

Abstract
Janus tyrosine kinase 2 (JAK2) mediates downstream signaling of cytokine receptors in all hematological lineages, yet constitutively active JAK2 mutants are able to drive selective expansion of particular lineage(s) in myeloproliferative neoplasm (MPN). The molecular basis of lineage specificity is unclear. Here we show that three activating JAK2 mutants with similar kinase activities in vitro elicit distinctive MPN phenotypes in mice by differentially expanding erythroid vs. granulocytic precursors. Molecularly, this reflects the differential binding of JAK2 mutants to cytokine receptors EpoR and GCSFR in the erythroid vs. granulocytic lineage and the creation of unique receptor/JAK2 complexes that generate qualitatively distinct downstream signals. Our results demonstrate that activating JAK2 mutants can differentially couple to selective cytokine receptors and change the signaling repertoire, revealing the molecular basis for phenotypic differences elicited by JAK2(V617F) or mutations in exon 12. Based on these findings, receptor-JAK2 interactions could represent new targets of lineage-specific therapeutic approaches against MPN, which may be applicable to other cancers with aberrant JAK-STAT signaling.
INTRODUCTION
The JAK/STAT pathway transduces signals from cytokine receptors to regulate diverse cellular processes, and this pathway is frequently activated in cancers 1-4. Constitutive activation of the JAK2 tyrosine kinase is causally linked to myeloproliferative neoplasm (MPN), a group of clonal hematopoietic stem cell diseases characterized by the overproduction of blood cells of the myelo-erythroid lineages, as reviewed in 5. A somatic activating JAK2 mutation, V617F, is identified in the majority of MPN patients and is sufficient to drive MPN 6-11. Other activating mutations in JAK2 exon 12 have also been identified in MPN, albeit with less frequency 12,13. Based on these findings, JAK2 inhibitors have been developed to treat MPN 14,15.
Despite these advances, the molecular basis for how JAK2 contributes to MPN is incompletely understood. It is puzzling that constitutive activation of JAK2, which mediates signaling of receptors in all hematological lineages, drives clonal disorders of only the myelo-erythroid lineage. It was initially postulated that this is due to lineage-specific expression of cytokine receptors. Receptors required to collaborate with JAK2(V617F) and enable cytokine-independent growth of hematopoietic cells are only expressed in myelo-erythroid but not lymphoid cells 16,17. However, it has since been shown that lymphoid receptor IL27Rα can also support JAK2(V617F)-mediated cytokine-independent growth in vitro 18, and that JAK2(V617F), when expressed alone, can induce signaling via endogenous receptors such as the IGF1R 8,19. In addition, other activating JAK2 mutations have been identified in patients with Down’s syndrome-associated acute lymphoblastic leukemia 20,21. Equally intriguing is that patients with the MPN, polycythemia vera (PV), can harbor either V617F or an exon 12 mutation and both present with erythrocytosis, but only those with V617F present with concomitant granulocytosis or thrombocytosis 12. We also do not fully understand why the same V617F mutation is present in patients with related, but distinct, MPN. Thus, the mechanisms underlying lineage specificity and phenotypic pleiotropy in MPN remain poorly understood.
Here, we report that, despite having similar kinase activities in vitro, three activating JAK2 mutants elicited distinctive MPN phenotypes in mice. We show that the different mutations cause JAK2 to differentially couple to selective lineage-specific cytokine receptors. Furthermore, unique receptor/JAK2 mutant complexes generate qualitatively distinct downstream signaling. Together, these differences account for the ability of JAK2 mutants to differentially drive selective lineage expansion. Our findings show that oncogenic JAK kinases can change signaling repertoire at the receptor level, providing a molecular basis for MPN pathology.
MATERIALS AND METHODS
Bone marrow transplantation
Bone marrow cells from Balb/c mice were transduced to express murine JAK2 or JAK2 mutants from the MSCV-IRES-GFP vector and injected into lethally irradiated recipients as described and in accordance with University guidelines 22.
Peripheral blood analysis and histology
Peripheral blood was analyzed on a Hemavet automated blood counter (Drew Scientific). Paraffin embedded sections were stained with hematoxylin and esoin (H&E), and with Chandler’s precision reticulum silver stain (American MasterTech Scientific Inc.) to detect reticulin fibers.
Murine colony-forming unit assay
Epo-independent endogenous CFU-E and cytokine-independent CFU-G were enumerated in methylcellulose media without exogenous cytokines (MethoCult M3234, StemCell Technologies). Sorted CMPs were plated with IL3, IL6, SCF, and Epo (MethoCult M3434, StemCell Technologies). Colonies were scored according to manufacturer manual.
Flow cytometry
HSCs were identified by SLAM markers 23 and CMP, MEP, and GMP progenitors were recognized as described 24. Erythroid precursors were identified using Ter119, CD71, and the FSC parameter 25. Granulocytic precursors were identified with Gr1 and Mac1. All data were acquired on a FACSCalibur, LSRII, or Aria (BD Biosciences) flow cytometers and analyzed with FlowJo software (Tree Star, CA).
BrdU labeling assay
For progenitor cells, 16 hrs post injection of BrdU (100mg/kg), BM and spleen cells were harvested, fixed and permeabilized with Cytofix/Cytoperm buffer (BD Biosciences), treated with DNaseI, and stained with Pacific Blue conjugated BrdU monoclonal antibody (Invitrogen). The percentages of cells with BrdU label were determined by flow cytometry. For precursors, cells were analyzed similarly 30 min post BrdU injection.
Annexin V binding assay
Apoptosis was quantified by staining cells with annexin V-APC (BD Pharmingen) for 15 min and a vital dye 7-AAD and analyzed by flow cytometry.
Cell culture
BaF3 cells (ATCC) stably expressing WT or mutant JAK2 in MSCV-neo vector with HA-EpoR, HA-EpoR(F8), HA-GCSFR, HA-GCSFR(F4) in pMX-IRES-GFP vector were sorted for GFP fluorescence, and maintained in media with 1mg/ml G418 and WEHI-3B cell supernatant as a source of IL3 17,22. To determine sensitivities to inhibitors, cells were grown in the presence of 10 μM of MEK inhibitor U0126, 10 μM of PI3K inhibitor LY294002, or 8 μM of STAT3/5 inhibitor pimozide.
Immunoprecipitation and immunoblotting
Cell lysates were immuno-blotted with HA, JAK2, phospho-STATA5 antibodies, or anti-phosphotyrosine antibody 4G10, and immunoprecipitated with HA agarose or JAK2 antibodies as indicated 26.
MTT assay
5,000 BaF3 cells were seeded per well of a 96 well plate and MTT assay was performed according to manufacturer’s instructions (Promega).
Human CD34+ and mononuclear cells isolation, lentiviral transduction and colony assays
Normal human CD34+ cells were purchased from Fred Hutchinson Cancer Center Hematopoietic Cell Processing and Repository. CD34+ cells were transduced with puromycin resistant lentiviruses expressing wild-type or mutant JAK2, then plated in methylcellulose media (H4230, StemCell Technologies) with puromycin (1μg/ml). For Epo-independent BFU-E, IL3 and SCF were added. Colonies were enumerated according to manufacturer manual. PV mononuclear cells were isolated from whole blood via Ficoll-Paque (GE Healthcare). Written informed consent was obtained according to Institutional Review Board guidelines.
Statistical analyses
Data are reported as means ± SEM. Statistical significance (P<0.05) of differences between groups was determined by Student’s t-test (2-tailed, unpaired) using the GraphPad Prism software.
Additional experimental procedures are detailed in Supplemental Methods
RESULTS
JAK2 mutants confer distinct MPN phenotypes in mice
From a functional screen for residues required for JAK2 auto-inhibition, we identified a panel of activating JAK2 mutations in addition to V617F 22. To determine their physiological consequences, a murine transplantation model was employed with bone marrow cells retrovirally expressing either wild-type JAK2 or JAK2 mutants, together with GFP. Specifically, we examined K539I, V617F and N622I; K539I lies in exon 12, whereas N622I had the highest mitogenic activity in hematopoietic BaF3 cells. These mutants exhibited similar in vitro kinase activity 22 and protein half-lives (supplemental Fig. 1). Cells were transduced at comparable efficiencies (and less than 30% to avoid doubly infected cells) prior to transplantation and expressed similar amounts of JAK2 protein (supplemental Fig. 2a-b). At two weeks post transplantation, GFP+ percentages and median fluorescence intensities, which correlate with JAK2 expression levels, were also comparable in total, Lin- as well as Lin-Sca1-Kit+ populations (supplemental Fig. 2c-d). Interestingly, we found that these mutants elicited drastically different phenotypes. Similar to MPN patients, JAK2 mutant mice exhibited splenomegaly (Fig. 1a). However, complete blood count (CBC) analyses showed that different lineage blood cells were overproduced (Fig. 1a). Mice receiving JAK2(K539I)-transduced cells showed primarily erythrocytosis with mild granulocytosis (Fig. 1a, red). Mice expressing JAK2(V617F) showed erythrocytosis and granulocytosis (Fig. 1a, green), whereas JAK2(N622I) animals displayed predominant granulocytosis (Fig. 1a, blue). Importantly, blood cell counts in mice overexpressing wild-type JAK2 were normal (Fig. 1a, black, referred to as WT mice hereafter).
Fig. 1.
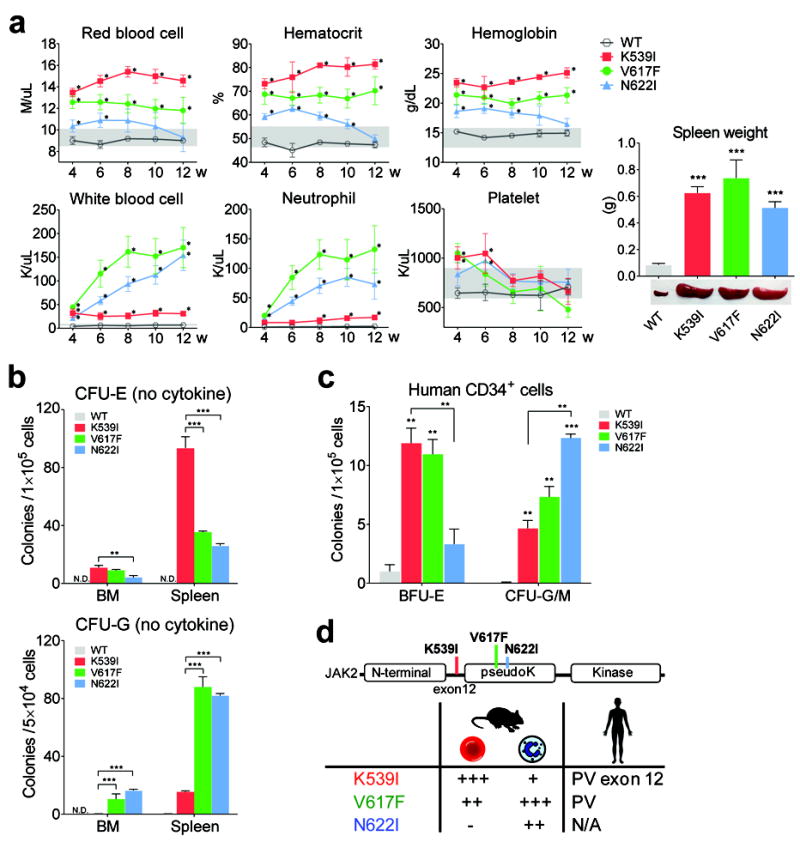
Activating JAK2 mutants confer distinct hematological phenotypes in mice. (a) Hematological parameters of mice assessed at indicated times post transplantation. Grey zones indicate normal range. w: weeks post transplantation. *p<0.01 (vs. WT). Mice expressing JAK2 mutants exhibited splenomegaly at 12 weeks post transplantation. (b) Cells from JAK2 mutant mice but not WT mice generated cytokine-independent erythroid and granulocytic colonies. *p<0.05, **p<0.01, ***p<0.001. n=3 in each group.ND: not detected. (c) Expression of JAK2 mutants in human CD34+ cells differentially affects erythroid vs. granulocytic lineages. Normal CD34+ cells were transduced with lentiviruses expressing WT or mutant JAK2 and plated in methylcellulose media with puromycin to select for transduced cells. Epo-independent BFU-E colonies and cytokine-independent CFU-G/M colonies, which include CFU-G, CFU-M and CFU-GM colonies are shown. Colonies were enumerated on day 14. n=3 in each group. *p<0.05, **p<0.01, ***p<0.001 (vs. WT unless specified). (d) Summary of blood phenotypes. The degree of erythrocytosis and granulocytosis are indicated by plus signs.Results represent at least three independent experiments.
Differential expansion of different lineages was confirmed in histological analyses of bone marrow (BM) and spleen sections, and as observed in MPN patients, reticulin fibrosis was observed in mice expressing JAK2 mutants (supplemental Fig. 3). At early time points, the low levels of erythrocytosis in JAK2(N622I) mice and the mild elevation of platelets in JAK2(K539I) and JAK2(V617F) mice, both transient, possibly reflected contribution from transduced progenitor cells (Fig. 1a). We thus focused on later time points after phenotypes were well established. Prior studies comparing different murine models have demonstrated that the level of JAK2(V617F) expression influences phenotype; expression level equal or lower than endogenous JAK2 is required to elicit thrombocytosis 11,27,28. The expression levels of JAK2 mutants are higher in our mice, which may have contributed to the lack of thrombocytosis.
Next we determined cytokine-independent colony formation, a hallmark of MPN. As shown in Fig. 1b, BM and spleen cells from mice expressing JAK2 mutants but not WT produced cytokine-independent erythroid (CFU-E) and/or granulocytic (CFU-G) colonies, in line with the constitutive activation of JAK2 mutants. Consistent with their distinct CBC phenotypes, JAK2(K539I) mice showed the highest number of cytokine-independent erythroid colonies, whereas JAK2(V617F) and JAK2(N622I) mice produced higher numbers of cytokine-independent granulocytic colonies (Fig. 1b).
We corroborated our findings in human CD34+ cells. CD34+ cells expressing JAK2(K539I) and JAK2(V617F) produced more Epo-independent erythroid colonies than cells expressing JAK2(N622I), whereas JAK2(N622I) and JAK2(V617F) cells produced more cytokine-independent granulocytic colonies than JAK2(K539I) cells (Fig. 1c).
Our results demonstrated that expression of three activating JAK2 mutants differentially drive erythrocytosis and/or granulocytosis in mice (Fig. 1d). We believe K539I is functionally equivalent to the K539L mutation found in MPN patients, and our results are consistent with clinical data showing that patients with the V617F mutation exhibit granulocytosis in addition to erythrocytosis, while those with exon 12 mutations (where K539 resides), exhibit isolated erythrocytosis 12. Thus, our three mutant models provided the opportunity to investigate differences in JAK2 signaling that might drive erythropoiesis and granulopoiesis.
JAK2 mutants differentially affect hematopoietic progenitor and precursor cells
To determine the cellular mechanisms underlying the differential expansion of erythrocytes and granulocytes in JAK2 mutant mice, we quantified erythroid and granulocytic precursors as well as earlier progenitors by flow cytometry. GFP+ cells were gated for each cell population.
Consistent with the erythrocytosis phenotype, total Ter119+ erythroid precursors per animal massively expanded over WT in JAK2(K539I) and JAK2(V617F) mice, especially in spleen (Fig. 2a). Mac1+/Gr1+ myeloid precursors increased in all three JAK2 mutant mice, and the overall Mac1+/Gr1+ cell numbers (7, 3, 1.5-fold over WT for JAK2(N622I), JAK2(V617F) and JAK2(K539I), respectively) are in line with their degree of granulocytosis (JAK2(N622I)> JAK2(V617F)> JAK2(K539I))(Fig. 2b).
Fig. 2.
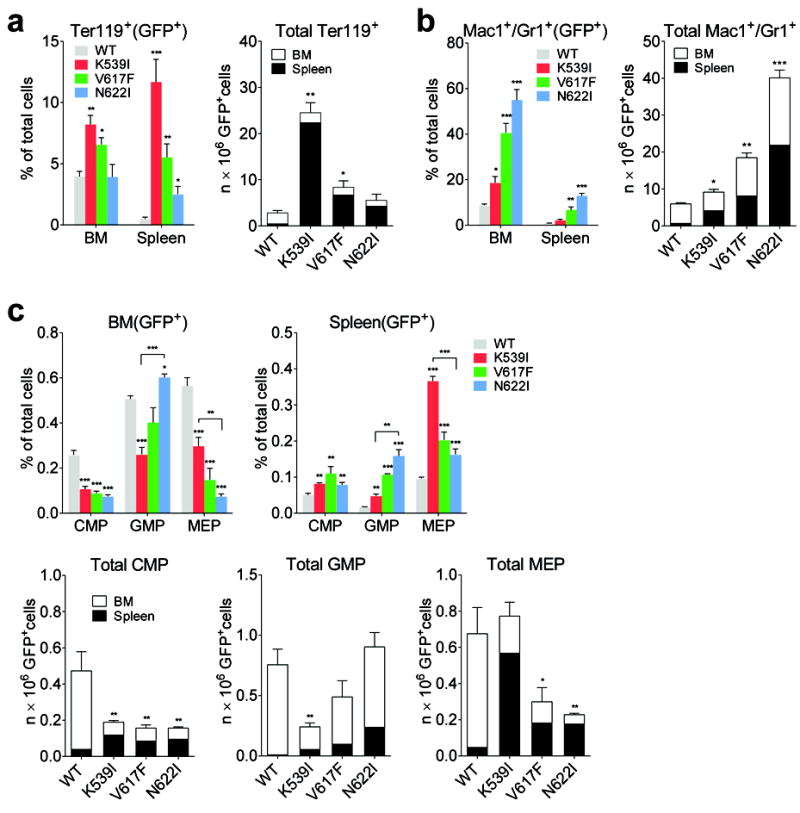
Erythroid and granulocytic precursors are differentially expanded in JAK2 mutant mice. (a-b)Frequency and total number of Ter119+ erythroid (a) and Mac1+/Gr1+ granulocytic (b) precursors were increased in JAK2 mutant mice. Because hind limbs BM represents approximately 25% of total bone marrow 32, total number of precursors per animal was calculated as the sum of the number from spleen and four fold the number from hind limbs BM. (c) Frequency and total number of BM and splenic CMP, MEP, and GMP progenitors from mice expressing JAK2 mutants or WT mice. In all panels, GFP+ indicates that GFP+ cells are gated for analyses. n=3 in each group. *p<0.05, **p<0.01, ***p<0.001 (vs. WT unless specified). Results represent at least three independent experiments.
Hematopoietic stem cells and progenitors were reduced in the BM of JAK2 mutant mice and increased in the spleen (Fig. 2c, supplemental Fig. 4). Similar numbers of CMP were observed in all three JAK2 mutant mice (Fig. 2c). JAK2(K539I) mice had the highest numbers of MEP, whereas JAK2(N622I) mice the highest numbers of GMP (Fig. 2c). Compared to WT mice, total CMP, GMP and MEP numbers per animal did not increase in JAK2 mutant mice (Fig. 2c). The degree of expansion is thus much greater in precursors than earlier progenitors.
JAK2 mutants differentially regulate apoptosis, proliferation and differentiation of progenitor and precursor populations
To further probe the mechanisms in different cell populations, we measured apoptosis and proliferation using Annexin V staining and in vivo bromodeoxyuridine (BrdU) incorporation, respectively.
Apoptosis in CMP, MEP and GMP progenitors was lower in JAK2 mutant mice compared to WT controls, but the differences between mutants were small (Fig. 3a, supplemental Fig. 5a-d). Expression of JAK2 mutants did not increase their proliferation compared to WT control (supplemental Fig. 5e). These effects could not explain the significant differences in BM GMP and MEP numbers among JAK2 mutant mice (Fig. 2c). We observed that with similar numbers of CMP cells, the ratio of GMP to MEP progeny populations differed greatly among JAK2 mutant mice (Fig. 3b). The GMP/MEP ratio in JAK2(V617F) and JAK2(N622I) mice was drastically greater than WT animals (3-10 fold in BM and spleen), but normal in JAK2(K539I) mice (Fig. 3b). We sorted BM CMP progenitors prospectively and after culture enumerated granulocytic and macrophage colonies vs. erythroid and megakaryocytic colonies. Sorted CMPs from JAK2(V617F) and JAK2(N622I) mice generated significantly more granulocytic and monocytic lineage colonies than erythroid and megakaryocytic lineage colonies compared to WT and JAK2(K539I) mice (Fig. 3c), consistent with the increased GMP/MEP ratios. Therefore, differentiation was skewed toward granulopoiesis in the early progenitors of JAK2(V617F) and JAK2(N622I) mice. Alternatively, JAK2(V617F) and JAK2(N622I) may selectively amplify GM-committed progenitors in the normal CMP gate.
Fig. 3.
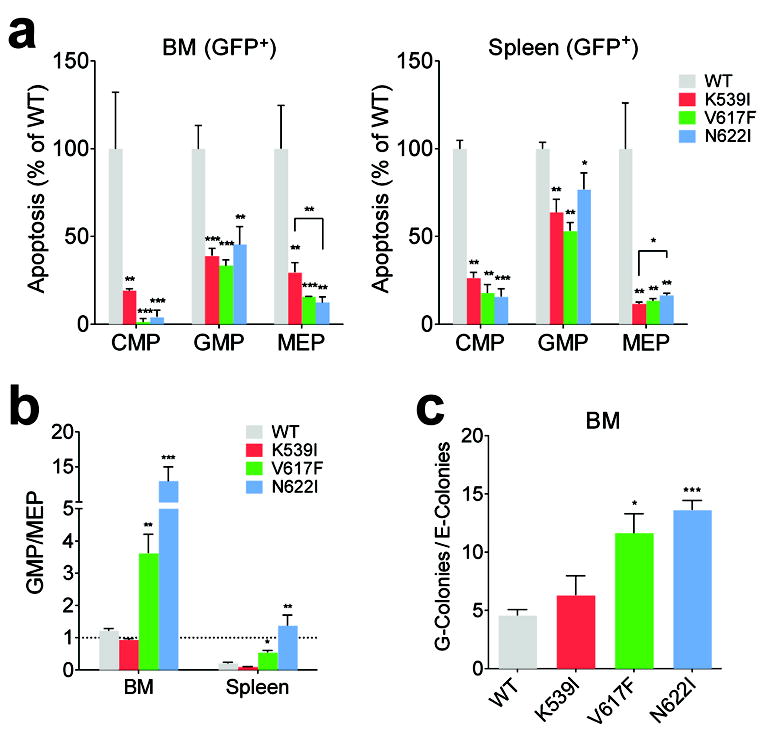
Survival and differentiation analyses of myeloid progenitors from JAK2 mutant mice. (a) Annexin V-binding analyses in CMP, MEP, and GMP progenitors normalized to WT controls.Apoptotic cells (GFP+AnnexinV+7AAD- plus GFP+AnnexinV+7AAD+) were quantified. (b) GFP+ GMP to MEP ratio is significantly higher in JAK2(V617F) and JAK2(N622I) mice compared to WT mice. (c) Sorted GFP+ CMP from JAK2(V617F) and JAK2(N622I) mice generated more granulocytic and macrophage colonies (G-colonies) than erythroid and megakaryocytic colonies (E-colonies) compared to WT mice. n=3 in each group, *p<0.05, **p<0.01, ***p<0.001 (vs. WT unless specified). Results represent at least three independent experiments.
Results in progenitors did not explain erythrocytosis presented in JAK2(K539I) and JAK2(V617F) mice. We next examined erythrocytic and granulocytic precursors. As Ter119+ and Mac1+/Gr1+ precursors are comprised of distinguishable subsets that differ in survival and proliferative potential, we further classified them into progressively differentiated subsets. Erythroid precursors were subdivided using Ter119, the transferrin receptor (CD71)and the “cell size” parameter (FSC), into progressively maturing proerythroblast (ProE), basophilic erythroblast (EryA), late basophilic and polychromatophilic erythroblasts (EryB), and orthochromatic erythroblasts (EryC) (supplemental Fig. 6a-c)25. Early apoptotic fractions (GFP+ AnnexinV+7AAD-) decreased significantly in splenic ProE, EryA and EryB erythroblasts in JAK2(K539I)and JAK2(V617F) mice to about 30% relative to WT controls (Fig. 4a). Similar observation was found in the 7AAD+ late apoptotic fractions (supplemental Fig. 6d). These results were consistent with the large expansion of JAK2(K539I) and JAK2(V617F) erythroblasts (Fig. 4a, supplemental Fig. 6e). Even though splenic EryA and EryB showed small reduction in apoptosis, massive increase in EryC accounted for the lack of erythrocytosis in JAK2(N622I) mice (Fig. 4a, supplemental Fig. 6d). For proliferation, none of the JAK2 mutants showed increased BrdU incorporation in ProE or EryA precursors compared to WT cells (supplemental Fig. 6f). Proliferation was not compared in EryB and EryC subsets because they did not tolerate the fixation/staining process. However, these later erythroblasts normally have low proliferative potential.
Fig. 4.

Survival and proliferation analyses of erythroid and myeloid precursor subsets from JAK2 mutant mice. (a) Quantitative analyses of Annexin V-binding in erythroblast subsets normalized to WT controls. Early apoptotic cells (GFP+AnnexinV+7AAD-), and frequency of BM and splenic erythroblast subsets were quantified. (b)Quantitative analyses of Annexin V-binding and BrdU incorporation in granulocytic precursor subsets normalized to WT controls. Early apoptotic cells (GFP+AnnexinV+7AAD-) were shown. Frequency of BM and splenic granulocytic precursor subsets were quantified. (c) Summary of cellular effects of JAK2 mutants.n=3 in each group. *p<0.05, **p<0.01, ***p<0.001 (vs. WT unless specified). Results represent two independent experiments.
Our results suggested that reduced apoptosis rather than increased proliferation of erythroid precursors likely contributed to their expansion in JAK2(K539I) and JAK2(V617F) mice. These results are consistent with the idea that JAK2 mutants exploit the high apoptotic rate in splenic ProE and EryA erythroblasts, which was shown as a main mechanism to ensure a reserve of erythroid precursors that can rapidly respond to acute erythropoietic stress 29.
Granulocyte precursors were further divided into immature Mac1+Gr1lo and mature Mac1+Gr1hi subsets (supplemental Fig. 7a-b). Decreased apoptosis was observed for both subsets in the spleens of JAK2 mutant mice, as well as in BM JAK2(V617F) and JAK2(N622I) Mac1+Gr1lo cells compared to WT controls (Fig. 4b, supplemental Fig. 7c). We also observed profoundly increased splenic precursor proliferation in both subsets in JAK2 mutant mice (Fig. 4b). Concomitantly, the decreased apoptosis and increased proliferation resulted in significantly increased granulocyte precursors, especially in JAK2(V617F) and JAK2(N622I) mice (Fig. 4b, supplemental Fig. 7d).
Together, these results identified the cellular mechanisms underlying the ability of JAK2 mutants to differentially affect hematopoiesis. We concluded that erythrocytosis-driving JAK2 mutants (K539I, V617F) cause a marked decrease in apoptosis of erythroid precursors, and that granulocytosis-driving mutants (V617F, N622I) reduce apoptosis and increase proliferation of granulocytic precursors. In addition, expression of JAK2(V617F) and JAK2(N622I) favors granulocyte-monocyte commitment of multipotent progenitors. (Fig. 4c).
JAK2 mutants differentially couple to EpoR and GCSFR
Having defined cellular mechanisms underlying the phenotypic differences, we next examined the molecular mechanisms. Because JAK2(V617F) signaling requires the co-expression of cytokine receptors that serves as scaffolds for downstream signaling 16-18, we surmised that differences in interaction between JAK2 mutants and cytokine receptors might account for the different phenotypic outcomes. Specifically, we hypothesized that JAK2 mutants differentially couple to EpoR and GCSFR, as functions of these two receptors mirror the effects seen in the specific progenitors/precursors 29-32.
We first tested this hypothesis in BaF3 cells, in which activating JAK2 mutants required the co-expression of either EpoR or GCSFR to grow in an IL3-independent manner. BaF3 cells stably expressing WT or mutant JAK2 with HA-tagged EpoR or GCSFR were maintained in media containing IL3. Here, instead of JAK2, HA-EpoR and HA-GCSFR were expressed bicistronically with GFP, so median GFP fluorescence intensity can be used to quantify total receptor expression levels. Cells were washed and cultured without IL3 and the amount of receptors necessary for each JAK2 mutant to grow IL3-independently was compared. Prior to IL3 withdrawal, all cells expressed similar levels of receptors, and IL3 withdrawal selected for cells with higher receptor levels (Fig. 5a). Interestingly, erythrocytosis-driving JAK2(K539I) required the least amount of EpoR, while granulocytosis-driving JAK2(N622I) the least amount of GCSFR. Levels of EpoR and GCSFR in cells expressing JAK2(V617F), which drives both erythrocytosis and granulocytosis, were medium (Fig. 5a). Receptor expression levels were confirmed by immunoblotting (supplemental Fig. 8a). These results suggest that JAK2 mutants may bind to receptors with different affinities, and/or differentially activate signaling in a receptor-JAK2 complex. Similar results were obtained in another hematopoietic cell line, 32D (supplemental Fig. 8b), except that JAK2(K539I) was not able to effectively promote IL3-independent growth in 32D cells co-expressing GCSFR, consistent with its inferior binding to GCSFR (discussed below).
Fig. 5.
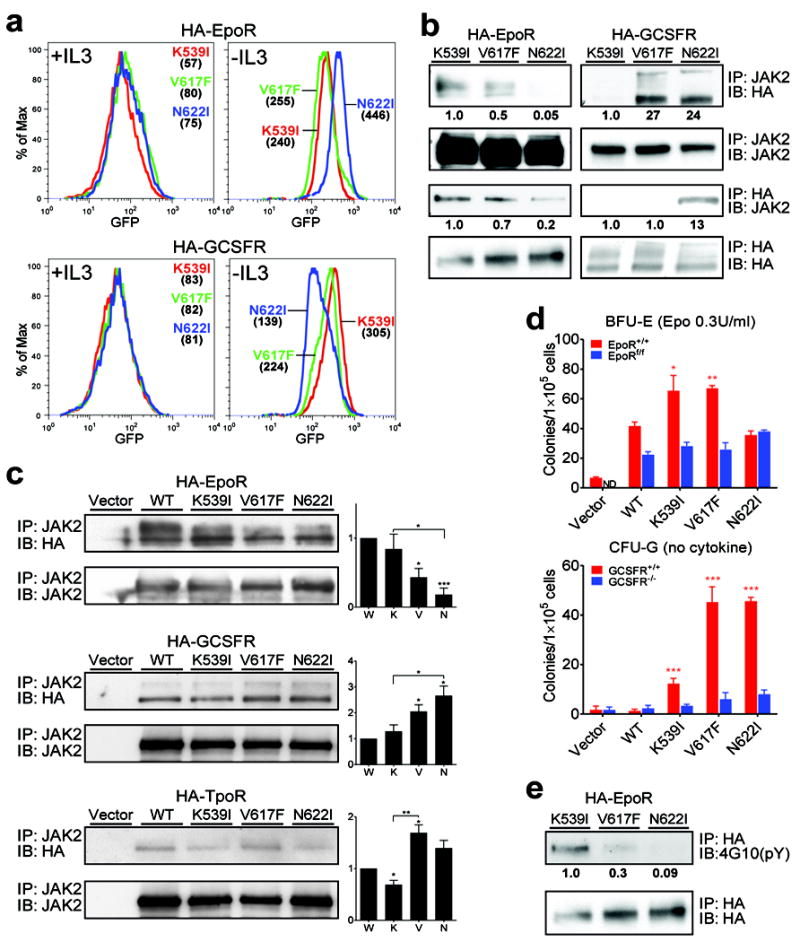
JAK2 mutants interact with cytokine receptors in different ways. (a) The amount of co-expressed receptors necessary for each JAK2 mutant to transform BaF3 is different. BaF3 cells stably expressing WT or mutant JAK2 with HA-tagged EpoR or GCSFR (bicistronically co-expressed with GFP) were selected in media without IL3. Median GFP fluorescence intensities (numbers in parentheses) indicate receptor expression level before and after IL3 withdrawal. (b) JAK2 mutants bind to receptors with different affinities. Co-immunoprecipitation analyses of JAK2 mutants and HA-tagged receptors in BaF3 cells using either JAK2 or HA antibodies.Immunoblot band intensity was quantified by ImageJ software and normalized to JAK2(K539I). (c) JAK2 mutants differentially bind to receptors in JAK2-deficient γ2A cells. JAK2 mutants were transiently expressed in γ2A cells in which HA-tagged EpoR, GCSFR, or TpoR were stably expressed. Interaction was examined by co-immunoprecipitation with JAK2 antibodies. Immunoblot band intensity was quantified by ImageJ software and normalized to loading control and wild-type JAK2. W:WT, K: K539I, V: V617F, N: N622I. (d) Deletion of EpoR or GCSFR impairs hypersensitive erythroid colonies and cytokine-independent granulocytic colonies produced by JAK2 mutants. Bone marrow cells from wild-type or EpoRflox/flox mice, transduced to express JAK2 mutants and GFPCre, and bone marrow cells from wild-type or GCSFR-/- mice, transduced to express JAK2 mutants, were plated in methylcellulose media (M3234) with 0.3U/ml Epo (for BFU-E) or with no cytokine (for CFU-G). Colonies were enumerated on day 5 and 7. (e) EpoR is tyrosine phosphorylated in cells expressing JAK2(K539I) and JAK2(V617F) but not JAK2(N622I). HA-EpoR immunoprecipitated via HA agarose was probed with anti-phosphotyrosine antibody 4G10. *p<0.05, **p<0.01, ***p<0.001 (vs. WT unless specified). ND: not detected. IP: immunoprecipitation. IB: immunoblot. Results represent at least three independent experiments.
The ability of JAK2 mutants to bind EpoR or GCSFR was examined using co-immunoprecipitation assays in BaF3 cells and γ2A cells that lack endogenous JAK2. As shown in Fig. 5b-c, more EpoR co-immunoprecipitated with JAK2(K539I) and JAK2(V617F) than JAK2(N622I), and more GCSFR co-immunoprecipitated with JAK2(V617F) and JAK2(N622I) than JAK2(K539I). Despite differences in receptor binding, all three mutants required the FERM domain to interact with EpoR and GCSFR, similar to wild-type JAK2, because a point mutation in JAK2 FERM domain (Y119E), previously shown to disrupt receptor binding 33,34, abolished their ability to cause IL3-independent growth in BaF3/EpoR and BaF3/GCSFR cells (supplemental Fig. 9).
To corroborate these results, we tested whether EpoR or GCSFR is required for JAK2 mutants to promote cytokine-independent colonies. BM cells from wild-type mice or EpoRflox/flox mice were transduced with retroviruses expressing JAK2 mutants and GFPCre (GFP-fused cre recombinase) bicistronically, so that transduced EpoRflox/flox cells but not wild-type cells will also be devoid of EpoR. We found that JAK2(K539I) and JAK2(V617F) exhibited Epo hypersensitivity in forming BFU-E colonies, which was normalized when EpoR was deleted (Fig 5d). To test for requirement of GCSFR, BM cells from GCSFR-/- or control (GCSFR+/+) mice were transduced to express JAK2 mutants. Cells from GCSFR-/- mice were strongly inhibited in forming JAK2 mutant mediated cytokine-independent granulocytic colonies, in contrast to control cells (Fig. 5d). These results are consistent with an essential role of EpoR and GCSFR in JAK2 mutant-mediated erythrocytosis and granulocytosis, respectively.
Because MPN patients with V617F but not exon 12 mutations exhibit thrombocytosis, we also compared interaction between JAK2 mutants and the thrombopoietin receptor (TpoR). JAK2(K539I) consistently co-immunoprecipitated less TpoR compared to JAK2(V617F) (Fig. 5c), suggesting a weaker interaction. Because our mice did not exhibit thrombocytosis, and we found that expression of TpoR alone or with WT JAK2 in BaF3 cells caused factor-independent growth that interfered with experimental interpretation, we focused on signaling from EpoR and GCSFR below.
We compared receptor tyrosine phosphorylation in cells expressing JAK2 mutants, the first step in canonical cytokine receptor signaling. As shown in Fig. 5e, EpoR was tyrosine-phosphorylated in JAK2(K539I)/EpoR and to a lesser extent JAK2(V617F)/EpoR cells but not in JAK2(N622I)/EpoR cells, indicating that EpoR was differentially phosphorylated by JAK2 mutants. Tyrosine phosphorylation of GCSFR was not observed using anti-phosphotyrosine antibodies (data not shown), which may be below detection limit. Together, these data established that JAK2 mutants differentially bind cytokine receptors and form unique signaling platforms (phosphorylated forms of receptors)to mediate IL3-independent growth in BaF3 cells.
Different receptor-JAK2 complexes elicit distinct downstream signaling
To interrogate downstream signaling generated by these unique receptor-JAK2 complexes, BaF3 cells expressing the different complexes were incubated with MEK inhibitor (U0126), PI3K inhibitor (LY294002), and STAT3/5 inhibitor (pimozide) 35 and their growth was monitored. These inhibitors blocked activities of their target proteins (supplemental Fig. 10). IL3-independent cells co-expressing EpoR with any of the three JAK2 mutants were sensitive to PI3K and STAT3/5 inhibitors, but only JAK2(N622I)/EpoR cells died in the presence of the MEK inhibitor (Fig. 6a). By contrast, IL3-independent cells co-expressing GCSFR and any of the three JAK2 mutants were sensitive to MEK and STAT3/5 inhibitors, but only JAK2(N622I)/GCSFR cells were also sensitive to PI3K inhibition (Fig. 6a). Therefore, receptor-JAK2 complexes formed with different JAK2 mutants relied on distinct downstream signaling pathways.
Fig. 6.
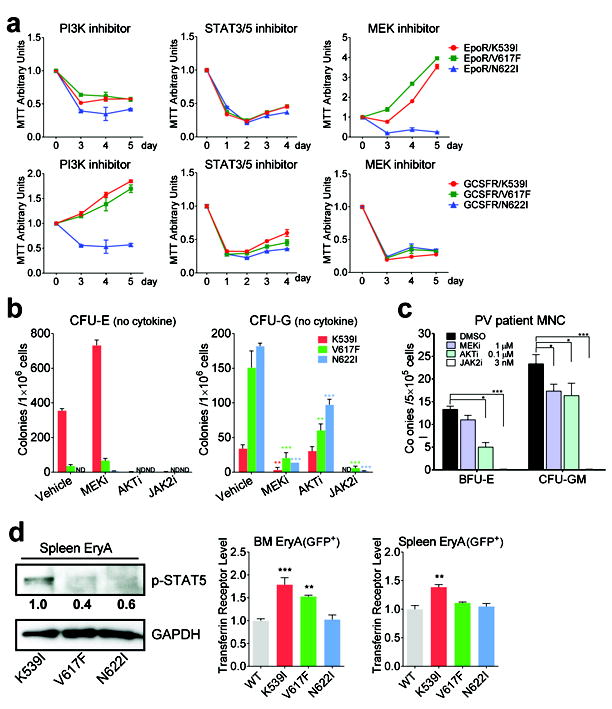
JAK2 mutants elicit distinct downstream signaling from receptor-JAK2 complex. (a) Sensitivity of BaF3 cells expressing JAK2 mutants with EpoR (top panels) or GCSFR (bottom panels) to inhibitors of PI3K (LY294002), STAT3/5 (pimozide) and MEK (U0126). (b) Cytokine-independent erythroid colonies and granulocytic colonies from JAK2 mutant mice grown in the presence of inhibitors to MEK (AZD6244), AKT (MK2206) or JAK2 (AZD1480) were enumerated.n=3 in each group. (c) Effects of inhibitors on the formation of Epo-independent BFU-E and cytokine-independent granulocytic colonies from JAK2(V617F)-positive PV patient mononuclear cells (MNC). Colonies were enumerated on day 14. n=3 in each group. (d) Levels of phospho-STAT5 and its target gene transferrin receptor are highest in sorted EryA cells expressing JAK2(K539I) among the three mutants. Immunoblot band intensity was quantified by ImageJ software and normalized to JAK2(K539I). *p<0.05, **p<0.01, ***p<0.001 (vs. vehicle (DMSO) in (b-c) or WT (d)). ND: not detected. Results represent at least three independent experiments.
We corroborated our findings using primary cells from JAK2 mutant mice as well as PV patients. Consistent with previous results, growth of cytokine-independent erythroid colonies from JAK2 mutant mice and from JAK2(V617F)-positive PV patients was sensitive to AKT inhibition, whereas growth of cytokine-independent granulocytic colonies from JAK2 mutant mice and from JAK2(V617F)-positive PV patients was sensitive to MEK inhibition (Fig. 6b-c). As controls, both types of colonies were sensitive to JAK2 inhibition. In line with data from BaF3 cells, MEK inhibitors did not curb cells from JAK2(K39I) and JAK2(V617F) mice and from JAK2(V617F) PV patients to form cytokine-independent erythroid colonies. MEK inhibitors actually caused an increase of cytokine-independent erythroid colonies from JAK2(K39I) mice, perhaps because constitutive MEK/ERK activation inhibits erythroid differentiation 36. Cytokine-independent erythroid colonies were too few in JAK2(N622I) mice to verify whether they were MEK inhibitor-sensitive. Consistent with our observations in BaF3 cells, cytokine-independent granulocytic colonies from JAK2(N622I) but not JAK2(K539I) mice were sensitive to AKT inhibitors (Fig. 6b). Different from results in BaF3 cells, cytokine-independent granulocytic colonies from JAK2(V617F) mice as well as from PV patients were also sensitive to AKT inhibitors, possibly reflecting differences between cell lines and primary cells.
Consistent with the high EpoR phosphorylation level, we observed high levels of activated STAT5 in EryA cells of JAK2(K539I) mice (Fig. 6d). Consequently, expression levels of one STAT5 target gene, the transferrin receptor (CD71), was also highest in JAK2(K539I) EryA cells (Fig. 6d). This is in line with findings that exon 12 mutations induce more robust activation of STAT5 than the V617F mutation 12, and a recent report of an exon 12 knock-in mouse model, showing higher levels of the transferrin receptor as well as enzymes in iron metabolism, which may optimize iron availability 13. We also observed that GFP+Lin-Sca1+Kit+ cells from JAK2(N622I) mice exhibited higher basal level activation of AKT, ERK and STAT5 than those from JAK2(K539I), JAK2(V617F) or WT mice (supplemental Fig. 11).
Together, our results demonstrated that interactions between JAK2 mutants and cognate cytokine receptors are fundamentally different, forming unique receptor/JAK2 complexes that elicit distinct downstream signaling, thereby dictating pathophysiological consequences in mice.
DISCUSSION
We report that selective coupling with cytokine receptors contributes to the different phenotypes elicited by three activating JAK2 mutants. Specifically, decreased binding of particular JAK2 mutants toward specific receptors impairs constitutive downstream signaling from that receptor-JAK2 complex. Moreover, unique receptor signaling platforms are created by different JAK2 mutants to elicit qualitatively distinct downstream pathways. These mechanisms, which act in concert and are not mutually exclusive, amplify single amino acid changes in JAK2 to yield immensely different disease phenotypes in vivo.
We show that JAK2 mutants bind to cytokine receptors differently. JAK2(N622I), which failed to elicit erythrocytosis in mice, binds less to EpoR than JAK2(K539I) and JAK2(V617F). JAK2(K539I), which generates the mildest granulocytosis among the 3 mutants, binds less to GCSFR than JAK2(N622I) and JAK2(V617F). Because JAK2 mutants still depend on their FERM domain for IL3-independent BaF3 cell growth, JAK2 mutations may directly or indirectly cause specific structural changes in the FERM domain to influence receptor binding.
Besides binding receptors differently, JAK2 mutants were able to form unique receptor-JAK complexes to activate qualitatively different downstream signaling. This is demonstrated by differences in receptor tyrosine phosphorylation, and downstream pathways required to cause IL3-independent BaF3 cell growth or to produce cytokine-independent colonies. We surmise that structural variations in JAK2 mutants might alter the position/orientation of the kinase domain relative to receptor tails, thus influencing the accessibility and efficiency to phosphorylate particular receptor tyrosines. The lowered affinity of specific JAK2 mutants towards a receptor may also impede phosphorylation.
Our study sheds light on some long-standing puzzles of phenotypic pleiotropy in MPN. First, polycythemia vera (PV) patients with either the JAK2 V617F mutant or exon 12 mutants present with erythrocytosis, but only those with V617F exhibit granulocytosis and thrombocytosis. Our results showed that JAK2(K539I) interacted less than JAK2(V617F) to GCSFR and TpoR, which may have limited its constitutive signaling from these receptors and prevented granulocytosis and thrombocytosis in MPN. Second, while most PV patients with the V617F mutation have at least a portion of mutation-homozygous clones, homozygosity is rare in exon 12 patients 37. We observed that JAK2 mutants bind less to the EpoR than WT JAK2, and JAK2(V617F) was inferior than JAK2(K539I) in binding and signaling from the EpoR. Therefore, bi-allelic expression of JAK2(V617F) may be required to overcome competition with WT JAK2 to cause erythrocytosis. Third, patients with the V617F mutation can present with PV, essential thrombocytosis (ET) or primary myelofibrosis. Because PV progenitors tend to be homozygous for the mutation while ET progenitors are nearly always heterozygous 38, a “mutational dosage” hypothesis was put forth 39. Our data suggest a new model in which activating mutations differentially affect JAK2 interaction with various receptors. This model aligns with the “dosage” hypothesis. As discussed, reduced coupling of JAK2(V617F) to the EpoR may necessitate bi-allelic expression for erythrocytosis, but the heterozygous state may be sufficient for JAK2(V617F) to couple to the TpoR to cause thrombocytosis. Our model is not confined to the receptors tested here, and does not preclude the possibility of signaling differences resulting from JAK2 mutants themselves not in complex with receptors. These may affect STAT1 activation, previously shown to differentiate JAK2(V617F)-heterozygous erythroid cells from ET or PV patients 40. It should also be noted that MPNs are clonal diseases, and other signaling events besides constitutive JAK-STAT signaling may be necessary for the expansion of a mutated HSC clone.
JAK2 inhibitors have been developed for MPN but toxicity is a common problem, calling for novel therapeutics that can target only the affected lineage while sparing others. We surmise that receptor-JAK2 interactions may represent targets of new lineage-specific therapeutic approaches. Alternatively, combining JAK inhibitors with inhibitors targeting aberrant downstream signaling that are specific to only the affected lineage may offer better specificity and reduced toxicity. Our results suggest that lineage-specific signaling nodes downstream of JAK2 mutants can be identified; oncogenic signaling from the EpoR depends on PI3K/AKT and STAT3/5 activity, whereas from the GCSFR depends on MEK and STAT3/5 activity. Consistent with these results, JAK2(V617F) expression fails to cause erythrocytosis and granulocytosis in STAT5-deficient mice 41,42, and PI3K inhibitors showed synergy with JAK inhibitors to curb cytokine-independent erythroid colony formation from JAK2(V617F) patient cells 43.
Our findings provide a molecular basis for MPN pathophysiology, and reveal a new way for constitutively active tyrosine kinases to generate oncogenic signaling. We suggest that previously unrecognized complexities in receptor-JAK interaction significantly impact signaling and pathology and may point to new therapeutic strategies in cancers with activated JAK/STAT pathway.
Supplementary Material
Acknowledgments
We thank Drs. Emery Bresnick, Saghi Ghaffari, Peter Michaely, Sandy Schmid, Jim Palis, Kathleen McGrath, Jian Xu, Srdan Verstovsek, Yumin Shen, Siayareh Rambally and Cheryl Lewis for helpful discussions and input. This study was supported by funding from the National Institute of Health (HL089966), Cancer Prevention Research Institute of Texas (CPRIT, RP110090) and the Ladies Leukemia League to L.J.H, from the National Natural Science Foundation of China (Grant No. 81200379) to Z.H., and a post-doctoral training grant from CPRIT to H.Y
Footnotes
AUTHOR CONTRIBUTION
H.Y., M.Y., Z.H., L.Z., M.H. and L.J.H. designed and performed research, and analyzed data. S.A.M. analyzed data. H.Y. and L.J.H. wrote the paper.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Thomas SJ, Snowden JA, Zeidler MP, Danson SJ. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br J Cancer. 2015 Jul 28;113(3):365–71. doi: 10.1038/bjc.2015.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene. 2013 May 23;32(21):2601–13. doi: 10.1038/onc.2012.347. [DOI] [PubMed] [Google Scholar]
- 3.Villarino AV, Kanno Y, Ferdinand JR, O’Shea JJ. Mechanisms of Jak/STAT signaling in immunity and disease. J Immunol. 2015 Jan 1;194(1):21–7. doi: 10.4049/jimmunol.1401867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stark GR, Darnell JE., Jr The JAK-STAT pathway at twenty. Immunity. 2012 Apr 20;36(4):503–14. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levine RL. JAK-mutant myeloproliferative neoplasms. Curr Top Microbiol Immunol. 2012 Aug 7;355:119–33. doi: 10.1007/82_2011_170. [DOI] [PubMed] [Google Scholar]
- 6.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005 Mar 19-25;365(9464):1054–61. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 7.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005 Apr 28;434(7037):1144–8. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 8.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005 Apr 28;352(17):1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 9.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005 Apr;7(4):387–97. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 10.Zhao R, Xing S, Li Z, Fu X, Li Q, Krantz SB, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005 Jun 17;280(24):22788–92. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mullally A, Lane SW, Brumme K, Ebert BL. Myeloproliferative neoplasm animal models. Hematol Oncol Clin North Am. 2012 Oct;26(5):1065–81. doi: 10.1016/j.hoc.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007 Feb 1;356(5):459–68. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grisouard J, Li S, Kubovcakova L, Rao TN, Meyer SC, Lundberg P, et al. JAK2 exon 12 mutant mice display isolated erythrocytosis and changes in iron metabolism favoring increased erythropoiesis. Blood. 2016 Aug 11;128(6):839–51. doi: 10.1182/blood-2015-12-689216. [DOI] [PubMed] [Google Scholar]
- 14.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012 Mar 1;366(9):799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012 Mar 1;366(9):787–98. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 16.Lu X, Huang LJ, Lodish HF. Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. J Biol Chem. 2008 Feb 29;283(9):5258–66. doi: 10.1074/jbc.M707125200. [DOI] [PubMed] [Google Scholar]
- 17.Lu X, Levine R, Tong W, Wernig G, Pikman Y, Zarnegar S, et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci U S A. 2005 Dec 27;102(52):18962–7. doi: 10.1073/pnas.0509714102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pradhan A, Lambert QT, Griner LN, Reuther GW. Activation of JAK2-V617F by components of heterodimeric cytokine receptors. J Biol Chem. 2010 May 28;285(22):16651–63. doi: 10.1074/jbc.M109.071191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Staerk J, Kallin A, Demoulin JB, Vainchenker W, Constantinescu SN. JAK1 and Tyk2 activation by the homologous polycythemia vera JAK2 V617F mutation: cross-talk with IGF1 receptor. J Biol Chem. 2005 Dec 23;280(51):41893–9. doi: 10.1074/jbc.C500358200. [DOI] [PubMed] [Google Scholar]
- 20.Bercovich D, Ganmore I, Scott LM, Wainreb G, Birger Y, Elimelech A, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008 Oct 25;372(9648):1484–92. doi: 10.1016/S0140-6736(08)61341-0. [DOI] [PubMed] [Google Scholar]
- 21.Malinge S, Ben-Abdelali R, Settegrana C, Radford-Weiss I, Debre M, Beldjord K, et al. Novel activating JAK2 mutation in a patient with Down syndrome and B-cell precursor acute lymphoblastic leukemia. Blood. 2007 Mar 1;109(5):2202–4. doi: 10.1182/blood-2006-09-045963. [DOI] [PubMed] [Google Scholar]
- 22.Zhao L, Dong H, Zhang CC, Kinch L, Osawa M, Iacovino M, et al. A JAK2 interdomain linker relays Epo receptor engagement signals to kinase activation. J Biol Chem. 2009 Sep 25;284(39):26988–98. doi: 10.1074/jbc.M109.011387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005 Jul 1;121(7):1109–21. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 24.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000 Mar 9;404(6774):193–7. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Pop R, Sadegh C, Brugnara C, Haase VH, Socolovsky M. Suppression of Fas-FasL coexpression by erythropoietin mediates erythroblast expansion during the erythropoietic stress response in vivo. Blood. 2006 Jul 1;108(1):123–33. doi: 10.1182/blood-2005-11-4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sulahian R, Cleaver O, Huang LJ. Ligand-induced EpoR internalization is mediated by JAK2 and p85 and is impaired by mutations responsible for primary familial and congenital polycythemia. Blood. 2009;113(21):5287–97. doi: 10.1182/blood-2008-09-179572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Kent DG, Chen E, Green AR. Mouse models of myeloproliferative neoplasms: JAK of all grades. Dis Model Mech. 2011 May;4(3):311–7. doi: 10.1242/dmm.006817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skoda RC. Less Jak2 makes more platelets. Blood. 2014 Oct 2;124(14):2168–9. doi: 10.1182/blood-2014-08-596361. [DOI] [PubMed] [Google Scholar]
- 29.Koulnis M, Porpiglia E, Hidalgo D, Socolovsky M. Erythropoiesis: from molecular pathways to system properties. Adv Exp Med Biol. 2014;844:37–58. doi: 10.1007/978-1-4939-2095-2_3. [DOI] [PubMed] [Google Scholar]
- 30.Koury MJ, Bondurant MC. Erythropoietin retards DNA breakdown and prevents programmed death in erythroid progenitor cells. Science. 1990 Apr 20;248(4953):378–81. doi: 10.1126/science.2326648. [DOI] [PubMed] [Google Scholar]
- 31.Panopoulos AD, Watowich SS. Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and ‘emergency’ hematopoiesis. Cytokine. 2008 Jun;42(3):277–88. doi: 10.1016/j.cyto.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richards MK, Liu F, Iwasaki H, Akashi K, Link DC. Pivotal role of granulocyte colony-stimulating factor in the development of progenitors in the common myeloid pathway. Blood. 2003 Nov 15;102(10):3562–8. doi: 10.1182/blood-2003-02-0593. [DOI] [PubMed] [Google Scholar]
- 33.Funakoshi-Tago M, Pelletier S, Matsuda T, Parganas E, Ihle JN. Receptor specific downregulation of cytokine signaling by autophosphorylation in the FERM domain of Jak2. Embo J. 2006 Oct 5; doi: 10.1038/sj.emboj.7601365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Funakoshi-Tago M, Pelletier S, Moritake H, Parganas E, Ihle JN. Jak2 FERM domain interaction with the erythropoietin receptor regulates Jak2 kinase activity. Mol Cell Biol. 2008 Mar;28(5):1792–801. doi: 10.1128/MCB.01447-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson EA, Sharma SV, Settleman J, Frank DA. A chemical biology approach to developing STAT inhibitors: molecular strategies for accelerating clinical translation. Oncotarget. 2011 Jun;2(6):518–24. doi: 10.18632/oncotarget.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J, Socolovsky M, Gross AW, Lodish HF. Role of Ras signaling in erythroid differentiation of mouse fetal liver cells: functional analysis by a flow cytometry-based novel culture system. Blood. 2003 Dec 1;102(12):3938–46. doi: 10.1182/blood-2003-05-1479. [DOI] [PubMed] [Google Scholar]
- 37.Scott LM. The JAK2 exon 12 mutations: a comprehensive review. Am J Hematol. 2011 Aug;86(8):668–76. doi: 10.1002/ajh.22063. [DOI] [PubMed] [Google Scholar]
- 38.Scott LM, Scott MA, Campbell PJ, Green AR. Progenitors homozygous for the V617F mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood. 2006 Oct 1;108(7):2435–7. doi: 10.1182/blood-2006-04-018259. [DOI] [PubMed] [Google Scholar]
- 39.Kilpivaara O, Levine RL. JAK2 and MPL mutations in myeloproliferative neoplasms: discovery and science. Leukemia. 2008 Oct;22(10):1813–7. doi: 10.1038/leu.2008.229. [DOI] [PubMed] [Google Scholar]
- 40.Chen E, Beer PA, Godfrey AL, Ortmann CA, Li J, Costa-Pereira AP, et al. Distinct clinical phenotypes associated with JAK2V617F reflect differential STAT1 signaling. Cancer Cell. 2010 Nov 16;18(5):524–35. doi: 10.1016/j.ccr.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walz C, Ahmed W, Lazarides K, Betancur M, Patel N, Hennighausen L, et al. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2(V617F) in mice. Blood. 2012 Apr 12;119(15):3550–60. doi: 10.1182/blood-2011-12-397554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan D, Hutchison RE, Mohi G. Critical requirement for Stat5 in a mouse model of polycythemia vera. Blood. 2012 Apr 12;119(15):3539–49. doi: 10.1182/blood-2011-03-345215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choong ML, Pecquet C, Pendharkar V, Diaconu CC, Yong JW, Tai SJ, et al. Combination treatment for myeloproliferative neoplasms using JAK and pan-class I PI3K inhibitors. J Cell Mol Med. 2013 Nov;17(11):1397–409. doi: 10.1111/jcmm.12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.