

Abstract
Objective
To investigate the association between levodopa‐induced dyskinesias and striatal cholinergic activity in patients with Parkinson's disease.
Methods
This study included 13 Parkinson's disease patients with peak‐of‐dose levodopa‐induced dyskinesias, 12 nondyskinetic patients, and 12 healthy controls. Participants underwent 5‐[123I]iodo‐3‐[2(S)‐2‐azetidinylmethoxy]pyridine single‐photon emission computed tomography, a marker of nicotinic acetylcholine receptors, [123I]N‐ω‐fluoropropyl‐2β‐carbomethoxy‐3β‐(4‐iodophenyl)nortropane single‐photon emission computed tomography, to measure dopamine reuptake transporter density and 2‐[18F]fluoro‐2‐deoxyglucose positron emission tomography to assess regional cerebral metabolic activity. Striatal binding potentials, uptake values at basal ganglia structures, and correlations with clinical variables were analyzed.
Results
Density of nicotinic acetylcholine receptors in the caudate nucleus of dyskinetic subjects was similar to that of healthy controls and significantly higher to that of nondyskinetic patients, in particular, contralaterally to the clinically most affected side.
Interpretation
Our findings support the hypothesis that the expression of dyskinesia may be related to cholinergic neuronal excitability in a dopaminergic‐depleted striatum. Cholinergic signaling would play a role in maintaining striatal dopaminergic responsiveness, possibly defining disease phenotype and progression.
Introduction
Levodopa is still the most effective drug in the management of Parkinson's disease (PD), but adverse events may appear along with disease progression. In particular, 40% of PD patients may develop enduring motor fluctuations1 such as levodopa‐induced dyskinesias (LIDs), which may be severely incapacitating. Young age at onset and high levodopa doses are known risk factors for its onset.2 At present, few therapeutic options exist for the treatment of LIDs.
The exact mechanisms underlying the development and maintenance of LIDs in vivo are not fully understood. Extensive evidence indicates that both pre‐ and postsynaptic components of the dopaminergic system contribute to the development of LIDs. Presynaptic mechanisms of particular relevance include nigrostriatal dopaminergic neuronal loss3 that results in decreased dopamine buffering capacity, and increased extracellular dopamine levels that arise with levodopa treatment.4, 5
Dopamine depletion and chronic levodopa create an imbalance in striatal neurotransmitter systems and glutamatergic, serotonergic, and peptidergic system changes have been noted in LIDs.6 Previous findings in animal models also support direct involvement of the nicotinic cholinergic system. In particular, elevated cholinergic signaling may contribute to motor complications arising from long‐term levodopa therapy in PD.7 Increasing evidence also suggests that nicotine and agonists of the nicotinic acetylcholine receptors (nAChRs) reduce LIDs via nAChRs desensitization.8
To further examine the role of striatal cholinergic and dopaminergic interplay in LIDs expression, as well as related brain glucose metabolic changes, we investigated PD patients with and without LIDs with three different radioligands, 5‐[123I]iodo‐3‐[2(S)‐2‐azetidinylmethoxy]pyridine (123I‐5‐IA) to determine nAChRs density, [123I]N‐ω‐fluoropropyl‐2β‐carbomethoxy‐3β‐(4‐iodophenyl)nortropane (123I‐FP‐CIT) to measure dopamine reuptake transporter (DAT) density, and 2‐[18F]fluoro‐2‐deoxyglucose (18F‐FDG) to assess regional cerebral metabolic activity.
Materials and Methods
Subjects
We enrolled consecutive patients with a diagnosis of PD according to the UK Parkinson Disease Brain Bank criteria. The PD dyskinesias group (PDSK) consisted of 13 patients with moderate to severe bilateral choreiform peak‐of‐dose LIDs, without off‐dyskinesias or biphasic dyskinesias, as repeatedly noted by investigators at clinical visits (>3 points at part IV of the Unified Parkinson's Disease Rating Scale (UPDRS), items 32 and 33). The PD control group (PD) consisted of 12 subjects with stable response to levodopa and who had never experienced LIDs despite four or more years of levodopa treatment.
Patients with significant comorbidity, previous history of other neurological conditions (e.g., stroke, head injury, epilepsy), with gait and balance problems, dementia, depression or sleep disorders (e.g., REM sleep behavior disorder) were not enrolled.
None of the patients were taking amantadine, antipsychotics, antidepressants, or other drugs possibly affecting the cholinergic systems (e.g., acetylcholinesterase inhibitor). Current smokers or patients with a history of smoking were excluded from this study.
Patients were assessed with the UPDRS, the Abnormal Involuntary Movement Scale (AIMS), the Mattis Dementia Rating Scale (MDRS), the Parkinson Neuropsychometric Dementia Assessment (PANDA), the Frontal Assessment Battery (FAB), and the Beck Depression Inventory (BDI). Neurological evaluations were performed about 2 h after the first daily dose of levodopa (100/25 mg of levodopa/benserazide), shortly before starting the cholinergic imaging study. We chose this protocol to investigate patients in similar clinical conditions and the PDSK subjects while presenting LIDs. All imaging studies, clinical and neuropsychological examinations were performed within 3 months.
Imaging procedures
All patients underwent a single‐photon emission computed tomography (SPECT) scan with 123I‐5‐IA to determine nAChRs density. SPECT scans were performed 240 min after an intravenous single bolus injection of 187.3 ± 7.2 MBq of 123I‐5‐IA.9, 10 These data were compared with a group of 12 healthy subjects (HC) age‐ (63.9 ± 11.7 years) and gender‐matched (5 males).11, 12 Nineteen patients (10 PDSK and 9 PD) also performed a second SPECT with 123I‐FP‐CIT to measure DAT density. 123I‐FP‐CIT scans were started 180 min after injection of 182.1 ± 3.9 MBq of 123I‐FP‐CIT.13 Both 40‐min scans were acquired on a dual‐headed integrated SPECT/CT system (Symbia T2; Siemens, Erlangen, Germany) with 60 projections of 40 sec each, photopeak window of 159 keV ± 15%, matrix 128 × 128, and zoom factor 1.23. Reconstruction was performed using OSEM 3D (8 subsets, 8 iterations, 8 mm Gaussian filtering) and CT‐based attenuation correction. PET with 18F‐FDG was performed in 18 subjects (nine PDSK and nine PD) to determine brain glucose metabolism. PET scans were obtained with a combined PET/CT scanner (Biograph mCT 64; Siemens Medical Solutions, Knoxville). All patients fasted overnight (>10 h) before injection and stayed in resting conditions for 30 min between the injection of 210.3 ± 24.4 MBq of 18F‐FDG and the scan. PET emission data were acquired in 3D mode for 10 min/one bed position using a 400×400 matrix with an axial resolution of 2 mm and an in‐plane resolution of 4.7 mm. PET data were reconstructed iteratively (24 subsets, 3 iterations, Gaussian filtering) using HD reconstruction mode and CT‐based attenuation correction.
Finally, we performed high‐resolution brain MRI scans (Siemens MAGNETOM Trio 3.0 T, 12 channels head coil, Erlangen, Germany) to exclude any structural brain pathologies and for anatomical co‐registration. The protocol included T2/PD‐weighted turbo spin echo, fluid‐attenuated inversion recovery, T1‐weighted gradient recalled echo and diffusion‐weighted imaging with apparent diffusion coefficient maps and an isotropic high‐resolution structural T1‐weighted MPRAGE‐Sequence (turbo‐FLASH (TFL) 1.0 × 1.0 × 1.0 mm³, TR: 2530 msec, TE: 3.37 msec, FA: 9°, TI: 1200 msec).
Image analysis
Volume‐of‐interest (VOI) analysis of the basal ganglia was performed with PMOD Version 3.6 (PMOD Technologies Ltd., Zurich, Switzerland). After partial volume correction, individual images were spatially normalized to tracer‐specific SPECT and PET brain templates9, 14, 15 and smoothed (8 mm full width half maximum). Each image was then delineated employing a whole brain VOI set with 67 predefined regions on the Automated Anatomical Labeling brain atlas.16 Average regional binding values were used to calculate regional estimates of the non‐displaceable binding potential (BP) for caudate nucleus and putamen, using as a reference the average global brain (for BP5‐IA)10, 17 and the average occipital lobe binding value (for BPFP‐CIT).13 Similarly, the standardized uptake value ratio (SUVRFDG) was calculated for each subject as the ratio of mean count per voxel of basal ganglia VOIs to mean count per voxel of the global18F‐FDG brain uptake. Additionally, for 123I‐FP‐CIT the asymmetry index (AI) and the putamen‐caudate ratio (PCIDX)18 were also calculated.
Statistical parametric mapping (SPM 8, Wellcome Department of Cognitive Neurology, University College, London) was additionally applied to compare the imaging data of 18F‐FDG and 123I‐5‐IA between PDSK and PD. 18F‐FDG PET images were co‐registered and spatially normalized to each individual MRI‐based template. After smoothing normalized images, significant differences in whole brain glucose metabolism were localized on a voxel‐by‐voxel basis applying the proportional global mean. PDSK and PD were compared using two‐sample t statistics covariated by age, disease duration, and levodopa equivalent daily dose (LEDD). The SPM {t} maps were obtained at a height threshold of P < 0.005 and an extent threshold of k ≥ 100 voxels. With regard to 123I‐5‐IA binding, spatial normalization was performed as described above (see VOI analysis). Then, the laterality of all 123I‐5‐IA datasets to the patient's side with more severe clinical motor symptoms was rearranged, so that the right side of the image referred to the patient's ipsilateral hemisphere. Voxel‐specific binding values (voxel/global cortical reference) were obtained using ImCalc in SPM. A two‐sample t‐test (PDSK vs. PD) was performed without global normalization or grand mean scaling, the masking threshold was set to zero. SPM {t} maps were computed with a height threshold of P < 0.005 and an extent threshold of k ≥ 100 voxels. SPM coordinates were finally converted into Talairach coordinates.19 .
Statistical analysis
We used nonparametric ANOVA (Kruskal–Wallis test) and Steel‐Dwass test to assess the differences in clinical and imaging measurements (BPFP‐CIT, BP5‐IA, and SUVRFDG) between groups and brain areas. Of note, BP5‐IA and SUVRFDG were compared after rearranging to the hemisphere with more and less severe clinical motor symptoms (i.e., lower or higher BPFP‐CIT). For HC, we used the mean value of left and right BP5‐IA. Fisher's exact test was used for categorical data. Correlations between BPs, SUVRFDG, and clinical data were analyzed with a multivariate analysis of variance and Spearman's rank correlation coefficient (Spearman's ρ) was used to measure dependencies between variables. We used JMP software version 12 (SAS Institute Inc., Car, North CA).
Standard protocol approvals, registration, and patient consents
The Institutional Review Board of the University Hospital Würzburg and the Governmental Radiation Protection Authority (Bundesamt für Strahlenschutz, Aktenzeichen: Z5‐22463/2‐2014‐022) approved this study. All patients gave written informed consent prior to participation in the study. Some of the scans have been performed as part of the clinical workup.
Results
Clinical and demographic characteristics
There were no significant differences in age and sex between HC, PD, and PDSK. Clinical severity and stage, LEDD, and neuropsychological evaluations were similar in PD and PDSK (Table 1).
Table 1.
Study population
| PD | PDSK | |
|---|---|---|
| Age (years) | 67.7 ± 7.4 | 63.4 ± 9.1 |
| Sex (F/M) | 2/10 | 5/8 |
| Age at onset (years) | 60.0 ± 7.9 | 53.9 ± 9.6 |
| Disease duration (years) | 7.9 ± 4.0 | 10.2 ± 4.1 |
| LEDD (mg/day) | 714.2 ± 458.5 | 900.1 ± 278.3 |
| UPDRS‐III (score: 0–108) | 21.8 ± 9.4 | 16.9 ± 9.2 |
| AIMS (score: 0–40) | – | 13.0 ± 6.4 |
| Hoehn and Yahr stage (1–5) | 2.4 ± 0.5 | 2.3 ± 0.8 |
| MDRS (score: 0–144) | 140.1 ± 3.0 | 138.0 ± 6.9 |
| PANDA (score: 0–30) | 24.2 ± 3.5 | 24.0 ± 4.8 |
| FAB (score: 0–18) | 15.2 ± 2.4 | 15.2 ± 2.5 |
| BDI (score: 0–63) | 5.9 ± 3.2 | 7.2 ± 3.7 |
Data are presented as mean ± SD.
PD, PD patients; PDSK, PD Patients with Dyskinesia; LEDD, Levodopa Equivalent Daily Dose; UPDRS, Unified Parkinson's Disease Rating Scale; AIMS, Abnormal Involuntary Movement Scale; MDRS, Mattis Dementia Rating Scale; PANDA, Parkinson Neuropsychometric Dementia Assessment; FAB, Frontal Assessment Battery; BDI, Beck Depression Inventory
Striatal DAT density
Striatal BPFP‐CIT did not differ between PDSK and PD (Table 2). In all patients, the putamen with lower BPFP‐CIT was contralateral to the clinically most affected side and showed a BPFP‐CIT significantly lower than the opposite one (P < 0.001). The AI did not differ between the two groups (AIPUTAMEN PDSK: 17.5 ± 17.3 and PD: 31.9 ± 25.0, P = 0.23; AICAUDATE PDSK: 12.8 ± 16.1 and PD: 21.8 ± 24.0, P = 0.39). Also the putamen‐caudate ratio did not differ between groups (PCIDXCONTRA PDSK: −0.15 ± 0.09 and PD: −0.13 ± 0.10, P = 1.00; PCIDXIPSI PDSK: −0.13 ± 0.07 and PD: −0.08 ± 0.07, P = 0.27).
Table 2.
Striatal dopamine reuptake transporters density and glucose metabolism
| PDIPSI | PDCONTRA | PDSKIPSI | PDSKCONTRA | |
|---|---|---|---|---|
| BPFP‐CIT Caudate | 1.80 ± 0.70 | 1.41 ± 0.50 | 1.83 ± 0.70 | 1.60 ± 0.57 |
| BPFP‐CIT Putamen | 1.54 ± 0.57 | 1.08 ± 0.36 | 1.43 ± 0.56 | 1.20 ± 0.54 |
| SUVRFDG Caudate | 0.60 ± 0.22 | 0.60 ± 0.20 | 0.72 ± 0.10 | 0.72 ± 0.10 |
| SUVRFDG Putamen | 1.16 ± 0.13 | 1.17 ± 0.15 | 1.15 ± 0.09 | 1.15 ± 0.09 |
Data are presented as mean ± SD.
PD, PD patients; PDSK, PD patients with dyskinesia; BP, binding potential; SUVR, standardized uptake value ratio.
Cerebral glucose metabolism
The SUVRFDG are listed in Table 2. SUVRFDG and SPM analysis did not show any differences in brain glucose metabolism in resting state between PDSK and PD.
nAChRs measurement
PDSK showed striatal BP5‐IA in the normal range and higher than nondyskinetic patients, in particular, contralaterally to the most affected side. Differences in BP5‐IA between PD and PDSK were significant also when weighting for demographic parameters (i.e., age and age at onset) as well as for disease duration and LEDD (Table 3). SPM analysis confirmed higher 123I‐5‐IA binding values in the ipsilateral and contralateral caudate nucleus of PDSK compared to PD (Figure 1 and Table 4).
Table 3.
nAChRs measurements
| PDIPSI | PDCONTRA | PDSKIPSI | PDSKCONTRA | HC | |
|---|---|---|---|---|---|
| BP5IA Caudate | 0.80 ± 0.29*# | 0.78 ± 0.22°§ | 1.00 ± 0.20# | 1.02 ± 0.16§ | 1.14 ± 0.10*° |
| BP5IA Putamen | 1.52 ± 0.14 | 1.47 ± 0.15 | 1.46 ± 0.23 | 1.45 ± 0.22 | 1.40 ± 0.06 |
| BP5IA Globus pallidus | 1.77 ± 0.18 | 1.73 ± 0.24 | 1.66 ± 0.40 | 1.71 ± 0.34 | 1.49 ± 0.09 |
| BP5IA Thalamus | 1.88 ± 0.38 | 1.83 ± 0.42 | 1.90 ± 0.50 | 2.06 ± 0.48 | 1.97 ± 0.08 |
Data are presented as mean (SD).
PD, PD patients; PDSK, PD patients with dyskinesia; BP, binding potential.
*and °P < 0.01 Steel‐Dwass all pairs, § P < 0.05 Steel‐Dwass all pairs, # P < 0.05 Wilcoxon each pair.
Figure 1.
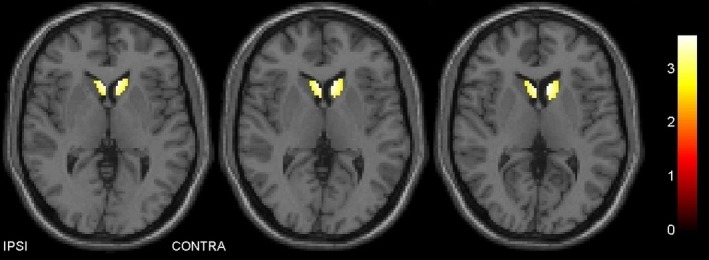
Voxelwise analysis of single‐photon emission computed tomography images with 123I‐5‐IA. In the caudate nucleus, dyskinetic patients show higher binding values than nondyskinetic PD patients. This difference is predominant in the hemisphere with lower dopamine transporter density (contralateral to the clinically most affected hemibody). The color bar on the right side indicates the corresponding t‐values.
Table 4.
Brain areas with significantly higher nAChRs density in dyskinetic versus nondyskinetic patients
| p FWE‐corr | k | Region | Coordinates (mm) | T | ||
|---|---|---|---|---|---|---|
| x | y | z | ||||
| 0.029 | 239 | Caudate body (contralateral) | −7 | 13 | 11 | 3.67 |
| 0.037 | 219 | Caudate body (ipsilateral) | 3 | 13 | 11 | 3.54 |
Regions and coordinates are in Talairach space.
In nondyskinetic patients only, the BP5‐IA of the ipsi‐ and the contralateral caudate nucleus negatively correlated with age (ipsilateral caudate ρ = −0.74, P = 0.006; contralateral caudate ρ = −0.67, P = 0.02) and age at onset (ipsilateral caudate ρ = −0.63, P = 0.03; contralateral caudate ρ = −0.61, P = 0.03) and positively correlated with SUVRFDG (ipsilateral caudate ρ = 0.92, P < 0.001; contralateral caudate ρ = 0.91, P < 0.001).
Discussion
This is the first in vivo study to investigate cholinergic activity in patients with Parkinson's disease with and without LIDs.
Neuronal nAChRs are pentameric ligand‐gated ion channels composed of α‐subunits (homomeric receptors) or of α‐ (α2 to α7) and β‐subunits (β2 to β4) (heteromeric receptors). The α4β2*‐nAChRs are the prominent subtype in the human brain. The striatum receives dense cholinergic innervation, and although striatal cholinergic interneurons are few in number, they have widespread axonal arborizations, which can directly influence motor behavior.20, 21 Among the validated ligands of nicotinic receptors, 123I‐5‐IA shows a high affinity for the β2 subunits22, thus providing in our case, in vivo evidence of cholinergic activity. Of note, in contrast to the general rule that chronic exposure to agonists (or a condition that increases the natural transmitter) results in downregulation of the target receptor, acetylcholine, nicotine, and other agonists (e.g., epibatidine) elicit an increase in the density of nAChRs.23
We found a normal density of nAChRs in the striatum of PDSK, which was significantly higher than nondyskinetic patients, especially in the caudate nucleus of the most dopamine‐depleted hemisphere, contralateral to the clinically most affected side. This finding can be explained by an overall preserved cholinergic activity in the striatum of dyskinetic PD patients or by a maladaptive sprouting (regenerative overgrowth) of cholinergic axon terminals along with disease progression.24
Dopamine and acetylcholine are proposed to have opposing functions, so that there is a relative increase in striatal cholinergic activity in the face of a deficit in dopamine.25 Acetylcholine directly promotes dopamine release via activation of presynaptic nAChRs at nigrostriatal terminals,26 dopamine acting via D2 receptors inhibits the release of acetylcholine from the cholinergic interneurons.27 However, dopamine, acting via D1/D5 receptors, potentiates the excitability of cholinergic neurons and their responsiveness to glutamatergic input.28 It has been also shown that the release of endogenous acetylcholine is sufficient to promote dopamine release via nAChR activity without activation of dopaminergic neurons.29 Therefore, presynaptic nAChRs on dopamine terminals appear to have different modulatory influences on dopamine release, one via activity of dopamine neurons and the other via thalamo‐striatal glutamate activity through cholinergic interneurons independent of the actual dopamine neuron activity.26, 28 Increased glutamatergic transmission has been shown in the striatum of dyskinetic PD patients following levodopa administration.30 Results of animal model studies also indicated that increased striatal glutamatergic activity is implicated in the development and maintenance of LIDs.31, 32 Recent studies showed that nAChR agonists (e.g., nicotine) attenuate LIDs, especially when striatal dopamine transmission is relatively intact.10, 33 This effect is believed to occur through desensitization of nAChRs, with a consequent decline in nAChR‐mediated function,34 and release of glutamate from cortical and thalamic afferent.35, 36 Notably, in comparison to the putamen, the dopaminergic innervation of the caudate nucleus is partially preserved in mild‐moderate subjects with PD, as in our study.
The importance of nondopaminergic transmitter systems that interact with dopaminergic projection in PD is apparent, and relevant to pharmacologic interventions directed not only toward acetylcholine, but also glutamate as well as serotonin, noradrenaline, adenosine, and cannabinoids for potential antidyskinetic action.25, 37 The serotonin (5‐HT) system has received much attention as 5‐HT fibers can convert levodopa into dopamine without the regulatory mechanisms required for dopaminergic transmission, resulting in dysregulated, nonphysiologic dopamine release, which may underlie LIDs.38, 39 Acetylcholine, nicotine, and nicotinic receptor agonists (i.e., epibatidine) can significantly increase 5‐HT release from striatal synaptosomes, an effect that was blocked by mecamylamine, a noncompetitive nicotinic receptor antagonist.40 In turn, 5‐HT tonically inhibits acetylcholine release from striatal cholinergic neurons by acting on a presynaptic receptor localized on cholinergic terminals.41 In vitro experiments have also demonstrated that the activation of α4β2 nAChRs by acetylcholine or nicotine provokes a long‐lasting enhancement of the glutamatergic input to 5‐HT neurons.42
We advance the hypothesis that, in predisposed PD patients, the cholinergic system might be involved in inducing serotonergic neurons toward a dopamine‐dependent activity. Such an acetylcholine‐dependent dopamine release from serotonin neurons would also explain the selective finding of higher nAChRs in PDSK than nondyskinetic patients in the caudate nucleus. Indeed, in PD the caudate nucleus presents the combination of mild to moderate dopamine loss and scarce serotonergic fibers. Postmortem as well as recent in vivo molecular imaging studies have suggested a more marked serotonin deficiency in the caudate nucleus than in the putamen.38, 39, 43 A sustained cholinergic signaling in the caudate nucleus would therefore be necessary to maintain a levodopa‐derived dopamine release from the remaining striatal serotonin neurons.
An alternative explanation for such a predominant cholinergic activity in the caudate nucleus could imply a specific role of this region in the onset of dyskinesias. Experimental findings in the rat show that a serotonin loss of 20–50% in the caudate nucleus is associated with abnormal behavior (increased spontaneous locomotor activity).44
Besides the serotonin system, a vast pharmacological literature points also to a causal involvement of the noradrenaline system in motor complications of PD therapy5, and a compensatory effect of the cholinergic system could be also confined within the catecholaminergic pathways.45, 46, 47
In agreement with previous findings,48 DAT binding measurements did not differ between PDSK and PD, thus further supporting that LIDs expression does not rely on the dopaminergic tone per se, but rather on compensatory attempts and derangements of different aminergic pathways and possibly on a disproportion in their degeneration.5, 38, 39 This finding, together with the usual young disease onset of dyskinetic patients2, might support the notion of a neuroprotective effect of the cholinergic system on nigrostriatal dopamine‐containing neurons.49, 50, 51 In this study, we were not able to properly address a putative neuroprotective effect of the cholinergic system on nigrostriatal dopamine‐containing neurons and disease progression. To this end, besides a clinical evaluation of medication, multiple FP‐CIT scans would have been required as DAT binding is influenced by age and do not follow a linear decline along with disease progression.52, 53
In this study, we also found a comparable resting state metabolism between PDSK and PD. However, PET measurements might have not captured subtle metabolic abnormalities or relative metabolic changes in small striatal areas. Still, in the caudate nucleus of nondyskinetic patients, we described a positive correlation between BP5IA and SUVRFDG, thus suggesting that within the caudate nucleus, cholinergic activity might enhance resting state metabolism. With regard to dyskinetic patients, a ceiling effect could be envisioned.
This study has several limitations. In particular, the lack of arterial input function prevented a quantitative assessment of α4β2*‐nAChRs. Patients were investigated only while on medication, but we were mostly interested in the cholinergic status in the dyskinetic state. Furthermore, none of the dyskinetic patients would have tolerated a prolonged suspension of all dopaminergic drugs.
In conclusion, this study supports the hypothesis that the expression of dyskinesias may result from a sustained cholinergic neuronal excitability in a dopaminergic‐depleted striatum. Further in vivo investigation into the role of acetylcholine, including different subtypes of receptors,50 is crucial for future development of new strategies for the pharmacological treatment of dyskinesias in patients with Parkinson's disease.
Author Contribution
The concept of the study was designed by IUI, KH, AKB, JV, and SS. Fundings were obtained by IUI and KH. EAM and SS took part in the development of the radioligands. The acquisition of the data was supervised by JB, GAH, KPC, CVD, and IUI. JB, SK, GM, GP, and IUI analyzed and interpretated the datasets. Statistical analysis was performed by JB, GM, and IUI. All authors contributed to drafting and critically revising the manuscript.
Conflicts of Interest
JV received grants and personal fees from Medtronic Inc., grants and personal fees from Boston Scientific, and personal fees from St. Jude, outside the submitted work. IUI received grants and personal fees from Medtronic Inc. outside the submitted work.
Acknowledgments
We are thankful to Drs. Stephan Klebe and Frank Steigerwald for patient referral. The study was sponsored by the Michael J. Fox Foundation for Parkinson Research, the Interdisziplinäres Zentrum für Klinische Forschung (IZKF) of the University Hospital Würzburg and the Fondazione Grigioni per il Morbo di Parkinson.
Funding Statement
This work was funded by Michael J. Fox Foundation grant ; The Interdisziplinäres Zentrum für Klinische Forschung (IZKF) of the University Hospital Würzburg Wuerzburg grant ; Fondazione Grigioni per il Morbo di Parkinson grant .
References
- 1. Ahlskog JE, Muenter MD. Frequency of levodopa‐related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001;16:448–458. [DOI] [PubMed] [Google Scholar]
- 2. Cilia R, Akpalu A, Sarfo FS, et al. The modern pre‐levodopa era of Parkinson's disease: insights into motor complications from sub‐Saharan Africa. Brain 2014;137:2731–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hong JY, Oh JS, Lee I, et al. Presynaptic dopamine depletion predicts levodopa‐induced dyskinesia in de novo Parkinson disease. Neurology 2014;82:1597–1604. [DOI] [PubMed] [Google Scholar]
- 4. Storch A, Wolz M, Beuthien‐Baumann B, et al. Effects of dopaminergic treatment on striatal dopamine turnover in de novo Parkinson disease. Neurology 2013;80:1754–1761. [DOI] [PubMed] [Google Scholar]
- 5. Cenci MA. Presynaptic mechanisms of L‐DOPA‐induced dyskinesia: the findings, the debate, and the therapeutic implications. Front Neurol 2014;5:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hout P, Johnstan TH, Koprich JB, et al. The pharmacology of L‐DOPA‐induced dyskinesia in Parkinson's disease. Pharmacol Rev 2013;65:171–222. [DOI] [PubMed] [Google Scholar]
- 7. Ding Y, Won L, Britt JP, et al. Enhanced striatal cholinergic neuronal activity mediates L‐DOPA‐induced dyskinesia in parkinsonian mice. Proc Natl Acad Sci USA 2011;108:840–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bordia T, Campos C, McIntosh MJ, Quik M. Nicotinic receptor‐mediated reduction in L‐DOPA‐induced dyskinesias may occur via desensitization. J Pharmacol Exp Ther 2010;333:929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oishi N, Hashikawa K, Yashida H, et al. Quantification of nicotinic acetylcholine receptors in Parkinson's disease with 123I‐5IA SPECT. J Neurol Sci 2007;256:52–60. [DOI] [PubMed] [Google Scholar]
- 10. Isaias IU, Spiegel J, Brumberg J, et al. Nicotinic acetylcholine receptor density in cognitively intact subjects at an early stage of Parkinson's disease. Front Aging Neurosci 2014;6:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cosgrove KP, Esterlis I, McKee SA, et al. Sex differences in availability of β 2*‐nicotinic acetylcholine receptors in recently abstinent tobacco smokers. Arch Gen Psychiatry 2012;69:418–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mitsis EM, Cosgrove KP, Staley JK, et al. Age‐related decline in nicotinic receptor availability with [(123)I]5‐IA‐85380 SPECT. Neurobiol Aging 2009;30:1490–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Isaias IU, Benti R, Cilia R, et al. [123I]FP‐CIT striatal binding in early Parkinson's disease patients with tremor vs. akinetic‐rigid onset. NeuroReport 2007;18:1499–1502. [DOI] [PubMed] [Google Scholar]
- 14. Isaias IU, Marotta G, Hirano S, et al. Imaging essential tremor. Mov Disord 2010;25:679–686. [DOI] [PubMed] [Google Scholar]
- 15. Della Rosa PA, Cerami C, Gallivanone F, et al. A standardized [18F]‐FDG‐PET template for spatial normalization in statistical parametric mapping of dementia. Neuroinformatics 2014;12:575–593. [DOI] [PubMed] [Google Scholar]
- 16. Tzourio‐Mazoyer N, Landeau B, Papathanassiou D, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single‐subject brain. NeuroImage 2002;15:273–289. [DOI] [PubMed] [Google Scholar]
- 17. Terrière E, Dempsey MF, Herrmann LL, et al. 5‐(123)I‐A‐85380 binding to the α4ß2‐nicotinic receptor in mild cognitive impairment. Neurobiol Aging 2010;31:1885–1893. [DOI] [PubMed] [Google Scholar]
- 18. Isaias IU, Canesi M, Benti R, et al. Striatal dopamine transporter abnormalities in patients with essential tremor. Nucl Med Commun 2008;29:349–353. [DOI] [PubMed] [Google Scholar]
- 19. Lancaster JL, Tordesillas‐Gutiérrez D, Martinez M, et al. Bias between MNI and Talairach coordinates analyzed using the ICBM‐152 brain template. Hum Brain Mapp 2007;28:1194–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Calabresi P, Picconi B, Parnetti L, Di Filippo M. A convergent model for cognitive dysfunctions in Parkinson's disease: the critical dopamine‐acetylcholine synaptic balance. Lancet Neurol 2006;5:974–983. [DOI] [PubMed] [Google Scholar]
- 21. Bonsi P, Cuomo D, Martella G, et al. Centrality of striatal cholinergic transmission in basal ganglia function. Front Neuroanat 2011;5:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fujita M, Ichise M, van Dyck CH, et al. Quantification of nicotinic acetylcholine receptors in human brain using [123I]5‐I‐A‐85380 SPET. Eur J Nucl Med Mol Imaging 2003;30:1620–1629. [DOI] [PubMed] [Google Scholar]
- 23. Govind AP, Vezina P, Green WN. Nicotine‐induced upregulation of nicotinic receptors: underlying mechanisms and relevance to nicotine addiction. Biochem Pharmacol 2009;78:756–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Spehlmann R, Stahl SM. Dopamine acetylcholine imbalance in Parkinson's disease: possible regenerative overgrowth of cholinergic axon terminals. Lancet 1976;1:724–726. [DOI] [PubMed] [Google Scholar]
- 25. Pisani A, Bernardi G, Ding J, Surmeier DJ. Re‐emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci 2007;30:545–553. [DOI] [PubMed] [Google Scholar]
- 26. Ding JB, Guzman JN, Peterson JD, et al. Thalamic gating of corticostriatal signaling by cholinergic interneurons. Neuron 2010;67:294–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yan Z, Song WJ, Surmeier J. D2 dopamine receptors reduce N‐type Ca2+ currents in rat neostriatal cholinergic interneurons through a membrane‐delimited, protein‐kinase‐C‐insensitive pathway. J Neurophysiol 1997;77:1003–1015. [DOI] [PubMed] [Google Scholar]
- 28. Aosaki T, Miura M, Suzuki T, et al. Acetylcholine–dopamine balance hypothesis in the striatum: an update. Geriatr Gerontol Int 2010;10(Suppl 1):148–157. [DOI] [PubMed] [Google Scholar]
- 29. Threlfell S, Lalic T, Platt NJ, et al. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron 2012;75:58–64. [DOI] [PubMed] [Google Scholar]
- 30. Ahmed I, Bose SK, Pavese N, et al. Glutamate NMDA receptor dysregulation in Parkinson's disease with dyskinesias. Brain 2011;134:979–986. [DOI] [PubMed] [Google Scholar]
- 31. Chase TN, Oh JD, Konitsiotis S. Antiparkinsonian and antidyskinetic activity of drugs targeting central glutamatergic mechanisms. J Neurol 2000;247(Suppl 2):36–42. [DOI] [PubMed] [Google Scholar]
- 32. Calon F, Morissette M, Ghribi O, et al. Alteration of glutamate receptors in the striatum of dyskinetic 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine‐treated monkeys following dopamine agonist treatment. Prog Neuropsychopharmacol Biol Psychiatry 2002;26:127–138. [DOI] [PubMed] [Google Scholar]
- 33. Quik M, Mallela A, Chin M, et al. Nicotine‐mediated improvement in L‐dopa‐induced dyskinesias in MPTP‐lesioned monkeys is dependent on dopamine nerve terminal function. Neurobiol Dis 2013;50:30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Piciotto MR, Addy NA, Mineur YS, Brunzell DH. It is not ‘‘either/or’’: activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol 2008;84:329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ding J, Peterson JD, Surmeier DJ. Corticostriatal and thalamostriatal synapses have distinctive properties. J Neurosci 2008;28:6483–6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Parikh V, Ji J, Decker MW, Sarter M. Prefrontal beta2 subunit‐containing and alpha7 nicotinic acetylcholine receptors differentially control glutamatergic and cholinergic signaling. J Neurosci 2010;30:3518–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Buck K, Ferger B. L‐DOPA‐induced dyskinesia in Parkinson's disease: a drug discovery perspective. Drug Discov Today 2010;15:867–875. [DOI] [PubMed] [Google Scholar]
- 38. Lee JY, Seo S, Lee JS, et al. Putaminal serotonergic innervation: monitoring dyskinesia risk in Parkinson disease. Neurology 2015;85:853–860. [DOI] [PubMed] [Google Scholar]
- 39. Roussakis AA, Politis M, Towey D, Piccini P. Serotonin‐to‐dopamine transporter ratios in Parkinson disease: relevance for dyskinesias. Neurology 2016;86:1152–1158. [DOI] [PubMed] [Google Scholar]
- 40. Reuben M, Clarke PB. Nicotine‐evoked [3H]5‐hydroxytryptamine release from rat striatal synaptosomes. Neuropharmacology 2000;39:290–299. [DOI] [PubMed] [Google Scholar]
- 41. Gillet G, Ammor S, Fillion G. Serotonin inhibits acetylcholine release from rat striatum slices: evidence for a presynaptic receptor‐mediated effect. J Neurochem 1985;45:1687–1691. [DOI] [PubMed] [Google Scholar]
- 42. Garduno J, Galindo‐Charles L, Jiménez‐Rodríguez J, et al. Presynaptic α4β2 nicotinic acetylcholine receptors increase glutamate release and serotonin neuron excitability in the dorsal raphe nucleus. J Neurosci 2012;32:15148–15157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kish S, Tong J, Hornykiewicz O, et al. Preferential loss of serotonin markers in caudate versus putamen in Parkinson's disease. Brain 2008;131:120–131. [DOI] [PubMed] [Google Scholar]
- 44. Carter CJ, Pycock CJ. The effects of 5,7‐dihydroxytryptamine lesions of extrapyramidal and mesolimbic sites on spontaneous motor behaviour, and amphetamine‐induced stereotypy. Naunyn Schmiedebergs Arch Pharmacol 1979;308:51–54. [DOI] [PubMed] [Google Scholar]
- 45. Kubo T, Amano H, Kurahashi K, Misu Y. Nicotine‐induced regional changes in brain noradrenaline and dopamine turnover in rats. J Pharmacobiodyn 1989. Feb;12:107–112. [DOI] [PubMed] [Google Scholar]
- 46. Rao TS, Correa LD, Adams P, et al. Pharmacological characterization of dopamine, norepinephrine and serotonin release in the rat prefrontal cortex by neuronal nicotinic acetylcholine receptor agonists. Brain Res 2003;990:203–208. [DOI] [PubMed] [Google Scholar]
- 47. Isaias IU, Marotta G, Pezzoli G, et al. Enhanced catecholamine transporter binding in the locus coeruleus of patients with early Parkinson disease. BMC Neurol 2011;11:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Linazasoro G, Van Blercom N, Bergaretxe A, et al. Ruiz Ortega JA. Levodopa‐induced dyskinesias in Parkinson disease are independent of the extent of striatal dopaminergic denervation: a pharmacological and SPECT study. Clin Neuropharmacol 2009;32:326–329. [DOI] [PubMed] [Google Scholar]
- 49. Quik M, O'Neill M, Perez XA. Nicotine neuroprotection against nigrostriatal damage: importance of the animal model. Trends Pharmacol Sci 2007;28:229–235. [DOI] [PubMed] [Google Scholar]
- 50. Srinivasan R, Henderson BJ, Lester HA, Richards CI. Pharmacological chaperoning of nAChRs: a therapeutic target for Parkinson's disease. Pharmacol Res 2014;83:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Egea J, Buendia I, Parada E, et al. Anti‐inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol 2015;97:463–472. [DOI] [PubMed] [Google Scholar]
- 52. Schwarz J, Storch A, Koch W, et al. Loss of dopamine transporter binding in Parkinson's disease follows a single exponential rather than linear decline. J Nucl Med 2004;45:1694–1697. [PubMed] [Google Scholar]
- 53. de la Fuente‐Fernández R, Schulzer M Kuramoto L, et al.Age‐specific progression of nigrostriatal dysfunction in Parkinson's disease. Ann Neurol 2011;69:803–810. [DOI] [PubMed] [Google Scholar]