusion increased (6). In order to gain a better understanding of the mechanism underlying ISAR regulation, we made a series of minigenes containing 2-bp substitutions in the 18 3′-most nucleotides of ISAR (designated hereafter as the ISAR core) in order to define regions of the core responsible for activation and repression (Fig. 2A). When we transfected cells with PI11-FS-SAR-20, we obtained essentially the same inclusion ratio (87% IIIb, 12% IIIc) as has been previously described (6) (Fig. 2B and C). Mutants 1 and 4–9 showed dramatically higher levels of exon IIIc inclusion and exon IIIb exclusion (all >26% exon IIIc inclusion) (Fig. 2B and C).
Mutations in ISAR affect both exon IIIb activation and exon IIIc repression
In order to determine whether the mutations affected exon IIIb activation or exon IIIc repression, two additional constructs were made that are similar to the previously described PI11-IIIb-plus and PI11-IIIc-plus (6) except for the addition of NotI and ClaI sites 5′ and 3′ of the ISAR sequence: these constructions were designated PI11-IIIb-NC and PI11-IIIc-NC (Fig. 3). We first performed experiments to ensure that the added NotI and ClaI sites did not alter the splicing profile of the single-exon minigenes (Fig. 4). In these experiments, we quantitated inclusion versus exclusion of single exons (IIIb or IIIc depending on minigene construction). The introduction of NotI and ClaI sites did not significantly alter exon inclusion in either the IIIb- or IIIc-containing minigene. We then introduced the same series of 2-bp mutations into the single-exon minigenes as done previously with the dual-exon PI11-FS minigenes. Interestingly, mutations introduced into the 5′ or 3′ ends of the ISAR core again had the most dramatic effect on IIIb inclusion (Fig. 5). Exon IIIb inclusion was 49% with PI11-IIIb-NC-SAR-20, the positive control, and 25% with PI11-IIIb-blue, the negative control. Exon IIIb inclusion decreased most significantly in mutants 1 and 4–9 with inclusion ratios of 15, 15, 28, 19, 6, 10 and 12% respectively. Similar to the dual-exon minigenes, mutants 2 and 3 displayed the least difference in exon inclusion with ratios of 37 and 36% respectively. The greatest effect on splicing regulation was observed in mutations disrupting the core of a previously proposed stem structure formed between ISAR and a previously reported human IAS2 sequence (Fig. 6). We designated the sequence rIAS2 (rat IAS2) to distinguish it from the human sequence (3).
Figure 4.
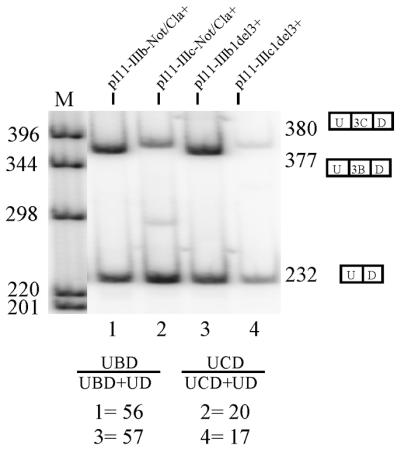
Quantitation of exon inclusion in DTE cells. RT–PCR following transfection of single-exon IIIb and IIIc minigenes into DTE cells revealed that introduction of NotI and ClaI sites flanking the ISAR sequence did not decrease exon IIIb usage or increase exon IIIc usage. pI11-IIIb1del3+ and pI11-IIIc1del3+ are the previously described single-exon minigenes containing either exon IIIb or exon IIIc respectively (6). pI11-IIIb-plus Not/Cla+ and pI11-IIIc-Not/Cla+ minigenes are identical to pI11-IIIb1del3+ and pI11-IIIc1del3+ except that they have NotI and ClaI sites flanking the ISAR sequence. U-3C-D represents the 380 bp exon IIIc included product. U-3B-D represents the 377 bp exon IIIb-included product. U-D represents the 232 bp PIP11 product in which the upstream and downstream PIP11 exons are spliced together without either exon IIIb or exon IIIc. Percentages under UBD/(UBD+UD) were calculated by quantitating the band migrating at 377 bp (U-3B-D) and dividing by the sum of band intensities of U-3B-D and the band migrating at 232 bp (U-D). Percentages under UCD/(UCD+UD) were calculated in the same manner except the intensity of the band migrating at 380 bp (U-3C-D) was quantitated and used for calculation instead of U-3B-D.
Figure 5.

Quantitation of exon inclusion in DTE cells from single-exon IIIb minigenes with 2-bp mutations in the ISAR sequence. Stable transfection of single-exon IIIb minigenes into DTE cells revealed that 2-nt substitutions in the terminal sequences of ISAR core (compare BSAR-20 to lanes 1, 4, 7, 8 and 9) decreased the ratio of exon IIIb inclusion from 49 to <15%. Quantitation of inclusion ratios and standard deviations shown were derived from phosphorimager analysis of at least three independent experiments. The phosphorimage shown is representative of one experiment. (A) Phosphorimage of electrophoresed RT–PCR of RNA from DTE cells following stable transfection with indicated single-exon IIIb minigenes. BBlue and BSAR-20 represent previously described single-exon IIIb-containing minigenes (6) (Fig. 3) utilized as negative and positive controls for exon IIIb inclusion respectively. U-3B-D and U-D represent exon IIIb-included and exon IIIb-excluded products respectively. UBD/(UBD+UD) inclusion percentages and standard deviations were calculated as described in Figure 4. (B) Normalized graphical representation of exon IIIb inclusion from data in (A) after arbitrarily setting the BSAR-20 minigene inclusion to 100% and the other minigenes to fractions thereof.
Figure 6.

A putative stem structure between ISAR and rIAS2. Structure is not drawn to scale. The four lines in the figure connecting the ISAR and rIAS2 regions represent the 21-nt stretch of ISAR and IAS2 involved in the putative stem structure. b represents exon IIIb. c represents exon IIIc. Nucleotides numbers 1–18 at the top of the figure denote ISAR nucleotides involved in the putative stem that were mutated in our study in 2-nt increments. Numbers 1–9 at the bottom of the figure correspond to minigene numbers containing the 2-bp substitutions. ISAR nucleotides are shown from 5′ to 3′ while rIAS2 nucleotides are shown from 3′ to 5′. The bulged UG made by nucleotides 5 and 6 of ISAR is noted.
Mutating the 18 nt of ISAR in the context of the exon IIIc minigenes had a significant although less dramatic effect on exon inclusion compared with the IIIb minigenes (Fig. 7). Inclusion of exon IIIc was 19% with PI11-IIIc-NC-SAR-20, the positive control, and 26% with PI11-IIIc-SAR5′, the negative control (Fig. 7). A significant loss in regulation of splicing (i.e. increase in exon IIIc inclusion) was again observed in mutants 1, 5, 6 and 7 with inclusion ratios of 24, 27, 28 and 29% respectively. This was consistent with an overlapping role for these nucleotides in exon IIIb activation and exon IIIc repression. Similar to dual-exon minigenes, the least effect on exon inclusion was observed in mutants 2 and 3, which contain nucleotide substitutions in or around the predicted bulge of the stem structure (Fig. 6).
Figure 7.

Quantitation of exon inclusion in DTE cells from single-exon IIIc minigenes with 2-bp mutations in the ISAR sequence. Transfection of single-exon IIIc minigenes stably transfected into DTE cells revealed that 2-nt substitutions in terminal sequences of the ISAR core increased the ratio of exon IIIc inclusion from 19 to 29% (Compare C-SAR-20 to lanes 5, 6 and 7). Quantitation of inclusion ratios and standard deviations shown were derived from phosphorimager analysis of at least three independent experiments. The phosphorimage shown is representative of one experiment. (A) Phosphorimage of electrophoresed RT–PCR of RNA from DTE cells following stable transfection with indicated single-exon IIIc minigenes. CSAR5′ and CSAR-20 represent previously described single-exon IIIc-containing minigenes (6) (Fig. 3) utilized as negative and positive controls for exon IIIc inclusion respectively. U-3C-D and U-D represent exon IIIb included and exon IIIb excluded products respectively. UCD/(UCD+UD) inclusion percentages and standard deviations were calculated as described in Figure 4. (B) Normalized graphical representation of exon IIIc inclusion from data in (A) after arbitrarily setting the CSAR-20 minigene inclusion ratio to 100% and adjusting the other minigenes inclusion ratios proportionately.
Construction of minigenes to test function of a rIAS2-ISAR stem structure
In order to address the role of the proposed rIAS2–ISAR stem structure, we constructed a single-exon IIIc-containing minigene that contained a 2-nt substitution in the position of rIAS2 compensatory to the seventh 2-bp substitution of ISAR, as this position had a dramatic effect on both exon IIIb activation and exon IIIc repression in all of our minigenes tested. This mutation is termed ‘compensatory’ because it is predicted to restore the base-pairing interaction disrupted by the seventh 2-bp mutation in ISAR. If the function of the seventh 2-bp region of ISAR derived from its ability to form a stem with the rIAS2 sequence, then mutation of the compensatory rIAS 2-bp region should have an equally dramatic effect on splicing regulation. Additionally, a minigene having compensatory mutations in both the seventh 2-bp positions of rIAS2 and ISAR should have restored splicing regulation. In order to construct the compensatory mutations in rIAS2, we engineered PacI and AscI restriction sites 5′ and 3′ to rIAS2 respectively, and annealed oligos containing either a wild-type sequence or a 2-bp mutation compensatory to the ISAR-7 mutation (Fig. 8A).
Mutation in rIAS2 causes loss in splicing regulation: compensatory mutation in ISAR restores regulation
Minigenes containing a WT rIAS2 and ISAR sequence were examined along with a minigene containing a mutation in the seventh 2-bp region of rIAS alone or with the compensatory mutation in ISAR. These minigenes were designated IIIcPA-WT, IIIcPA-MUT7 and IIIcPA-MUT7-ISAR7 respectively. Minigene IIIcPA-WT containing the PacI and AscI site flanking the IAS2 sequence showed an exon IIIc inclusion ratio of 23%, slightly higher than PI11-IIIc-SAR-20 possibly due to the flanking PacI and AscI restriction sites (Fig. 8B). Minigene IIIcPA-MUT7, containing a 2-bp mutation in the IAS2 sequence, showed a dramatically increased exon IIIc inclusion of 33%, slightly higher than that of PI11-IIIc-ISAR7 of 29% (Fig. 7). Minigene IIIcPA-MUT7-ISAR7, containing 2-bp mutations in ISAR compensatory to rIAS2 displayed a restoration of exon IIIc repression (25%), consistent with the restoration of an rIAS2–ISAR stem required for exon IIIc repression (Fig. 8B). Normalized exon inclusion ratios displayed in the graph in Figure 8B illustrate the degree of loss of splicing regulation (i.e. increase in exon IIIc inclusion) of the rIAS mutant compared to the WT minigene and the restoration of splicing regulation from the minigene containing compensatory mutations in both rIAS and ISAR regions.
DISCUSSION
In this study, we provide evidence that terminal sequences in the ISAR core are responsible for both exon IIIb activation and exon IIIc repression. We demonstrated with dual-exon minigenes that a 2-bp mutation in the 5′ or 3′ end of the core was sufficient to change the ratio of exon IIIb to exon IIIc inclusion in DTE cells. We then demonstrated with single-exon minigenes that the same mutations causing loss of exon IIIb activation caused loss of exon IIIc repression. We then showed that mutations in rIAS2 caused a loss in splicing regulation that is restored by compensatory mutations in ISAR. Based on this evidence, we propose a model, similar to that proposed by Del Gatto et al. (3) for the human FGFR2 sequence. In our model, a stem structure formed between rIAS2 and ISAR is required not only for activation of exon IIIb splicing, as proposed by Del Gatto et al. (3), but also for repression of exon IIIc splicing.
The proposed stem between rIAS2 and ISAR contains one bulge in which 2 nt of the ISAR core do not base-pair with rIAS2 sequences. The analogous stem structure of the human FGFR2 sequence contains a bulge in which 4 nt of ISAR do not base-pair with IAS2 sequences. Additionally, in the human FGFR2 minigene, there is 1 nt of IAS2 that occurs in this bulged region. The conservation of this bulge from rat to human suggests that the bulge does have a function. What this function might be is unclear as none of the substitutional mutations that occurred in the bulged region had an effect on the regulation of exon IIIb or IIIc splicing (Figs 2B, 5 and 7). The bulge may allow for a degree of flexibility in the middle of the stem that is essential for function. However, a preliminary experiment in which the bulged region was deleted from a rat FGFR2 minigene failed to indicate a significant effect (R.P.Carstens, unpublished results). Del Gatto and colleagues demonstrated that the stem structure does not act simply by bringing ISAR closer to exon IIIb (3) since deleting intronic sequence between exon IIIb and ISAR did not activate exon IIIb splicing. How then might the secondary structure affect simultaneous exon IIIb activation and exon IIIc repression? Examples have been described whereby secondary structures promote splice site usage (19). In addition, examples have been described whereby secondary structures repress the use of splice sites presumably by hiding them (20–22). As pointed out by Del Gatto et al. (3), most secondary structures that influence splice site selection have very short loops connecting the stems. The loop connecting IAS2 and ISAR is ∼800 nt in length. Therefore additional protein factors are probably required for maintenance of this secondary structure.
Our studies and those of others indicate that the cell-specific mutually-exclusive expression of FGFR2 exons IIIb or IIIc is a complex, multi-factoral phenomenon at splicing and post-transcriptional levels. Several additional cis-acting elements responsible for the regulation of FGFR2 exon selection have been described from rat and human (Fig. 9). These elements include ISS1, ISS2 (7) and the TAGG sequence that act to repress exon IIIb inclusion, and IAS1, which acts to activate exon IIIb inclusion. This is in addition to the stem described here formed by rIAS2 and ISAR (IAS3 in human) that acts to simultaneously activate exon IIIb and repress exon IIIc inclusion.
Figure 9.


Summary of the regulation of exon IIIb and IIIc splicing in FGFR2. (A) Cis-acting elements shown to control exon IIIb and IIIc inclusion are the following: ISS1 and ISS2 (7), the exonic TAGG (2) sequence, IAS1, IAS2 and IAS3 (ISAR) (1,5). (B) Impact of each cis-acting regulatory element in DTE and AT3 cells. + indicates a known role in the activation of exon inclusion while – indicates a known role in the repression of exon inclusion. NF indicates no demonstrated function in the indicated cell type.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Mariano Garcia-Blanco (Duke University) for helpful advice, sharing reagents and data and general support during the course of this work. We also thank Drs Mikio Kan and Fen Wang (Texas A&M) for helpful discussions. This work was supported by NIH Grants DK35310, DK47039 and CA59971 to W.L.M.
References
- 1.Del Gatto F. and Breathnach,R. (1995) Exon and intron sequences, respectively, repress and activate splicing of a fibroblast growth factor receptor 2 alternative exon. Mol. Cell. Biol., 15, 4825–4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Del Gatto F., Gesnel,M.C. and Breathnach,R. (1996) The exon sequence TAGG can inhibit splicing. Nucleic Acids Res., 24, 2017–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Del Gatto F., Plet,A., Gesnel,M.C., Fort,C. and Breathnach,R. (1997) Multiple interdependent sequence elements control splicing of a fibroblast growth factor receptor 2 alternative exon. Mol. Cell. Biol., 17, 5106–5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Del Gatto-Konczak F., Olive,M., Gesnel,M.C. and Breathnach,R. (1999) hnRNP A1 recruited to an exon in vivo can function as an exon splicing silencer. Mol. Cell. Biol., 19, 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carstens R.P., Eaton,J.V., Krigman,H.R., Walther,P.J. and Garcia-Blanco,M.A. (1997) Alternative splicing of fibroblast growth factor receptor 2 (FGF-R2) in human prostate cancer. Oncogene, 15, 3059–3065. [DOI] [PubMed] [Google Scholar]
- 6.Carstens R.P., McKeehan,W.L. and Garcia-Blanco,M.A. (1998) An intronic sequence element mediates both activation and repression of rat fibroblast growth factor receptor 2 pre-mRNA splicing. Mol. Cell. Biol., 18, 2205–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carstens R.P., Wagner,E.J. and Garcia-Blanco,M.A. (2000) An intronic splicing silencer causes skipping of the IIIb exon of fibroblast growth factor receptor 2 through involvement of polypyrimidine tract binding protein. Mol. Cell. Biol., 20, 7388–7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan G., Fukabori,Y., McBride,G., Nikolaropolous,S. and McKeehan,W.L. (1993) Exon switching and activation of stromal and embryonic fibroblast growth factor (FGF)-FGF receptor genes in prostate epithelial cells accompany stromal independence and malignancy. Mol. Cell. Biol., 13, 4513–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan G., Fukabori,Y., Nikolaropoulos,S., Wang,F. and McKeehan,W.L. (1992) Heparin-binding keratinocyte growth factor is a candidate stromal-to-epithelial-cell andromedin. Mol. Endocrinol., 6, 2123–2128. [DOI] [PubMed] [Google Scholar]
- 10.McKeehan W.L., Wang,F. and Kan,M. (1998) The heparan sulfate-fibroblast growth factor family: diversity of structure and function. Prog. Nucleic Acid Res. Mol. Biol., 59, 135–176. [DOI] [PubMed] [Google Scholar]
- 11.Feng S., Wang,F., Matsubara,A., Kan,M. and McKeehan,W.L. (1997) Fibroblast growth factor receptor 2 limits and receptor 1 accelerates tumorigenicity of prostate epithelial cells. Cancer Res., 57, 5369–5378. [PubMed] [Google Scholar]
- 12.Matsubara A., Kan,M., Feng,S. and McKeehan,W.L. (1998) Inhibition of growth of malignant rat prostate tumor cells by restoration of fibroblast growth factor receptor 2. Cancer Res., 58, 1509–1514. [PubMed] [Google Scholar]
- 13.Wang J. and Manley,J.L. (1997) Regulation of pre-mRNA splicing in metazoa. Curr. Opin. Genet. Dev., 7, 205–211. [DOI] [PubMed] [Google Scholar]
- 14.McKeown M. (1992) Alternative mRNA splicing. Annu. Rev. Cell Biol., 8, 133–155. [DOI] [PubMed] [Google Scholar]
- 15.Caceres J.F., Stamm,S., Helfman,D.M. and Krainer,A.R. (1994) Regulation of alternative splicing in vivo by overexpression of antagonistic splicing factors. Science, 265, 1706–1709. [DOI] [PubMed] [Google Scholar]
- 16.Chandler S.D., Mayeda,A., Yeakley,J.M., Krainer,A.R. and Fu,X.D. (1997) RNA splicing specificity determined by the coordinated action of RNA recognition motifs in SR proteins. Proc. Natl Acad. Sci. USA, 94, 3596–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanamura A., Caceres,J.F., Mayeda,A., Franza,B.R.,Jr and Krainer,A.R. (1998) Regulated tissue-specific expression of antagonistic pre-mRNA splicing factors. RNA, 4, 430–444. [PMC free article] [PubMed] [Google Scholar]
- 18.Jones R.B., Wang,F., Luo,Y., Yu,C., Jin,C., Suzuki,T., Kan,M. and McKeehan,W.L. (2001) The nonsense-mediated decay pathway and mutually exclusive expression of alternatively spliced FGFR2IIIb and IIIc mRNAs. J. Biol. Chem., 276, 4158–4167. [DOI] [PubMed] [Google Scholar]
- 19.Domenjoud L., Gallinaro,H., Kister,L., Meyer,S. and Jacob,M. (1991) Identification of a specific exon sequence that is a major determinant in the selection between a natural and a cryptic 5′ splice site. Mol. Cell. Biol., 11, 4581–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balvay L., Libri,D. and Fiszman,M.Y. (1993) Pre-mRNA secondary structure and the regulation of splicing. Bioessays, 15, 165–169. [DOI] [PubMed] [Google Scholar]
- 21.Clouet d’Orval B., d’Aubenton Carafa,Y., Sirand-Pugnet,P., Gallego,M., Brody,E. and Marie,J. (1991) RNA secondary structure repression of a muscle-specific exon in HeLa cell nuclear extracts. Science, 252, 1823–1828. [DOI] [PubMed] [Google Scholar]
- 22.Clouet d’Orval B., d’Aubenton-Carafa,Y., Brody,J.M. and Brody,E. (1991) Determination of an RNA structure involved in splicing inhibition of a muscle-specific exon. J. Mol. Biol., 221, 837–856. [DOI] [PubMed] [Google Scholar]