

Abstract
N-Terminal self-cleavage (autoprocessing) of the HIV-1 protease precursor is crucial for liberating the active dimer. Under drug pressure, evolving mutations are predicted to modulate autoprocessing, and the reduced catalytic activity of the mature protease (PR) is likely compensated by enhanced conformational/dimer stability and reduced susceptibility to self-degradation (autoproteolysis). One such highly evolved, multidrug resistant protease, PR20, bears 19 mutations contiguous to sites of autoproteolysis in retroviral proteases, namely clusters 1–3 comprising residues 30–37, 60–67, and 88–95, respectively, accounting for 11 of the 19 mutations. By systematically replacing corresponding clusters in PR with those of PR20, and vice versa, we assess their influence on the properties mentioned above and observe no strict correlation. A 10–35-fold decrease in the cleavage efficiency of peptide substrates by PR20, relative to PR, is reflected by an only ~4-fold decrease in the rate of Gag processing with no change in cleavage order. Importantly, optimal N-terminal autoprocessing requires all 19 PR20 mutations as evaluated in vitro using the model precursor TFR-PR20 in which PR is flanked by the transframe region. Substituting PR20 cluster 3 into TFR-PR (TFR-PRPR20-3) requires the presence of PR20 cluster 1 and/or 2 for autoprocessing. In accordance, substituting PR clusters 1 and 2 into TFR-PR20 affects the rate of autoprocessing more drastically (>300-fold) compared to that of TFR-PR™20-3 because of the cumulative effect of eight noncluster mutations present in TFR-PR20PR-12. Overall, these studies imply that drug resistance involves a complex synchronized selection of mutations modulating all of the properties mentioned above governing PR regulation and function.
Graphical abstract
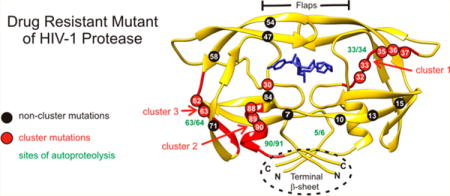
The human immunodeficiency virus type 1 protease (HIV-1 PR) mediates the processing of the Gag and Gag-Pol polyproteins into mature structural and functional proteins essential for assembly and maturation into infective progeny virus.1–4 The HIV-1 genome encodes a single copy of the 99-amino acid protease. PR is translated in the pol open-reading frame and catalyzes its own release at its termini (autoprocessing) from the Gag-Pol polyprotein via transient dimerization (Figure 1A).1–3 Each subunit of the dimer contributes one of the two active-site Asp25 residues. Cleavage between the transframe region (TFR) and N-terminus of PR (TFR/PR site) is concomitant with the appearance of mature-like catalytic activity and stable dimer formation (Kdimer < 10 nM) and precedes the cleavage at the C-terminus of the protease.2 These results are consistent with studies showing that HIV-1 particles from four different strains obtained from different cell lines contain only the 11 kDa mature protease and no TFR-PR precursor.5 Further, a mutation at the TFR/PR cleavage site leads to the production of an N-terminally extended 17 kDa protease species, resulting in a severe defect in Gag polyprotein processing and a complete loss of viral infectivity.5
Figure 1.
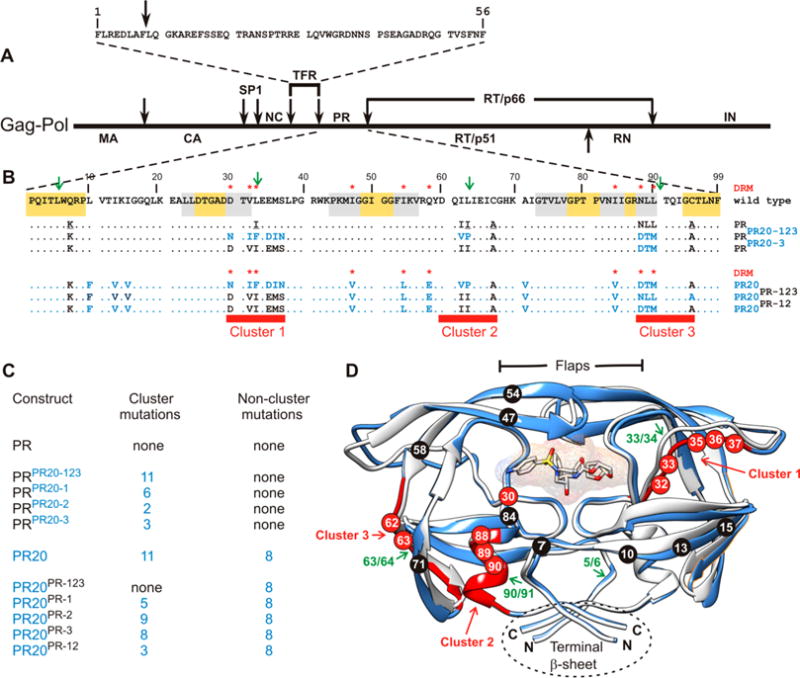
(A) Domain organization of the HIV-1 Gag-Pol polyprotein. Abbreviations: MA, matrix; CA, capsid; SP1, spacer peptide 1; NC, nucleocapsid; TFR, transframe region; PR, protease; RT, reverse transcriptase; RN, ribonuclease; IN, integrase. (B) Sequence alignment of wild-type protease, pseudo-wild-type protease (PR), and the multidrug resistant variant PR20. PR bears mutations Q7K, L33I, L63I, C66A, and C95A (colored black and underlined) to restrict autoproteolysis at sites indicated by green arrows and avoid cysteine-thiol oxidation. Introduced mutations common to PR and PR20 are Q7K, C67A, and C95A. The 19 mutations, both naturally occurring and selected under drug pressure, in PR20 are colored blue. Dots denote identical residues. Highly conserved regions in PR52 are highlighted in yellow, and regions where major drug resistance mutations (indicted by red asterisks; http:/hivdb.stanford.edu/cgi-bin/PIResiNote.cgi and ref31) occur are highlighted in gray. The three clusters that contain mutations in PR20 next to sites of autoproteolysis in the wild type are indicated by red stripes. (C) List of PR, PR20, and derived mutants showing the number of cluster and noncluster mutations. (D) Superimposition of ribbon representations of PR20 in blue (PDB entry 3UCB) and PR in white (PDB entry 2IEN) bound to the inhibitor darunavir (stick representation) with the regions corresponding to the three clusters colored red as well as indicated by the red arrows corresponding to red stripes in panel B. Positions of mutations in cluster 1 (residues 30–37), cluster 2 (residues 60–67), and cluster 3 (residues 88–94) are labeled in white on red circles. The remaining eight of 19 mutations in PR20 are shown in white on black circles. Sites of autoproteolysis are denoted by the green arrows.
The active site of the mature PR dimer has been the subject of intervention strategies for almost 25 years since its discovery, and clinical protease inhibitors form a crucial part of combination antiretroviral therapy.6 However, the short replication cycle (1.2 days7) of HIV-1, together with the error-prone reverse transcriptase, contributes to the rapidly evolving selection of mutations in PR leading to drug resistance. Major and accessory mutations involving 37 of the 99 PR residues contribute resistance to clinical protease inhibitors in current use by several mechanisms.8,9 Selection of primary drug resistance mutations under drug pressure can result in structural changes that may compromise autoprocessing and the catalytic efficiency of the released mature PR.1,2,4 These adverse effects are likely to be offset by accessory mutations,10,11 which do not directly affect interactions with protease inhibitors but restore conformational/dimer stability and catalytic function. For example, drug resistance mutation I84V in mature PR decreases the melting temperature (Tm), a measure of the stability of the tertiary fold, by ~2.8 °C. Mutations L10I and A71V individually restore the Tm of the I84V mutant to that of the wild type, and the L63P/I84V mutant exhibits a Tm slightly higher than that of the wild type.12 Combinations of drug resistance mutations with side-chain positions in the mature enzyme that are relatively close spatially, such as the frequently occurring pair D30N and N88D, may provide stabilization through polar or electrostatic interactions.13 The general consensus points to evolving resistance mutations proximal and remote from the active site and flaps that act synergistically by influencing the structure and dynamics of the protease and its affinity for clinical inhibitors.14–16 Co-evolving mutations under drug pressure spanning protease cleavage sites of the Gag have also been reported to offset the reduced catalytic activity of drug resistant mutants and contribute to fitness recovery.17–19 Statistical methods have been developed to identify selection pressure by protease inhibitors for specific mutations in PR, and for evaluating the effect of mutations at one site on selection of mutations at other sites.20,21 In addition, exponentially growing databases in recent years have led to the development of machine learning and computational methods for the evaluation and optimization of combination antiretroviral therapy, with respect to virological outcomes in HIV-1-infected patients.22 However, to the best of our knowledge, molecular mechanisms of drug resistance pertaining to autoprocessing, a pivotal step in PR regulation, have not been thoroughly explored.
We recently characterized an extreme multidrug resistant clinical isolate of PR, termed PR20,23–25 bearing 19 mutations. Relative to PR, mature PR20 has a similar catalytic constant (kcat), an ~13-fold higher Km for the hydrolysis of a peptide substrate, and an affinity for the clinical protease inhibitors darunavir and saquinavir decreased by >3 orders of magnitude. Even though the mature PR20 dissociates (Kdimer ~ 30 nM) more readily than PR, the Tm of PR20 is 6 °C higher than that of PR. A crystal structure of PR20 with water-coordinated yttrium in the active site showed that PR20 has an expanded active site and wide-open flaps, instead of the usual closed conformation observed for PR.25 In spite of these differences, the precursor form of this variant, TFR-PR20, undergoes efficient autoprocessing24 even in the presence of >150 μM darunavir or saquinavir, which otherwise completely inhibits the autoprocessing of the TFR-PR precursor at concentrations of 1–3 μM. Another characteristic feature of PR20 is its resistance to self-degradation (autoproteolysis). We proposed that mutations at or near these sites, which significantly limit autoproteolysis, can prolong the lifetime of mature PR20, thereby compensating for its reduced catalytic activity during the viral replication cycle.26
In earlier studies, we had shown that inhibition of autoprocessing of TFR-PR20 by darunavir or saquinavir could not be restored by substituting either one or two specific drug resistance mutations at various positions by the corresponding wild-type residue(s).24 As PR20 represents a highly evolved drug resistant mutant, it provides a template for investigating the interrelationship of groups of contiguous mutations in different regions accounting for properties of the mature PR as well as autoprocessing of its precursor. Major drug resistance mutations in PR occur (Figure 1B, gray highlight) between regions of natural variation (no highlight) and a high degree of conservation (yellow highlight) that are indispensable for optimal folding and catalytic activity. Two clusters of mutations in PR20 flank these regions. Cluster 1 is adjacent to the active site, and cluster 3 is located in the single α-helix following highly conserved residues G86 and R87 (Figure 1B,C).2,27 Specific residues (D29 and R87) contiguous to these two clusters, together with R8, stabilize inter- and intrasubunit contacts as well as the C-terminal β-sheet interface, critical for dimer formation (see Figure 1C).28 Surface-exposed residues in cluster 2 may influence folding due to the L63P substitution that coselects with the I84V drug resistance mutation, leading to enhanced conformational stability.12 Coincidentally, each of these clusters spans a site of autoproteolysis in PR (green arrows in Figure 1B,D), such that 11 of the 19 mutations in PR20 occur vicinal to these sites, cluster 3 site being the least susceptible to autoproteolysis in HIV-1 group M PR as compared to proteases from group N29 and HIV-2.30 Interestingly, no major drug resistance mutations in PR20 are contiguous to the major site of autoproteolysis in PR between L5 and W6. Four individual substitutions, D30N, V32I, and L33F/I/V in cluster 1 and N88D/S/T/G and L90M in cluster 3, are known drug resistance mutations (http:/hivdb.stanford.edu/cgi-bin/PIResiNote.cgi and ref31).
Here we address the effects of clustered drug resistance mutations on a comprehensive set of properties, for the first time, such as the dimer dissociation constant (Kdimer), kinetic parameters (kcat/Km and Ki), thermal stability in the absence and presence of protease inhibitor, Gag processing, autoproteolysis, and autoprocessing governing PR function. Initially, we substituted all PR20 clusters (or cassettes) into PR and its precursor mimetic and, conversely, all PR clusters into PR20 and its corresponding precursor and examined the effect of these mutations on the mature protease. Kinetic parameters of PR-mediated hydrolysis of synthetic substrates were compared with the rate and order of Gag processing. Subsequently, constructs bearing either single or dual cassette substitutions were examined to further elucidate the role of cooperativity among the clusters in promoting autoprocessing and to determine whether the rate of autoprocessing correlates with specific properties of the corresponding mature proteases.
MATERIALS AND METHODS
Protease Constructs
Wild-type, pseudo-wild-type (PR2,32), and PR20 sequences are shown in Figure 1. Precursor constructs with the N-terminal transframe region (TFR, 56 residues) are designated as TFR-PR and TFR-PR20 (Figure 1). The TFR sequence is identical in both constructs. Constructs modified by substitution of all PR20 clusters into the PR template (Figure 1B,C) are termed PRPR20-123 and TFR-PRPR20-123. When substituted with only one or two clusters, they are denoted with the corresponding cluster designation. Constructs bearing PR clusters in the PR20 template are designated similarly.
Expression and Purification
Previously reported constructs used in this study are PR,32 PR20,24,25 TFR-PR,32 and TFR-PR20.24 The rest of the constructs were used for the first time. Inserts encoding some mutant precursors as well as the corresponding mature proteases were custom synthesized (DNA 2.0, Menlo Park, CA), cloned either in the pJ414 vector or in the pET11a vector flanked by NdeI and BamHI sites, and transformed into Escherichia coli BL-21(DE3). Substitution mutations in some constructs were introduced by QuikChange mutagenesis (Agilent Technologies, Santa Clara, CA). Cells bearing the appropriate plasmid were grown at 37 °C in Luria-Bertani medium. Total cell extracts were mixed with sample loading buffer and subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) on 20% homogeneous PhastGels (GE Healthcare, Piscataway, NJ). Induction for protein expression, isolation of inclusion bodies, and protein purification followed previously described protocols.29,30 The composition of purified proteins was verified by electrospray ionization mass spectrometry (ESI-MS). Uniformly 15N-labeled or 15N- and 13C-labeled mature PRPR20-3 and PR20PR-12 were prepared by growing cultures in M9 minimal medium containing 15NH4Cl and [13C6]glucose as the sole nitrogen and carbon sources, respectively.
Time Course of Autoproteolysis
PRPR20-123 and PR20PR-123 were folded at room temperature by the quench protocol as described previously33 to give a final concentration of 0.7 μM in 50 mM sodium acetate buffer (pH 5.0) (buffer A). Activity was measured as a function of time for 0–96 h by assaying aliquots of the reaction mixture in 50 mM sodium acetate (pH 5.0) containing 250 mM sodium chloride (buffer B) at an enzyme concentration of 0.6 μM and 400 μM chromogenic substrate [Lys-Ala-Arg-Val-Nle-[4-nitrophenylalanine]-Glu-Ala-Nle-NH2 (California Peptide Research, Napa, CA)]34 in a total volume of 120 μL. Autoproteolysis was also monitored by SDS-PAGE under the same conditions at final concentrations of 12–15 μM. Aliquots (10 μL) were withdrawn at the specified times, mixed with 4 μL of sample loading buffer to terminate the reaction, and subjected to electrophoresis on 20% homogeneous PhastGels (GE Healthcare) using 6 lane × 4 μL applicators. Gels were stained with PhastGel Blue R, destained, and digitized.
Dimer Dissociation Constant (Kdimer) and Kinetic Parameters (Km and kcat)
Protease activity was assayed in buffer B at 28 °C by following the decrease in absorbance at 310 nm upon cleavage of chromogenic substrate (Δ ε = 1797 Abs M−1 cm−1). Assays were conducted at varying protein concentrations and curve fitted using a previously described equation to determine Kdimer.29 Kinetic parameters for substrate cleavage were measured at enzyme concentrations from 0.3 to 0.5 μM. When possible, Km and kcat were estimated by fitting the data to the Michaelis–Menten equation using the Enzyme Kinetics module of SigmaPlot 10.0 (Systat Software Inc.). When the apparent Km was too high to permit accurate determination, the catalytic efficiency (kcat/Km) was approximated by the initial slope of rate versus substrate concentration under first-order conditions.
Gag Processing
Assays (100 μL) for monitoring the order and rate of cleavage of ΔGag, a truncated Gag construct spanning the matrix, capsid, SP1, and nucleocapsid domains, were conducted as described previously.35 ΔGag (50 μM) in 20 mM sodium phosphate (pH 6.5), 300 mM NaCl, 0.1 mM ZnCl2, and 1 mM tris(2-carboxyethyl)phosphine was mixed with the appropriate protease to give a final concentration of either 0.5 μM PR or 1 μM PR20, PRPR20-123, and PR20PR-123 at room temperature. Aliquots (5 μL) were drawn at the indicated times and mixed with 48 μL of the assay buffer described above and 50 μL of 2× SDS–PAGE sample loading buffer, and 5 μL of this mixture was subjected to electrophoresis on 18% Trisglycine gels (Life Technologies, Grand Island, NY).
Inhibitor Binding
Inhibition (dissociation) constants (Ki) for the binding of inhibitors to PRPR20-123, PRPR20-1, PR20PR-123, and PR20PR1 were measured using a MicroCal iTC200 microcalorimeter (GE Healthcare). The enzyme (~2 mg/mL in 12 mM HCl) was folded33 to give a final concentration of ~14 μM as dimer in 350 μL of buffer A. Samples were titrated with 120 μM darunavir or saquinavir in the same buffer at 28 °C. PR20PR-123 was titrated with 80 μM reduced peptide bond inhibitor [H-Arg-Val-Leu-()-Phe-Glu-Ala-Nle-NH2, where () denotes the reduced peptide bond (Bachem Americas, Inc., Torrance, CA)]. Data were analyzed using the instrument’s Origin software. Titration of PR20 or PRPR20-123 with reduced peptide bond inhibitor, and PR20PR-123 with saquinavir, did not give adequate thermal responses.
When Ki values could not be accurately measured by ITC, kinetic methods were used. Values of Ki for the binding of reduced peptide bond inhibitor to PR20 and PRPR20-123 were determined in buffer B by use of Dixon plots36 of 1/(initial rate) versus inhibitor concentration at two or three substrate concentrations. Because tight binding to the enzyme at low inhibitor concentrations depletes the free inhibitor, IC50 values were determined by fitting the quadratic Morrison equation37,38 to data (typical plots shown in Figure S5) relating initial rates to the added concentration of darunavir (with PRPR20-3), saquinavir (with PR20PR-123), or reduced peptide bond inhibitor (with PRPR20-1, PRPR20-3, and PR20PR-1). The Ki was calculated from the IC50 values using the equation
Differential Scanning Calorimetry (DSC)
Proteins were freshly folded to give a final dimer concentration of ~14 μM in buffer A.33 For experiments in the presence of inhibitors, a 2fold molar excess of inhibitor relative to protease dimer concentration was added during folding. DSC scans were performed at a rate of 90 °C/h using a MicroCal VP-DSC microcalorimeter (GE Healthcare) as previously described.39 Tm values were determined by curve fitting using the instrument’s Origin software or from the maxima of the thermal transition peaks.
Autoprocessing of Protease Precursor Constructs
Precursor proteins were expressed in the presence of a protease inhibitor to facilitate their accumulation and purified as described previously.24 The time course of in vitro autoprocessing of purified precursor proteins was determined by diluting the stock solution (typically 2 mg/mL) maintained in 12 mM HCl with 2.3 volumes of 5 mM sodium acetate buffer (pH 6) either with or without inhibitor, followed immediately by the addition of 3.3 volumes of 100 mM acetate buffer (pH 5). Aliquots (10 μL) were drawn at the indicated times, mixed with 4 μL of sample loading buffer, and subjected to SDS–PAGE on PhastGels as described above.
Nuclear Magnetic Resonance (NMR) Experiments
Two-dimensional 1H–15N transverse relaxation optimized (TROSY) correlation spectra were recorded at 20 °C on uniformly 15N-labeled or 15N- and 13C-labeled samples at a protein concentration of ~14 μM in 20 mM sodium phosphate buffer (pH 5.7) using a 600 MHz Bruker Avance spectrometer equipped with a triple-resonance z-axis gradient cryogenic probe.
RESULTS
Rationale and Construct Definition
The 19 drug resistance mutations in PR20 are grouped as clusters (1–3), based on their proximity to sites of autoproteoysis, and nonclusters (Figure 1B). This grouping permits substituting the clusters of the opposite template, namely PR20, into the PR template or vice versa either individually or in combination to assess the role of cooperativity in defining the various properties of the protease precursor and the corresponding mature enzyme. Each construct is named on the basis of the cluster with which it is substituted (shown in superscript); for example, PRPR20-123 denotes the construct bearing PR20 clusters 1–3 in the PR template. Therefore, this construct contains the 11 cluster mutations and none of the noncluster mutations (Figure 1C). The reverse construct PR20PR-123 consists of only the eight noncluster mutations and no cluster mutations, as all of the PR20 clusters are substituted with PR clusters (Figure 1C). When expressed as a precursor, each construct is designated with TFR preceding the name of the protease construct.
Effect of Substituting All Clusters into a Mature Protease: Properties of PRPR20-123 and PR20PR-123
Autoproteolysis
The wild-type protease catalyzes its own cleavage at four specific sites (L5/W6, L33/E34, L63/I64, and L90/I91) in the absence of added substrate or inhibitor.29,30,40,41 Autoproteolysis also occurs, although at a reduced rate, in the pseudo-wild-type protease (PR) designed with mutations Q7K, L33I, and L63I to restrict autoproteolysis and permit reliable kinetic and biophysical studies. As an optimal length of seven residues is required for binding specificity and cleavage of substrates,42 we chose the clusters to match the sequence spanning four residues on either side of the cleavage sites (Figure 1B, red bars).
In stark contrast to PR, PR20 shows no degradation products for as long as 115 h at pH 5 and room temperature (Figure S1).26 The time course of autoproteolysis of PRPR20-123 and PR20PR-123 was monitored over 96 h by activity assays (Figure 2A,B), and the appearance of degradation products was followed by SDS–PAGE (Figure 2C,D). The catalytic activity of PRPR20-123 is virtually unchanged even after 96 h, similar to that of PR20,26 whereas the activity of PR20PR-123 is reduced by ~50% after 48 h, similar to that of PR, and continues to decrease at longer incubation times. Thus, substituting the three clusters reverses their susceptibility to autoproteolysis such that autoproteolysis of PRPR20-123 and PR20PR-123 occurs at a rate similar to that of PR20 and PR, respectively (compare with Figure S1).26,41
Figure 2.
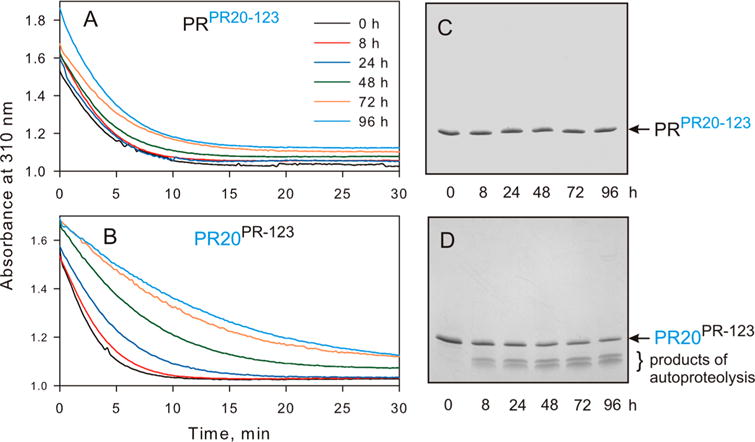
Time course of autoproteolysis of PRPR20-123 and PR20PR-123. Hydrolysis of the chromogenic substrate in 50 mM sodium acetate (pH 5.0) containing 250 mM sodium chloride as a function of time of incubation of (A) PRPR20-123 and (B) PR20PR-123 at room temperature (in hours, prior to assaying). Aliquots of (C) PRPR20-123 and (D) PR20PR-123 were withdrawn at the indicated times, mixed with gel loading buffer, subjected to SDS-PAGE, and stained. For comparison with parent constructs, PR and PR20, see Figure S1.
Dimer Dissociation Constant and Conformational Stability
Kdimer increases >30- and >26-fold for PRPR20-123 and PR20PR-123, respectively, relative to that of PR, shifting the equilibrium in favor of dissociation (Table 1 and Figures S1 and S2). This indicates that either the 11 cluster mutations or 8 noncluster mutations in the PR backbone exert an approximately equal effect with Kdimer values of ~0.17 and ~0.13 μM for PRPR20-123 and PR20PR-123, respectively. Interestingly, all 19 mutations in PR20 exert a much smaller increase of only ~6-fold, relative to that of PR (<0.005 μM), indicating a concerted role in the selection of cluster and noncluster mutations in defining Kdimer.
Table 1.
Properties of PR, PR20, and Selected Cluster Mutants
| PR | PRPR20-123 | PRPR20-1 | PRPR20-3 | PR20 | PR20PR-123 | PR20PR-1 | |
|---|---|---|---|---|---|---|---|
| ~Kdimer (μM) | <0.005a | 0.17 | 0.53 | 0.67 | 0.029b | 0.13 | 0.031 |
| Tm (°C) (DSC) | |||||||
| no inhibitor | 65.7c | 66.5 | 71.7b | 61.8 | |||
| ΔTm (with darunavir) | 22.4c | 11.5 | 5.3b | 16.6 | |||
| ΔTm (with saquinavir) | 19.3d | 8.1 | 2.9 | ≥11 | |||
| kinetic parameters | |||||||
| Km (μM) | 48 ± 3e | >1000 | >1071 | 124 ± 16 | 617 ± 84b | 180 ± 26 | 220 ± 30 |
| kcat (min−1) | 174 ± 3e | >450 | 213 ± 123 | 118 ± 5 | 215 ± 19b | 234 ± 15 | 112 ± 7 |
| kcat/Km (μM−1 min−1) | 3.63 ± 0.29e | 0.27 ± 0.01 | 0.16 ± 0.07 | 0.95 ± 0.15 | 0.35 ± 0.08 | 1.3 ± 0.27 | 0.51 ± 0.1 |
| inhibition constant Ki (nM) | |||||||
| darunavir | ≤0.01f | 5 ± 2.1ITC | 34.8 ± 9.7ITC | ≤1M | 41 ± 10b | 10 ± 3.2ITC | 16 ± 4ITC |
| saquinavir | 0.28 ± 0.02g | 34 ± 12ITC | 930 ± 93b | 50 ± 25M | |||
| reduced peptide bond inhibitor | 54 ± 22c | 1050 ± 50D | ≥300M | 10.8 ± 3.6M | 811 ± 52D | 48 ± 24ITC | 50 ± 14M |
| vitality | |||||||
| darunavir | 1 | 37 | 153 | 26 | 395 | 358 | 225 |
| saquinavir | 1 | 9 | 64 | 320 | |||
| Gag processingh | 1 | ~0.125 | ~0.25 | ~0.17 |
From ref32.
From ref24.
From ref39.
From ref36.
From ref47.
From refs and 50.
From ref51.
This value represents the approximate rate of processing relative to that of PR. References to published data are cited for comparison only; all other data are reported here for the first time. Ki values determined by ITC (1/Kassociation) and by kinetics using Morrison (M) and Dixon (D) plots are indicated.
The Tm (71.7 °C) of PR20 is 6 °C higher than for PR,24,26 indicative of a more stable fold (Figure S1, Table 1, and Figure S2). Substituted clusters of PR20 in PR increase the Tm minimally by ~1 °C, whereas substituted clusters of PR in PR20 decrease the Tm by ~10 °C. These observations suggest that the eight mutations outside of the clusters, together with the 11 clustered mutations in PR20, contribute to its Tm being higher than that of PR. There appears to be no direct relationship between Kdimer and overall stability of the fold as monitored by Tm.
Kinetic Parameters
The Km of PR20 for binding of the chromogenic substrate is ~13-fold higher than that of PR. Thus, replacing PR clusters with PR20 (PRPR20-123) increases the Km for which only a lower limit of ~1 mM could be estimated (Figure S3, Table 1, and Figure S2). The latter value indicates >20-fold weaker substrate binding as a result of introduction of the three PR20 clusters. Conversely, the introduction of PR clusters in the PR20 template decreases Km and restores kcat/Km for PR20PR-123 by ~4-fold. These observations indicate that the 11 mutations in the three clusters of PR20 are primarily responsible for the lower catalytic efficiency of PR20. The eight remaining mutations outside of the clusters in PR20 contribute to its lower catalytic activity relative to that of PR by a factor of ~3 [compare PR20PR-123 with PR (Table 1 and Figure S2)].
Inhibitor Binding and Drug Resistance
PR20 contains nine mutations associated with resistance to currently used clinical protease inhibitors (Figure 1B, red asterisks), five in clusters 1 and 3 and four outside the clusters, two of which are located in the flaps (Figure 1). Values of Ki for the binding of darunavir, saquinavir, and reduced peptide bond inhibitor are listed in Table 1 and shown in Figure S2. Inhibitor dissociation constants (l/Kassociation, equivalent to Ki for competitive inhibitors) were measured by ITC (Figure S4) or by kinetic methods (see Materials and Methods and Figure S5). For the sake of consistency, Ki is used throughout the text, and the method for each determination is identified in Table 1. PR20 clusters in PR (PRPR20-123) decrease its affinity for darunavir (increase in Ki) by ~500-fold. This trend is the same but significantly smaller than the ~4000-fold difference in Ki for PR20 relative to that of PR, due to additional effects of noncluster mutations, three of which are known darunavir resistance mutations (I47V, I54L, and I84V). Three mutations in PR20 are associated with resistance to saquinavir (I54L, I84V, and L90M). The Ki for saquinavir is increased by ~3000-fold for PR20 relative to that of PR. However, this Ki is increased by only 120-fold for the L90M mutation in PRPR20-123 lacking the two noncluster drug resistance mutations in PR20 associated with both darunavir and saquinavir resistance.
Substitution of PR clusters in PR20PR-123 decreases Ki, consistent with tighter binding of darunavir (~4-fold) and saquinavir (~l9-fold). The weaker effects on inhibitor binding with PR clusters in PR20, relative to PR20 clusters in PR, indicate that noncluster drug resistance mutations in PR20, namely, I54L and I84V, partially offset the effect of the PR clusters. In contrast, the Ki for the reduced peptide bond inhibitor, a substrate analogue based on the capsid/SP1 cleavage site in Gag, is increased by only ~20-fold relative to that of PR, comparable to the increase in Km for the chromogenic substrate.
The difference (ΔTm) between the Tm values in the presence and absence of an inhibitor provides a semiquantitative assessment of the relative strength of inhibitor binding.36 Thus, DSC (Table 1 and Figure S1E,F) was used to complement ITC and kinetic data for darunavir and saquinavir binding. Larger ΔTm values correlate with stronger inhibitor binding affinity (Table 1 and Figure S2) as observed for PR relative to PRPR20-123 (ΔTm increased by 10.9 °C for darunavir and 11.2 °C for saquinavir). Similarly, PR20 exhibits ΔTm values upon binding of darunavir or saquinavir that are 9–11 °C smaller than those of PR20PR-123, consistent with their Ki values. Thus, the three clustered mutations together contribute significantly to the drug resistance of PR20.
Order of Cleavage and Rate of Gag Polyprotein Processing
Gag processing occurs by an ordered sequence of steps as shown schematically in Figure 3.4 To verify if the kinetic parameters estimated using synthetic peptide substrates reflect the rates and order of cleavage of the native Gag precursor, a truncated recombinant Gag lacking the SP2 and p6 domains (ΔGag) and encompassing three protease cleavage sites was used as a substrate to evaluate processing catalyzed by PR PR20, PRPR20-123, and PR20PR-123. The time course for the disappearance of full-length ΔGag and the appearance of products matrix (MA), capsid (CA), and nucleocapsid (NC) was followed by SDS–PAGE (Figure 3, four gel panels). In spite of the differences in Kdimer and kinetic parameters observed for mature PR, PR20, and constructs bearing the three sets of cluster mutations, the order in which Gag cleavages occur is clearly the same for all four enzymes. The apparent rates of cleavage do not differ widely, being approximately 1.5–2-fold slower with PR20 than with PR, consistent with ~3–4-fold lower enzymatic activity when taking into account the fact that concentrations of PR20, PRPR20-123, and PR20PR-123 used are 2 times higher than that of PR. This contrasts with the ~10-fold difference in catalytic efficiency between mature PR and PR20 when assayed with the chromogenic substrate and the ~14–35-fold differences for cleavage of short (10-mer) peptides with sequences corresponding to the SP1/NC, MA/CA, and CA/SP1 sites.26 Rates of Gag cleavage catalyzed by PR20PR-123 are comparable to those of PR20, suggesting that the PR clusters have little effect on the catalytic efficiency of PR20 for Gag cleavages. In contrast, substitution of PR20 clusters in PR decreases the cleavage rates by ~50% relative to that of PR20. This suggests that the PR20 cluster mutations, along with the eight noncluster mutations, contribute to the slightly slower rate of Gag processing by PR20 relative to that of PR.
Figure 3.
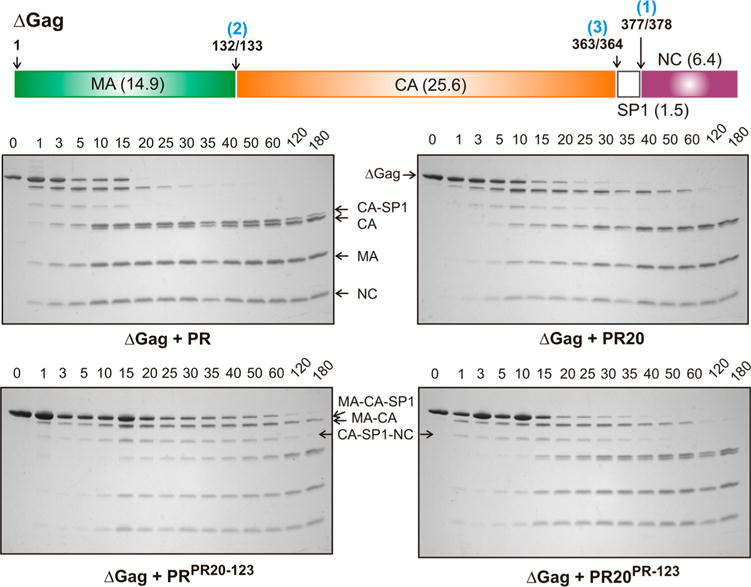
Processing of ΔGag by the protease. Schematic representation of the sequential cleavages of ΔGag with the order of cleavage (blue), cleavage-site positions, and calculated molecular weight (in kDa) of the products indicated. The four bottom panels show the time course of processing of ΔGag by mature protease monitored by SDS–PAGE on 18% Tris-glycine gels. Reaction mixtures contained 50 μM ΔGag and either 0.5 μM PR or 1 μM PR20PR-123, PR20, and PRPR20-123 as indicated. Cleavage of full-length 48.4 kDa ΔGag between SP1 and NC (step 1) yields MA-CA-SP1 and NC, followed by cleavage between MA and CA (step 2) to generate the mature MA and the intermediate CA-SP1. The spacer polypeptide SP1 is cleaved subsequently (step 3) from the C-terminus of CA-SP1, some of which is still detectable just above mature CA at up to 180 min. No full-length ΔGag remains after reaction for 20 min with PR at half the concentration of the other three constructs, and no MA-CA-SP1 is detectable after 35 min. SP1, because of its small size, is predicted to have run out of the gel. A faint band between the MA-CA-SP1 and CA-SP1 bands corresponds to CA-SP1-NC formed by a minor MA/CA cleavage pathway.
The relative rates listed in Table 1 (and shown in Figure S2) are based on the first cleavage (SP1/NC). We note that even though the rate of cleavage of the SP1/NC site mediated by PR20PR-123 is slower than that for PR20, comparison of the relative ratios of CA-SP1 and CA products indicates slightly faster cleavage of this site when PR clusters are introduced into PR20. The ~2-fold slower rate of SP1/NC cleavage by PR20PR123 does not lead to a corresponding slowing of the cleavage at the CA/SP1 site, the ratios of CA–SP1 and CA being roughly the same.
Effect of Substituting Cluster Mutations on Autoprocessing
Similar to the parent precursor TFR-PR and the drug resistant precursor TFR-PR20, the mutant TFR-PRPR20-123 precursor bearing the three clusters of PR20, accounting for 11 of the 19 mutations present in PR20, and the reverse construct TFR-PR20PR-123 bearing only the eight remaining mutations outside the clusters undergo efficient autoprocessing in E. coli to release mature PR (Figure 4A,B, black arrows). Thus, substituting the clusters collectively does not adversely affect autoprocessing of this model precursor.
Figure 4.
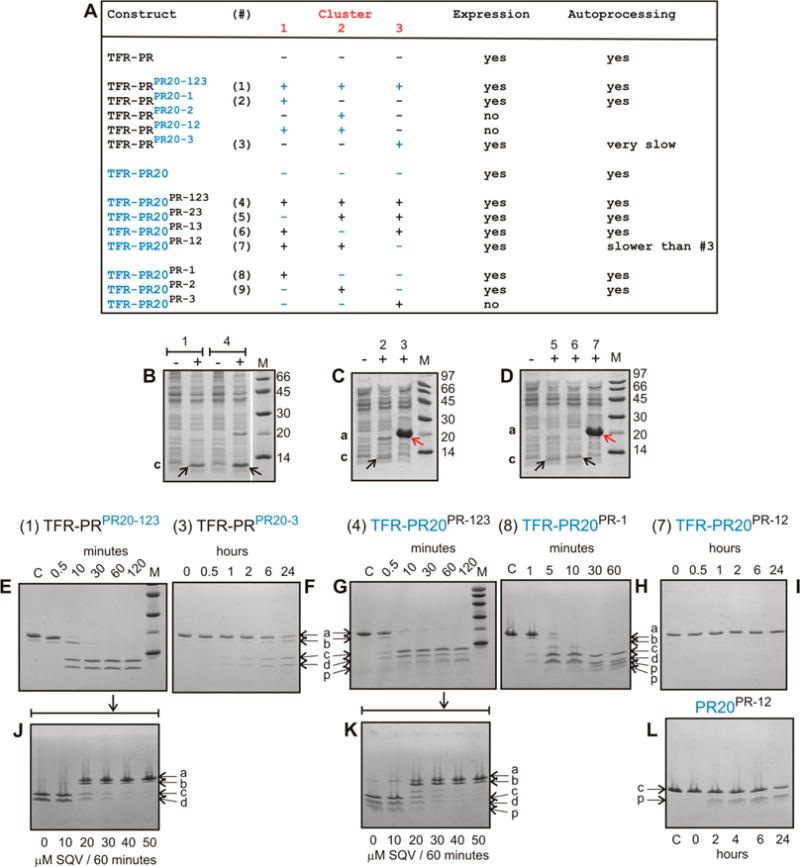
Expression and autoprocessing of TFR-PR precursors. (A) Identification of precursor constructs shown in panels B–I. Individual clusters and various combinations are shown with PR clusters in black and PR20 clusters in blue. (B–D) Expression and autoprocessing of precursor constructs containing one or more substituted clusters. Total cell extracts were analyzed by SDS–PAGE. Numbers above the lanes correspond to the designated construct shown in panel A, and lanes indicated by minus (−) and plus (+) represent total extracts of uninduced and induced cultures, respectively. (E–I) Time course of N-terminal autoprocessing of purified precursors in vitro. (F and I) TFR-PRPR20-3 and TFR-PR20PR-12 exhibit very slow or no discernible autoprocessing, respectively, as indicated in panel A. (J and K) Inhibition of autoprocessing of TFR-PRPR20-123 and TFR-PR20PR-123 conducted for 60 min in the presence of increasing concentrations of saquinavir. (L) Time course of autoproteolysis of mature PR20PR-12. Lowercase letters a–d and p indicate full-length precursor, an intermediate cleavage product of TFR-PR, TFR9–56-PR (cleavage occurs between residues 8 and 9 of TFR), mature PR, TFR and products of mature PR autoproteolysis, respectively. The pathway to release the minor intermediate product TFR9–56-PR precedes the cleavage at the N-terminus of PR. We have shown previously that this cleavage is pH-dependent and becomes pronounced at pH 4.2 M denotes molecular weight markers in kilodaltons. SQV denotes saquinavir.
We also analyzed a series of precursor constructs containing either single or dual cluster mutations as listed in Figure 4A Of 10 such mutant precursor constructs, seven showed expression and three failed to express. Of the seven clones showing expression, the precursor invariably accumulated in two as shown by analysis of total extracts by SDS–PAGE [Figure 4C (construct 3) and Figure 4D (construct 7), red arrows]. Thus, to obtain a more accurate assessment of relative rates of autoprocessing, we isolated six precursor proteins, including constructs TFR-PRPR20-123 (construct 1) and TFR-PR20PR-123 (construct 4). Precursors were subjected to autoprocessing in vitro (Figure 4E–I) and the results compared to previously published results on TFR-PR32 and TFR-PR20.24 Note that when the total extract (Figure 4C,D) is monitored, the mature PR product derived by autoprocessing of TFR-PRPR20-1 (construct 2), TFR-PR20PR-23 (construct 5), and TFR-PR20PR-13 (construct 6) during expression in E. coli is only weakly visible (indicated by black arrows) because of autoproteolysis of the corresponding released mature PR, in contrast to TFR-PRPR20-3 (construct 3) and TFR-PR20PR-12 (construct 7) precursors. Autoprocessing of precursors (constructs 2, 5, and 6) during expression was verified by isolating the accumulated protein in inclusion bodies using established protocols24 and subjecting them to SDS–PAGE and ESI-MS, showing the presence of the released mature PR.
At pH ≥5, TFR-PR undergoes maturation in a single step via cleavage at the N-terminus of PR (TFR/PR site) concomitant with the appearance of the products, TFR and PR, and mature-like catalytic activity.32 Similarly, as shown in panels E and G of Figure 4, both precursors bearing three cluster substitutions, TFR-PRPR20-123 and TFR-PR20PR-123, upon folding undergo rapid autoprocessing at the N-terminus of PR with more than half of the full-length protein converted to products in ~10 min. This pattern of cleavage is clearly evident for TFR-PRPR20-123 [Figure 4E, products c (PR) and d (TFR)] as the cluster mutations restrict the autoproteolysis of the released PR, in contrast to TFR-PR20PR123, which shows products (denoted by p) of autoproteolysis consistent with the results shown in Figure 2 for the cluster mutants of mature proteases. The rates of autoprocessing of TFR-PRPR20-123 and TFR-PR20PR-123 are similar to that of the wild-type construct, TFR-PR43 Although the TFR-PR20 precursor could not be isolated in quantities that are adequate for in vitro studies, even in the presence of 150 μM darunavir, a small amount of TFR-PR20PR-1 could be isolated, permitting its analysis (Figure 4H). The rate of autoprocessing of TFR-PR20PR1 is similar to that of TFR-PR20PR-123 and TFR-PRPR20-123. In contrast, autoprocessing of the two constructs, TFR-PRPR20-3 (Figure 4F) and TFR-PR20PR-12 (Figure 4I), is significantly compromised, with only 50% of the former converted to products in 24 h and no visible products observed for the latter after 24 h. Interestingly, both precursors bear the same combination of PR clusters (1 and 2) with PR20 cluster 3 and differ only in the eight substitution mutations present in the PR20 template (see Figures 1B,C and 4A).
Comparison of the Rate of Autoprocessing with the Catalytic Efficiency of the Mature Protease
We next addressed the question of whether there is a correlation between N-terminal autoprocessing of the precursors and the properties of the mature proteases. We therefore compared the two precursors that are defective in autoprocessing (TFR-PRPR20-3 and TFR-PR20PR-12) with their corresponding mature enzymes. As mature PR20PR-12 exhibits irreproducible kinetics with the chromogenic substrate, we monitored its intrinsic autoproteolysis as an alternative measure of its catalytic efficiency. Surprisingly, this trend resembles that of its closest parent PR20PR-123 (compare Figure 4L with Figure 2D). Accurate determination of Kdimer was not possible, because its observed catalytic activity with the chromogenic substrate decreases with an increasing enzyme concentration, likely related to rapid autoproteolysis even in the presence of substrate. In contrast, PRPR20-3, which perturbs autoprocessing by >150-fold, is a catalytically competent dimer with a Kdimer of 0.67 μM and a favorable kcat/Km of 0.95 μM−1 min−1 (Table 1 and Figure S2). PRPR20-3 binds tightly to darunavir (Ki ≤ 1 nM) as well as to the substrate analogue/reduced peptide bond inhibitor with a Ki of ~11 nM, ~5-fold better than that of PR. Thus, there is no clear correlation between defective autoprocessing and the catalytic activity of the mature protease. Further, Kdimer values for the mature proteases do not consistently correlate with rates of precursor autoprocessing. As an example, the Kdimer values for the corresponding mature proteases PRPR20-1 and PRPR20-3 (Table 1 and Figure S2) are very similar; TFR-PRPR20-1 autoprocesses efficiently at a rate comparable to that of TFR-PRPR20-123, while TFR-PRPR20-3 does so only extremely slowly (Figure 4F).
Tertiary Fold of Cluster Mutants Defective in Autoprocessing
The folding/dimerization potential of mature PR20PR-12 was further verified by acquiring a 1H–15N correlation NMR spectrum and comparing this to that of PRPR20-3 (Figure S6). Acquiring a spectrum of inhibitor-free PR20PR-12 was not feasible because of rapid degradation resulting in signals corresponding to only fragments of the protease (Figure S6B, inset lane C) in a narrow region of the spectrum. However, the spectra of PRPR20-3 and PR20PR-12 folded in the presence of the symmetric inhibitor DMP32344 are well-resolved and characteristic of the folded dimer (Figure S6A,B). Additional cross-peaks are observed for PR20PR-12 because of the presence of some unbound form of the dimer as a result of weak DMP323 binding.
Inhibition of Autoprocessing of Cluster Mutants by Saquinavir
We had previously shown that the two most potent inhibitors of autoprocessing of TFR-PR precursor are darunavir and saquinavir with IC50 values of 1–2 μM.24 Here we used saquinavir because of its better solubility under the experimental conditions employed. Inhibition of the TFR-PR20 precursor could not be studied in vitro because protease inhibitor added to the culture medium during expression does not block autoprocessing, making its isolation impossible.24 However, isolation of both TFR-PRPR20-123 and TFR-PR20PR-123 was possible. On the basis of dose–response experiments, IC50 values were estimated to be ≥15 μM for the inhibition of autoprocessing of both precursors by saquinavir (Figure 4J,K). This value is ≥300-fold higher than for the inhibition of the corresponding mature enzymes by saquinavir (Table 1 and Figure S2). The 10-fold improved binding of saquinavir to TFR-PRPR20-123 and TFR-PR20PR-123 precursors, relative to TFR-PR20 (IC50 > 150 μM24), suggests that both cluster and noncluster residues play a collective role in defining saquinavir binding to the precursor.
DISCUSSION
PR20 presents an excellent example for evaluating the interrelationships among kinetic parameters (kcat, Km, and Ki), Kdimer, and the conformational stability of mature PR as it evolves under drug pressure and how these properties relate to N-terminal autoprocessing and autoproteolysis. The latter two events define the spatiotemporal regulation of protease activity late in the HIV-1 replication cycle. Our studies indicate that selected mutations within three clusters, defined by their proximity to sites of autoproteolysis, and outside the clusters exert a cumulative effect on the kinetic parameters of the mature PR.
Vitality (V) defined as V = [(Kikcat/Km)mutant/(Kikcat/Km)PR] is a measure of the enzymatic fitness of the mature PR as it evolves in the presence of a given inhibitor.45 By definition, vitality is 1 for PR, the surrogate of the wild-type protease. In spite of substantial variations in kcat/Km for the set of cluster mutants presented here, vitality scores (Table 1 and Figure S2) in the presence of darunavir increase in the order PR (1) < PRPR20-123 (37) < PR20PR-123 (358) ≤ PR20 (395), roughly paralleling the increase in Ki for darunavir inhibition and consistent with the importance of selection for drug resistance in the evolution of these mutants. A similar trend is observed with the inhibitor saquinavir with vitality scores in the following order: PR (1) < PRPR20-123 (9) < PR20PR-123 (64) < PR20 (320).
Regardless of the large differences observed in kinetic parameters and Kdimer relative to those of PR, the rates of processing of Gag for PR20 and the cluster mutants PRPR20-123 and PR20PR-l23 differ by 4–8-fold, but display no alteration in cleavage order with cleavage at the SP1/NC junction occurring first, followed by MA/CA and CA/SP1. Thus, monitoring the rate of Gag processing may represent a more realistic approach than using synthetic peptide substrates to assess the competence of drug resistant mutants because an ~10-fold decrease in catalytic efficiency (kcat/Km) for cleaving the chromogenic substrate (analogue of CA/SP1 site) and an ~35-fold decrease in kcat/Km for cleaving an unmodified decapeptide spanning the same site26 correspond to an only ~4-fold decrease in the rate of cleavage of the CA/SP1 site in Gag mediated by PR20 relative to PR. Similarly, PR20 cleaves the SPl/NC peptide substrate ~34-fold slower than PR does. This suggests that slight decreases in Gag processing efficiency are tolerated to yield viable progeny virus as long as N-terminal autoprocessing is not impaired, as exemplified by the TFR-PR20 precursor.
Although we were unable to analyze a complete set of mutant cluster combinations because of a lack of expression of some constructs, substitution analysis of nine cluster mutants reveals important insights into co-selection for optimal autoprocessing. Substituting all PR20 clusters in PR or vice versa does not perturb autoprocessing. However, substituting cluster 3 of PR20 by itself into PR without cosubstituting either cluster 1 or 2 of PR20 significantly slows autoprocessing. The decrease is more drastic when PR20 is being reverted with only clusters 1 and 2 of PR. These results clearly suggest that coevolution of cluster and noncluster mutations as well as coordination of cluster 3 mutations with either cluster 1 or 2 is required for optimal autoprocessing.
For the mature protease, we find no relationship between Kdimer and fold stability, as measured by Tm. Also, no strict relationship exists between the estimated Kdimer for the mature PR and N-terminal autoprocessing. A lack of correlation between autoprocessing of TFR-PR precursors and catalytic efficiency (kcat/Km) or Kdimer of mature protease suggests that different catalytic environments exist for the initial N-terminal maturation of the precursor and hydrolysis of substrates mediated by mature protease. This may point to conformations of the transient TFR-PR dimer that require specific long-range coordination between residues in clusters 3 collectively selected with clusters 1 and 2 and differing significantly from the corresponding mature protease, an effect possibly also related to the binding of protease inhibitors to precursors that is poorer by 3–5 orders of magnitude relative to that of mature PR. It is worth noting that mutations of highly conserved residue G86 or R8746 located at the N-terminus of the α-helix and contiguous to cluster 3 perturb the internally placed C-terminal strands (residues 96–99) of the dimer interface.47 Both G86A and R87K mutations drastically affect dimer formation.27 We have also shown that the C-terminal interface is indispensible for dimer formation of the mature protease.2,47 In the context of PR maturation, this feature is particularly important because prior to cleavage at the N-terminus of PR, the precursor lacks the characteristic four-stranded β-sheet, because of fraying of the N-terminal residues when they are flanked by TFR.48 Thus, interactions of the C-terminal strands will be a crucial requirement for transient dimerization, consistent with the observation that deletion of residues 96–99 in TFR-PR blocks N-terminal autoprocessing.47 Overall, these studies imply that evolution of PR20 under drug pressure modulates its kinetic parameters and Kdimer by synchronized selection for mutations in specific regions to allow N-terminal autoprocessing and minimally affect the rate and order of cleavage of the Gag polyprotein by the mature enzyme.
Supplementary Material
Acknowledgments
Clinical protease inhibitors used in this study were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health. We acknowledge support from the National Institute of Diabetes and Digestive and Kidney Diseases Advanced Mass Spectrometry Core.
Funding
This research was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (NIH), and the Intramural AIDS-Targeted Program of the Office of the Director, NIH (to G.M.C.).
ABBREVIATIONS
- HIV-1
human immunodeficiency virus type 1
- PR
mature HIV-1 protease containing the Q7K, L33I, L63I, C67A, and C95A mutations
- PR20
PR bearing 19 drug resistance mutations in addition to Q7K, C67A, and C95A
- DSC
differential scanning calorimetry
- ITC
isothermal titration calorimetry
- Kdimer
protease dimer dissociation constant
- Ki
inhibitor dissociation constant
- PDB
Protein Data Bank
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00759.
Six additional figures (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Louis JM, Weber IT, Tozser J, Marius Clore G, Gronenborn AM. HIV-1 protease: maturation, enzyme specificity, and drug resistance. Adv Pharmacol. 2000;49:111–146. doi: 10.1016/s1054-3589(00)49025-3. [DOI] [PubMed] [Google Scholar]
- 2.Louis JM, Ishima R, Torchia DA, Weber IT. HIV-1 protease: structure, dynamics, and inhibition. Adv Pharmacol. 2007;55:261–298. doi: 10.1016/S1054-3589(07)55008-8. [DOI] [PubMed] [Google Scholar]
- 3.Tang C, Louis JM, Aniana A, Suh JY, Clore GM. Visualizing transient events in amino-terminal autoprocessing of HIV-1 protease. Nature. 2008;455:693–696. doi: 10.1038/nature07342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee SK, Potempa M, Swanstrom R. The choreography of HIV-1 proteolytic processing and virion assembly. J Biol Chem. 2012;287:40867–40874. doi: 10.1074/jbc.R112.399444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tessmer U, Krausslich HG. Cleavage of human immunodeficiency virus type 1 proteinase from the N-terminally adjacent p6* protein is essential for efficient Gag polyprotein processing and viral infectivity. J Virol. 1998;72:3459–3463. doi: 10.1128/jvi.72.4.3459-3463.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hammer SM, Squires KE, Hughes MD, Grimes JM, Demeter LM, Currier JS, Eron JJ, Jr, Feinberg JE, Balfour HH, Jr, Deyton LR, Chodakewitz JA, Fischl MA. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N Engl J Med. 1997;337:725–733. doi: 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- 7.Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:15821586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 8.Weber IT, Agniswamy J. HIV-1 Protease: Structural Perspectives on Drug Resistance. Viruses. 2009;1:1110–1136. doi: 10.3390/v1031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weber IT, Kneller DW, Wong-Sam A. Highly resistant HIV-1 proteases and strategies for their inhibition. Future Med Chem. 2015;7:1023–1038. doi: 10.4155/fmc.15.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qurñones-Mateu ME, Weber J, Rangel HR, Chakraborty B. HIV-1 Fitness and Antiviral Drug Resistance. AIDS Rev. 2001;3:223–234. [Google Scholar]
- 11.Nijhuis M, Schuurman R, de Jong D, Erickson J, Gustchina E, Albert J, Schipper P, Gulnik S, Boucher CA. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS. 1999;13:2349–2359. doi: 10.1097/00002030-199912030-00006. [DOI] [PubMed] [Google Scholar]
- 12.Chang MW, Torbett BE. Accessory mutations maintain stability in drug-resistant HIV-1 protease. J Mol Biol. 2011;410:756–760. doi: 10.1016/j.jmb.2011.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haq O, Andrec M, Morozov AV, Levy RM. Correlated electrostatic mutations provide a reservoir of stability in HIV protease. PLoS Comput Biol. 2012;8:e1002675. doi: 10.1371/journal.pcbi.1002675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ragland DA, Nalivaika EA, Nalam MN, Prachanronarong KL, Cao H, Bandaranayake RM, Cai Y, Kurt-Yilmaz N, Schiffer CA. Drug resistance conferred by mutations outside the active site through alterations in the dynamic and structural ensemble of HIV-1 protease. J Am Chem Soc. 2014;136:11956–11963. doi: 10.1021/ja504096m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foulkes-Murzycki JE, Rosi C, Kurt Yilmaz N, Shafer RW, Schiffer CA. Cooperative effects of drug-resistance mutations in the flap region of HIV-1 protease. ACS Chem Biol. 2013;8:513–518. doi: 10.1021/cb3006193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cai Y, Myint W, Paulsen JL, Schiffer CA, Ishima R, Kurt Yilmaz N. Drug Resistance Mutations Alter Dynamics of Inhibitor-Bound HIV-1 Protease. J Chem Theory Comput. 2014;10:34383448. doi: 10.1021/ct4010454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fun A, Wensing AM, Verheyen J, Nijhuis M. Human Immunodeficiency Virus Gag and protease: partners in resistance. Retrovirology. 2012;9:63. doi: 10.1186/1742-4690-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giandhari J, Basson AE, Coovadia A, Kuhn L, Abrams EJ, Strehlau R, Morris L, Hunt G. Genetic Changes in HIV-1 Gag-Protease Associated with Protease Inhibitor-Based Therapy Failure in Paediatric Patients. AIDS Res Hum Retroviruses. 2015;31:776. doi: 10.1089/aid.2014.0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolli M, Ozen A, Kurt-Yilmaz N, Schiffer CA. HIV-1 protease-substrate coevolution in nelfinavir resistance. J Virol. 2014;88:7145–7154. doi: 10.1128/JVI.00266-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen L, Perlina A, Lee CJ. Positive selection detection in 40,000 human immunodeficiency virus (HIV) type 1 sequences automatically identifies drug resistance and positive fitness mutations in HIV protease and reverse transcriptase. J Virol. 2004;78:3722–3732. doi: 10.1128/JVI.78.7.3722-3732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen L, Lee C. Distinguishing HIV-1 drug resistance, accessory, and viral fitness mutations using conditional selection pressure analysis of treated versus untreated patient samples. Biol Direct. 2006;1:14. doi: 10.1186/1745-6150-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prosperi MC, De Luca A. Computational models for prediction of response to antiretroviral therapies. AIDS Rev. 2012;14:145–153. [PubMed] [Google Scholar]
- 23.Dierynck I, De Wit M, Gustin E, Keuleers I, Vandersmissen J, Hallenberger S, Hertogs K. Binding kinetics of darunavir to human immunodeficiency virus type 1 protease explain the potent antiviral activity and high genetic barrier. J Virol. 2007;81:13845–13851. doi: 10.1128/JVI.01184-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Louis JM, Aniana A, Weber IT, Sayer JM. Inhibition of autoprocessing of natural variants and multidrug resistant mutant precursors of HIV-1 protease by clinical inhibitors. Proc Natl Acad Sci U S A. 2011;108:9072–9077. doi: 10.1073/pnas.1102278108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agniswamy J, Shen CH, Aniana A, Sayer JM, Louis JM, Weber IT. HIV-1 protease with 20 mutations exhibits extreme resistance to clinical inhibitors through coordinated structural rearrangements. Biochemistry. 2012;51:2819–2828. doi: 10.1021/bi2018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Louis JM, Tozser J, Roche J, Matuz K, Aniana A, Sayer JM. Enhanced stability of monomer fold correlates with extreme drug resistance of HIV-1 protease. Biochemistry. 2013;52:76787688. doi: 10.1021/bi400962r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishima R, Gong Q, Tie Y, Weber IT, Louis JM. Highly conserved glycine 86 and arginine 87 residues contribute differently to the structure and activity of the mature HIV-1 protease. Proteins: Struct Funct Genet. 2010;78:1015–1025. doi: 10.1002/prot.22625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Louis JM, Ishima R, Nesheiwat I, Pannell LK, Lynch SM, Torchia DA, Gronenborn AM. Revisiting monomeric HIV-1 protease. Characterization and redesign for improved properties. J Biol Chem. 2003;278:6085–6092. doi: 10.1074/jbc.M209726200. [DOI] [PubMed] [Google Scholar]
- 29.Sayer JM, Agniswamy J, Weber IT, Louis JM. Autocatalytic maturation, physical/chemical properties, and crystal structure of group N HIV-1 protease: relevance to drug resistance. Protein Sci. 2010;19:2055–2072. doi: 10.1002/pro.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Louis JM, Ishima R, Aniana A, Sayer JM. Revealing the dimer dissociation and existence of a folded monomer of the mature HIV-2 protease. Protein Sci. 2009;18:2442–2453. doi: 10.1002/pro.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wensing AM, Calvez V, Gunthard HF, Johnson VA, Paredes R, Pillay D, Shafer RW, Richman DD. 2014 Update of the drug resistance mutations in HIV-1. Top Antivir Med. 2014;22:642–650. [PMC free article] [PubMed] [Google Scholar]
- 32.Louis JM, Clore GM, Gronenborn AM. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat Struct Biol. 1999;6:868–875. doi: 10.1038/12327. [DOI] [PubMed] [Google Scholar]
- 33.Ishima R, Torchia DA, Louis JM. Mutational and structural studies aimed at characterizing the monomer of HIV-1 protease and its precursor 1. J Biol Chem. 2007;282:17190–17199. doi: 10.1074/jbc.M701304200. [DOI] [PubMed] [Google Scholar]
- 34.Richards AD, Phylip LH, Farmerie WG, Scarborough PE, Alvarez A, Dunn BM, Hirel PH, Konvalinka J, Strop P, Pavlickova L. Sensitive, soluble chromogenic substrates for HIV-1 proteinase. J Biol Chem. 1990;265:7733–7736. [PubMed] [Google Scholar]
- 35.Deshmukh L, Ghirlando R, Clore GM. Conformation and dynamics of the Gag polyprotein of the human immunodeficiency virus 1 studied by NMR spectroscopy. Proc Natl Acad Sci U S A. 2015;112:3374–3379. doi: 10.1073/pnas.1501985112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sayer JM, Louis JM. Interactions of different inhibitors with active-site aspartyl residues of HIV-1 protease and possible relevance to pepsin. Proteins: Struct, Funct, Genet. 2009;75:556568. doi: 10.1002/prot.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Furfine ES. Identifying and Characterizing HIV Protease Inhibitors. Methods Mol Med. 2000;24:313–326. doi: 10.1385/1-59259-245-7:313. [DOI] [PubMed] [Google Scholar]
- 38.Murphy DJ. Determination of accurate Ki values for tight-binding enzyme inhibitors: an in silico study of experimental error and assay design. Anal Biochem. 2004;327:61–67. doi: 10.1016/j.ab.2003.12.018. [DOI] [PubMed] [Google Scholar]
- 39.Sayer JM, Liu F, Ishima R, Weber IT, Louis JM. Effect of the active site D25N mutation on the structure, stability, and ligand binding of the mature HIV-1 protease. J Biol Chem. 2008;283:13459–13470. doi: 10.1074/jbc.M708506200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rose JR, Salto R, Craik CS. Regulation of autoproteolysis of the HIV-1 and HIV-2 proteases with engineered amino acid substitutions. J Biol Chem. 1993;268:11939–11945. [PubMed] [Google Scholar]
- 41.Mildner AM, Rothrock DJ, Leone JW, Bannow CA, Lull JM, Reardon IM, Sarcich JL, Howe WJ, Tomich CS, Smith CW. The HIV-1 protease as enzyme and substrate: mutagenesis of autolysis sites and generation of a stable mutant with retained kinetic properties. Biochemistry. 1994;33:9405–9413. doi: 10.1021/bi00198a005. [DOI] [PubMed] [Google Scholar]
- 42.Tozser J. Comparative studies on retroviral proteases: substrate specificity. Viruses. 2010;2:147–165. doi: 10.3390/v2010147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Louis JM, Wondrak EM, Kimmel AR, Wingfield PT, Nashed NT. Proteolytic processing of HIV-1 protease precursor, kinetics and mechanism. J Biol Chem. 1999;274:23437–23442. doi: 10.1074/jbc.274.33.23437. [DOI] [PubMed] [Google Scholar]
- 44.Lam PY, Ru Y, Jadhav PK, Aldrich PE, DeLucca GV, Eyermann CJ, Chang CH, Emmett G, Holler ER, Daneker WF, Li L, Confalone PN, McHugh RJ, Han Q, Li R, Markwalder JA, Seitz SP, Sharpe TR, Bacheler LT, Rayner MM, Klabe RM, Shum L, Winslow DL, Kornhauser DM, Hodge CN. Cyclic HIV protease inhibitors: synthesis, conformational analysis, P2/P2′ structure-activity relationship, and molecular recognition of cyclic ureas. J Med Chem. 1996;39:3514–3525. doi: 10.1021/jm9602571. [DOI] [PubMed] [Google Scholar]
- 45.Gulnik SV, Suvorov LI, Liu B, Yu B, Anderson B, Mitsuya H, Erickson JW. Kinetic characterization and cross-resistance patterns of HIV-1 protease mutants selected under drug pressure. Biochemistry. 1995;34:9282–9287. doi: 10.1021/bi00029a002. [DOI] [PubMed] [Google Scholar]
- 46.Pearl LH, Taylor WR. A structural model for the retroviral proteases. Nature. 1987;329:351–354. doi: 10.1038/329351a0. [DOI] [PubMed] [Google Scholar]
- 47.Ishima R, Torchia DA, Lynch SM, Gronenborn AM, Louis JM. Solution structure of the mature HIV-1 protease monomer: insight into the tertiary fold and stability of a precursor. J Biol Chem. 2003;278:43311–43319. doi: 10.1074/jbc.M307549200. [DOI] [PubMed] [Google Scholar]
- 48.Agniswamy J, Sayer JM, Weber IT, Louis JM. Terminal interface conformations modulate dimer stability prior to amino terminal autoprocessing of HIV-1 protease. Biochemistry. 2012;51:1041–1050. doi: 10.1021/bi201809s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.King NM, Prabu-Jeyabalan M, Nalivaika EA, Wigerinck P, de Bethune MP, Schiffer CA. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J Virol. 2004;78:12012–12021. doi: 10.1128/JVI.78.21.12012-12021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brower ET, Bacha UM, Kawasaki Y, Freire E. Inhibition of HIV-2 protease by HIV-1 protease inhibitors in clinical use. Chem Biol Drug Des. 2008;71:298–305. doi: 10.1111/j.1747-0285.2008.00647.x. [DOI] [PubMed] [Google Scholar]
- 51.Ohtaka H, Schon A, Freire E. Multidrug resistance to HIV-1 protease inhibition requires cooperative coupling between distal mutations. Biochemistry. 2003;42:13659–13666. doi: 10.1021/bi0350405. [DOI] [PubMed] [Google Scholar]
- 52.Ceccherini-Silberstein F, Erba F, Gago F, Bertoli A, Forbici F, Bellocchi MC, Gori C, d’Arrigo R, Marcon L, Balotta C, Antinori A, Monforte AD, Perno CF. Identification of the minimal conserved structure of HIV-1 protease in the presence and absence of drug pressure. AIDS. 2004;18:F11–F19. doi: 10.1097/01.aids.0000131394.76221.02. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.