

Abstract
The impact of Hepatitis C virus (HCV) RNA levels on immune status in chronically HCV mono-infected when compared to HIV/HCV co-infected on antiretroviral therapy (ART) remains poorly understood. A total of 78 African American subjects HCV viremic/naïve to HCV treatment (33 HCV genotype 1 mono-infected, 45 HCV genotype 1/HIV co-infected on ART) were studied. Clinical and liver enzymes measurements were performed. Whole blood was analyzed for immune subset changes by flow-cytometry. Peripheral blood mononuclear cells (PBMC) were used for same-day constitutive and in vitro Interferon (IFN)-α-induced Signal Transducer and Activator of Transcription (STAT) phosphorylation, K562 target cell lysis and K562 target cell recognition-mediated IFN-γ production. Statistical analysis was done using R (2.5.1) or JMP Pro 11. While both groups did not differ in the level of liver enzymes, HIV/HCV had higher T cell activation/exhaustion, and constitutive STAT-1 phosphorylation compared to HCV. In contrast, CD4+FoxP3+CD25+ frequency, IFN-αR expression on NK cells, as well as constitutive and IFN-α-induced direct cytotoxicity were lower in HIV/HCV. Linear regression models further supported these results. Finally, increase in HCV viral load (vl) and CD4+ T cell count had an opposite effect between the two groups on NK cell activity, and T cell activation respectively. HCV viraemia in antiretroviral -treated HIV/HCV co-infection was associated with greater immune activation/exhaustion and NK dysfunction than HCV viral load alone in HCV mono-infection. The more pronounced immune modulation noted in antiretroviral treated HIV co-infected / untreated HCV viremic subjects may impact HCV disease progression and/or response to immunotherapy.
Keywords: HCV, HIV/HCV, HCV viral load, NK, T cells
INTRODUCTION
Hepatitis C virus (HCV) infects nearly 200 million people worldwide with over 80% of infected people progressing to chronic infection (1–4). Due to shared routes of transmission, HCV and human immunodeficiency virus (HIV) co-infection is common (5), affecting 60–95% of subjects with parenteral exposure [e.g. intravenous (IV) drug users, blood products recipients]. Sexual transmission of HCV is less common, but may be facilitated by concurrent HIV infection. It is estimated that approximately 4 to 5 million people are living with chronic HIV/HCV co-infection (6).
A number of studies support that HCV viremia is associated with both immune activation and subsequent gradual loss of immune function (7). A number of reports show that in chronic HCV infection HCV-specific cytotoxic T lymphocytes have impaired or exhausted proliferative, cytokine, and cytotoxic effector functions (8–12). Natural killer (NK) cell activity has also been described to be decreased (13) as a result of several mechanisms including direct interaction of the viral protein E2 with surface CD81 on NK cells (14), hepatocyte reduction of type 1 interferon (IFN) production via protein kinase R inactivation (15, 16), greater stabilization of hepatocyte major histocompatiblity complex-I molecules (17), and early inhibition of activation and IFN-γ production by NK cells (14). Other mechanisms of immune dysfunction include decreased dendritic cells (DC) frequencies, impairment of the antigen-presenting function of DCs (18–21), direct impairment of plasmacytoid DC (PDC) function by HCV core protein via increased interleukin-10, and reduced production of interleukin-12, and IFN-α (22, 23).
Similar to HCV infection, HIV infection also leads to increased T cell activation (24), and functional impairment (25), which is partially restored by antiretroviral therapy (ART) (26, 27). The impact of HIV on the natural course of HCV infection is deleterious with higher HCV viral load (vl), higher rate of HCV persistence, and a higher risk of mortality or co-morbidities than HCV mono-infected patients (5, 28). On the other hand, some studies also support a significant effect of HCV infection on the progression of HIV to acquired immune deficiency syndrome-defining illness and related mortality (29–34). HIV/HCV co-infected subjects have been described to retain high levels of immune activation that persist after ART-mediated HIV suppression (35–38), yet the role of HCV viremia in driving persistent immune activation and/or its relationship to innate immune reconstitution after ART remains unknown.
Little is known about the additive effect of ART-treated HIV co-infection and HCV vl on innate and adaptive immune status in subjects with untreated chronic HCV infection. To address these questions, we used freshly obtained blood and peripheral blood mononuclear cells (PBMCs) for the characterization of adaptive and innate cells subsets, activation/exhaustion, and innate signaling and function in HIV/HCV co-infected and HCV mono-infected subjects.
MATERIALS AND METHODS
Participants
33 untreated HCV viremic mono-infected (HCV mono-infected) and 45 ART-treated HIV co-infected / untreated HCV viremic (HIV/HCV co-infected) subjects were studied. Clinical parameters [complete blood count differential, CD4 count, HCV and HIV vl], liver enzymes [alanine aminotransferase, aspartate aminotransferase], and immune markers were assessed in all subjects by Quest Diagnostics (NJ, USA). All participants were chronically infected with HCV, had HCV single or mixed genotype 1, were HCV viremic and naïve to HCV treatment. HCV infection was diagnosed by detection of antibodies (Abs) against HCV and confirmed by two PCR-based determinations of HCV RNA (limit of detection: 43 copies/ml) with time elapsed 4–6 months. Subjects with established non-compensated cirrhosis, current IV drug use or usage of IV drugs within 3 months prior to enrollment, or current alcohol abuse [>40g/day (two drinks/day) or average of >80g/day (4 drinks a day)] anytime in 3 months prior to enrollment were excluded. HIV infection in HIV/HCV co-infected subjects was confirmed by western blot or PCR (limit of detection: 20 copies/ml). Presence of ART treatment at the time of study was necessary for inclusion of HIV/HCV co-infected subjects in the study, yet no minimum time of ART or minimum CD4 T cells/mm3 were established as criteria for inclusion. Informed, written consent was obtained from all participants. The study was performed according to the World Medical Association Declaration of Helsinki. The study protocol was approved by the Institutional Review Boards of the authors’ institutions.
Whole blood-based phenotypic characterization of immune subsets
To assess adaptive and innate cell subsets, same day whole blood 7-color staining was performed as previously described (39) by using the combinations of directly fluorochrome-conjugated anti-human cell surface monoclonal Abs shown in Supplementary Table 1. All antibodies were from Becton Dickinson (BD) Biosciences (San Diego, CA, USA) except blood dendritic cells antigen (BDCA) 2-allophycocyanin (APC), BDCA4-APC and IgG1-APC which were purchased from Miltenyi Biotec (San Diego, CA, USA). Stainings “a-c” allowed for the assessment of activation/exhaustion markers [CD25, CD38, CD94, CD95, HLA-DR, programmed cell death 1 (PD1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), B and T lymphocyte attenuator (BTLA), CD160] on T cells (CD3+CD8−, CD3+CD8+). Stainings “d-h” allowed for the identification of exhaustion [programmed death-ligand (PDL) 1, PDL2, herpes virus entry mediator (HVEM)], costimulatory (CD86) and apoptosis [tumor necrosis factor-alpha-related apoptosis-Inducing ligand (TRAIL)] markers on DC (40) [BDCA2+BDCA4+ (PDC), and CD19−BDCA1+CD11c+ (myeloid DC, MDC)] and monocytes (CD14+). Staining “i” allowed for the assessment of CD81, and CD69 expression on T cells (CD3+), as well as for the assessment of NK cell subsets (41) (CD3−CD56dimCD16−, CD3−CD56dimCD16+, CD3−CD56bright, and CD3−CD56−CD16+), and of CD81, CD69, and IFN-α receptor (IFN-αR) expression on NK cell subsets.
Briefly, 200 μl of whole blood were incubated for 15 min at room temperature with the appropriate Ab combinations, lysed for 10 min at room temperature with 3 ml of FACS Lysis solution (BD Biosciences), and centrifuged for 5 min at 1200 rpm. Cells were then washed for 5 min with 3 ml of FACS washing buffer at 1500 rpm, and re-suspended in 200 μl of FACS washing buffer. Regulatory T cells (Tregs, CD4+CD25hiFoxP3+) staining was performed by using the BD Biosciences FoxP3 staining kit according the manufacturer’s instructions. Cells were analyzed on LSRII cytometer (BD Biosciences) by collecting >200000 events. Data were analyzed using FloJo software (Version 8.8.4, Tree Star, Ashland, OR, USA). Gating was originally done on singlets, and then on “live lymphocyte” (for T, NK, and Tregs) or “all live cell” (for DC, and monocytes) gates defined by size and granularity in forward scatter and side scatter. Thresholds were set by isotype-matched negative controls and unstained cells. Results were expressed as mean fluorescent intensity (MFI), percent positive (%) and cells/mm3.
Assessment of the in vitro role of IFN-α on STAT-1 phosphorylation within PBMC cell subsets
To assess constitutive and in vitro induced signal transducer and activator of transcription (STAT) phosphorylation, fresh PBMC (2×106/ml), isolated from whole blood as previously described by standard Ficollhypaque density gradient centrifugation (39), were stained for: a) CD3-fluorescein isothiocyanate (FITC), CD14-FITC, CD19-APC, CD20-APC, CD16-Pacific Blue, CD56-phycoerythrinCy7 (PECy7), and b) CD14-FITC, BDCA2-APC, BDCA4-APC, CD3-Pacific Blue, or c) corresponding isotypes (IgG1k-FITC, IgG2ak-FITC, IgG1-APC, IgG1k-Pacific Blue, IgG1k-PECy7) for 30 min at 4°C, washed with 1xPBS at 1500 rpm for 5 min and re-suspended in warm 1xPBS. PBMCs were then treated for 10 min at 37°C with media alone, or in vitro IFN-α (5000 U/ml, PBL). Cells were then fixed with paraformaldehyde (final concentration 5%) for 10 min at 37°C, washed and permeabillized with PhosFlow buffer (BD Biosciences) for 30 min at RT. Subsequently, PBMCs were washed in FACS washing buffer at 2200 rpm for 10 min, stained with an Ab against phosphorylated (p)-STAT-1 [p-STAT-1-peridinin chloropyll Cy5.5 (PerCP-Cy5.5)] or corresponding isotype IgG2ak-PerCP-Cy5.5 for 1 hr at RT, washed with FACS washing buffer and analyzed in the Cyan cytometer as described above. Staining “a” allowed for the assessment of NK cell subsets (identified as: Lin3−CD56+CD16+, Lin3−CD56+CD16−, or Lin3−CD56−CD16+, with Lin3 consisting of CD3, CD14, CD19, and CD20), while staining “b” allowed for the identification of monocytes (CD3−CD14+), and PDC (CD3−CD14−BDCA2+BDCA4+). All antibodies were from BD Biosciences except BDCA2-APC, BDCA4-APC and IgG1-APC which were purchased from Miltenyi Biotec. Constitutive STAT-1 phosphorylation for all the above described cell subsets was expressed as MFI of p-STAT-1 in the absence of in vitro IFN-α stimulation. In vitro IFN-α-induced STAT-1 phosphorylation for all the above described cell subsets was calculated by dividing in vitro IFN-α-induced MFI of p-STAT-1 by the constitutive MFI of p-STAT-1.
Assessment of intracellular IFN-γ cytokine production in NK cells following effector cells/target interactions
To study functional NK-target cell interaction within the context of cell-specific measures on NK subsets, constitutive and target-induced cytokine production (IFN-γ) in the presence or absence of in vitro stimulation were measured using flow cytometry. Briefly, fresh PBMC (1×106 cells per condition) were incubated with or without in vitro IFN-α (5000 U/ml, PBL, Piscataway, NJ, USA) and in the absence or presence of K562 target cells (2×105 cells per condition) for 2 hrs at 37ºC, followed by addition of Brefeldin (10 μg/ml), and further incubation for 16 hrs at 37ºC. At the end of the incubation, cells were stained for 15 min at RT with NK cell surface Ab combinations (Lin3-FITC, CD16-Pacific Blue, CD56-PECy7), washed with 2 ml FACS washing buffer at 1500 rpm for 5 min and fixed/permeabilized for 10 min at RT with 250 μl Cytofix/Cytoperm. After added washes, cells were stained for intracellular IFN-γ-APC or corresponding isotype IgG1k-APC, washed again with 2 ml Perm washing buffer at 1500 rpm for 5 min and re-suspended in 200 μl of FACS washing buffer. All Abs were from BD Biosciences. Cells were analyzed in the LSRII as described above. This staining allowed for the assessment of IFN-γ-producing NK cell subsets (identified as: Lin3−CD56−CD16+, Lin3−CD56dimCD16−, Lin3−CD56dimCD16+, and Lin3−CD56bright). Constitutive IFN-γ production was calculated by subtracting the percentage of cells producing IFN-γ constitutively in the absence of targets from the percentage of cells producing IFN-γ constitutively in the presence of targets. In vitro induced IFN-γ-production was calculated by subtracting the percentage of cells producing IFN-γ constitutively in the presence of targets from the percentage of cells producing IFN-γ after in vitro stimulation with IFN-α in the presence of targets.
Assessment of direct cytotoxicity against a MHC-cell null cancer target cell line
The standard 51Cr release assay was used as previously described to assess constitutive and in vitro induced NK cell-mediated cytotoxicity, using fresh PBMC preparations as effectors cells against the tumor derived erythroblastoid MHC-null cell line K562 (42).
Briefly, fresh PBMC were treated for 18 hrs at 37C° with media alone or in vitro IFN-α (5000 U/ml, PBL). K562 cells, which served as targets, were labeled with Na251CrO4 (~ 50μCi) for 1.30 hr at 37C°, washed and re-suspended at a concentration of 1×105 cells/ml in media. Effectors and labeled-K562 targets were cultured in triplicate to yield the desired effector:target (E:T) ratios in 0.2 ml volume (usually 50:1, 25:1, 12.5:1, and 6.25:1) in round bottomed 96-well plates and incubated for 4 hrs at 37C°. Percent lysis was determined as [(experimental counts-spontaneous released counts)/(total counts-spontaneous released counts)] × 100. Results were expressed as area under the curve (AUC) for E:T ratios of 50:1, 25:1, 12.5:1 and 6.25:1 for both constitutive and in vitro IFN-α-induced NK function.
Statistical Analysis
Data were described as medians, 25th and 75th quartiles. Group comparisons were done by t-test or Wilcoxon rank sum test depending on data distributions. Unadjusted p-values that were less than 0.05 are reported, along with adjusted p-values, based on the approach of Benjamini and Yekutieli (BY), that were less than 0.01. Multivariate linear regression models were used to explore the effect of HCV vl (log10 IU/ml) and HIV co-infection, as well as of CD4+ T cell count and HIV co-infection on a subset of the variables with unadjusted p-values <0.05 between the two groups. Analysis was performed using R version 2.5.1 (R Core Team, R Foundation for Statistical Computing, Vienna, Austria), or JMP Pro 11 (SAS Institute, Cary, NC, USA).
RESULTS
Study subjects demographics
Demographic and clinical characteristics of the study subjects are shown in Table 1. A total of 5/45 HIV/HCV co-infected subjects had HIV vl >400 copies/ml suggesting lack of ART-mediated suppression in a minority of the HIV/HCV co-infected subjects. Interestingly, 22/45 HIV/HCV co-infected subjects had CD4+ T cell count <400 cells/mm3, with only 2/22 having HIV vl >400 copies/ml, suggesting lack of complete immune re-constitution in about 1/2 of the HIV/HCV co-infected subjects, that was not associated with lack of HIV suppression. Finally, no significant difference was observed between the two groups for plasma levels of liver enzymes (alanine aminotransferase, aspartate aminotransferase).
Table 1.
Subjects characteristics
| HCV | HIV/HCV | |
|---|---|---|
| Total number of subjects | 33 | 45 |
| Gender* | 23M, 10F | 28M, 16F, 1M to F |
| Age (years), median (25th, 75th quartiles) | 54 (50, 59.5) | 50 (46, 54) |
| Race | 33 African American | 45 African American |
| Ethnicity | 33 Non-Hispanic | 45 Non-Hispanic |
| Log10 HCV vl (IU/ml), median (25th, 75th quartiles) | 6.29 (5.9, 6.74) | 6.08 (5.42, 6.51) |
| HIV vl (copies/ml), median (25th, 75th quartiles) | N/Aᵼ | 48 (48, 73) |
| CD4+ T cell count (cells/mm3), median (25th, 75th quartiles) | 963 (743.5, 1272.5) | 412 (232.5, 563.5) |
Gender: M:Male, F:Female.
N/A:Not Applicable.
Higher levels of T cell activation/exhaustion and lower levels of Tregs in HIV/HCV co-infected subjects
HIV infection leads to increased T cell activation (24), and functional impairment (25), which is partially restored by ART (26, 27, 43, 44). In equal manner, in HIV/HCV co-infected subjects, we detected higher levels of T cell activation [e.g. MFI of CD38 on CD8+ (p<0.001) and CD8− (p<0.001) T cells], together with increased expression of exhaustion marker (45) CD160 on CD8+ T cells (p=0.007) when compared to HCV mono-infected subjects. Unadjusted data were also consistent with higher expression of exhaustion markers (i.e. CD160, BTLA/CD160, and CTLA-4) on CD8+ T cells. In contrast, HIV/HCV co-infected subjects had lower frequencies of CD3+CD4+ T cells (p<0.001) and Tregs (p=0.02) when compared to HCV mono-infected subjects (Table 2, Fig. 1, Supplementary Fig. 1).
Table 2.
T cell activation/exhaustion, and NK and myeloid cells activation and function in HCV and HCV/HCV subjects before and after multiple testing adjustment
| Category | Variable Name | HCV Median (25th, 75th quartile) |
N (HCV) |
HIV/HCV Median (25th, 75th quartile) |
N (HIV/HCV) |
P (HCV vs HIV/HCV) |
P (BY*: HCV vs HIV/HCV) |
|---|---|---|---|---|---|---|---|
| T cells | CD3+CD4+ % | 40.2 (36.1, 47.2) | 33 | 18.7 (13.7, 25.3) | 44 | <0.001 | <0.001 |
| CD3+CD8+ % | 24 (19.5, 27.5) | 33 | 45 (38.9, 51.6) | 44 | <0.001 | <0.001 | |
| T cell activation | CD3+CD8+CD38+ % | 7 (5.9, 9.4) | 33 | 24.8 (14.9, 30) | 44 | <0.001 | <0.001 |
| CD3+CD8+HLADR+ % | 0 (0, 0.1) | 33 | 0.1 (0.1, 0.3) | 44 | <0.001 | 0.01 | |
| CD3+CD8−CD38+ % | 15.9 (10.5, 17.7) | 33 | 11 (7.2, 14.7) | 44 | 0.03 | nsᵼ | |
| CD3+CD8+CD38+ % of CD3+CD8+ | 34.6 (27.1, 43.7) | 33 | 55.8 (38.1, 68.5) | 44 | <0.001 | <0.001 | |
| CD3+CD8−CD38+ % of CD3+CD8− | 36.6 (28.4, 40.6) | 33 | 54.6 (45.8, 63) | 44 | <0.001 | <0.001 | |
| CD3+CD8−HLA-DR+ % of CD3+CD8− | 0.3 (0.2, 0.5) | 33 | 0.8 (0.4, 1.1) | 44 | 0.02 | ns | |
| MFI of CD38 on CD3+CD8+ | 156 (118, 201) | 33 | 299.5 (178, 444) | 44 | <0.001 | <0.001 | |
| MFI of HLA-DR on CD3+CD8+ | 46.8 (41.9, 51.4) | 33 | 50.4 (42.3, 62.3) | 44 | 0.04 | ns | |
| MFI of CD38 on CD3+CD8− | 166 (131, 206) | 33 | 338 (259, 418) | 44 | <0.001 | <0.001 | |
| MFI of HLA-DR on CD3+CD8− | 44.6 (40.3, 51.2) | 33 | 52.6 (45.9, 64.1) | 44 | 0.009 | ns | |
| Tregs | CD4+FoxP3+CD25+ % | 0.8 (0.6, 0.9) | 33 | 0.4 (0.3, 0.7) | 44 | <0.001 | 0.02 |
| T cell exhaustion | CD3+CD8+BTLA+CD160+ % of CD3+CD8+ | 0.5 (0.1, 1.2) | 33 | 1.5 (0.5, 3.6) | 44 | 0.01 | ns |
| MFI of CD160 on CD3+CD8− | 24.4 (20.2, 26.9) | 33 | 29.1 (22.3, 33.8) | 44 | 0.01 | ns | |
| MFI of CD160 on CD3+CD8+ | 35 (27.9, 46.6) | 33 | 52.7 (44.5, 71) | 44 | <0.001 | 0.007 | |
| MFI of CTLA-4 on CD3+CD8+ | 63.4 (44.1, 72.5) | 33 | 69.3 (59.8, 91.1) | 44 | 0.02 | ns | |
| Myeloid cells activation/apoptosis | MFI of CD86 on CD3−CD14+ | 325 (287,364) | 33 | 333.5 (306.8, 389) | 44 | 0.03 | ns |
| MFI of CD86 on CD19−BDCA1+CD11c+ | 279 (261, 296) | 33 | 296.5 (267, 321.5) | 44 | 0.004 | ns | |
| MFI of TRAIL on CD3−CD14+ | 53.1 (28.8, 69) | 33 | 61.4 (50.2,80.4) | 44 | 0.004 | ns |
| Category | Variable Name | HCV Median (25th, 75th quartile) |
N (HCV) |
HIV/HCV Median (25th, 75th quartile) |
N (HIV/HCV) |
P (HCV vs HIV/HCV) |
P (* BY: HCV vs HIV/HCV) |
|---|---|---|---|---|---|---|---|
| Myeloid cells activation/apoptosis | MFI of TRAIL on BDCA2+BDCA4+ | 75.5 (66.7,86.3) | 33 | 86.5 (77,94.7) | 44 | 0.02 | ns |
| NK cell activation | MFI of CD69 on ǂ Lin3−CD56dimCD16+ | 70.2 (59.8, 73.7) | 32 | 72.8 (65, 83.8) | 43 | 0.04 | ns |
| MFI of CD69 on Lin3−CD56bright | 75.2 (70, 80) | 32 | 82.4 (72.2, 90.7) | 43 | 0.04 | ns | |
| IFN-αR on NK cells | MFI of IFN-αR on Lin3−CD56dimCD16+ | 526 (456, 627.5) | 32 | 480 (367, 520.5) | 43 | 0.01 | ns |
| MFI of IFN-αR on Lin3−CD56dimCD16− | 465.5 (427, 560.2) | 32 | 410 (353, 475.5) | 43 | 0.02 | ns | |
| Signaling (Constitutive STAT-1) | MFI of p-STAT-1 on Lin3−CD56+CD16+ | 57.7 (47.2, 66) | 33 | 68 (59.3, 78.7) | 43 | <0.001 | 0.05 |
| MFI of p-STAT-1 on Lin3−CD56+CD16− | 101 (76.1, 110) | 33 | 116 (93.6, 136.5) | 43 | 0.006 | ns | |
| MFI of p-STAT-1 on Lin3−CD56−CD16+ | 57.4 (45.1, 70.2) | 33 | 68.8 (60.1, 88.5) | 43 | 0.002 | ns | |
| MFI of p-STAT-1 on CD3−CD14+ | 145 (129, 157) | 33 | 171 (145.5, 194) | 43 | <0.001 | 0.02 | |
| MFI of p-STAT-1 on BDCA2+BDCA4+ | 116 (83.9, 135) | 33 | 134 (113, 151) | 43 | 0.01 | ns | |
| NK cell direct cytotoxicity | AUC§ Constitutive | 1080.3 (631.9, 1716.2) | 29 | 889.9 (401.2, 1188.2) | 40 | 0.04 | N/A¶ |
| AUC IFNα-induced - AUC constitutive | 320.1 (173.4, 419) | 29 | 157.5 (73.7, 277.6) | 40 | 0.02 | N/A |
BY: Benjamini and Yekutieli adjusted p.
ns: non-significant.
Lin3: CD3, CD14, CD19, CD20.
AUC: Area under the curve.
N/A: not applicable.
Fig. 1.
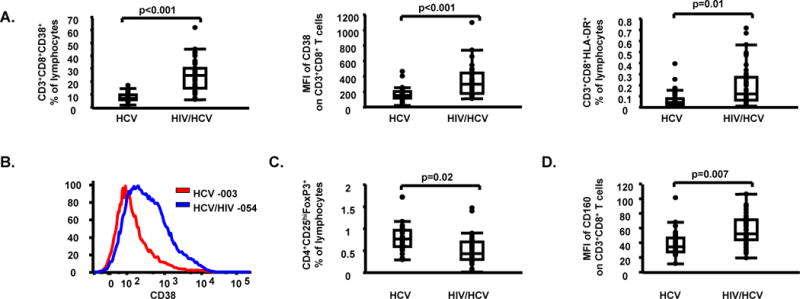
Higher levels of T cell activation and lower levels of Tregs in HIV/HCV co-infected subjects when compared to HCV mono-infected subjects. (A) CD3+CD8+CD38+ percentages (%) of lymphocytes, mean fluorescent intensity (MFI) of CD38 on CD3+CD8+ T cells, and CD3+CD8+HLA-DR+ % of lymphocytes in HCV mono-infected and HIV/HCV co-infected subjects; (B) MFI of CD38 on CD3+CD8+ T cells in one representative HCV mono-infected subject [subject 003, MFI CD38 on CD3+CD8+ T cells = 156 (red line)] and one representative HIV/HCV co-infected subject [subject 054, MFI of CD38 on CD3+CD8+ T cells = 360 (blue line)] are shown; (C) Tregs % of lymphocytes in HCV mono-infected and HIV/HCV co-infected subjects; (D) MFI of CD160 on CD3+CD8+ T cells, in HCV mono-infected and HIV/HCV co-infected subjects. Data in panels (A), (C) and (D) are shown as inter-quartile box plots with median and outliers for each group, and significant (<0.1) P values.
Higher levels of innate activation and constitutive STAT-1 phosphorylation and lower NK function in HIV/HCV co-infected subjects
While total frequency of NK and myeloid cell subsets examined (MDC, PDC, CD14 subsets) did not differ between groups, we document that constitutive STAT-1 phosphorylation was significantly higher on NK cells (p=0.05), and monocytes (p=0.02) in HIV/HCV co-infected subjects when compared to HCV mono-infected subjects (Table 2, Fig. 2). NK function was assessed by measurement of direct cytotoxicity against K562 target cells and NK cell-associated IFN-γ production following in vitro stimulation with IFN-α (Supplementary Fig. 2). HIV/HCV co-infected subjects had lower constitutive (p=0.04) and in vitro IFN-α-induced direct cytotoxicity against K562 target cells (p=0.02) (Table 2, Fig. 2). Consistent with group comparison results obtained after multiple testing adjustment in support of an increased immune activation state in HIV/HCV co-infected subjects, it should be noted that (1) a lower IFN-αR expression on NK cells together with higher expression of CD69 on NK cells, and (2) a higher expression of CD86 (46), and TRAIL (47) on myeloid cells were also detected in HIV/HCV co-infected subjects when compared to HCV mono-infected subjects before multiple testing adjustment (Table 2).
Fig. 2.
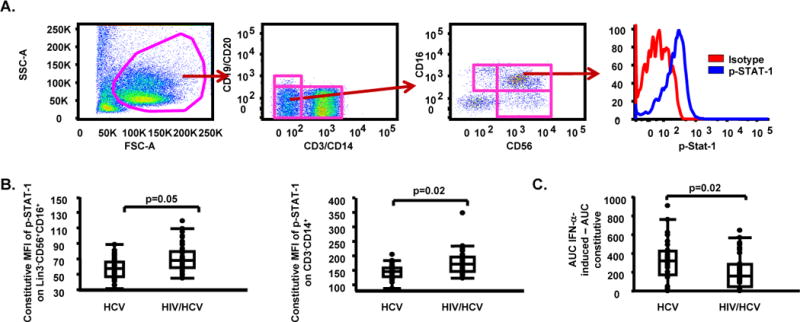
Higher levels of constitutive STAT-1 phosphorylation and lower levels of in vitro IFN-α-induced direct cytotoxicity against K562 target cells in HIV/HCV co-infected subjects when compared to HCV mono-infected subjects. (A) Gating approach for phosphorylated (p)-STAT-1 on NK cell subsets [(isotype (red line), p-STAT-1 (blue line)]; (B) Constitutive MFI of p-STAT-1 on Lin3−CD56+CD16+, and CD3+CD14+ cells in HCV mono-infected and HIV/HCV co-infected subjects; (C) In vitro IFN-α-induced area under the curve (AUC) - constitutive AUC for effector:target (E:T) ratios of 50:1, 25:1, 12.5:1 and 6.25:1 in HCV mono-infected and HIV/HCV co-infected subjects. Data in panels (B) and (C) are shown as inter-quartile box plots with median and outliers for each group, and significant (<0.1) P values.
Overall, innate markers of activation and function as listed in Table 2 support the interpretation of higher constitutive STAT-1-mediated activation in both monocyte and NK cells in spite of lower NK cytotoxicity in HIV/HCV co-infected subjects as compared to HCV mono-infected.
HIV co-infection affects the relationship between HCV vl and NK cell function
We assessed the effect of HCV vl and HIV co-infection on selected variables using multivariate linear regression models. The results of this analysis are summarized in Table 3 and Fig. 3. Briefly, when the effect of HCV vl and patient group was assessed independently (model 1), of all the variables tested, only CD81 expression on NK cells (Supplementary Fig. 3) was positively associated with HCV vl (p=0.02). The same model indicated no significant effect of HIV co-infection on CD81 expression. On the other hand, consistent with group comparisons described above, HIV co-infection, but not HCV vl, had a significant effect on T cell activation/exhaustion, as CD38 (p<0.0001), CTLA-4 (p=0.04), and CD160 (p<0.0001) expression on CD8+ T cells were higher, while Tregs levels (p=0.0002) were lower in HIV/HCV co-infected subjects than in HCV mono-infected subjects (Table 3, Fig. 3). HIV co-infection had also an effect on NK cell activity, as CD69 (p=0.04) and p-STAT-1 expression (p=0.002) were higher, while IFN-αR expression (p=0.01) and IFN-α-induced direct cytotoxicity (p=0.02) were lower in HIV/HCV co-infected subjects than in HCV mono-infected subjects (Table 3, Fig. 3). Models assessing the effect of CD4+ T cell count showed that HIV co-infection, but not CD4+ T cell count, had a significant effect on T cell activation/exhaustion, with CD38 (p=0.001), BTLA (p=0.03), and CD160 (p=0.01) expression on CD8+ T cells being higher, and Tregs levels (p=0.01) being lower in HIV/HCV co-infected subjects than in HCV mono-infected subjects (Supplementary Table 2, Supplementary Fig. 4).
Table 3.
Linear models using HCV vl (log10 copies/ml) and patients group as predictors of immune variables
| Response | Terms ▸ | HCV vl | Group* | ||
|---|---|---|---|---|---|
|
| |||||
| Model ▾ | Estimate | p | Estimate | p | |
| Log10 MFI of CD38 on CD3+CD8+ | 1 | −0.04 | nsᵼ | −0.14 | <0.0001 |
| CD4+FoxP3+CD25+ % | 1 | −0.008 | ns | 0.14 | 0.0002 |
| MFI of PD1 on CD3+CD8+ | 1 | −1.99 | ns | −3.85 | ns |
| MFI of CTLA-4 on CD3+CD8+ | 1 | 2.83 | ns | −11.48 | 0.04 |
| MFI of BTLA on CD3+CD8+ | 1 | −11.22 | ns | −23.75 | ns |
| MFI of CD160 on CD3+CD8+ | 1 | 0.36 | ns | −8.86 | <0.0001 |
| MFI of CD69 on Lin3−CD56dimCD16+ | 1 | −1.65 | ns | −2.78 | 0.04 |
| MFI of CD81 on Lin3−CD56dimCD16+ | 1 | 590.17 | 0.02 | −98.78 | ns |
| MFI of IFN-αR on Lin3−CD56dimCD16+ | 1 | −5.03 | ns | 39.33 | 0.01 |
| Constitutive MFI of p-STAT-1 on Lin3−CD56+CD16+ | 1 | −4.18 | ns | −5.92 | 0.002 |
| AUC IFNα-induced - AUC constitutive | 1 | 26.11 | ns | 58.39 | 0.02 |
| Log10 (IFNα-induced - constitutive) Lin3−CD56dimCD16+IFN-γ+ % of Lin3−CD56dimCD16+ | 1 | −0.07 | ns | 0.11 | ns |
| Log10 (IFN-α-induced - constitutive) Lin3−CD56brightIFN-γ+ % of Lin3−CD56bright | 1 | 0.06 | ns | −0.03 | ns |
| Response | Terms ▸ | HCV vl | Group | HCV vl: Group | |||
|---|---|---|---|---|---|---|---|
| Model ▾ | Estimate | p | Estimate | p | Estimate | p | |
| MFI of CD69 on Lin3−CD56dimCD16+ | 2 | −2.04 | ns | −2.7 | ns | −3.93 | 0.03 |
| Constitutive MFI of p-STAT-1 on Lin3−CD56+CD16+ | 2 | −3.64 | ns | −6.01 | 0.001 | 4.78 | 0.04 |
| Log10 (IFNα-induced - constitutive) Lin3−CD56dimCD16+IFN-γ+ % of Lin3−CD56dimCD16+ | 2 | −0.03 | ns | 0.1 | ns | 0.21 | 0.008 |
Group: HCV mono-infected, HIV/HCV co-infected.
ns: non-significant
Fig. 3.
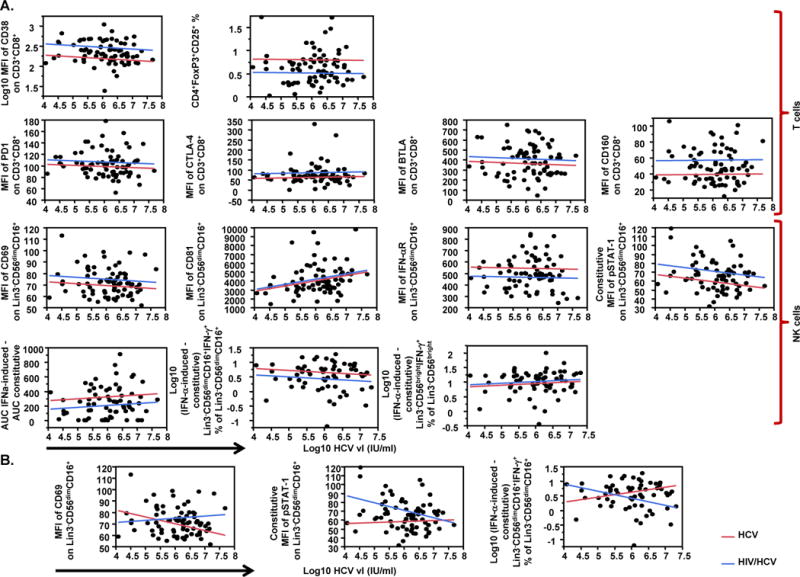
Multivariate linear regression models of HCV vl and patient group as predictors of immune variables levels. (A) Effect of HCV vl (log10 IU/ml) and patient group (HCV or HIV/HCV) as independent predictors (model 1) on T cell markers of activation/exhaustion [MFI of CD38 on CD3+CD8+, CD4+FoxP3+CD25+ percentage (%) of lymphocytes, MFI of PD1 on CD3+CD8+, MFI of CTLA-4 on CD3+CD8+, MFI of BTLA on CD3+CD8+, MFI of CD160 on CD3+CD8+] (top panel), and NK cell markers of activation and function {MFI of CD69 on Lin3−CD56dimCD16+, MFI of CD81 on Lin3−CD56dimCD16+, MFI of IFN-αR on Lin3−CD56dimCD16+, constitutive MFI of phosphorylated (p) STAT-1 on Lin3−CD56dimCD16+, in vitro IFN-α-induced direct cytotoxicity against K562 target cells [expressed as in vitro IFN-α-induced area under the curve (AUC) - constitutive AUC for effector:target (E:T) ratios of 50:1, 25:1, 12.5:1 and 6.25:1], (in vitro IFNα-induced - constitutive) Lin3−CD56dimCD16+IFN-γ+ % of Lin3−CD56dimCD16+, and (in vitro IFNα-induced - constitutive) Lin3−CD56brightIFN-γ+ % of Lin3−CD56bright} (bottom panel); (B) Effect of HCV vl (log10 IU/ml) and patient group (HCV or HIV/HCV) including the addition of interaction term (model 2) on MFI of CD69 on Lin3−CD56dimCD16+, constitutive MFI of pSTAT-1 on Lin3−CD56dimCD16+, and (in vitro IFNα-induced - constitutive) Lin3−CD56dimCD16+IFN-γ+ % of Lin3−CD56dimCD16+ NK cells. Data in panels (A) and (B) are shown as plots of each variable (axis y) against log10 HCV vl (IU/ml) (axis x). Lines in plots represent the perfect fit for each group of patients (red line for HCV mono-infected and blue line for HIV/HCV co-infected subjects).
The effect of HIV co-infection on the association between the markers described above and HCV vl was assessed by adding an interaction term to the already described models. Significant interactions were found for NK cells-associated variables, but not for T cells. More precisely, the model suggests that the association between HCV vl and NK cells expression of CD69 (p=0.03), constitutive STAT-1 phosphorylation (p=0.04) and frequency of IFN-α-induced IFN-γ+ cells (p=0.008) is significantly different between HIV/HCV co-infected and HCV mono-infected subjects (Table 3, Fig. 3). In contrast, addition of interaction term in the model assessing the effect of HIV co-infection on the association between the markers described above and CD4+ T cell count showed that of the variables tested only the association between CD4+ T cell count and CD38 expression on CD8+ T cells was significantly different between HIV/HCV co-infected and HCV mono-infected subjects. More precisely, increasing CD4+ T cell count was associated with low levels of CD38 expression in HIV/HCV co-infected in contrast to HCV mono-infected (Supplementary Table 2, Supplementary Fig. 4), possibly as a result of ART-mediated immune reconstitution.
Taken together, these findings support that increasing HCV viremia is associated with a greater degree of NK cell activation and dysfunction in ART-treated HIV co-infected / untreated HCV subjects than in untreated HCV viremic mono-infected subjects.
DISCUSSION
We assessed the effects of HCV viremia in ART-treated HIV/HCV co-infected versus HCV mono-infected subjects where ethnic distribution, HCV genotype, liver enzymes, and a lack of de-compensating disease are comparable between subjects. Based on the deleterious effect of HIV on the natural course of HCV infection, we expected to observe greater immune activation and dysfunction in HIV/HCV co-infected subjects, with HCV vl levels playing a determinant role in this immunodeficiency. We confirmed this hypothesis showing that HCV viremia associates with greater NK cell activation and dysfunction in HIV/HCV co-infected than in HCV mono-infected subjects, yet we found no difference on the association between HCV vl and T cell activation between groups.
HIV/HCV co-infected subjects have been reported to have high levels of immune activation, particularly T cell activation (i.e. CD38, HLA-DR), even in the presence of suppressive ART (35–38). In this study, we confirmed higher levels of T cell activation, and also observed higher levels of markers of T cell exhaustion in ART-treated HIV/HCV co-infected subjects, as noted by the higher expression of CD160 and other exhaustion markers on CD8+ T cells. Linear regression models further supported these results by showing an effect of patient group but not of HCV vl on T cell activation/exhaustion markers, with HIV/HCV co-infected subjects having higher levels of T cell activation/exhaustion. These results are overall in agreement with the findings by Feuth et al (48) suggesting an increased exhaustion in HCV viremic / HIV co-infected subjects on ART, despite the fact that in our study, subjects with established non-compensated cirrhosis were excluded.
Recent studies have shown that Treg cell activity is increased in patients with chronic HCV infection when compared to those who clear infection (49–51) and that Tregs contribute to HCV persistence by suppressing the proliferation and IFN-γ production of HCV-specific CD8+ T cells (8, 49, 52). There is limited and conflicting information available on Treg cells in HIV/HCV co-infection (53, 54). Consistent with Roe et al (54) who showed lower Tregs levels in ART untreated HIV/HCV co-infection when compared to HCV mono-infection, our observations on ART-treated HIV/HCV co-infected subjects showed lower Tregs frequency when compared to HCV mono-infected, suggesting the possible presence of an association between Tregs frequency and HIV co-infection maintained even after ART. This finding was supported by linear regression models showing an effect of patient group, but not of HCV vl, on lower Tregs levels in HIV/HCV co-infected subjects.
In both HCV and HIV infection a quantitative and qualitative impairment of DC subsets have been reported, which is partially restored in HIV-infected subjects after therapy (18–23, 27, 54–56). In our study, myeloid and NK cells were detected to have higher levels of constitutive STAT-1 phosphorylation. While a higher stringency of analysis (adjusted p values) did not detect a difference in the frequency of DC subsets or the expression of co-stimulatory or inhibitory molecules on MDC or PDC between study groups, unadjusted p values did suggest the potential for higher expression of CD86 and TRAIL in HIV/HCV co-infected subjects.
IFN-αR is involved in anti-HCV responses as noted by the positive association of its expression on NK cells and a favorable response to Peg-IFN-α/RBV (57, 58). We show that in ART-treated HIV/HCV co-infection a lower IFN-αR expression may still indicate a lack of innate immune reconstitution by a sustained state of chronic NK activation with lower functionality. Group comparisons and linear regression models suggested lower expression of IFN-αR on NK cells together with higher constitutive STAT-1 phosphorylation and lower cytotoxic potential in HIV/HCV co-infected subjects irrespective of HCV vl. We interpret that the latter is consistent with the retention of an activated, yet dysfunctional (“exhausted”) NK response in the presence of HCV viremia. Although no difference was observed between study groups for the expression of CD81 on NK cells, we do show that HCV vl was associated to an increase in CD81 expression irrespective of patient group. The direct interaction of the HCV protein E2 with surface CD81 on NK cells (14) has been suggested to be one of the mechanisms resulting in the decreased NK cell activity observed during HCV infection (13), while our data show that in the context of HCV viremia HIV co-infection-associated additional mechanisms may contribute to greater NK dysfunction. More precisely, HCV vl showed an opposite effect in both groups for NK cell activation, constitutive STAT-1 phosphorylation and IFN-α-induced IFN-γ production, suggesting an increase in NK activation and loss of NK function with HCV vl in the context of HIV/HCV co-infection. Further studies with greater numbers in each group are needed to confirm these findings. Higher STAT-1 phosphorylation levels in NK cells together with decreased IFN-αR expression raises the hypothesis that this dysfunction may help explain prior reports of lower HCV sustained responses in HIV/HCV co-infected when compared to HCV mono-infected when an older approach to HCV therapy was taken by using pegylated-IFN-α / ribavirin (59, 60).
A strength of our study is that all assays were performed on fresh blood or freshly isolated PBMCs, thereby eliminating any influence of cryopreservation on immune cells and markers of activation/exhaustion, signaling and function. Another strength is the homogeneity of the study groups limiting the secondary effects of ethnicity or non-compensated cirrhosis. On the other hand, the lack of HCV specific T cell responses restricting our findings as non-HCV specific is a limitation of our study. However, it has been suggested that non-specific activation may affect disease progression, as liver infiltrates are largely composed of HCV non-specific T cells (61, 62). Another limitation of our study is the lack of an HIV ART-treated / HCV uninfected control group to address direct differences in ART-mediated immune reconstitution with HIV/HCV co-infected subjects in the presence of HCV viremia. However, while the association between CD4+ T cell count and T cell activation was significantly different between the two groups, no association between CD4+ T cell count and NK markers was observed in either group suggesting that the level of CD4+ T cell immune reconstitution did not affect the HCV viremia effects on NK cells noted here. Future longitudinal studies should address the impact of HCV vl on ART-mediated immune reconstitution, where pre-ART CD4+ T cell count is matched between HIV/HCV co-infected and HIV mono-infected groups.
Overall, in HIV ART-treated / HCV viremic co-infected subjects, we found evidence to support higher levels of activation on T, NK and myeloid cells together with lower levels of IFN-αR expression on NK cells and NK cell direct cytotoxicity than in HCV mono-infected subjects. Importantly, HCV vl was found to have opposite effects on NK activation and function between groups suggesting the presence of greater detrimental effects for HCV vl on NK cells in HIV/HCV co-infected subjects despite ART. This data show a greater state of immune dysfunction or lack of functional innate immune reconstitution in HIV/HCV co-infected subjects when compared to HCV mono-infected, which may impact HCV disease progression, comorbidities, and/or response to therapy between HCV mono-infected and HIV/HCV ART-treated co-infected subjects.
Supplementary Material
Supplementary Fig. 1 Gating approach for CD4+CD25+FoxP3+ (Tregs).
Supplementary Fig. 2 Gating approach for IFN-γ+ NK cells in the presence of K562 target cells.
Supplementary Fig. 3 Gating approach for CD81 on NK cell subsets (blue line: isotype, red line: CD81).
Supplementary Fig. 4 Multivariate linear regression model of CD4+ T cell count and patient group as predictors of CD38 levels on CD8+ T cells. (A) Effect of CD4+ T cell count (cells/mm3) and patient group (HCV or HIV/HCV) as independent predictors (model 1) on T cell markers of activation/exhaustion [MFI of CD38 on CD3+CD8+, CD4+FoxP3+CD25+ percentage (%) of lymphocytes, MFI of PD1 on CD3+CD8+, MFI of CTLA-4 on CD3+CD8+, MFI of BTLA on CD3+CD8+, MFI of CD160 on CD3+CD8+] (top panel), and NK cell markers of activation and function {MFI of CD69 on Lin3−CD56dimCD16+, MFI of CD81 on Lin3−CD56dimCD16+, MFI of IFN-αR on Lin3−CD56dimCD16+, constitutive MFI of phosphorylated (p) STAT-1 on Lin3−CD56dimCD16+, in vitro IFN-α-induced direct cytotoxicity against K562 target cells [expressed as in vitro IFN-α-induced area under the curve (AUC) - constitutive AUC for effector:target (E:T) ratios of 50:1, 25:1, 12.5:1 and 6.25:1], (in vitro IFNα-induced - constitutive) Lin3−CD56dimCD16+IFN-γ+ % of Lin3−CD56dimCD16+, and (in vitro IFNα-induced - constitutive) Lin3−CD56brightIFN-γ+ % of Lin3−CD56bright} (bottom panel); (B) Effect of CD4+ T cell count (cells/mm3) and patient group (HCV or HIV/HCV) including the addition of interaction term (model 2) on MFI of CD38 on CD3+CD8+ T cells. Data in panels (A) and (B) are shown as plots of each variable (axis y) against CD4+ T cell count (cells/mm3) (axis x). Lines in plots represent the perfect fit for each group of patients (red line for HCV mono-infected and blue line for HIV/HCV co-infected subjects).
Supplementary Table 1 Staining combinations for real-time whole blood-based phenotypic characterization of immune subsets.
Supplementary Table 2. Linear models using CD4+ T cell count (cells/mm3) and patients group as predictors of immune variables.
Acknowledgments
We thank the subjects who participated in the study and their providers. We thank the subjects who participated in the study and their providers. We acknowledge support for this work by G. Reynolds, N. Opsitnick, C. Calloway, and M. Pistilli.
DISCLOSURES
This work was primarily supported by a grant to L.J.M. by the National Institutes of Health (R01AI073219), the Philadelphia Foundation (Robert I. Jacobs Fund), The Stengel-Miller family, as well as AIDS funds from the Commonwealth of Pennsylvania and from the Commonwealth Universal Research Enhancement Program, Pennsylvania Department of Health. The funding sources had no involvement in the study design; collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
Abbreviations
- HCV
hepatitis C virus
- HIV
human immunodeficiency virus
- IV
intravenous
- NK
natural killer
- IFN
interferon
- DC
dendritic cells
- PDC
plasmacytoid DC
- ART
antiretroviral therapy
- vl
viral load
- PBMCs
peripheral blood mononuclear cells
- Abs
antibodies
- BD
becton dickinson
- BDCA
blood dendritic cells antigen
- APC
allophycocyanin
- PD1
programmed cell death 1
- CTLA-4
cytotoxic T-lymphocyte-associated protein 4
- BTLA
B and T lymphocyte attenuator
- PDL
programmed death-ligand
- HVEM
herpes virus entry mediator
- TRAIL
tumor necrosis factor-alpha-related apoptosis-inducing ligand
- MDC
myeloid DC
- IFN-αR
IFN-α receptor
- Tregs
regulatory T cells
- MFI
mean fluorescent intensity
- %
percent positive
- STAT
signal transducer and activator of transcription
- FITC
fluorescein isothiocyanate
- PECy7
phycoerythrinCy7
- p-STAT-1
phosphorylated-STAT-1
- PerCP-Cy5.5
peridinin chlorophyll Cy5.5
- E
T, effector:target
- AUC
area under the curve
- BY
Benjamini and Yekutieli
Footnotes
Conflict of interest: The authors declare that no conflict of interest exists.
References
- 1.Alter MJ. Epidemiology of hepatitis C virus infection. World J Gastroenterol. 2007;13(17):2436–41. doi: 10.3748/wjg.v13.i17.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang KM. Immunopathogenesis of hepatitis C virus infection. Clin Liver Dis. 2003;7(1):89–105. doi: 10.1016/s1089-3261(02)00068-5. [DOI] [PubMed] [Google Scholar]
- 3.Mohsen AH, Easterbrook P, Taylor CB, Norris S. Hepatitis C and HIV-1 coinfection. Gut. 2002;51(4):601–8. doi: 10.1136/gut.51.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5(9):558–67. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 5.Sherman KE, O’Brien J, Gutierrez AG, et al. Quantitative evaluation of hepatitis C virus RNA in patients with concurrent human immunodeficiency virus infections. J Clin Microbiol. 1993;31(10):2679–82. doi: 10.1128/jcm.31.10.2679-2682.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Operskalski EA, Kovacs A. HIV/HCV co-infection: pathogenesis, clinical complications, treatment, and new therapeutic technologies. Curr HIV/AIDS Rep. 2011;8(1):12–22. doi: 10.1007/s11904-010-0071-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deignan T, Curry MP, Doherty DG, et al. Decrease in hepatic CD56(+) T cells and V alpha 24(+) natural killer T cells in chronic hepatitis C viral infection. J Hepatol. 2002;37(1):101–8. doi: 10.1016/s0168-8278(02)00072-7. [DOI] [PubMed] [Google Scholar]
- 8.Boettler T, Spangenberg HC, Neumann-Haefelin C, et al. T cells with a CD4+CD25+ regulatory phenotype suppress in vitro proliferation of virus-specific CD8+ T cells during chronic hepatitis C virus infection. J Virol. 2005;79(12):7860–7. doi: 10.1128/JVI.79.12.7860-7867.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowen DG, Walker CM. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature. 2005;436(7053):946–52. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 10.Gruener NH, Lechner F, Jung MC, et al. Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J Virol. 2001;75(12):5550–8. doi: 10.1128/JVI.75.12.5550-5558.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radziewicz H, Ibegbu CC, Fernandez ML, et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol. 2007;81(6):2545–53. doi: 10.1128/JVI.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen T, Chen X, Chen Y, Xu Q, Lu F, Liu S. Increased PD-L1 expression and PD-L1/CD86 ratio on dendritic cells were associated with impaired dendritic cells function in HCV infection. J Med Virol. 2010;82(7):1152–9. doi: 10.1002/jmv.21809. [DOI] [PubMed] [Google Scholar]
- 13.Corado J, Toro F, Rivera H, Bianco NE, Deibis L, De Sanctis JB. Impairment of natural killer (NK) cytotoxic activity in hepatitis C virus (HCV) infection. Clin Exp Immunol. 1997;109(3):451–7. doi: 10.1046/j.1365-2249.1997.4581355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crotta S, Stilla A, Wack A, et al. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J Exp Med. 2002;195(1):35–41. doi: 10.1084/jem.20011124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gale M, Jr, Blakely CM, Kwieciszewski B, et al. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanisms of kinase regulation. Mol Cell Biol. 1998;18(9):5208–18. doi: 10.1128/mcb.18.9.5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gale MJ, Jr, Korth MJ, Katze MG. Repression of the PKR protein kinase by the hepatitis C virus NS5A protein: a potential mechanism of interferon resistance. Clin Diagn Virol. 1998;10(2–3):157–62. doi: 10.1016/s0928-0197(98)00034-8. [DOI] [PubMed] [Google Scholar]
- 17.Moradpour D, Grabscheid B, Kammer AR, et al. Expression of hepatitis C virus proteins does not interfere with major histocompatibility complex class I processing and presentation in vitro. Hepatology. 2001;33(5):1282–7. doi: 10.1053/jhep.2001.23793. [DOI] [PubMed] [Google Scholar]
- 18.Anthony DD, Yonkers NL, Post AB, et al. Selective impairments in dendritic cell-associated function distinguish hepatitis C virus and HIV infection. J Immunol. 2004;172(8):4907–16. doi: 10.4049/jimmunol.172.8.4907. [DOI] [PubMed] [Google Scholar]
- 19.Auffermann-Gretzinger S, Keeffe EB, Levy S. Impaired dendritic cell maturation in patients with chronic, but not resolved, hepatitis C virus infection. Blood. 2001;97(10):3171–6. doi: 10.1182/blood.v97.10.3171. [DOI] [PubMed] [Google Scholar]
- 20.Bain C, Fatmi A, Zoulim F, Zarski JP, Trepo C, Inchauspe G. Impaired allostimulatory function of dendritic cells in chronic hepatitis C infection. Gastroenterology. 2001;120(2):512–24. doi: 10.1053/gast.2001.21212. [DOI] [PubMed] [Google Scholar]
- 21.Murakami H, Akbar SM, Matsui H, Horiike N, Onji M. Decreased interferon-alpha production and impaired T helper 1 polarization by dendritic cells from patients with chronic hepatitis C. Clin Exp Immunol. 2004;137(3):559–65. doi: 10.1111/j.1365-2249.2004.02550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dolganiuc A, Chang S, Kodys K, et al. Hepatitis C virus (HCV) core protein-induced, monocyte-mediated mechanisms of reduced IFN-alpha and plasmacytoid dendritic cell loss in chronic HCV infection. J Immunol. 2006;177(10):6758–68. doi: 10.4049/jimmunol.177.10.6758. [DOI] [PubMed] [Google Scholar]
- 23.Lee CH, Choi YH, Yang SH, Lee CW, Ha SJ, Sung YC. Hepatitis C virus core protein inhibits interleukin 12 and nitric oxide production from activated macrophages. Virology. 2001;279(1):271–9. doi: 10.1006/viro.2000.0694. [DOI] [PubMed] [Google Scholar]
- 24.Resino S, Navarro J, Bellon JM, Gurbindo D, Leon JA, Munoz-Fernandez MA. Naive and memory CD4+ T cells and T cell activation markers in HIV-1 infected children on HAART. Clin Exp Immunol. 2001;125(2):266–73. doi: 10.1046/j.1365-2249.2001.01612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Younes SA, Yassine-Diab B, Dumont AR, et al. HIV-1 viremia prevents the establishment of interleukin 2-producing HIV-specific memory CD4+ T cells endowed with proliferative capacity. J Exp Med. 2003;198(12):1909–22. doi: 10.1084/jem.20031598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Azzoni L, Chehimi J, Zhou L, et al. Early and delayed benefits of HIV-1 suppression: timeline of recovery of innate immunity effector cells. AIDS. 2007;21(3):293–305. doi: 10.1097/QAD.0b013e328012b85f. [DOI] [PubMed] [Google Scholar]
- 27.Chehimi J, Campbell DE, Azzoni L, et al. Persistent decreases in blood plasmacytoid dendritic cell number and function despite effective highly active antiretroviral therapy and increased blood myeloid dendritic cells in HIV-infected individuals. J Immunol. 2002;168(9):4796–801. doi: 10.4049/jimmunol.168.9.4796. [DOI] [PubMed] [Google Scholar]
- 28.Bonacini M, Govindarajan S, Blatt LM, Schmid P, Conrad A, Lindsay KL. Patients co-infected with human immunodeficiency virus and hepatitis C virus demonstrate higher levels of hepatic HCV RNA. J Viral Hepat. 1999;6(3):203–8. doi: 10.1046/j.1365-2893.1999.00153.x. [DOI] [PubMed] [Google Scholar]
- 29.Cacoub P, Geffray L, Rosenthal E, Perronne C, Veyssier P, Raguin G. Mortality among human immunodeficiency virus-infected patients with cirrhosis or hepatocellular carcinoma due to hepatitis C virus in French Departments of Internal Medicine/Infectious Diseases, in 1995 and 1997. Clin Infect Dis. 2001;32(8):1207–14. doi: 10.1086/319747. [DOI] [PubMed] [Google Scholar]
- 30.Lesens O, Deschenes M, Steben M, Belanger G, Tsoukas CM. Hepatitis C virus is related to progressive liver disease in human immunodeficiency virus-positive hemophiliacs and should be treated as an opportunistic infection. J Infect Dis. 1999;179(5):1254–8. doi: 10.1086/314720. [DOI] [PubMed] [Google Scholar]
- 31.Martin-Carbonero L, Soriano V, Valencia E, Garcia-Samaniego J, Lopez M, Gonzalez-Lahoz J. Increasing impact of chronic viral hepatitis on hospital admissions and mortality among HIV-infected patients. AIDS Res Hum Retroviruses. 2001;17(16):1467–71. doi: 10.1089/08892220152644160. [DOI] [PubMed] [Google Scholar]
- 32.Piroth L, Duong M, Quantin C, et al. Does hepatitis C virus co-infection accelerate clinical and immunological evolution of HIV-infected patients? AIDS. 1998;12(4):381–8. doi: 10.1097/00002030-199804000-00006. [DOI] [PubMed] [Google Scholar]
- 33.Rosenthal E, Pialoux G, Bernard N, et al. Liver-related mortality in human-immunodeficiency-virus-infected patients between 1995 and 2003 in the French GERMIVIC Joint Study Group Network (MORTAVIC 2003 Study) J Viral Hepat. 2007;14(3):183–8. doi: 10.1111/j.1365-2893.2006.00791.x. [DOI] [PubMed] [Google Scholar]
- 34.Rosenthal E, Poiree M, Pradier C, et al. Mortality due to hepatitis C-related liver disease in HIV-infected patients in France (Mortavic 2001 study) AIDS. 2003;17(12):1803–9. doi: 10.1097/00002030-200308150-00009. [DOI] [PubMed] [Google Scholar]
- 35.Kottilil S, Yan MY, Reitano KN, et al. Human immunodeficiency virus and hepatitis C infections induce distinct immunologic imprints in peripheral mononuclear cells. Hepatology. 2009;50(1):34–45. doi: 10.1002/hep.23055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rempel H, Sun B, Calosing C, Abadjian L, Monto A, Pulliam L. Monocyte activation in HIV/HCV coinfection correlates with cognitive impairment. PLoS One. 2013;8(2):e55776. doi: 10.1371/journal.pone.0055776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gonzalez VD, Falconer K, Blom KG, et al. High levels of chronic immune activation in the T-cell compartments of patients coinfected with hepatitis C virus and human immunodeficiency virus type 1 and on highly active antiretroviral therapy are reverted by alpha interferon and ribavirin treatment. J Virol. 2009;83(21):11407–11. doi: 10.1128/JVI.01211-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kovacs A, Al-Harthi L, Christensen S, Mack W, Cohen M, Landay A. CD8(+) T cell activation in women coinfected with human immunodeficiency virus type 1 and hepatitis C virus. J Infect Dis. 2008;197(10):1402–7. doi: 10.1086/587696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papasavvas E, Ortiz GM, Gross R, et al. Enhancement of human immunodeficiency virus type 1-specific CD4 and CD8 T cell responses in chronically infected persons after temporary treatment interruption. J Infect Dis. 2000;182(3):766–75. doi: 10.1086/315748. [DOI] [PubMed] [Google Scholar]
- 40.Collin M, McGovern N, Haniffa M. Human dendritic cell subsets. Immunology. 2013;140(1):22–30. doi: 10.1111/imm.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caligiuri MA. Human natural killer cells. Blood. 2008;112(3):461–9. doi: 10.1182/blood-2007-09-077438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chehimi J, Azzoni L, Farabaugh M, et al. Baseline viral load and immune activation determine the extent of reconstitution of innate immune effectors in HIV-1-infected subjects undergoing antiretroviral treatment. J Immunol. 2007;179(4):2642–50. doi: 10.4049/jimmunol.179.4.2642. [DOI] [PubMed] [Google Scholar]
- 43.Hammer SM, Squires KE, Hughes MD, et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N Engl J Med. 1997;337(11):725–33. doi: 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- 44.Rinaldo CR, Jr, Liebmann JM, Huang XL, et al. Prolonged suppression of human immunodeficiency virus type 1 (HIV-1) viremia in persons with advanced disease results in enhancement of CD4 T cell reactivity to microbial antigens but not to HIV-1 antigens. J Infect Dis. 1999;179(2):329–36. doi: 10.1086/314599. [DOI] [PubMed] [Google Scholar]
- 45.Wherry EJ, Ha SJ, Kaech SM, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27(4):670–84. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 46.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–48. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 47.Herbeuval JP, Grivel JC, Boasso A, et al. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood. 2005;106(10):3524–31. doi: 10.1182/blood-2005-03-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feuth T, Arends JE, Fransen JH, et al. Complementary role of HCV and HIV in T-cell activation and exhaustion in HIV/HCV coinfection. PLoS One. 2013;8(3):e59302. doi: 10.1371/journal.pone.0059302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cabrera R, Tu Z, Xu Y, et al. An immunomodulatory role for CD4(+)CD25(+) regulatory T lymphocytes in hepatitis C virus infection. Hepatology. 2004;40(5):1062–71. doi: 10.1002/hep.20454. [DOI] [PubMed] [Google Scholar]
- 50.Chase AJ, Yang HC, Zhang H, Blankson JN, Siliciano RF. Preservation of FoxP3+ regulatory T cells in the peripheral blood of human immunodeficiency virus type 1-infected elite suppressors correlates with low CD4+ T-cell activation. J Virol. 2008;82(17):8307–15. doi: 10.1128/JVI.00520-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Garra A, Vieira P. Regulatory T cells and mechanisms of immune system control. Nat Med. 2004;10(8):801–5. doi: 10.1038/nm0804-801. [DOI] [PubMed] [Google Scholar]
- 52.Rushbrook SM, Ward SM, Unitt E, et al. Regulatory T cells suppress in vitro proliferation of virus-specific CD8+ T cells during persistent hepatitis C virus infection. J Virol. 2005;79(12):7852–9. doi: 10.1128/JVI.79.12.7852-7859.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rallon NI, Lopez M, Soriano V, et al. Level, phenotype and activation status of CD4+FoxP3+ regulatory T cells in patients chronically infected with human immunodeficiency virus and/or hepatitis C virus. Clin Exp Immunol. 2009;155(1):35–43. doi: 10.1111/j.1365-2249.2008.03797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roe B, Coughlan S, Dean J, et al. Phenotypic characterization of lymphocytes in HCV/HIV co-infected patients. Viral Immunol. 2009;22(1):39–48. doi: 10.1089/vim.2008.0074. [DOI] [PubMed] [Google Scholar]
- 55.Tsubouchi E, Akbar SM, Horiike N, Onji M. Infection and dysfunction of circulating blood dendritic cells and their subsets in chronic hepatitis C virus infection. J Gastroenterol. 2004;39(8):754–62. doi: 10.1007/s00535-003-1385-3. [DOI] [PubMed] [Google Scholar]
- 56.Gompels M, Patterson S, Roberts MS, Macatonia SE, Pinching AJ, Knight SC. Increase in dendritic cell numbers, their function and the proportion uninfected during AZT therapy. Clin Exp Immunol. 1998;112(2):347–53. doi: 10.1046/j.1365-2249.1998.00590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marcellin P, Boyer N, Gervais A, et al. Long-term histologic improvement and loss of detectable intrahepatic HCV RNA in patients with chronic hepatitis C and sustained response to interferon-alpha therapy. Ann Intern Med. 1997;127(10):875–81. doi: 10.7326/0003-4819-127-10-199711150-00003. [DOI] [PubMed] [Google Scholar]
- 58.Conry SJ, Meng Q, Hardy G, et al. Genetically associated CD16(+)56(-) natural killer cell interferon (IFN)-alphaR expression regulates signaling and is implicated in IFN-alpha-induced hepatitis C virus decline. J Infect Dis. 2012;205(7):1131–41. doi: 10.1093/infdis/jis027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ghany MG, Strader DB, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49(4):1335–74. doi: 10.1002/hep.22759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gonzalez VD, Landay AL, Sandberg JK. Innate immunity and chronic immune activation in HCV/HIV-1 co-infection. Clin Immunol. 2010;135(1):12–25. doi: 10.1016/j.clim.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 61.Minutello MA, Pileri P, Unutmaz D, et al. Compartmentalization of T lymphocytes to the site of disease: intrahepatic CD4+ T cells specific for the protein NS4 of hepatitis C virus in patients with chronic hepatitis C. J Exp Med. 1993;178(1):17–25. doi: 10.1084/jem.178.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neumann-Haefelin C, Timm J, Spangenberg HC, et al. Virological and immunological determinants of intrahepatic virus-specific CD8+ T-cell failure in chronic hepatitis C virus infection. Hepatology. 2008;47(6):1824–36. doi: 10.1002/hep.22242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1 Gating approach for CD4+CD25+FoxP3+ (Tregs).
Supplementary Fig. 2 Gating approach for IFN-γ+ NK cells in the presence of K562 target cells.
Supplementary Fig. 3 Gating approach for CD81 on NK cell subsets (blue line: isotype, red line: CD81).
Supplementary Fig. 4 Multivariate linear regression model of CD4+ T cell count and patient group as predictors of CD38 levels on CD8+ T cells. (A) Effect of CD4+ T cell count (cells/mm3) and patient group (HCV or HIV/HCV) as independent predictors (model 1) on T cell markers of activation/exhaustion [MFI of CD38 on CD3+CD8+, CD4+FoxP3+CD25+ percentage (%) of lymphocytes, MFI of PD1 on CD3+CD8+, MFI of CTLA-4 on CD3+CD8+, MFI of BTLA on CD3+CD8+, MFI of CD160 on CD3+CD8+] (top panel), and NK cell markers of activation and function {MFI of CD69 on Lin3−CD56dimCD16+, MFI of CD81 on Lin3−CD56dimCD16+, MFI of IFN-αR on Lin3−CD56dimCD16+, constitutive MFI of phosphorylated (p) STAT-1 on Lin3−CD56dimCD16+, in vitro IFN-α-induced direct cytotoxicity against K562 target cells [expressed as in vitro IFN-α-induced area under the curve (AUC) - constitutive AUC for effector:target (E:T) ratios of 50:1, 25:1, 12.5:1 and 6.25:1], (in vitro IFNα-induced - constitutive) Lin3−CD56dimCD16+IFN-γ+ % of Lin3−CD56dimCD16+, and (in vitro IFNα-induced - constitutive) Lin3−CD56brightIFN-γ+ % of Lin3−CD56bright} (bottom panel); (B) Effect of CD4+ T cell count (cells/mm3) and patient group (HCV or HIV/HCV) including the addition of interaction term (model 2) on MFI of CD38 on CD3+CD8+ T cells. Data in panels (A) and (B) are shown as plots of each variable (axis y) against CD4+ T cell count (cells/mm3) (axis x). Lines in plots represent the perfect fit for each group of patients (red line for HCV mono-infected and blue line for HIV/HCV co-infected subjects).
Supplementary Table 1 Staining combinations for real-time whole blood-based phenotypic characterization of immune subsets.
Supplementary Table 2. Linear models using CD4+ T cell count (cells/mm3) and patients group as predictors of immune variables.