

Abstract
Most of the studies on air pollution focus on emissions from fossil fuel burning in urban centers. However, approximately half of the world's population is exposed to air pollution caused by biomass burning emissions. In the Brazilian Amazon population, over 10 million people are directly exposed to high levels of pollutants resulting from deforestation and agricultural fires. This work is the first study to present an integrated view of the effects of inhalable particles present in emissions of biomass burning. Exposing human lung cells to particulate matter smaller than 10 µm (PM10), significantly increased the level of reactive oxygen species (ROS), inflammatory cytokines, autophagy, and DNA damage. Continued PM10 exposure activated apoptosis and necrosis. Interestingly, retene, a polycyclic aromatic hydrocarbon present in PM10, is a potential compound for the effects of PM10, causing DNA damage and cell death. The PM10 concentrations observed during Amazon biomass burning were sufficient to induce severe adverse effects in human lung cells. Our study provides new data that will help elucidate the mechanism of PM10-mediated lung cancer development. In addition, the results of this study support the establishment of new guidelines for human health protection in regions strongly impacted by biomass burning.
Introduction
Most of the overwhelming amount of research on exposure to air pollution is focused on urban centers and on the role of fossil fuels as the most important source of atmospheric pollutants. However, approximately 3 billion people in the world are exposed to air pollution from biomass burning, originating from using wood or coal as cooking fuel in simple stoves, home heating with open fires, deforestation, and agricultural practices1.
Biomass burning emits significant quantities of known pollutants hazardous to health, including several carcinogenic compounds2. World Health Organization (WHO) reported that in 2012, approximately 7 million people - one in eight total global deaths - as a result of exposure to air pollution3. Fire is a global phenomenon, and is an integral part of the earth’s ecosystem4, 5.
In particular, the Brazilian Amazon region contains world’s largest tropical forest and is considered, during the rainy season, one of the continental regions least affected by human activities6, 7. However, during the dry season, high concentrations of aerosol particles from biomass burning (mainly agricultural practices and deforestation) have been documented in this region7, 8. The combination of forest fires and human occupation has turned biomass burning into a serious public health threat. The majority of forest fires occur in the deforestation arc, a belt in the southern and western regions of the forest, directly impacting over 10 million people in the area9. Many studies in the area have identified severe effects on human health, such as increased incidences of asthma, morbidity and mortality, mainly in the most vulnerable populations such as children and elderly10, 11.
The smoke plume extends over millions of km2, covering large areas of South America, with significant health impacts extending far from the Amazon region12, 13. A recent study has estimated that reduction in the rate of deforestation in the Amazon in previous years has been preventing approximately 400 to 1,700 premature adult deaths annually, throughout South America13.
Studies show that inhabitants in the deforestation arc breathe air with high concentrations of particulate matter smaller than 10 µm (PM10). The problem is aggravated during the dry season, when high concentrations of PM10 have been measured (ranging from 400 up to 600 µg.m−3)14, exceeding the upper limits of concentration established by WHO (24 h exposure to PM10 – 50 µg.m−3) by 8 to 12 times. These inhalable particles have been classified as class 1 cancer-causing agents in 2013 by the International Agency for Research on Cancer (IARC)15. They can penetrate the alveolar regions of the lung, pass through the cell membrane, reach the blood and can accumulate in other human organs16.
Although epidemiological studies on the effects of urban PM on human health are numerous, there are relatively few that focused on the impact of air pollution resulting from biomass burning2, 17. Even scarcer are the studies that investigate the cellular and molecular mechanisms underlying PM toxicity. In one of these studies, Borgie and collaborators observed that PM increased the histone H2AX phosphorylation (γ-H2AX) (a DNA damage marker), telomerase activity, and induced epigenetic changes in bronchial epithelial cells18. Several groups reported that PM induces cell cycle alterations19, oxidative stress20, 21 and cell death22. Most of these studies focused on PM in urban areas. Recently, our group showed that organic PM10 in Amazon biomass burning emissions had mutagenic effect on plant cells and human lung cells23, 24. The objective of the present study was to investigate these effects in depth and provide a thorough analysis of the toxic cellular and molecular effects of relevant concentrations of PM10 resulting from Amazon biomass burning, on human lung. In fact, this manuscript is a sequel to an article published recently by our group characterizing in detail the chemical composition of the inhalable material whose health effects are investigated here25.
Our findings raise concern regarding human health of affected people, since we investigated the effects of PM on human lung cells using concentrations within plausible range of in vitro assays.
Results
PM10 from Amazon region reduces cell viability and increases ROS generation in human lung cells
We first evaluated the viability of human lung epithelial cells (A549 cell line) exposed to 200 and 400 µg.mL−1 of organic PM10 (Fig. 1A). There was no significant difference in the viability of cells exposed to PM10 for 24 h compared to that of untreated cells (Fig. 1A). However, significant loss of viability was observed in cells exposed to 400 µg.mL−1 of PM10 for 72 h (Fig. 1B). We also evaluated the cytotoxicity of retene (RET) (Fig. 1A and B). The concentration of RET used in this study was 30 ng.mL−1, which corresponds to the contribution of RET to the organic PM extract at 400 µg.mL−1. Our data showed that treatment with this compound for 72 h was cytotoxic to human lung cells (Fig. 1B). Polycyclic aromatic hydrocarbons (PAHs) were used as positive control (10 ng.mL−1) for cytotoxicity analysis (Fig. 1A and B)24.
Figure 1.

Cell viability (XTT) analysis of A549 human lung cells after 24 h (A) or 72 h (B) continuous treatment with organic PM10 (200 or 400 µg.mL-1). ROS induction under the same conditions was also evaluated, using the DCFDA (C and D) or the Mitosox (E and F) fluorescence probes. *p < 0.05 and **p < 0.01 are statistically significant compared to negative control (DMEM), according to Dunnett's test. DC: DMSO control (0.1%).
Since PM collected with different source characteristics was implicated in ROS generation26, 27, we investigated the ROS levels in A549 cells exposed to PM10. Intracellular ROS generation was significantly higher in cells exposed to PM10 for 24 and 72 h compared to the control cells (Fig. 1C and D). The effect was also dose and time-dependent. In particular, there was an 8.6-fold increase in fluorescence intensity in cells treated with PM10 for 72 h (Fig. 1D).
Mitochondrial superoxide indicator (MitoSOX) was used to evaluate the possible involvement of mitochondria in ROS formation. Flow cytometry analysis revealed that treatment with organic PM10 (200 and 400 μg.mL−1) markedly increased mitochondrial ROS generation in A549 cells in a dose and time-dependent manner, starting just after 24 h (Fig. 1E), and continuing till after exposure 72 h, specially at the higher concentration (Fig. 1F). These data suggest an important role of mitochondria in ROS generation upon exposure to PM10.
PM10 induces the secretion of inflammatory cytokines by human lung cells
Since PM10 known is to induce the secretion of inflammatory cytokines28, 29, we next investigated the concentrations of TNF-α and IL−1β in the culture media of cells exposed to PM10. IL-1β levels were significantly elevated in cells treated with 200 or 400 μg.mL−1 of PM10, for 24 or 72 h, in comparison to those in control cultures (Fig. 2A and B). Similar results were observed for TNF-α (Fig. 2C and D), indicating the induction of pro-inflammatory response in human lung cells exposed to PM10.
Figure 2.

PM10 in biomass burning emissions induces the secretion of inflammatory cytokines by human lung cells. IL-1β release 24 h (A) and 72 h (B) after PM10 exposure (200 and 400 µg.mL−1); (B) TNF-α release 24 h (C) and 72 h (D) after PM10 exposure. *p < 0.05, **p < 0.01 and ***p < 0.001 statistically significant compared to negative control (NC) (DMEM), according to Bonferroni test.
PM10 induces autophagosome accumulation
The increase in ROS generation prompted us to investigate autophagy30, a pathway that is induced by ROS and also can mitigate the generation of ROS. For investigating the autophagy response, we chose 400 μg.mL−1 of PM10 concentration, since this dose caused a more dramatic increase in ROS levels after 72 h treatment (Fig. 1D and F). As shown in Fig. 3A,B and C, the number of A549 cells engaged in autophagy (as measured by the formation of cytoplasmic, fluorescent round-shape GFP-LC3 structures) increased after exposure to 400 μg.mL−1 of PM10 in a time-dependent manner.
Figure 3.

PM10 induces autophagosome accumulation in a time-dependent manner in human lung cells. (A) Quantification of autophagosome accumulation at 1, 6, 24 and 72 h after treatment with 400 μg.mL−1 of PM10. Chloroquine (CQ) 10µM was used as a positive control for autophagosome accumulation. (B) Comparison of number of autophagosome after 24 h exposure to PM10 or CQ. (C) Representative images of the formation of autophagosome in human lung cells with negative control (NC), 400 μg.mL−1 of PM10 or CQ (10µM). (D) Expression levels of LC3I and LC3-II as measured by Western blotting. The blots were cropped. *p < 0.05, **p < 0.01 and ***p < 0.001 statistically significant compared to NC according to Bonferroni test.
To confirm these results, we measured, by western blotting, the conversion of unprocessed cytosolic LC3-I to the LC3-II (autophagosome-associated) form, following PM10 treatment. Our results show an increase in LC3-II in A549 cells exposed to 200 and 400 μg.mL−1 of PM10 for 24 h, demonstrating LC3-processing (Fig. 3D). These results confirmed that PM10 exposure induced autophagy in A549 cells.
PM10 induces DNA damage and cell cycle alterations in human lung cells
An increase in ROS levels can result in DNA damage and mutagenesis31. Therefore, we evaluated the induction of DNA lesions using comet assay. Analysis was performed 24 h after treatment with PM10 since, at later time points, DNA fragmentation induced by cell death could interfere with the results. Results of this analysis showed that organic PM10 induced significant DNA damage, as evidenced by tail formation at all doses tested. We used the PAHs mix as positive control in this experiment and confirmed its genotoxic effect (Fig. 4A). We also assessed the genotoxic potential of RET, since there are very few studies investigating the genetic damage caused by this compound generated during biomass combustion. RET induced DNA damage in human lung cells, and the difference in the extent of damage between treatment group and negative control was significant (Fig. 4A).
Figure 4.

PM10 from biomass burning emissions induces DNA damage in human lung cells. (A) Image of comet test after treating A549 human lung cells with PM10; mean of comet tail moment after treatment with 200 and 400 μg.mL−1 of PM10, RET (30 ng. mL−1) and PAHs mix (10 ng.mL−1) for 24 h. (B) γ-H2AX formation after 24 and 72 h treatment with PM10. *p < 0.05 and **p < 0.01 statistically significant compared to negative control (NC) (DMEM) according to Dunnett's test. DC: DMSO control (0.1%).
Additionally, we evaluated the formation of γ-H2AX, a widely used DNA damage marker32. The nuclei of human lung cells exposed to PM10 for 24 h stained positive for γ-H2AX, although the degree of staining was not significant. The number of γ-H2AX-positive cells increased in a time and dose-dependent manner, and a significant increase in γ-H2AX formation in A549 cells was observed at higher concentration of PM10 (400 µg.mL−1) after 72 h treatment (Fig. 4B).
We then investigated whether these inhalable particles can alter the cell cycle of human lung cells. Figure 5A shows the cell cycle profile of the A549 cells exposed to organic PM10 for 24 h. The results showed an increase in the number of cells in G1 phase, although only in cells treated with 400 μg.mL−1 of PM10, consistent with the induction of γ-H2AX formation at this dose.
Figure 5.

Organic PM10 from Amazon region induces the expression of p53 and p21 and alters cell cycle. (A) Cell cycle profile of the A549 cells treated with organic PM10 for 24 h. (B) Expression of p53 and p21 proteins in cells treated with organic PM10 for 24 h. Quantification from three independent experiments is shown. Western blots were cropped. *p < 0.05 statistically significant compared to negative control (NC) according to Dunnett’s test.
The mechanism underlying the cell cycle alterations was investigated by analyzing the expression of two key proteins, p53 and p21, by western blotting. There was an increase in the levels of these proteins in cells treated with organic PM10 for 24 h (Fig. 5B), suggesting that DNA damage induced by PM10 leads to cell cycle alterations.
PM10 induces apoptosis and necrosis in human lung cells
We, next investigated if PM10 can induce cell death in human lung cells, possibly as a consequence of DNA damage. For these assays, we chose the highest concentration of organic PM10 (400 µg.mL−1) used in this study.
To evaluate the cell death response precisely, we quantified the sub-G1 cell population, which represents cells with fragmented nuclei, an indicator of apoptosis induction33. After 72 h exposure to organic PM10, we detected an increase in sub-G1 cells (Fig. 6A). To complement this analysis, we stained the cells with PI and Hoescht, and observed nuclear morphology, as well as PI uptake, using a fluorescence microscope. Figure 6B shows the occurrence of early (6.8%) and late apoptosis (10%), as well as necrosis (30.8%) after 72 h continuous treatment with PM10. Interesting, the RET induced cell death mainly by necrosis in human lung cells with a significant difference compared to negative control.
Figure 6.
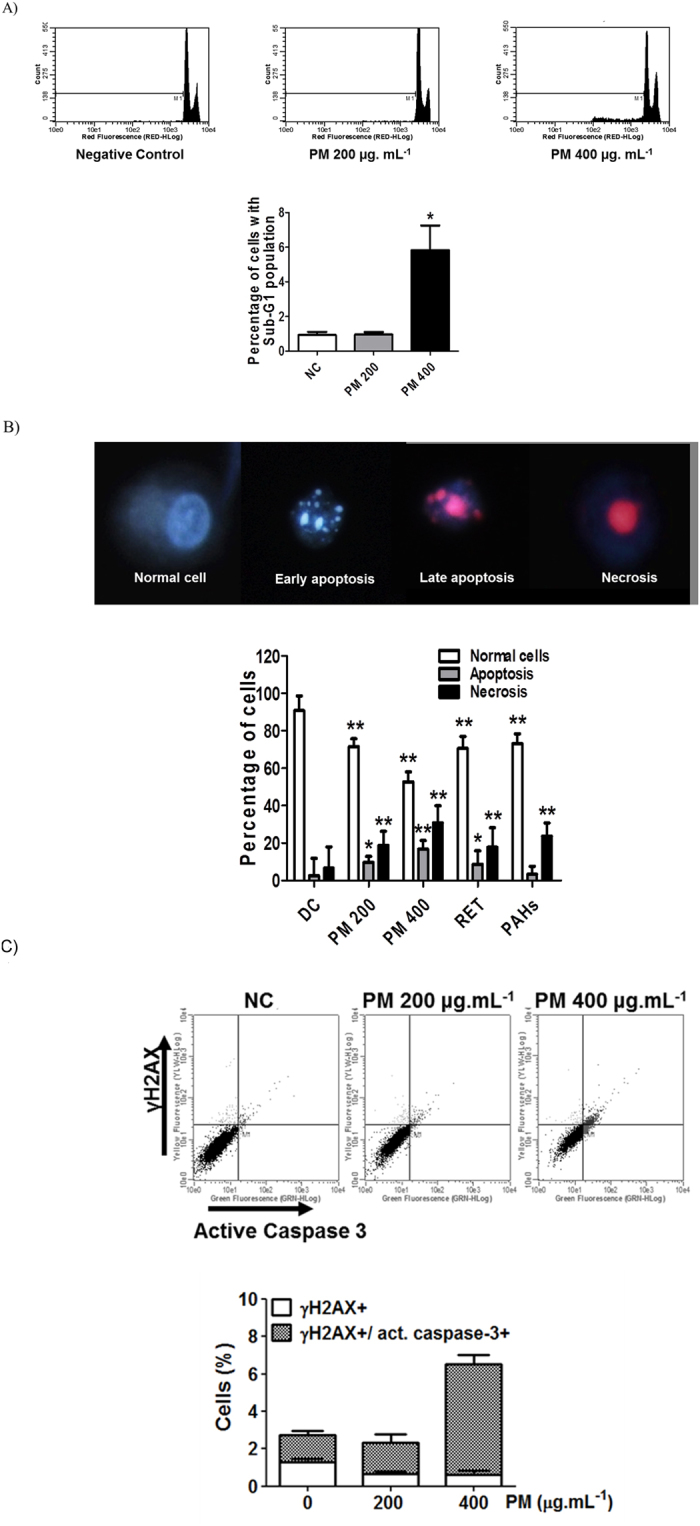
PM10 from biomass burning emissions induces cell death. (A) Nuclear fragmentation in human lung cells after treating the cells with the organic fraction of PM10 for 72 h. (B) Cell death assay by fluorescence microscopy that distinguishes apoptosis (early and late) from necrosis. (C) γ-H2AX and active caspase-3 analyses by flow cytometry after 72 h exposure to PM10. NC: negative control; DC: DMSO control (0.1%) *p < 0.05 and **p < 0.01 statistically significant compared to NC according to Dunnett’s test.
Based on these results, we hypothesized that the DNA fragmentation that occurs during apoptosis increases the levels of γ-H2AX staining. Therefore, we analyzed the active caspase-3 content, concomitantly with γ-H2AX staining and observed a clear association between these two markers (Fig. 6C). To validate these results, we performed this assay with ultraviolet-C (UVC) light, which induces two types of γ-H2AX staining, moderate and high. Only the latter correlates with cell death34. Strikingly, PM-induced γ-H2AX differs from that induced by UVC, and is exclusively related to apoptosis (Figure S1).
Discussion
It is well recognized that PM from urban air pollution induces lung cancer15. However, the exact mechanisms underlying this effect are still unclear. The present study revealed that the PM10 emitted during biomass burning in the Amazon region caused toxic effects at molecular and cellular levels in human lung cells. Here, we present for the first time, an integrated analysis of the effects of particulate emissions from the forest fires of Amazonian deforestation arc, on oxidative stress, inflammatory response, autophagy induction, cell cycle arrest, DNA damage, and cell death of human lung cells. It has been proposed that these pathways represent important toxicological mechanisms associated with PM-induced lung cancer35, 36.
It is clear that the effects of PM depend, not only on the particle size, but also on the composition of PM. The identification of all the toxic components within the PM ensemble is an extremely challenging task, given the complexity of the composition and number of compounds37. PAHs are known to be largely responsible for the toxicity of PM generated from a variety of sources15, 38. An earlier study quantified the PAH content in the organic extract used in this study, identifying that 40% of these compounds were known carcinogens25. One key compound identified in this work was dibenzo[a,h]anthracene. Another study of our group, focusing on nitrated and oxygenated PAHs (de Oliveira Galvão, manuscript accepted)39, also established the contribution of nitrated and oxygenated-PAHs to human carcinogenic risk from the same material, indicating the identity of the components responsible for the effects studied here.
We investigated in vitro, the effects of PM10 on human lung cells using concentrations within the plausible range. Our results show that ROS generation occurs as an early event. We hypothesized that ROS-dependent oxidative stress caused by PM10 would lead to the release of inflammatory cytokines. Consistent with the results of previous studies, organic fractions from urban emissions40, 41 and wildfire emissions42, 43 strongly induced the secretion of IL-1β and TNF-α. In addition, the impact of long-range transport episodes of wildfire smoke was investigated in the mouse cell line, RAW 264.7. The cytokine production observed in this study44 was substantially lower, compared to that induced by corresponding particles of the seasonal average period. It is suggested that this reduction could be due to the chemical transformation of the organic fraction during aging44. Besides, it is interesting to note that 1-nitropyrene, a compound identified in the PM extract used here (de Oliveira Galvão, manuscript accepted)39, induced oxidative DNA damage by generation of ROS in A549 cells45, which may explain the previously identified mutagenic and carcinogenic potential of this compound46.
Moreover, mitochondria are also responsible for the increase in ROS, as evidenced by the formation of significant quantities of mitochondrial superoxide after exposure to PM10. An earlier study showed the involvement of mitochondria in ROS production in rat alveolar type II and human lung cells exposed to urban PM47. Interestingly, in another study using urban pollution samples, low dose PM increased ROS production in bronchial epithelial cells, but not the formation of mitochondrial superoxide. Thus, besides superoxide, those other mitochondria-independent pathways may also drive ROS generation in certain cell types19.
Several studies have reported that polycyclic aromatic quinones cause severe adverse biological effects such as allergic inflammation via induction of the ROS pathway48–50. Shang et al.26, for example, showed that some quinones (1,2-naphthoquinone, 2-methylanthraquinone, 9,10-phenanthrenequinone, 2-methyl-1,4-naphthoquinone, and acenaphthenequinone) caused viability decrease, cellular LDH release, DNA damage and ROS production in A549 cells. Correspondingly, 2-methylanthraquinone was the most abundant compound among oxygenated-PAHs in our samples (de Oliveira Galvão, manuscript accepted)39. Therefore, it is likely that this compound also contributed significantly, because of its high redox potency, to the adverse health effects reported here.
Furthermore, excessive levels of ROS can cause severe damage to DNA and proteins. In this respect, autophagy has been conventionally considered a pathway that is activated in response to these insults, to maintain cellular homeostasis51. Our results clearly showed the accumulation of autophagosomes in PM10-treated cells. In agreement, Deng and collaborators showed that PM-induced oxidative stress probably plays a key role in autophagy induction in A549 cells (same cellular model used in this work), which may contribute to PM-induced impairment of pulmonary function52. These authors also reported that PM could elicit oxidative stress by decreasing the activity of antioxidant enzymes activity, finally leading to cell death52.
Another interesting finding of this study was the cell cycle arrest at G1-phase and formation of DNA strand breaks in response to PM10 treatment. In this context, Longhin and collaborators showed that PM altered cell cycle significantly in the human epithelial cells just after 3 h of exposure19. DNA damage has also been reported to result from exposure to PM in coke oven53, vehicular54, and industrial emissions55. Different mechanisms have been proposed to explain these effects, such as DNA strand breaks caused by the oxidative stress exerted by different agents (including PAHs)56 and formation of bulky DNA adducts by the direct action of reactive PAHs57.
An appropriate response of cells to DNA damage involves activation of checkpoint proteins that regulate cell cycle progression. In fact, we noticed an increase in the expression of p53 and its downstream target, p21, which is capable of inducing cell cycle arrest. This is an early event after PM10 treatment. Similarly, a recent study reported that urban PM induced G1 arrest in alveolar epithelial cells with p21 playing a critical role in the regulation of cell cycle and preventing apoptosis58. These effects have been observed when the cells were exposed to carcinogenic PAHs. For example, dibenzo[a,l]pyrene and benzo[a]pyrene are able to induce the formation of DNA adducts, increases in the levels of p53 and p21, and G1 or G2/M arrests in human mammary carcinoma cells (MCF-7)59.
These early responses are not sufficient to counteract the DNA damage effects, if the exposure to PM10 continues, which will lead to the activation of cell death processes. Our results showed cell death by apoptosis and necrosis after 72 h of exposure to PM10. Interestingly, we observed a positive correlation between the activation of caspase-3, an apoptosis protease effector, and γ-H2AX formation, probably due to apoptotic DNA fragmentation60. A previous study also reported that urban aerosols during a biomass burning episode in Indonesia caused cell death in A549 cells and that this effect could be related to the cytotoxic effects of metals and PAHs contained in the ambient PM61.
Real-world biomass burning emissions contain myriad compounds, including those recognized as carcinogenic and toxic. These compounds were also present in the PM samples used in this study. Nonetheless, there are many other compounds whose health effects have not yet been studied. For example, RET is a PAH normally associated with wood combustion62 and was present in high concentrations in the samples used in this study25. RET has not been included in the risk assessment of PAHs by US-EPA63. Given its high concentrations in the Amazon samples, we decided to investigate its genotoxic effect. We found that it is highly genotoxic, and also caused cell death by necrosis. These results provide new evidence for the effect of RET on human health, especially in regions impacted by biomass burning. Further studies focusing on the role of RET in lung cancer development are therefore strongly encouraged.
Considering all these data together, we propose a mechanism of action of PM10 present in the emissions from biomass burning in the Amazon region, in human lung cells (Fig. 7). DNA damage is an early molecular hallmark of exposure to PM. Organic matter present in PM10 extracts can transpose human lung cells and generate oxidative stress by mitochondrial ROS generation. ROS induces cytokine release contributing to the oxidative stress, leading to DNA damage. This situation can activate autophagy for reestablishing normal cellular homeostasis as well as causing cell cycle arrest for dealing with this damage. If the cells are not able to counteract these effects, cell death pathways are activated, increasing DNA fragmentation by apoptosis and also necrosis. DNA fragmentation also induces the formation of γ-H2AX. The resultant loss of cells may explain some acute effects of respiratory diseases. However, if the cells are not able to deal with DNA damage and not progress to cell death, mutagenesis can occur leading to the development of lung cancer64.
Figure 7.

Proposed mechanism of action of PM10 from biomass burning emissions in the Amazon region. As early events, formation of DNA strand breaks, increased production of ROS and cytokines, mitochondrial alterations, and autophagy could be observed clearly. PM10 also caused G1 cell cycle arrest and an increase in the expression of p53 and p21 proteins. In the event of continued exposure, this early response is not sufficient to counteract the effects of PM10. During late events, both apoptosis and necrosis are activated. DNA fragmentation induces γ-H2AX formation. The genetic instability driving tumorigenesis is fueled by DNA damage that can cause molecular alterations (e.g., mutations) and can play an important role in lung cancer development.
To summarize, this study investigated the acknowledged adverse health effects of deforestation fires in the Amazon Basin, using several analytical techniques. The PM in the emissions from biomass burning in the Amazon region has been observed to cause DNA damage and cell death. The results described here could be used for detailed chemical speciation, which will provide clues to compounds involved in the negative health effects. Our study provides new data on the mechanism of action of PM10 in contributing to the development of lung cancer. A better understanding of the pathways by which particulate pollutants contribute to the burden of lung diseases, is an important step in reducing the risk of adverse respiratory effects resulting from exposure to biomass burning emissions and to develop individual and community-level interventions. It is also an important contribution to the establishment of new guidelines for human health protection in regions strongly impacted by biomass burning.
Methods
Particle collection and PM10 sample preparation
The aerosol sampling was conducted in the Brazilian Amazon region, located about 5 km north of Porto Velho. A total of 19 PM10 samples were collected with quartz fiber filters using a high-volume sampler during the dry season (August–October/2011). The samples were extracted as previously described25. The organic extract was dissolved in dimethyl sulfoxide (DMSO) and was used for cell treatment.
Cell culture, PM10 treatment and cell proliferation assay
A549 cells were routinely grown at 37 °C, in a humidified 5% CO2 in Dulbecco’s Modified Eagle’s Medium (DMEM) containing phenol red and supplemented with 10% fetal bovine serum (FBS) and antibiotics (0.1 mg.mL−1 penicillin, 0.1 mg.mL−1 streptomycin and 0.25 mg.mL−1 fungizone).
Human lung cells were exposed to organic PM10 (200 µg.mL−1 and 400 µg.mL−1). It is important to note that, despite allowing detailed analysis, in vitro cell exposure studies require pre-treatment of inhalable material such as liquid extraction and thus could limit/modify the effects of real-world exposure.
The exposure doses to be used in the present study were difficult to determine, since there were no previous studies on the in vitro mechanisms of particles emitted during the Amazon fires. We used as reference, the estimated alveolar mass deposition accumulating over 24 h in human adults exposed to air concentrations identified in the Amazon region. PM10 concentrations used for mass calculation were 60 µg.m−3 (peak identified during forest fire in Porto Velho - 2011) and 118 µg.m−3 (peak identified during forest fire in Porto Velho - 2010)7. The average daily inhalation rate used in this study was 16 m3.day−1 for adults65 and the alveolar deposition rate of PM10 in the human respiratory tract is approximately 20%66. It was, of course, not possible to directly extrapolate the real-world conditions, since we did not correct the dose with respect to alveolar surface. In addition, we used liquid extracts, which eliminated the effect of surface/reactivity properties of small particles. However, we consider reasonable to say that our concentrations were within a plausible range.
RET, the most abundant PAH in Amazon samples, was also evaluated in this study. A mix of PAHs (acenaphthylene, phenanthrene, anthracene, fluorene, pyrene, benz[a]anthracene, chrysene, benzo[b]fluoranthene, benzo[k]fluoranthene, benzo[a]pyrene, indene[1,2,3-c,d]pyrene, dibenz[a,h]anthracene and benzo[g,h,i]pyrene; EPA 525 PAH Mix A (48953-U, Sigma)) was also used as positive control based on previous studies24, 67. Cell viability was performed 24 and 72 h after PM10 treatment using Cell Proliferation Kit II (XTT) (Roche, Basel, Switzerland) as described elsewhere68. The absorbance was measured at 492 nm (650 nm as reference). Cell viability was expressed as percentage of control cells.
Alkaline comet assay
DNA strand breaks were detected using alkaline comet assay described in an earlier study34. A549 cells exposed to PM10 were trypsinized and resuspended in 180 µL 0.5% low melting point agarose at 37 °C. The cells were then spread homogenously on two microscope slides pre-coated with 1.5% agarose, immediately covered with coverslips and kept at 4 °C for 10 min. Cells were then lysed overnight in chilled lysis solution at 4 °C. The slides were then placed horizontally in an electrophoresis chamber with cold alkaline buffer (300 mM NaOH, 1 mM EDTA, pH > 13) for 25 min at 25 V and 300 mA. The slides were then neutralized (0.4 M Tris, pH 7.5) and fixed with ice-cold 100% ethanol. Finally, the slides were stained with ethidium bromide and imaged with a fluorescence microscope (Axiovert 200, Zeiss) at the magnification of 400x. At least 100 comets per slide were scored, on duplicate slides, using the Komet 6.0 software. The results were plotted by tail moment.
ROS formation
ROS formation were determined using DCFH-DA (2′,7′-dichlorofluorescein diacetate, Sigma-Aldrich,35845). After exposure to PM10, A549 cells were detached with trypsin and incubated with DCFHA-DA dye for 30 min at 37 °C in DMEM without phenol red and supplemented with 0.2% FBS. The green fluorescence intensity in each well was quantified using flow cytometry (Guava® easyCyte, USA). Results are shown in relative fluorescence units.
Mitochondrial superoxide formation
Mitochondrial superoxide production was measured by flow cytometry following staining with MitoSOX Red mitochondrial superoxide probe (Life Technologies, M36008). A549 cells treated with PM10 were detached by trypsin digestion. The cells were resuspended in DMEM without phenol red and supplemented with 0.2% FBS and incubated with 5 µM MitoSOX reagent for 20 min at 37 °C in the dark. The cells were washed twice with PBS and assessed using flow cytometry. Results are shown as relative fluorescence units.
Cytokines release analysis
In order to evaluate the release of pro-inflammatory cytokines (IL-1β and TNF-α), we exposed A549 cells to organic PM10. The supernatant was snap frozen and stored at −80 °C until analysis by Cytometric Bead Array assay, using the Human Inflammatory Cytokines Kit (BD Biosciences, USA). We used the BD Accuri C6 (BD Biosciences) flow cytometer along with the FCAP 3.0 software to analyze the fluorescence of each bead in the FL2 and FL3 channels, which correspond to the concentration of the different cytokines.
Analysis of autophagosome formation using GFP-LC3-expressing cells
Autophagosome formation, a hallmark of autophagy69, was monitored to establish the dynamics of autophagic process in PM10-exposed cells. For this purpose, we generated A549 cells stably expressing LC3 (a critical component of autophagosome formation) fused to GFP (GFP-LC3), and observed autophagosome formation under a fluorescence microscope, as described elsewhere69. Chloroquine treatment was used as a positive control for autophagosome visualization33.
Sub-G1 and cell cycle analysis by flow cytometry
Sub-G1 and cell cycle analysis were performed as previously described70. A549 cells treated with PM10 were trypsinized, washed with PBS, and fixed with chilled 70% ethanol for at least 24 h, at −20 °C. Staining was performed at room temperature for 30 min in filtered PBS containing 200 µL propidium iodide (PI) solution (200 mg. mL−1 RNase A, 20 mg. mL−1 PI, and 0.1% Triton X-100 in PBS) and each sample was analyzed in a flow cytometer. Guava Express Plus software was used to quantify sub-G1, and cell cycle analysis was performed using ModFit LT software.
Double staining of γ-H2AX and active caspase-3 by FACS
For concomitant analyses of γ-H2AX and active caspase-3 by FACS, the protocol was adapted from Quinet et al.34. Briefly, adherent cells were combined and fixed with 1% formaldehyde in ice, washed in PBS, and stored in 70% ethanol for at least overnight at −20 °C. Cells were blocked and permeabilized with BSA-T buffer (0.2% Triton X-100, 1% BSA in PBS). For γ-H2AX staining, samples were incubated with 1/500 anti-γ-H2AX antibody (05–636 Millipore) for 3 h at room temperature followed by two washes with BSA-T buffer and incubation with 1/200 anti-mouse TRITC (tetramethylrhodamine) secondary antibody (T5393 Sigma-Aldrich) for 1 h at room temperature. After washing twice with BSA-T buffer, samples were incubated with 1/50 FITC- (fluorescein iso-thiocyanate) conjugated anti-active caspase-3 antibody (559341 BD Pharmigen) overnight at 4 °C. Fluorescence spectral overlap was compensated using positive controls for both γ-H2AX or active caspase-3 staining. Data was acquired on a Guava Flow Cytometer and analyzed using CytoSoft Data Acquisition and Analysis Software (Millipore).
Assessment of cell death
Cell death assessment was performed as described71. Briefly, A549 cells were collected by trypsinization and resuspended in 200 μL PBS. Ten microliters of this suspension was incubated for 5 min with 2 μL dye mixture containing 1 mg.mL−1 PI (Sigma-Aldrich), 1.5 mg.mL−1 diacetate fluorescein (Invitrogen, Life Technologies), and 1 mg.mL−1 Hoechst 33342 (Invitrogen, Life Technologies) at 37 °C, and transferred to a microscope slide. For each treatment, the presence of live cells, early apoptosis, late apoptosis, and necrosis were evaluated in 500 cells (in triplicate) using a Zeiss microscope Bloom. Healthy cells and early apoptotic cells are refractory to PI staining, and show intact or fragmented nuclei, respectively, in blue (Hoechst staining). Late apoptotic cells and necrotic cells display fragmented or intact nuclei, respectively and stain red, since they are permeable to PI.
Protein extraction, quantification, and western blot analysis
Total protein extracts were analyzed as previously described70. Quantification was performed using the Pierce BCA Protein Assay kit (Thermo Scientific, USA). Fifty micrograms of total protein from cell extracts was separated on a 10% SDS polyacrylamide gel, and blotted onto a nitrocellulose membrane. Membranes were blocked for 1 h in 5% (w/v) milk powder in PBS-Tween 20 (PBST), incubated overnight at 4 °C with primary antibody, washed three times with PBST, and incubated for 1 h with secondary antibody. After final washes with PBST, the blots were developed using an enhanced chemiluminescence detection system (Immobilon Western, Merck Millipore). Protein expression was detected using anti-p53 (1:500, M7001, Dako Denmark A/S); anti-p21 (1:200, sc-56335, Santa Cruz, Santa Cruz, CA, USA); anti-LC3 (1:1000 dilution, L8918, Sigma-Aldrich); anti-GAPDH (1:1000 dilution, Santa Cruz, Santa Cruz, CA, USA) antibodies.
Statistical analysis
Statistical analysis was performed using the Statistical Package for Social Sciences (SPSS) version 15.0 and Prism 5, GraphPad Software Inc. Statistical significance was assessed using one-way ANOVA followed by the Dunnet’s test or Bonferroni test. The mean differences were considered significant at p ≤ 0.05.
Electronic supplementary material
Acknowledgements
The authors thank: Ana Lúcia Loureiro (IF-USP), Maria Cristina N. de Oliveira (CETESB-SP) and Giácomo Cuoco (CETESB-SP) by weighing the filters; Fernando Morais (IF-USP) by the installation of high volume sampler; Leonardo Lima, Ligia Castro and Veridiana Munford (ICBUSP- DNA Repair Laboratory) by helping with flow cytometry; Clarissa Ribeiro (ICBUSP- DNA Repair Laboratory) by helping with cell culture; Letícia Koch Lerner (ICBUSP- DNA Repair Laboratory) by helping with comet assay; and Wanderley Bastos from University of Rondônia (UNIR) (Wolfgang C. Pfeiffer Environmental Biogeochemistry Laboratory). This work was supported by the National Council of Technological and Scientific Development (CNPq, Brazil, Process Nº 471033/2011-1), by the Brazilian Network on Global Climate Change Research funded by CNPq (Nº 550022/2014-7), FINEP (Nº 01.13.0353.00) and São Paulo Research Foundation (FAPESP, Sao Paulo, Brazil, Process Nº 2014/02297-3, Process Nº 2014/15982-6 and Process Nº 2013/05014-0).
Author Contributions
N.O.A. had the conception of the research project, performed the experimental work, analyzed data and wrote the manuscript. A.T.V., A.Q., R.S.F., G.S.K. and M.S.P. helped in the experimental work and participated in the evaluation of the results and contributed to the writing of the manuscript. S.R.B.M., C.F.M.M., S.S.H., P.A. and P.S. were involved in the conception and design of the experiments and analyzed the data. All the authors have read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-11024-3
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.WHO. Household air pollution and health. World Health Organization Available at: http://www.who.int/mediacentre/factsheets/fs292/en/ (2016).
- 2.Naeher LP, et al. Woodsmoke health effects: a review. Inhal. Toxicol. 2007;19:67–106. doi: 10.1080/08958370600985875. [DOI] [PubMed] [Google Scholar]
- 3.WHO. Burden of disease from the joint effects of Household and Ambient Air Pollution for 2012. Available at: www.who.int/phe (2014).
- 4.Artaxo, P. et al. Physical and chemical properties of aerosols in the wet and dry seasons in Rondônia, Amazonia. J. Geophys. Res. Atmos. 107, LBA 49-1–LBA 49-14 (2002).
- 5.Bowman DMJS, et al. Fire in the Earth System. Science (80-.). 2009;324:481–484. doi: 10.1126/science.1163886. [DOI] [PubMed] [Google Scholar]
- 6.Andreae MO, et al. The Amazon Tall Tower Observatory (ATTO): overview of pilot measurements on ecosystem ecology, meteorology, trace gases, and aerosols. Atmos. Chem. Phys. 2015;15:10723–10776. doi: 10.5194/acp-15-10723-2015. [DOI] [Google Scholar]
- 7.Artaxo P, et al. Atmospheric aerosols in Amazonia and land use change: from natural biogenic to biomass burning conditions. Faraday Discuss. 2013;165:203. doi: 10.1039/c3fd00052d. [DOI] [PubMed] [Google Scholar]
- 8.Brito J, et al. Ground-based aerosol characterization during the South American Biomass Burning Analysis (SAMBBA) field experiment. Atmos. Chem. Phys. 2014;14:12069–12083. doi: 10.5194/acp-14-12069-2014. [DOI] [Google Scholar]
- 9.IBGE. Instituto Brasileiro de Geografia e Estatística. Available at: www.ibge.gov.br (2014).
- 10.Ignotti, E., Hacon, S. D., Artaxo, P., Carlos, A. & Ponce, M. Air pollution and hospital admissions for respiratory diseases in the subequatorial Amazon : a time series approach Poluição do ar e admissões hospitalares por doenças respiratórias na Amazônia subequatorial: abordagem de séries temporais. 26, 747–761 (2010). [DOI] [PubMed]
- 11.Jacobson LDSV, et al. Association between fine particulate matter and the peak expiratory flow of schoolchildren in the Brazilian subequatorial Amazon: a panel study. Environ. Res. 2012;117:27–35. doi: 10.1016/j.envres.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Andreae MO, et al. Transport of biomass burning smoke to the upper troposphere by deep convection in the equatorial region. Geophys. Res. Lett. 2001;28:951–954. doi: 10.1029/2000GL012391. [DOI] [Google Scholar]
- 13.Reddington CL, et al. Air quality and human health improvements from reductions in deforestation-related fire in Brazil. Nat. Geosci. 2015;8:768–771. doi: 10.1038/ngeo2535. [DOI] [Google Scholar]
- 14.Artaxo P, et al. Physical and chemical properties of aerosols in the wet and dry seasons in Rondônia, Amazonia. J. Geophys. Res. 2002;107:8081. doi: 10.1029/2001JD000666. [DOI] [Google Scholar]
- 15.International Agency for Research on Cancer, I. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: Outdoor Air Pollution. 109 (2016). [PMC free article] [PubMed]
- 16.Guan W-J, Zheng X-Y, Chung KF, Zhong N-S. Impact of air pollution on the burden of chronic respiratory diseases in China: time for urgent action. Lancet. 2016;388:1939–1951. doi: 10.1016/S0140-6736(16)31597-5. [DOI] [PubMed] [Google Scholar]
- 17.Reid CE, et al. Critical Review of Health Impacts of Wildfire Smoke Exposure. Environ. Health Perspect. 2016;124:1334–1343. doi: 10.1289/ehp.1409277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borgie M, et al. Genotoxic and epigenotoxic effects of fine particulate matter from rural and urban sites in Lebanon on human bronchial epithelial cells. Environ. Res. 2015;136:352–62. doi: 10.1016/j.envres.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 19.Longhin E, et al. Cell cycle alterations induced by urban PM2.5 in bronchial epithelial cells: characterization of the process and possible mechanisms involved. Part. Fibre Toxicol. 2013;10:63. doi: 10.1186/1743-8977-10-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verma V, et al. Physicochemical and toxicological profiles of particulate matter in Los Angeles during the October 2007 Southern California wildfires. Environ. Sci. Technol. 2009;43:954–960. doi: 10.1021/es8021667. [DOI] [PubMed] [Google Scholar]
- 21.Lakey PSJ, et al. Chemical exposure-response relationship between air pollutants and reactive oxygen species in the human respiratory tract. Sci. Rep. 2016;6:32916. doi: 10.1038/srep32916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gualtieri M, et al. Airborne urban particles (Milan winter-PM2.5) cause mitotic arrest and cell death: Effects on DNA, mitochondria, AhR binding and spindle organization. Mutat. Res. 2011;713:18–31. doi: 10.1016/j.mrfmmm.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 23.de Oliveira Alves N, et al. Genotoxicity and composition of particulate matter from biomass burning in the eastern Brazilian Amazon region. Ecotoxicol. Environ. Saf. 2011;74:1427–33. doi: 10.1016/j.ecoenv.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 24.de Oliveira Alves N, et al. Genetic damage of organic matter in the Brazilian Amazon: a comparative study between intense and moderate biomass burning. Environ. Res. 2014;130:51–8. doi: 10.1016/j.envres.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 25.de Oliveira Alves N, et al. Biomass burning in the Amazon region: Aerosol source apportionment and associated health risk assessment. Atmos. Environ. 2015;120:277–285. doi: 10.1016/j.atmosenv.2015.08.059. [DOI] [Google Scholar]
- 26.Shang Y, Zhang L, Jiang Y, Li Y, Lu P. Airborne quinones induce cytotoxicity and DNA damage in human lung epithelial A549 cells: the role of reactive oxygen species. Chemosphere. 2014;100:42–9. doi: 10.1016/j.chemosphere.2013.12.079. [DOI] [PubMed] [Google Scholar]
- 27.Chan JKW, et al. Combustion-derived flame generated ultrafine soot generates reactive oxygen species and activates Nrf2 antioxidants differently in neonatal and adult rat lungs. Part. Fibre Toxicol. 2013;10:34. doi: 10.1186/1743-8977-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ekstrand-Hammarström B, et al. Oxidative stress and cytokine expression in respiratory epithelial cells exposed to well-characterized aerosols from Kabul, Afghanistan. Toxicol. In Vitro. 2013;27:825–33. doi: 10.1016/j.tiv.2012.12.022. [DOI] [PubMed] [Google Scholar]
- 29.Becker S, Mundandhara S, Devlin RB, Madden M. Regulation of cytokine production in human alveolar macrophages and airway epithelial cells in response to ambient air pollution particles: further mechanistic studies. Toxicol. Appl. Pharmacol. 2005;207:269–75. doi: 10.1016/j.taap.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 30.Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen s1. Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid. Redox Signal. [Internet]. 2009 [cited 2015 Nov 10];11:777–90. Antioxid. Redox Signal. 2009;11:777–90. doi: 10.1089/ars.2008.2270. [DOI] [PubMed] [Google Scholar]
- 31.Waris G, Ahsan H. Reactive oxygen species: role in the development of cancer and various chronic conditions. J. Carcinog. 2006;5:14. doi: 10.1186/1477-3163-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonner WM, et al. γH2AX and cancer. Nat. Rev. Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vessoni AT, et al. Chloroquine-induced glioma cells death is associated with mitochondrial membrane potential loss, but not oxidative stress. Free Radic. Biol. Med. 2016;90:91–100. doi: 10.1016/j.freeradbiomed.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 34.Quinet A, et al. Gap-filling and bypass at the replication fork are both active mechanisms for tolerance of low-dose ultraviolet-induced DNA damage in the human genome. DNA Repair (Amst). 2014;14:27–38. doi: 10.1016/j.dnarep.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 35.Møller P, et al. Air pollution, oxidative damage to DNA, and carcinogenesis. Cancer Lett. 2008;266:84–97. doi: 10.1016/j.canlet.2008.02.030. [DOI] [PubMed] [Google Scholar]
- 36.Donaldson K, Poland CA. Inhaled nanoparticles and lung cancer - what we can learn from conventional particle toxicology. Swiss Med. Wkly. 2012;142:w13547. doi: 10.4414/smw.2012.13547. [DOI] [PubMed] [Google Scholar]
- 37.Kelly FJ, Fussell JC. Size, source and chemical composition as determinants of toxicity attributable to ambient particulate matter. Atmos. Environ. 2012;60:504–526. doi: 10.1016/j.atmosenv.2012.06.039. [DOI] [Google Scholar]
- 38.Teixeira EC, Pra D, Idalgo D, Henriques JAP, Wiegand F. DNA-damage effect of polycyclic aromatic hydrocarbons from urban area, evaluated in lung fibroblast cultures. Environ. Pollut. 2012;162:430–8. doi: 10.1016/j.envpol.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 39.De Oliveira Galvão, M. et al. Biomass burning particles in the Brazilian Amazon region: Mutagenic effects of nitro and oxy-PAHs and assessment of health risks. Manuscr. accepted to Environ. Pollut. (2017). [DOI] [PubMed]
- 40.Shang Y, et al. Genotoxic and inflammatory effects of organic extracts from traffic-related particulate matter in human lung epithelial A549 cells: the role of quinones. Toxicol. In Vitro. 2013;27:922–31. doi: 10.1016/j.tiv.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 41.Oh SM, Kim HR, Park YJ, Lee SY, Chung KH. Organic extracts of urban air pollution particulate matter (PM2.5)-induced genotoxicity and oxidative stress in human lung bronchial epithelial cells (BEAS-2B cells) Mutat. Res. 2011;723:142–51. doi: 10.1016/j.mrgentox.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 42.Wegesser TC, Franzi LM, Mitloehner FM, Eiguren-Fernandez A, Last JA. Lung antioxidant and cytokine responses to coarse and fine particulate matter from the great California wildfires of 2008. Inhal. Toxicol. 2010;22:561–570. doi: 10.3109/08958370903571849. [DOI] [PubMed] [Google Scholar]
- 43.Williams KM, Franzi LM, Last JA. Cell-specific oxidative stress and cytotoxicity after wildfire coarse particulate matter instillation into mouse lung. Toxicol. Appl. Pharmacol. 2013;266:48–55. doi: 10.1016/j.taap.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jalava PI, et al. In vitro inflammatory and cytotoxic effects of size-segregated particulate samples collected during long-range transport of wildfire smoke to Helsinki. Toxicol. Appl. Pharmacol. 2006;215:341–353. doi: 10.1016/j.taap.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 45.Kim YD, Ko YJ, Kawamoto T, Kim H. The effects of 1-nitropyrene on oxidative DNA damage and expression of DNA repair enzymes. J. Occup. Health. 2005;47:261–266. doi: 10.1539/joh.47.261. [DOI] [PubMed] [Google Scholar]
- 46.International Agency for Research on Cancer, I. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Diesel and Gasoline Engine Exhausts and Some Nitroarenes (2014). [PMC free article] [PubMed]
- 47.Soberanes S, et al. Mitochondrial complex III-generated oxidants activate ASK1 and JNK to induce alveolar epithelial cell death following exposure to particulate matter air pollution. J. Biol. Chem. 2009;284:2176–86. doi: 10.1074/jbc.M808844200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Role of Quinones in Toxicology †. Chem. Res. Toxicol. 2000;13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- 49.Chung MY, et al. Aerosol-Borne Quinones and Reactive Oxygen Species Generation by Particulate Matter Extracts. Environ. Sci. Technol. 2006;40:4880–4886. doi: 10.1021/es0515957. [DOI] [PubMed] [Google Scholar]
- 50.Sheng K, Lu J. Typical airborne quinones modulate oxidative stress and cytokine expression in lung epithelial A549 cells. J. Environ. Sci. Heal. Part A. 2017;52:127–134. doi: 10.1080/10934529.2016.1237127. [DOI] [PubMed] [Google Scholar]
- 51.Vessoni aT, Filippi-Chiela EC, Menck CF, Lenz G. Autophagy and genomic integrity. Cell Death Differ. 2013;20:1444–54. doi: 10.1038/cdd.2013.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deng X, et al. PM2.5-induced oxidative stress triggers autophagy in human lung epithelial A549 cells. Toxicol. In Vitro. 2013;27:1762–70. doi: 10.1016/j.tiv.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 53.Zhai Q, et al. Genetic damage induced by organic extract of coke oven emissions on human bronchial epithelial cells. Toxicol. In Vitro. 2012;26:752–8. doi: 10.1016/j.tiv.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 54.DeMarini DM. Genotoxicity biomarkers associated with exposure to traffic and near-road atmospheres: a review. Mutagenesis. 2013;28:485–505. doi: 10.1093/mutage/get042. [DOI] [PubMed] [Google Scholar]
- 55.de Brito KCT, et al. Comparative genotoxicity of airborne particulate matter (PM2.5) using Salmonella, plants and mammalian cells. Ecotoxicol. Environ. Saf. 2013;94:14–20. doi: 10.1016/j.ecoenv.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 56.Liping Jiang, et al. Ambient particulate matter on DNA damage in HepG2 cells. Toxicol. Ind. Health. 2011;27:87–95. doi: 10.1177/0748233710387001. [DOI] [PubMed] [Google Scholar]
- 57.Abbas, I. et al. Polycyclic aromatic hydrocarbons within airborne particulate matter (PM(2.5)) produced DNA bulky stable adducts in a human lung cell coculture model. J. Appl. Toxicol., doi:10.1002/jat.1722 (2011). [DOI] [PubMed]
- 58.Reyes-Zárate E, et al. Atmospheric particulate matter (PM10) exposure-induced cell cycle arrest and apoptosis evasion through STAT3 activation via PKC?? and Src kinases in lung cells. Environ. Pollut. 2016;214:646–656. doi: 10.1016/j.envpol.2016.04.072. [DOI] [PubMed] [Google Scholar]
- 59.Binková, B., Giguère, Y. Jr., P. R. Dostál, M. & Šrám, R. J. The effect of dibenzo [a, l] pyrene and benzo [a] pyrene on human diploid lung fibroblasts: the induction of DNA adducts, expression of p53 and p21 WAF1 proteins and cell cycle distribution. 471, 57–70 (2000). [DOI] [PubMed]
- 60.Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 2000;275:9390–5. doi: 10.1074/jbc.275.13.9390. [DOI] [PubMed] [Google Scholar]
- 61.Pavagadhi S, Betha R, Venkatesan S, Balasubramanian R, Hande MP. Physicochemical and toxicological characteristics of urban aerosols during a recent Indonesian biomass burning episode. Environ. Sci. Pollut. Res. 2013;20:2569–2578. doi: 10.1007/s11356-012-1157-9. [DOI] [PubMed] [Google Scholar]
- 62.Ramdahl T. Retene—a molecular marker of wood combustion in ambient air. Nature. 1983;306:580–582. doi: 10.1038/306580a0. [DOI] [Google Scholar]
- 63.Hawliczek A, et al. Developmental toxicity and endocrine disrupting potency of 4-azapyrene, benzo[b]fluorene and retene in the zebrafish Danio rerio. Reprod. Toxicol. 2012;33:213–23. doi: 10.1016/j.reprotox.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 64.Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer. 2015;16:20–33. doi: 10.1038/nrc.2015.2. [DOI] [PubMed] [Google Scholar]
- 65.U.S. Environmental Protection Agency. in Exposure factors handbook 6.1–6.90 (2011).
- 66.Heyder J. Deposition of Inhaled Particles in the Human Respiratory Tract and Consequences for Regional Targeting in Respiratory Drug Delivery. Proc. Am. Thorac. Soc. 2004;1:315–320. doi: 10.1513/pats.200409-046TA. [DOI] [PubMed] [Google Scholar]
- 67.Roubicek Da, Gutiérrez-Castillo ME, Sordo M, Cebrián-García ME, Ostrosky-Wegman P. Micronuclei induced by airborne particulate matter from Mexico City. Mutat. Res. 2007;631:9–15. doi: 10.1016/j.mrgentox.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 68.Moraes MCS, et al. Both XPA and DNA polymerase eta are necessary for the repair of doxorubicin-induced DNA lesions. Cancer Lett. 2012;314:108–18. doi: 10.1016/j.canlet.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 69.Gomes LR, Vessoni AT, Menck CFM. Three-dimensional microenvironment confers enhanced sensitivity to doxorubicin by reducing p53-dependent induction of autophagy. Oncogene. 2015;34:5329–40. doi: 10.1038/onc.2014.461. [DOI] [PubMed] [Google Scholar]
- 70.Andrade-Lima LC, Andrade LN, Menck CFM. ATR suppresses apoptosis after UVB irradiation by controlling both translesion synthesis and alternative tolerance pathways. J. Cell Sci. 2015;128:150–9. doi: 10.1242/jcs.161596. [DOI] [PubMed] [Google Scholar]
- 71.Cortat B, et al. The relative roles of DNA damage induced by UVA irradiation in human cells. Photochem. Photobiol. Sci. 2013;12:1483–95. doi: 10.1039/c3pp50023c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.