

Abstract
A metathesis reaction occurs when a diaryliodonium triflate is heated with an aryl iodide, resulting in formation of a new diaryliodonium triflate.
Keywords: hypervalent iodine, iodonium salts, metathesis
Graphical Abstract
Aryl iodides undergo I-arylation upon treatment with diaryliodonium triflates
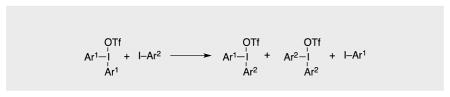
This paper describes a thermally induced, metal-free metathesis reaction between diaryliodonium triflates and certain aryl iodides according to Eq. 1 (Scheme 1). While DiMagno observed fluoride-catalyzed aryl ligand exchange between diaryliodonium species (Eq. 2),[1] and Koser reported oxygen ligand exchange between PhI(OH)OTf and aryl iodides (Eq. 3),[2] the chemistry of Eq. 1 appears to be undocumented.
Scheme 1.
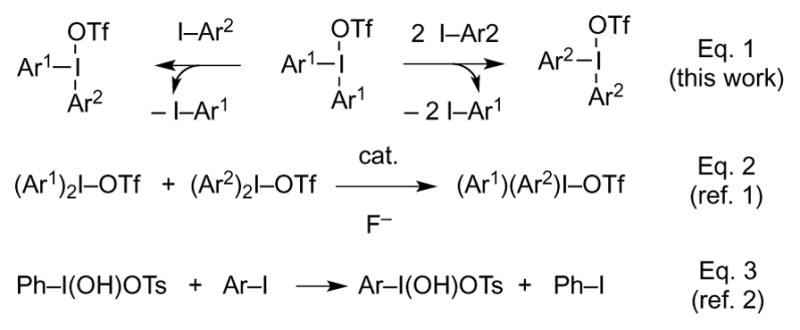
Iodonium metathesis reactions.
By way of background, diaryliodonium species are carriers of electrophilic aryl synthons, hence they arylate a diversity of nucleophiles,[3] even very weak ones.[4] For that reason, they are of significant interest in organic chemistry.[5] On the other hand, Yamamoto has shown that organic iodides possess appreciable I-nucleophilicity.[6] This raises the question of whether diaryliodonium agents might be able to I-arylate aryl iodides; i.e., whether an iodonium metathesis reaction might be possible. The ability to prepare diaryliodonium complexes by this method would have a favorable impact in organic, materials, and medicinal chemistry;[3–5] however, the prognosis for the feasibility the transformation seemed poor. For instance, Olofsson, detected no aryl group exchange between bis(4-tolyl)iodonium triflate and 2,6-dimethyl-iodobenzene upon heating in DMF.[7]
In contrast with the foregoing, we found that 4-iodotoluene, 4-iodoanisole, and 1-iodonaphthalene displace PhI from Ph2IOTf, leading to mixtures of products of mono- and bis-substitution. With low-melting[8] aryl iodides, the reaction may be induced by melting a mixture of the reactants.[9] However, incomplete consumption of Ph2IOTf was consistently observed under these conditions. Generally better results were obtained by operating in 1,2-dichloroethane (DCE) solution, in which case most of the starting Ph2IOTf was consumed (Table 1). The solution protocol is thus the method of choice. Interestingly, such metathesis reactions failed in donor solvents such as (CF3)2CHOH, MeCN, DMF, or DMSO. This stands in contrast with DiMagno’s report that the chemistry of Eq.2 takes place in MeCN solution,[1] but it is consistent with Olofsson’s observation that no aryl group exchange between diaryliodonium triflates and aryl iodides occurs in DMF.[7] A rationale for all this segues from a possible mechanism for the metathesis reaction proposed herein.
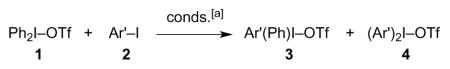
Table 1.
Solution metathesis reactions of Ph2IOTf.
| ||||
|---|---|---|---|---|
| entry | Ar’ | time (h) | ratio 3:4:1[b] | yield[c] |
| a | 4-Me-C6H4 | 24 | 9.8:28.5:1.0 | 48 |
| b | 4-MeO-C6H4 | 25 | 27.3:23.1:1.0 | 78 |
| c | 1-naphthyl | 26 | 2.0:1.0:0.0 | 14 |
Conditions: 0.2 M solution of Ph2IOTf in (CH2Cl)2, 5 equiv aryl iodide, thick-walled glass tube sealed with a Teflon screwcap and immersed in an oil bath kept at 120–125 °C.
Molar ratios calculated by integration of 1H NMR spectra.
Percent yield after silica gel column chromatography (gradient 10% → 40% acetone-CH2Cl2) to remove nonpolar byproducts. The stated value is the sum of the yields of individual compounds present in the product mixture. A comparison of 1H NMR spectra of crude and purified reaction mixtures indicated that insignificant changes in the ratio of iodonium triflate products had occurred upon chromatography.
As seen in Table 1, the reaction tends to afford mixtures of products. In an effort to improve selectivity, we examined the metathesis chemistry of mixed iodonium triflates, in which one of the aryl ligands is electron-poor, and the other, electron-rich.[10] Such species react with common nucleophiles by what appears to be an SNAr mechanism, resulting in transfer of the more electron-deficient aryl group[11] — at least in the absence of transition metal catalysts.[12] Greater aryl group transfer selectivity might obtain if such a preference were maintained in the context of the metathesis reaction.
The foregoing complexes did react selectively, but contrary to expectations, they preferentially transferred the more electron-rich group to 1-iodonaphthalene, 4-I-C6H4Me, 4-I-C6H4OMe, and Ph-I, (Tables 2–3), indicating that the metathesis reaction is unlikely to proceed by an SNAr mechanism.[13] On the other hand, thienyliodonium triflate 8 transferred the 4-MeOOCC6H4 group selectively (Table 4), leading to mixtures of products 9 and 4, but only traces of 10. In light of precedent,[11] Coenen, who also observed preferential expulsion of 2-iodothiophene in SNAr reactions of mixed 2-thienyliodonium salts,[14] ascribed this selectivity to the electron-rich character of the thienyl group. In the present case, such an explanation would be untenable, given the results of Tables 2–3. A more satisfactory rationale for aryl group transfer selectivity in iodonium metathesis emerged from a computational estimate of the fractional positive charge on the I-atom of various aryl iodides (Table 5). Halides at the top of the list exhibit a smaller (+)-charge on the I-atom, and may thus be expected to be better I-nucleophiles than those at the bottom of the table, for which, increased I-(+)-charge translates into greater nucleofugality. A diaryliodonium triflate based on an aryl iodide with a greater extent of (+)-charge on the I-atom is likely to be destabilized relative to one centered on a less I-positive iodide, because the I-atom must acquire additional (+)-character in the hypervalent state.[17] Such an accumulation of (+)-charge on an already positive I-atom is surely energetically unfavorable. Therefore, iodonium metathesis occurs so that the extent of (+)-charge on the hypervalent I-atom decreases; i.e., so that a more I-nucleophilic iodide displaces a less I-nucleophilic one. Table 5 seems to rationalize the formation of mixtures in reactions of Ph2IOTf with 4-I-C6H4Me, 4-I-C6H4OMe, and 1-iodonaphthalene, given the similar charges on the I-atom of nucleophiles and nucleofuge (PhI). The selective transfer of the 4-MeOOCC6H4 group from 5 is consistent with the greater extent of (+)-charge on the I-atom of 2-iodothiophene (better nucleofuge) relative to methyl 4-iodobenzoate. Finally, the fact that no metathesis occurred between Ph2IOTf and 5-iodouridine triacetate[18–19] or 4-Br-C6H4-I may be ascribed to the unfavorable change in (+)-charge on the hypervalent I-atom that would be incurred during the reaction.[20] On the basis of the foregoing, a possible mechanism for the iodonium metathesis reaction may be ventured as outlined in Scheme 2. Reversible dissociation of starting complex 11 and addition of a nucleophilic aryl iodide to iodonium ion 12 yields 13. The latter would form only in polar, nonnucleophilic media such as DCE: donor solvents (DMF, MeCN, etc.), surely more nucleophilic than aryl iodides, would outcompete Ar3I for 12, thus retarding / suppressing metathesis. Kinetically faster expulsion of the more nucleofugal aryl iodide from 13, perhaps via transition state 14,[7] leads to iodonium ion 15, which then combines with TfO− to give the final 16.
Table 2.
Metathesis reactions of iodonium triflate 5.
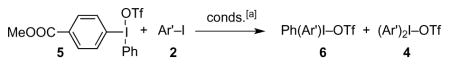
Table 3.
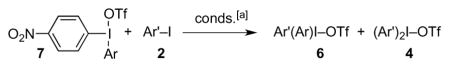
Metathesis reactions of iodonium triflates 7.
| ||||
|---|---|---|---|---|
| entry | Ar | Ar’ | ratio 6:4:7:(other)[b] | yield[c] |
| a[d] | Ph | Ph | 12.9[e]:1.0:(−)[f] | 65 |
| b[d] | “ | 4-MeC6H4 | 1.0:1.3:0.0:(−)[f] | 73 |
| c[d] | “ | 4-MeOC6H4 | 60.1:35.1:1.0:(−)[f] | 76 |
| d[d] | “ | 1-naphthyl | 33.3:5.9:1.0:(−)[f] | 59 |
| e[d] | 4-MeOC6H4 | Ph | 11.5:8.2:1.0:(1.0)[g] | 60 |
| f[d] | “ | 4-MeC6H4 | 1.3:1.0:0.0:(−)[f] | 79 |
| g[d] | “ | 4-MeOC6H4 | 6.9[e]:1.0:(−)[f] | 89 |
| h[d] | “ | 1-naphthyl | 8.4:1.2:1.2:(1.0+0.6)[h] | 64 |
| i[d] | mesityl | 4-MeC6H4 | 1.0:7.0:0.0:(−)[f] | 26 |
| j[d] | “ | 4-MeOC6H4 | 1.0:13.0:0.0:(−)[f] | 37 |
| k[d] | “ | 1-naphthyl | 1.0:8.4:8.4:(−)[i] | 27 |
: same as in Table 1.
Reaction time: 12 h.
Compounds 7 and 4 are identical in this case.
Other products — if at all detectable — were present in insignificant amounts.
This other product was bis-(4-anisyl)iodonium triflate (1H-NMR, MS), perhaps arising as per ref. 1.
The major other product was 1-naphthyl-4-nitrophenyl-iodonium triflate; the minor other product was bis-(4-anisyl)iodonium triflate (1H-NMR, MS).
A trace amount of 1-naphthyl-4-nitrophenyl-iodonium triflate, too small to be measured accurately, was apparent by 1H-NMR and MS.
Table 4.
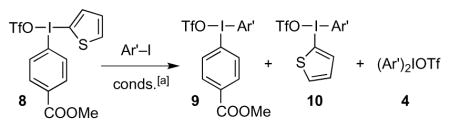
Metathesis reactions of thienyliodonium triflate 8.
| |||
|---|---|---|---|
| entry | Ar’ | ratio 9:10:4:8:(other)[b] | yield[c] |
| a[d] | Ph | 1.0:trace:15.1:0.0:(−)[e] | 40 |
| b[d] | 4-MeC6H4 | 1.0:trace:7.0:0.0:(−)[e] | 66 |
| c[d] | 4-MeOC6H4 | 1.3:trace:1.0:trace:(−)[e] | 91 |
| d[d] | 1-naphthyl | 1.0:trace:4.1:trace:(−)[e] | 38 |
: same as in Table 1.
reaction time: 12h.
Other products — if at all detectable — were present in insignificant amounts.
Table 5.
Calculated charge on the I-atom of various aryl iodides.
| substance | charge on the I-atom[a] | |
|---|---|---|
| 4-iodotoluene | + 0.068 |
|
| iodobenzene | + 0.068 | |
| 1-iodonaphthalene | + 0.070 | |
| 4-iodoanisole | + 0.071 | |
| 4-bromo-1-iodobenzene | + 0.088 | |
| methyl 4-iodobenzoate | + 0.093 | |
| 2-chloro-5-iodopyridine | + 0.120 | |
| 4-nitro-1-iodobenzene | + 0.120 | |
| 5-iodo-1-methyluracil | + 0.162 | |
| 2-iodothiophene | + 0.166 | |
Scheme 2.
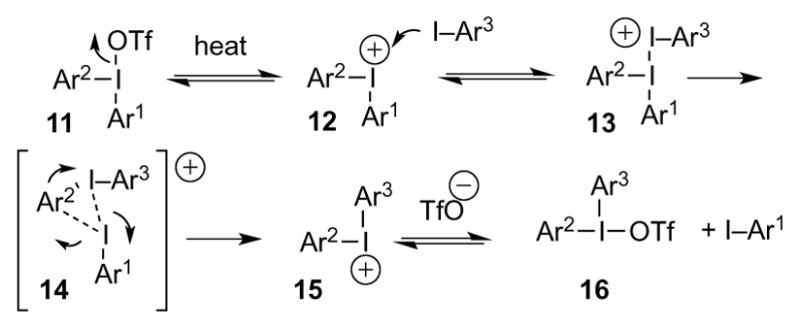
Mechanistic hypothesis for the iodonium metathesis reaction.
In summary, iodonium metathesis reactions are feasible. This engenders numerous opportunities that are actively being researched. Pertinent results will be disclosed in due time.
Supplementary Material
Acknowledgments
We thank the University of British Columbia, the Canada Research Chair Program, NSERC, CFI, and BCKDF for support. T. K. is a recipient of a UBC FYF Fellowship; L. R., of a Vanier Fellowship; D. P., of an Onassis Foundation Scholarship.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Wang B, Cerny RL, Uppaluri S, Kempinger JJ, DiMagno SG. J Fluorine Chem. 2010;131:1113. doi: 10.1016/j.jfluchem.2010.04.004. See also ref. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koser GF, Wettach RH. J Org Chem. 1980;45:1542. [Google Scholar]
- 3.Reviews: Yusubov MS, Maskaev AV, Zhdankin VV. ARKIVOC. 2011:370.Silva LF, Jr, Olofsson B. Nat Prod Rep. 2011;28:1722. doi: 10.1039/c1np00028d.Merritt EA, Olofsson B. Angew Chem Int Ed. 2009;48:9052. doi: 10.1002/anie.200904689.Angew Chem. 2009;121:9214.Recent example: Ghosh R, Olofsson B. Org Lett. 2014;16:1830. doi: 10.1021/ol500478t.
- 4.Ochiai M, Okubo T, Miyamoto K. J Am Chem Soc. 2011;133:3342. doi: 10.1021/ja200479p. [DOI] [PubMed] [Google Scholar]
- 5.In addition to refs 2–3 above, see: Zhdankin VV, Stang PJ. Chem Rev. 2002;102:2523. doi: 10.1021/cr010003+.Wirth T. Top Curr Chem. 2003;224:1.Zhdankin VV. Top Curr Chem. 2003;224:99.
- 6.Organoiodine compounds can function as nucleophilic catalysts: Albert BJ, Yamamoto H. Angew Chem, Int Ed. 2010;49:2747. doi: 10.1002/anie.200907076.Angew Chem. 2010;122:2807.Brady BP, Albert BJ, Akakura M, Yamamoto H. Chem Sci. 2013;4:3223., and refs. cited therein.
- 7.Malmgren J, Santoro S, Jalalian N, Himo F, Olofsson B. Chem Eur J. 2013;19:10334. doi: 10.1002/chem.201300860. See especially p. 10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The aryl iodide should melt below ca. 120 °C, in that most diaryliodonium triflates are stable at or below this temperature: Lancer KM, Wiegard GH. J Org Chem. 1976;41:3360.McEwen WE, DeMassa JW. Heteroatom Chem. 1996;7:349.
- 9.Two examples are provided as Supporting Information.
- 10.The diaryliodonium triflates employed in this study were prepared as per: Bielawski M, Zhu M, Olofsson B. Adv Synth Catal. 2007;349:2610., and recrystallized before use (See Supporting Information for details).
- 11.Beringer FM, Forgione PS, Yudis MD. Tetrahedron. 1960;8:49.Martin-Santamaria S, Carrol MA, Carroll CM, Carter CD, Pike VW, Rzepa HS, Widdowson DA. Chem Commun. 2000:649.Pinto de Magalhaes H, Lüthi HP, Togni A. Org Lett. 2012;14:3830. doi: 10.1021/ol3014039.. See also ref. 7.
- 12.Ref. 3 as well as: Deprez NR, Sanford MS. Inorg Chem. 2007;46:1924. doi: 10.1021/ic0620337.
- 13.Diaryliodonium species with two electron-rich aryls were less reactive. Thus, reaction of bis-(4-anisyl)iodonium triflate with 4-iodotoluene went to only 65% conversion after 16 h to give a 1.6:2.0:1.0 mixture of starting complex, 4-anisyl-4-tolyliodonium triflate, and bis-4-tolyliodonium triflate. Therefore, no further work was done with such electron-rich complexes.
- 14.Ross TL, Ermert J, Hocke C, Coenen HH. J Am Chem Soc. 2007;129:8018. doi: 10.1021/ja066850h.Similar results were previously reported in ref. 11b
- 15.The geometric mean better reflects the “true” value (the actual (+)-charge on the I-atom) toward which a set of numbers (calculated charges) tend: Fleming PJ, Wallace JJ. Commun ACM. 1986;29:218.
- 16.MNDO, MNDO-d, AM1, PM3 as provided in the Hyperchem® package.
- 17.This finds support in semiempirical calculations (Supporting Information), which, however, tend to be imprecise for hypervalent halogen compounds (e.g.: Stewart JJP. J Comput Chem. 1989;10:221.). Calculated values should be taken only as reflecting a trend.
- 18.The significance of this reaction derives from the fact that aryl(5-uridinyl)iodonium complexes are precursors of 5-18F-uridine, which is of considerable interest in PET imaging: ref. 14 as well as Yasubov MS, Svitich DV, Larkina MS, Zhdankin VV. ARKIVOC. 2013:364.
- 19.Iodouridine triacetate was computationally modeled with the structurally simpler 5-iodo-1-methyluracil.
- 20.Metathesis also failed with highly electron-rich aryl iodides such as 4-iodoveratrole, N-(4-iodophenyl)phthalimide, and N, N-dimethyl-4-iodo-aniline, which upon contact diaryliodonium complexes developed blue-green colors and yielded intractable mixtures of products, perhaps due to the occurrence of competing SET processes.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.