


Abstract
The enriched PCR widely used for detection of mutant K-RAS in either tumor tissues or circulating DNA was modified so that abundant wild-type K-RAS alleles are cleaved prior to PCR. We took advantage of an AluI recognition site located immediately upstream of the K-RAS codon 12. The site was reconstituted upon DNA denaturation followed by annealing with a ‘stencil’, a 16-bp synthetic oligonucleotide complementary to the wild-type sequence. As opposed to normal K-RAS, the mutant allele forms, upon annealing with the stencil, a mismatch at the codon 12 which lies within the AluI enzyme binding site and partially inhibits its activity. The mismatch also lowers the melting temperature of the stencil-mutant K-RAS double helix as compared to stencil–wild-type duplex, so that only the latter is double stranded and selectively digested by AluI at elevated temperatures. The proposed method of stencil-aided mutation analysis (SAMA) based on selective pre-PCR elimination of wild-type sequences can be highly advantageous for detection of mutant K-RAS due to: (i) an enhanced sensitivity because of reduced competition with a great excess of normal K-RAS, and (ii) a decrease in a number of false-positive results from Taq polymerase errors. Application of SAMA for generalized detection of DNA mutations is discussed.
INTRODUCTION
Screening for oncogene and suppressor gene mutations is of great importance for early diagnosis of tumors still small enough to be amenable to curative treatment (1–14). The K-RAS gene [mutations in codon 12 or 13 of which occur in approximately half of all colorectal cancers (1), 75–100% of pancreatic cancer (15) and ∼50% of lung adenocarcinoma (16)] is especially interesting. Detection of these mutations against a predominant background of normal DNA requires highly sensitive assays.
One of the most effective methods widely used for the detection of K-RAS mutations is enriched PCR, which is based on the introduction of artificial restriction sites and employs a two-stage procedure with selective removal of only wild-type sequences between two steps (8,9). However, several drawbacks are inherent in this technique.
First, false-positive results are possible (16). Because the error rate of Taq polymerase is about 10–4–10–5, the expected mutant frequency after 20 template doublings of a 1000-bp sequence is >40% (17,18). After enrichment for mutant sequences between two rounds of PCR, even very small quantities of this artificially mutated DNA can result in a false positive signal. Such occasional emergence of false positive signals, i.e. mutated bands in negative control DNA samples from healthy donors, has been observed by many researchers (18, and our unpublished results).
Secondly, the competition between mutant sequences and a large excess of wild-type sequences that takes place during the first stage of enriched PCR can, presumably, lead to underestimation of mutant K-RAS. It should also be taken into consideration that only PCR products (i.e., those sequences with artificially introduced recognition site) are substrates for the restriction endonuclease, while the original templates are not. Therefore, competition during the second stage of enriched PCR should also occur, though evidently it is not as intense as during the first stage.
Thirdly, due to the persistence of the original templates, which cannot be removed during the enrichment step (see above), the wild-type sequences are almost always co-amplified together with the mutant allele. The heterogeneity of PCR products necessitates their electrophoretical separation and precludes application of various very efficient real-time approaches for PCR product detection.
It is evident that elimination of wild-type sequences, or at least significant reduction in their numbers in the original DNA sample prior to PCR, would be beneficial for detection of mutant sequences since this can minimize the consequences of Taq polymerase errors as well as the mutant/wild-type sequences competition problems. Here we propose a so-called stencil-aided mutation analysis (SAMA) procedure aimed at selective cleavage of wild-type sequences in an original DNA sample. The method takes advantage of the presence of AluI restriction sites in the K-RAS gene, one of which is located immediately upstream of codon 12. Oligonucleotide stencil complementary to the wild-type sequence, but having a mismatch with a mutant allele, is added to a DNA sample and, after a denaturation–annealing step, digestion with the AluI restriction endonuclease is performed at conditions that prevent formation of a double-stranded structure with the mutant sequence. As a result, the wild-type but not mutant K-RAS is cleaved.
MATERIALS AND METHODS
DNA samples
DNA from the human colon adenocarcinoma cell line SW480, which carries two mutated alleles at codon 12 of the K-RAS oncogene (7), and DNA from peripheral lymphocytes of healthy donors were used as positive and negative controls for K-RAS mutation, respectively. Samples of ‘normal’ tissues surrounding colon cancer were obtained during surgery on patients in the Cancer Research Center (Moscow). The samples were stored frozen before use. DNA was extracted using the standard phenol–chloroform procedure, precipitated by ethanol and dissolved in TE buffer.
Pre-PCR treatment
DNA samples were treated in Eppendorf tubes in a thermocycler. At the first stage, the minus strand of wild-type K-RAS was cleaved using a synthetic short oligonucleotide (termed stencil). The plus-stencil is a 16-bp oligonucleotide (5′-AGTTGGAGCTGGTGGC-3′) spanning the wild-type immediately upstream from codon 12. Six nucleotides flanking both sides of the AluI recognition site were required for the enzyme to bind and cleave efficiently. The DNA sample (1–1000 ng) in 60 µl of AluI restriction endonuclease buffer was mixed with 20 pmol of plus-stencil, heated at 95°C for 10 min, annealed at 47°C for 10 min and digested by AluI (5 U) at 42.5°C for 2 h (see Results).
At the second stage, the plus strand of wild-type K-RAS was cleaved similarly using the minus-stencil (5′-GCCACCAGCTCCAACT), the latter being complementary to the plus-stencil, specified above. The 2-fold amount (i.e., 40 pmol) of minus-stencil in AluI buffer was added to the DNA sample and heated at 95°C for 10 min (the excess of minus-stencil is needed to ensure complete hybridization with template DNA despite the presence of the complementary plus-stencil). Denaturation, annealing and digestion steps were repeated as described. Finally, the restriction enzyme was inactivated by heating at 95°C for 10 min, DNA was precipitated with ethanol, washed with 70% ethanol, dissolved in water and subjected to PCR.
PCR assay
K-RAS mutations were detected by a two-stage enriched PCR using selective restriction enzyme digestion of an artificially created BstNI site to enrich for mutant K-RAS sequence (8). An additional BstNI site was also created downstream of codon 12 to serve as an internal control for completion of the digestion. As a result, non-restricted PCR product was 157 bp long, while that restricted at both sites (wild-type sequence) was 113 bp long and product restricted only at the right site (the left site is modified by mutation) was 142 bp long.
The reaction mixture (25 µl) was cycled 15 times at 94°C for 48 s, 56°C for 90 s and 72°C for 155 s. An aliquot of 10 µl adjusted to 1× BstNI reaction buffer was digested with 10 U of BstNI at 60°C for 90 min. Ten microliters of the digested PCR mixture was transferred to a new tube and a new reaction mixture was set up for the second amplification step (35 cycles of 94°C for 48 s, 56°C for 90 s and 72°C for 60 s) using identical constituents. The second BstNI digestion was performed using 25 µl of the second-step PCR product at 60°C for 90 min.
Some experiments were performed with the modifications proposed by Kopreski et al. (9) and gave essentially the same results.
The final digestion products were separated by electrophoresis in a 7% polyacrylamide gel.
RESULTS
We took advantage of an AluI restriction site located immediately upstream of codon 12 in K-RAS (Fig. 1) to selectively destroy the wild-type K-RAS sequence. The ‘stencil’ is a short (16 bp in this particular case) oligonucleotide specifically designed to discriminate between wild and mutant sequences. Its size is determined by minimal requirements for effective binding and cleavage by the AluI restriction nuclease, namely the 4-bp recognition site and the flanking non-specific 6 bp on both sides of the AluI restriction site (New England Biolabs; http://www.neb.com/neb/tech/tech_resource/restriction/properties/cleave_oligo.html). The stencil spans wild-type codon 12 with 10 nt upstream and 3 nt downstream. Two stencils (‘plus’ and the complementary ‘minus’) are used to cleave the ‘minus’ and ‘plus’ wild-type K-RAS strands, respectively.
Figure 1.

Schematic presentation of stencil-aided selective cleavage of wild-type K-RAS. (A) The fragment (157 bp) of K-RAS gene amplified by PCR. Positions of AluI recognition sites as well as the left and right primers are indicated. The primer K-RAS-L is immediately upstream of codon 12. (B) Denaturation-annealing of wild-type DNA (plus-strand) with the excess of complementary minus-stencil spanning AluI recognition site plus 6 bp on both sides restores an adequate substrate for the enzymatic cleavage. (C) Denaturation-annealing of mutant DNA with the same stencil restores the duplex with a mismatch at codon 12 (mutant nucleotides at either first or second positions are indicated as X and Y).
The rationale for the proposed approach is based on the fact that mismatches in short oligonucleotides cause significant changes in the duplex stability. Using the equation
Tm = 81.5 – 16.6 (log10[Na+]) + 0.41(%G+C) – (600/N)
(19) one can calculate that duplexes formed by K-RAS sequences with perfectly matched complementary stencils, described above, in AluI digestion buffer have Tm ∼ 48°C. It is known that for every 1% of mismatching bases in a double-stranded DNA, there is a reduction in Tm of 1–1.5°C, mismatches in the middle of the oligonucleotide being far more deleterious than mismatches at the ends (19). This means that a mismatch formed by mutant K-RAS and the stencil lowers the Tm by 6.5–10°C, i.e. to 38–41.5°C. Thus, at the intermediate temperature, i.e. between 42 and 48°C, only wild-type complexes would have double-stranded secondary structure and be amenable to AluI cleavage.
This idea was tested in model experiments with perfectly matched and mismatched complementary stencils annealed and digested by AluI endonuclease at various temperatures. Oligonucleotides used were the plus-stencil and minus-stencil specified above, which were perfectly complementary to each other, as well as a mutated minus-stencil (5′-GCCATCAGCTCCAACT) with a substitution (C→T, underlined) forming a mismatch with the plus-stencil at the second nucleotide of codon 12. Figure 2 shows the electrophoretic mobility of single-stranded oligonucleotides (lanes 1–3) as well as double-stranded perfect and mismatched oligonucleotides (lanes 4 and 5, respectively). Panels I, II and III demonstrate electrophoretic patterns of intact oligonucleotides, and of those digested by AluI at 37 and 42.5°C, respectively. Figure 2 shows that the perfectly matched oligonucleotides were cleaved almost completely at both temperatures (II and III, lanes 4). Mismatched oligonucleotides were partially cleaved at 37°C (II, lane 5), presumably due to the duplex destabilization at this temperature, but were not cleaved at 42.5°C (III, lane 5), thus proving the validity of the approach. We postulated that the heavily stained band of mismatched stencils locates between positions of single-stranded and double-stranded oligonucleotides (Fig. 2, panel III, lane 5) because they are single stranded during the incubation procedure but begin to hybridize as the temperature lowers during electrophoresis. In any case, under the conditions chosen there is no sign of mismatched stencil digestion at 42.5°C. The fact that mismatched duplexes are digested to some extent by AluI restriction endonuclease at 37°C (Fig. 2, panel II, lane 5) implies that this enzyme is relatively insensitive to the structure of sequences adjacent to the recognition site.
Figure 2.

AluI digestion of perfectly matched and mismatched 16 bp oligonucleotides. Oligonucleotides (50 pmol in 50 µl of AluI buffer) used in this experiment were wild-type plus-stencil (5′-AGTTGGAGCTGGTGGC), wild-type minus-stencil (5′-GCCACCAGCTCCAACT) and mutant minus-stencil (5′-GCCATCAGCTCCAAC) with a substitution (C→T, underlined) of the second nucleotide in codon 12. Oligonucleotides were taken either alone (lanes 1–3), or in pairs, perfectly matched (lanes 4) or mismatched (lanes 5). They were denatured at 94°C for 5 min, annealed at 47°C for 10 min and incubated for 60 min under different conditions: at 37°C with no enzyme added (panel I); at 37 and 42.5°C, with 5 U of AluI (panels II and III, respectively). Aliquots of the samples were subjected to electrophoresis in 20% PAAG and stained with ethidium bromide (0.5 µg/ml). Positions of single-stranded, double-stranded and cleaved oligonucleotides are indicated by arrows. Single-stranded and double-stranded oligonucleotides are distinguished by electrophoretic mobility and intensity of ethidium bromide staining.
The results obtained with short oligonucleotides formed the basis for the procedure of selective pre-PCR cleavage of wild-type K-RAS (see Materials and Methods). This procedure efficiently eliminated wild-type K-RAS sequences tested either alone (Fig. 3) or in a combination with mutant alleles (Fig. 4) with no appreciable adverse effect on the latter. Figure 3 shows that the signal of mutant K-RAS even taken in low amounts remains unchanged after AluI digestion, while the signal of wild-type K-RAS is not seen, except for a 1000 ng sample where it is significantly reduced. Experiments with mixtures of wild-type and mutant K-RAS (Fig. 4) led to the same conclusion. After AluI treatment the band specific for mutant K-RAS was not reduced, while the band of wild-type K-RAS disappeared. At the highest K-RAS(W)/K-RAS(M) ratio used (1000/1) the competition of mutant K-RAS with the large excess of wild-type sequences in the control sample considerably weakened the mutant signal. However, the stencil-aided pre-PCR digestion of the wild-type sequences made the mutant K-RAS band much stronger (Fig. 4).
Figure 3.
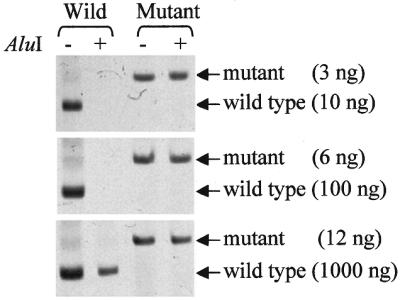
Pre-PCR digestion of wild-type or mutant K-RAS followed by enriched PCR. Normal DNA from blood cells of healthy donors (10–1000 ng) and mutant DNA from the SW480 cell line (3–12 ng) were separately subjected to the procedure of stencil-aided AluI cleavage. The target sequences were thereafter amplified by the enriched PCR. The samples with or without AluI restriction enzyme treatment are indicated above the lanes. Note that the signal of mutant K-RAS even taken in low amounts remains unchanged after AluI digestion, while wild-type K-RAS is fully destroyed, except for 1000 ng sample (see text).
Figure 4.
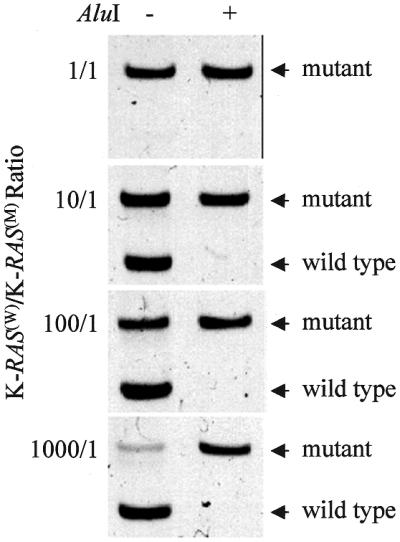
SAMA of mutant K-RAS. Mixtures of normal DNA (ranging from 1 to 1000 ng) and constant amount (1 ng) of mutant SW480 cell DNA were subjected to the stencil-aided AluI cleavage (untreated mixtures were used as controls). The target sequences were amplified by the enriched PCR. Note that wild-type sequences are completely removed by the stencil-aided treatment and the signal of mutant K-RAS becomes stronger at the highest K-RAS(W)/K-RAS(M) ratio used.
Higher sensitivity of SAMA compared to the standard enriched PCR was also demonstrated in another set of experiments when both techniques were used to detect mutant K-RAS in ‘normal’ tissues surrounding colorectal adenocarcinoma, in which a low, if any, concentration of mutant K-RAS sequences could be envisaged. Figure 5 shows that mutant K-RAS was detected by SAMA, but not by enriched PCR alone, in one of three such DNA samples. Besides, major bands of wild-type sequences amplified by enriched PCR were absent when SAMA was used for detection. This evidently excludes the suggestion that the mutant allele revealed by SAMA is due to Taq polymerase errors, since artificially created and amplified mutant sequences should be quantitatively proportional to (and much lesser than) those of wild-type.
Figure 5.
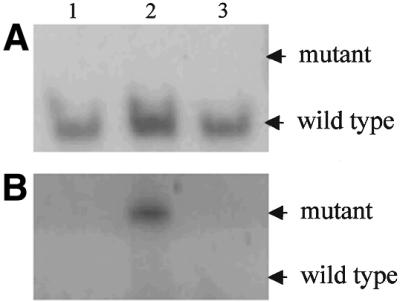
Detection of mutant K-RAS in colorectal cancers. DNA samples isolated from ‘normal’ tissues surrounding a tumor and removed during surgery (Materials and Methods) were subjected to enriched PCR (A) or SAMA (B). Lanes 1, 2 and 3, DNA samples from individual patients.
DISCUSSION
The data presented demonstrate that the stencil-aided pre-PCR digestion of the wild-type K-RAS improves detection of mutant K-RAS. In our experiments SAMA has been tested at the practically useful ratios (from 1/1 to 1/103) of mutant to wild-type K-RAS sequences. In some reports much higher ratio values (1/104 and even 1/106) were successfully tested (8,12), but they obviously required some specific reaction setup. As a matter of fact, ∼1 ng of the tumor genome DNA can be considered as a minimal amount required for reliable detection of mutant K-RAS (assuming, first, that a cancer cell usually contains a single mutant K-RAS allele, secondly, that DNA content of the human cell is ∼6 pg, and thirdly, that a sensitivity limit of the standard PCR using ethidium bromide staining is about 100–1000 copies of a unique sequence) (20). In reality, lower amounts of tumor genome DNA (10–100 pg) did not give, in our hands, a clear signal. Under these conditions, at the higher mutant/wild-type ratios (1/104–1/106), the amount of wild-type DNA added to the standard 25–50 µl PCR probe should be impractically high (10 µg–1 mg). In this case, some unpleasant problems, such as high viscosity, non-specific amplification, or chelation of magnesium ions, could arise. As a result, we preferred to evaluate our approach by side-by-side comparison of standard enriched PCR and SAMA at the commonly used mutant/wild-type ratios.
The stencil-aided digestion of the wild-type K-RAS prior to PCR helps to resolve some problems associated with detection of mutant alleles in the presence of a huge excess of wild-type sequences. First, decreasing the concentration of wild-type alleles in DNA sample SAMA should minimize the possibility of false-positive results due to Taq polymerase errors during the amplification process and, thus, give more credibility to mutant K-RAS determinations. This is the main advantage of this approach over the existing methods (8,9,16–18). Secondly, side-by-side comparison of SAMA with the standard technique of enriched PCR (Figs 3–5) demonstrated higher sensitivity of the former in mutant K-RAS detection. Though the described procedure was applied here to improve the detection of mutant codon 12, it is evident that mutations of codon 13 would also affect the stability of the stencil–DNA complex and therefore discriminate between wild and mutant sequences. Finally, the homogeneity of the product obtained by the SAMA creates the possibility of applying real-time PCR technology for quantitative analysis of mutant sequences in clinical practice (21).
To apply the SAMA procedure for detection of K-RAS mutations we took advantage of the AluI restriction site located immediately upstream of codon 12. This does not mean, however, that SAMA is applicable only to such exclusive situations, because a slightly modified method can be applied to detection of any known mutation. It was demonstrated earlier that single-stranded DNA can be cleaved at any predetermined site by interaction with a specially designed oligodeoxyribonucleotide adaptor and the class-IIS or the class-IIN restriction endonuclease, FokI or XcmI, respectively (22,23). This adaptor used as the stencil could discriminate between the wild-type and mutant sequences in single-stranded DNA in accordance with the principle described above.
At the same time, it should be noted that the procedure described was most efficient if pre-PCR cleavage of wild-type sequences was followed by enriched PCR (i.e., a two-stage amplification procedure with a BstNI digestion step in between). It was observed in the model experiments with synthetic oligonucleotides that stencil-aided AluI cleavage of wild-type sequences was very significant (>95%, as revealed by densitometric measurements) but not quantitative (Fig. 2, and our unpublished data). A negligible amount of wild-type sequences survives such a treatment. Though the reduced amount of wild-type allele competes with the mutant allele much less effectively, a band corresponding to the PCR product of the wild-type sequence is sometimes visible in a gel if the intermediate BstNI digestion step is omitted. Therefore, pre-PCR cleavage of wild-type alleles, on one hand, and enrichment of mutant alleles by BstNI digestion, on the other, should be considered as complementary steps, rather than mutually exclusive.
Recently, new PCR variants aimed at detection of mutant K-RAS have been described. One of them is restriction endonuclease-mediated selective (REMS)–PCR capable of detecting one mutant allele (codon 12 of the K-RAS gene) in the presence of 1000 wild-type alleles (24). Reaction requires the concurrent activity of a restriction endonuclease and a DNA polymerase, both of which must be sufficiently thermostable to retain activity during thermocycling. Inclusion of the restriction endonuclease in REMS–PCR inhibits amplification of sequences containing the recognition site, thus producing selective amplification of sequences that lack the recognition site. Application of this approach to detection of various mutations is restricted by a limited number of highly thermostable restriction endonucleases, which must endure repeated thermocycling up to 94°C.
Another recently proposed two-step protocol combines an allele-specific PCR followed by a PCR–RFLP (restriction fragment length polymorphism) confirmatory step (25). The method resembles a nested PCR technique starting directly from genomic DNA material and allows detection of one mutant allele in 103 normal alleles. This protocol, being similar to the standard enriched PCR, suffers from the same drawback, namely the possibility of false positive results due to the inherent error rate of Taq polymerase.
In conclusion, we believe that the method proposed is capable of increasing the sensitivity of mutant gene detection and decreasing the probability of false-positive results, and can be applied to various areas of molecular diagnostics and genetic testing.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr M. Jakubovskaya for critical discussions, Dr M. Kiefer for valuable advice, and anonymous reviewers for helpful suggestions. This work was supported by a Russian Ministry of Science ‘Human Genome’ grant and Russian Fund for Fundamental Research grant 01-04-48463 to A.V.L.
References
- 1.de Kok J.B., van Solinge,W.W., Magnenat,J.-L., Ruers,T.J.M., Roelofs,R.W.H.M., van Muijen,G.N.P., Willems,J.L. and Swinkels,D.W. (1997) Detection of tumor DNA in serum of colorectal cancer patients. Scand. J. Clin. Lab. Invest., 57, 601–604. [DOI] [PubMed] [Google Scholar]
- 2.Anker P., Mulcahy,H., Chen,X.Q. and Stroun,M. (1999) Detection of circulating tumour DNA in the blood (plasma/serum) of cancer patients. Cancer Metastasis Rev., 18, 65–73. [DOI] [PubMed] [Google Scholar]
- 3.Chen X.Q., Stroun,M., Magnenat,J.L., Nicod,L.P., Kurt,A.M., Lyautey,J., Lederrey,C. and Anker,P. (1996) Microsatellite alterations in plasma DNA of small cell lung cancer patients. Nature Med., 2, 1033–1035. [DOI] [PubMed] [Google Scholar]
- 4.Mulcahy H.E., Lyautey,J., Lederrey,C., Chen,X.Q., Lefort,F., Vasioukhin,V., Anker,P., Alstead,E.M., Farthing,M.J. and Stroun,M. (2000) Plasma DNA K-ras mutations in patients with gastrointestinal malignancies. Ann. N. Y. Acad. Sci., 906, 25–28. [DOI] [PubMed] [Google Scholar]
- 5.Nawroz H., Koch,W., Anker,P., Stroun,M. and Sidransky,D. (1996) Microsatellite alterations in serum DNA of head and neck cancer patients. Nature Med., 2, 1035–1037. [DOI] [PubMed] [Google Scholar]
- 6.Botezatu I., Serdyuk,O., Potapova,G., Shelepov,V., Alechina,R., Molyaka,Y., Ananev,V., Bazin,I., Garin,A., Narimanov,M., Knysh,V., Melkonyan,H., Umansky,S. and Lichtenstein,A. (2000) Genetic analysis of DNA excreted in urine: a new approach for detecting specific genomic DNA sequences from cells dying in an organism. Clin. Chem., 46, 1078–1084. [PubMed] [Google Scholar]
- 7.Haliassos A., Chomel,J.C., Grandjouan,S., Kruh,J., Kaplan,J.C. and Kitsis,A. (1989) Detection of minority point mutations by modified PCR technique: a new approach for a sensitive diagnosis of tumor-progression markers. Nucleic Acids Res., 17, 8093–8099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kahn S.M., Jiang,W., Culbertson,T.A., Weinstein,I.B., Williams,G.M., Tomita,N. and Ronai,Z. (1991) Rapid and sensitive nonradiactive detection of mutant K-ras genes via ‘enriched’ PCR amplification. Oncogene, 6, 1079–1083. [PubMed] [Google Scholar]
- 9.Kopreski M.S., Benko,F.A., Kwee,C., Leitzel,K.E., Eskander,E., Lipton,A. and Gocke,C. (1997) Detection of mutant K-ras DNA in plasma or serum of patients with colorectal cancer. Br. J. Cancer, 76, 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mulcahy H. and Farthing,M.J. (1999) Diagnosis of pancreatico-biliary malignancy: detection of gene mutations in plasma and stool. Ann. Oncol., 10 [Suppl. 4], 114–117. [PubMed] [Google Scholar]
- 11.Mulcahy H.E., Lyautey,J., Lederrey,C., Chen,X.Q., Anker,P. and Alstead,E.M. (1998) A prospective study of K-ras mutations in the plasma of pancreatic cancer patients. Clin. Cancer Res., 4, 271–275. [PubMed] [Google Scholar]
- 12.Schimanski C., Linnemann,U. and Berger,M.R. (1999) Sensitive detection of K-ras mutations augments diagnosis of colorectal cancer metastases in the liver. Cancer Res., 59, 5169–5175. [PubMed] [Google Scholar]
- 13.Yamada T., Nakamori,S., Ohzato,H., Oshima,S., Aoki,T., Higaki,N., Sugimoto,K., Akagi,K., Fujiwara,Y., Nishisho,I., Sakon,M., Gotoh,M. and Monden,M. (1998) Detection of K-ras gene mutations in plasma DNA of patients with pancreatic adenocarcinoma: correlation with clinicopathological features. Clin. Cancer Res., 4, 1527–1532. [PubMed] [Google Scholar]
- 14.Yamaguchi Y., Watanabe,H., Yrdiran,S., Ohtsubo,K., Motoo,Y., Okai,T. and Sawabu,N. (1999) Detection of mutations of p53 tumor suppressor gene in pancreatic juice and its application to diagnosis of patients with pancreatic cancer: comparison with K-ras mutation. Clin. Cancer Res., 5, 1147–1153. [PubMed] [Google Scholar]
- 15.Smit V.T., Boot,A.J., Fleuren,G.J., Cornelisse,C.J. and Bos,J.L. (1989) K-ras codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res., 16, 7773–7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahrendt S.A., Chow,J.T., Xu,L.-H., Yang,S.C., Eisenberger,C.F., Esteller,M., Herman,J.G., Wu,L., Decker,P.A., Jen,J. and Sidransky,D. (1999) Molecular detection of tumor cells in bronchoalveolar lavage fluid from patients with early stage lung cancer. J. Natl Cancer Inst., 91, 332–339. [DOI] [PubMed] [Google Scholar]
- 17.Smith J. and Modrich,P. (1997) Removal of polymerase-produced mutant sequences from PCR products. Proc. Natl Acad. Sci. USA, 94, 6847–6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobs G., Tscholl,E., Sek,A., Pfreundschuh,M., Daus,H. and Trumper,L. (1999) Enrichment polymerase chain reaction for the detection of Ki-ras mutations: relevance of Taq polymerase error rate, initial DNA copy number and reaction conditions on the emergence of false-positive mutant bands. J. Cancer Res. Clin. Oncol., 125, 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sambrook, J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 20.McPherson M.J. and Møller,S.G. (2000) PCR (The Basics series). BIOS Scientific Publishers, Oxford, UK.
- 21.Xu J., Stolk,J.A., Zhang,X., Silva,S.J., Houghton,R.L., Matsumura,M., Vedvick,T.S., Leslie,K.B., Badaro,R. and Reed,S.G. (2000) Identification of differentially expressed genes in human prostate cancer using subtraction and microarray. Cancer Res., 60, 1677–1682. [PubMed] [Google Scholar]
- 22.Shaw P.-C. and Mok,Y.-K. (1993) XcmI as a universal restriction enzyme for single-stranded DNA. Gene, 133, 85–89. [DOI] [PubMed] [Google Scholar]
- 23.Podhajska A.J., Kim,S.C. and Szybalski,W. (1992) Conferring new specificities on restriction enzymes: cleavage at any predetermined site by combining adapter oligodeoxynucleotide and class-IIS enzyme. Methods Enzymol., 216, 303–309. [DOI] [PubMed] [Google Scholar]
- 24.Fuery C.J., Impey,H.L., Roberts,N.J., Applegate,T.L., Ward,R.L., Hawkins,N.J., Sheehan,C.A., O’Grady,R. and Todd,A.V. (2000) Detection of rare mutant alleles by restriction endonuclease-mediated selective-PCR: assay design and optimization. Clin. Chem., 46, 620–624. [PubMed] [Google Scholar]
- 25.Behn M., Thiede,C., Neubauer,A., Pankow,W. and Schuermann,M. (2000) Facilitated detection of oncogene mutations from exfoliated tissue material by a PNA-mediated ‘enriched PCR’ protocol. J. Pathol., 190, 69–75. [DOI] [PubMed] [Google Scholar]