

Summary
Gene fusions are increasingly recognized as important cancer drivers. KIF5B-RET gene was recently identified as a primary driver in a subset of lung adenocarcinomas. Targeting human KIF5B-RET to epithelia in Drosophila directed multiple aspects of transformation including hyperproliferation, epithelial-to-mesenchymal transition, invasion, and extension of striking invadopodia-like processes. KIF5B-RET-transformed human bronchial cell line showed similar aspects of transformation including invadopodia-like processes. Through a combination of genetic and biochemical studies we demonstrate that the kinesin and kinase domains of KIF5B-RET act together to establish an emergent microtubule and RAB vesicle-dependent RET-SRC-EGFR-FGFR ‘signaling hub’. We demonstrate that drugs designed to inhibit RET alone work poorly in KIF5B-RET-transformed cells. However, combining the RET inhibitor sorafenib with drugs that target EGFR or microtubules or FGFR led to strong efficacy in both Drosophila and human cell line KIF5B-RET models. This work demonstrates the utility in exploring the full biology of fusions to identify rational therapeutic strategies.
Keywords: KIF5B-RET, NSCLC, kinase-fusions, Drosophila, EGFR, FGFR
Graphical abstract
Das and Cagan find that each portion of the KIF5B-RET fusion oncoprotein recruits different components to assemble a multi-kinase oncogenic signaling hub that promotes invadopodia formation. This suggests that multiple kinase components of this KIF5B-RET hub need to be simultaneously targeted therapeutically.
Introduction
Approximately 1.3 million new cases of non-small-cell lung cancer (NSCLC) patients are identified each year, comprising 80% of primary lung cancers (McCoach and Doebele, 2014). The prognosis for advanced and metastatic NSCLC patients is poor (Jemal et al., 2010). The identification of specific genetic alterations in large cohorts of NSCLC patients (e.g., KRAS and EGFR) has yielded a potential avenue for matching treatments (e.g., RAS and EGFR pathway inhibitors) to specific groups of patients. Inhibition of these pathways using tyrosine kinase inhibitors (TKIs) have led to some clinical benefits, indicating the importance of stratifying patient populations based on driving genetic alterations (Pao et al., 2004).
Multiple gene fusions involving the Rearranged-during-transfection (RET) kinase were recently identified in lung adenocarcinoma patients, including KIF5B-RET, NCOA4-RET, and CCDC6-RET (Ju et al., 2012; Kohno et al., 2012; Lipson et al., 2012; Takeuchi et al., 2012). KIF5B-RET fusions account for approximately 2% of all NSCLC patients, primarily nonsmokers whose tumors exhibit few other genetic changes in known cancer drivers (Takeuchi et al., 2012). Current efforts to treat patients with KIF5B-RET fusion driver oncogenes are focused on RET pathway inhibition with eight active ongoing NSCLC clinical trials (McCoach and Doebele, 2014). However, our previous Drosophila studies indicated that the RET fusions NCOA4-RET and CCDC6-RET act through different signaling pathways and respond to different anti-cancer drugs, indicating functional differences that may impact patient treatment (Levinson and Cagan, 2016). The nature of the differences between activating point mutant RET isoforms including RET fusions is unknown.
In this study we find that KIF5B-RET’s C-terminal RET kinase domain activates canonical signaling pathways while its N-terminal KIF5B domain activates multiple RTK’s including EGFR and FGFR signaling. The result is an emergent network that best responds to multi-targeting therapeutic cocktails. Therapeutics for other kinase-fusion oncogenes may benefit from understanding the signaling pathways activated by each portion of the protein and how they act in concert to direct a unique transformation network.
Results
Human KIF5B-RET activates EGFR signaling in a Drosophila model
We cloned a patient-derived KIF5B-RET cDNA (Kohno et al., 2012) into a UAS-based expression vector, allowing us to target expression of the UAS-KIF5B-RET transgene to various fly tissues using the binary GAL4-UAS system (Suppl. Fig. 1E). Uniform KIF5B-RET expression in the third instar larval wing epithelium (‘wing disc’) with a 765-GAL4 driver (765>KIF5B-RET; Suppl. Fig. 1B) led to adult wings with multiple ectopic veins, which is indicative of elevated RAS/MAPK signaling (Karim and Rubin, 1998). 765>KIF5B-RET’s ectopic venation phenotype was stronger than similarly targeted expression of wild type human RET (see below) or expression of the Drosophila oncogenic ortholog dRETM955T (765>dRETM955T), a constitutively active point mutant RET variant commonly observed in medullary thyroid carcinoma patients (Beldjord et al., 1995). Control flies displayed normal wing venation (Suppl. Fig. 1A–C). We conclude that human KIF5B-RET is capable of strongly activating the RAS/MAPK signaling pathway, a primary downstream effector of signaling by RET and other receptor tyrosine kinases (RTKs). Consistent with this view, RET activity was elevated in western blot analyses using an antibody to phosphorylated RET (pRET; Fig. 1E).
Figure 1. KIF5B-RET activates and localizes EGFR to filopodia/invadopodia-like processes.

(A–B) Third instar larval wing epithelia, en face view. Expression of human KIF5B-RET in a central stripe of cells (ptc>hKIF5B-RET, marked by eGFP expression) resulted in upregulation of pEGFR levels. Some KIF5B-RET expressing cells migrated away (B; asterisk) from the ptc region where the oncogene was expressed. Immunofluorescence images are composite overlays of z-stacks spanning the full depth of the epithelia. In subsequent figures hKIF5B-RET is referred to as KIF5B-RET. Dotted line shows region where lateral view of z-series is shown in (F, G). (C–D) Expression of ptc>hKIF5B-RET led to strong expression of EGFR activity reporter aos-lacZ compared to controls. Anti-β-galactosidase antibody was used to detect reporter activity. (E) Western blot of developing wing epithelia of the indicated genotypes; syntaxin was used as loading control. Four independent KIF5B-RET transgenic lines (765>KIF5B-RET) showed upregulation of pRET, pEGFR, total EGFR, and pERK levels. Expression of activating point mutant RET (765>dRETM955T) did not show upregulation of pEGFR. (F–G) Many ptc>KIF5B-RET cells extruded basally and showed basal enrichment of pEGFR signal (G. solid arrow). Arrowhead indicates invagination of epithelia as cells extrude basally. Lateral view of z-series showing the full depth of epithelia. E-cadherin marks apical region. (H–I) ptc>KIF5B-RET cells displayed filopodia-like processes enriched with pRET signal. These processes can extend laterally beyond KIF5B-RET-expressing cell bodies marked by GFP (H′ and H″, I′ and I″; asterisk). I′–I″: examples of especially long processes (asterisks). (J–K) In KIF5B-RET cells, the pRET signal is localized throughout the filopodia-like processes (K; asterisks). Processes are visualized by cytoskeletal marker Moesin fused to GFP (ptc>moesinGFP, KIF5B-RET). (L) High resolution microscopy shows pEGFR signal is also enriched in these laterally projecting filopodia-like processes. A higher magnification view (L′) shows that the pEGFR signal extends to the distal ends.
EGFR signaling is an axis of the RAS/MAPK cascade that regulates wing vein development (Martin-Blanco et al., 1999). Expressing the KIF5B-RET transgene in a stripe of cells at the center of the wing disc (ptc>KIF5B-RET) led to a strong upregulation of EGFR activity in vivo as assessed with an antibody to activated, phosphorylated EGFR (pEGFR; Fig. 1B). Control and ptc>dRETM955T (Suppl. Fig. 1D) flies did not show similar upregulation of pEGFR signal. In Drosophila, EGFR signaling activates transcription of downstream targets including argos and pointed. Wing discs expressing ptc>KIF5B-RET displayed in vivo upregulation of β–Galactosidase reporters for each of these genes (aos-lacZ, Fig. 1C, D; pnt-lacZ, Suppl. Fig. 1F, G), indicating that upregulation of EGFR activity led to activation of its canonical downstream signaling. In addition, 765>KIF5B-RET wing discs showed strong upregulation of pEGFR by western blot analysis (Fig. 1E). Knockdown of Drosophila EGFR can suppress the vein thickening phenotype (ptc>KIF5B-RET; EGFRRNAi; Suppl. Fig. 1I, K) in KIF5B-RET cells indicating EGFR activation by KIF5B-RET has functional outcomes. Together, this data indicates that KIF5B-RET—in contrast to RETM918T—directs a complex signaling mechanism that includes activation of EGFR. To understand how KIF5B-RET and EGFR work together to promote transformation, we explored their subcellular localization.
KIF5B-RET localizes EGFR to filopodia, invadopodia-like processes
Expression of KIF5B-RET in wing epithelial cells led to these ptc>KIF5B-RET cells shifting basally, an indication that they are undergoing an epithelial-to-mesenchymal transition similar to our previous SRC- and RET-based models (Fig. 1G; (Dar et al., 2012; Vidal et al., 2006). Distinct from these models, however, ptc>KIF5B-RET cells extended long processes into the neighboring region of wild type cells (Fig. 1H, I), a phenomenon not observed in control tissue (Fig. 1F). Interestingly pRET immunofluorescence signal was detected along the length of these processes (Fig. 1H, I; asterisks).
To more precisely analyze distribution of KIF5B-RET and EGFR, we co-expressed GFP-tagged Moesin (ptc>moesin-GFP,KIF5B-RET), an Actin-binding protein that outlines cellular processes. Using high resolution fluorescence microscopy, we found that pRET, pEGFR, and pSRC (Fig. 1J–L′, Fig. 4E below) were present throughout the filopodia-like processes. Confocal z-stacks indicated that pEGFR protein was also enriched basally in ptc>moesin-GFP,KIF5B-RET cells that had moved basally (Fig. 1G).
Figure 4. KIF5B-RET regulates pEGFR through RAB GTPases, GRIP1.

(A) Quantitative viability tests to assess whether reducing specific KIF5B-RET pathway components suppresses pupal lethality induced by ptc>KIF5B-RET. Percent viability (eclosion) represents number of adults that eclose after 12–14 days divided by the total number of embryos originally present (n). Asterisks indicate significance at P<0.05 for each genotype compared to DMSO control using student-T test with Welch’s correction; error bars represent SEM here and in subsequent figures; see experimental procedures. For each datapoint, >KIF5B-RET + “genotype (n=total number of embryos analyzed)”: KIFB-RET_DMSO(202); +Rab5RNAi(255); +Rab6RNAi(242); +Rab7RNAi(218); +Rab8RNAi(301); +Rab9RNAi;(218); +Rab11RNAi(318); +Rab21RNAi(257); +Rab23RNAi(234), +GRIP1RNAi(273), +GRIP#2RNAi(136), +Arp66BRNAi(234), +CortactinRNAi(239), +Src64BRNAi(313), +PvrRNAi(91), +RetRNAi(140), +FGFR#1(btl)RNAi (366), +FGFR#2(htl)RNAi(336), +InRRNAi(201). (B–C) Low magnification immunofluorescence images: in ptc>eGFP,KIF5B-RET wing discs, almost all discs showed strong activation of pEGFR (B′, asterisks). Simultaneous knockdown of Rab9 (ptc>eGFP, KIF5B-RET, Rab9RNAi) reduced the number of discs showing high pEGFR activation (C′). Representation of relative number of discs showing high, low, and no detectable pEGFR expression (B″, C″), where (n) represents number of discs analyzed. Examples of wing discs in inset marked by white arrowhead; dotted line outlines entire tissue. (D–K) Immunofluorescence images of wing discs showing effect of GRIP1RNAi knockdown on phospho-protein marker activation by KIF5B-RET. ptc>KIF5B-RET cells displayed high levels of pRET, pSRC, pEGFR, and pFGFR. Including knockdown of kinesin cofactor protein GRIP1, (ptc>KIF5B-RET,GRIP1RNAi) did not alter pRET, pSRC, or pFGFR(H, I, K) but strongly suppressed pEGFR activation(J). (L–M) Knockdown of GRIP1 suppressed loss of polarity induced by KIF5B-RET expression. ptc>KIF5B-RET cells showed loss of localized apical E-Cadherin and high levels of basally localized pSRC in distinct cellular processes (L, asterisk). Including knockdown of GRIP1 (ptc>KIF5B-RET,GRIP1RNAi) maintained E-cadherin primarily in the apical region of the epithelia and showed low levels of pSRC that failed to strongly enrich in basal processes (M, asterisk).
Thus, KIF5B-RET expressing cells displayed the presence of cellular processes indicative of migratory and invasive properties, including filopodia and invadopodia-like structures enriched with active EGFR and RET receptors. We next investigated whether KIF5B-RET activated canonical RET pathways and whether other features of cellular transformation were present.
KIF5B-RET activates canonical RET-signaling through SRC
Previous work including our own has shown that activating point mutant RET isoforms promote signaling in part through the SRC signal transduction pathway (Dar et al., 2012; Liu et al., 2004; Read et al., 2005). Wing cells expressing ptc>KIF5B-RET strongly and cell-autonomously upregulated pSRC activity as assessed with a pSRC-specific antibody (Fig. 2B, 2J); pSRC was localized to the basal region of the epithelium (Fig. 2D). EGFR and SRC are associated with invadopodia-like structures in migrating or metastatic cancer cells (Mader et al., 2011). Indeed KIF5B-RET expressing cells showed upregulation and basal localization of Arp3 (Fig. 2E, F), a key structural component of invadopodia (Clark et al., 2007). Thus, KIF5B-RET activated and localized pSRC, pEGFR, and ARP3—central components of invadopodia—to basal regions of the epithelium. We next investigated if other aspects of transformation were altered including degradation of basal lamina and cell polarity. Using a fly strain harboring a Collagen-GFP fusion transgene (Buszczak et al., 2007), we found that the basal lamina of KIF5B-RET expressing cells was strongly degraded in contrast to controls or adjacent wild type cells (Fig. 2G, H). KIF5B-RET expressing cells also showed loss of polarity: E-cadherin which is normally present primarily in the apical regions of epithelia, was delocalized (Fig. 2I). Western blot analysis of wing epithelium expressing KIF5B-RET showed upregulation of cell motility regulators pJNK, RAC1, and RHO1 levels (Fig. 2J; Suppl. Fig. 1L, M).
Figure 2. KIF5B-RET activates canonical RET-signaling through SRC.

(A–B) ptc>KIF5B-RET cells (B) showed strong upregulation of pSRC signal compared to controls (A), in third instar larval wing epithelium. (C–D) In control cells pSRC was localized to apical regions of epithelia, overlapping E-cadherin. In ptc>KIF5B-RET cells, pSRC re-localized to strongly accumulate in basal regions (arrowheads) of epithelia. Lateral z-series view. (E–F) ptc>KIF5B-RET cells showed localized upregulation (E) or uniform upregulation (F) of Arp3 in basal regions. Arrowhead indicates basal region, bracket indicates region expressing KIF5B-RET. (G–H) ptc>KIF5B-RET cells showed strong reduction of basal lamina signal compared to control cells as visualized with Collagen-GFP and MyR-RFP. Bracket indicates RFP-positive ptc>KIF5B-RET cells associated with reduced basal lamina. Collagen-GFP signal in the apical region is from overlying peripodial epithelia. (I–I′) ptc>moesinGFP,KIF5B-RET cells showed loss of polarity: E-cadherin was no longer localized to apical adherens junctions and instead was distributed uniformly within cells. Compare with (C). pSRC was enriched in processes extending beyond basal lamina (asterisks, I). Bracket indicates region of KIF5B-RET cells. (J) Western blot of developing wing epithelia of the indicated genotypes. Compared to control, four independent KIF5B-RET transgenic lines (765>KIF5B-RET) showed upregulation of pJNK, pSRC, Rac1, Rho1.
Taken together, KIF5B-RET signaling alters key aspects linked to cellular transformation. KIF5B-RET expression led to elevated activity of canonical RET signaling effectors including SRC, but also upregulated at least one additional pathway, EGFR. We therefore explored how different KIF5B-RET structural domains contributed to each of these activities.
The KIF5B-RET motor domain regulates pEGFR and pFGFR activation
We used deletion and point mutation based structure/function studies to explore how each domain contributes to the complex mechanisms by which KIF5B-RET promotes transformation. The KIF5B-RET fusion protein consists of three major structural domains: motor, coiled-coil, and kinase. We generated three KIF5B-RET variant fly models: ΔMD (kinesin motor domain deletion), ΔCC (coiled-coil domain deletion), and 3Y-3F (tyrosine to phenylalanine changes of the three key residues 905, 1015, 1062 within the kinase domain of RET (Plaza-Menacho et al., 2014); Fig. 3I). Each domain was fused to the inducible UAS promoter, and each UAS-transgene—including KIF5B-RET and RET controls—was targeted to the same genomic site using the attp system to ensure similar expression levels (Groth et al., 2004).
Figure 3. KIF5B-RET motor domain regulates pEGFR and pFGFR activation.

(A–A‴) Control cells showed basal levels of pRET, pEGFR, pSRC, and pFGFR signal in developing wing epithelia. (B–B‴) ptc>KIF5B-RET cells showed strong upregulation of all four markers. (C–C‴) KIF5B-RET(ΔMD) construct activates pRET (C) but cannot activate pEGFR, pSRC, and pFGFR. (D–D‴) KIF5B-RET(ΔCC) and, (E–E‴) KIF5B-RET(3Y-3F) variant failed to activate all four markers.(F–F‴) Expression of the intact human RET gene RET(WT) activates the three markers, pRET, pSRC, and pFGFR only slightly above basal levels. It fails to activate pEGFR. (G–G‴) Expression of CCDC6-RET activated pRET, and weakly activated pSRC and pFGFR, but did not activate pEGFR. (H–H‴) Expression of NCOA4-RET activated all four markers moderately. (I) Schematic of altered versions of the human KIF5B-RET transgene used in our structure-activity-relationship (SAR) studies. ΔMD = deletion of motor domain, aa’s 2-324; ΔCC = deletion of coiled-coil domain, aa’s 324-582; 3Y-3F = tyrosine residues 905, 1016, 1062 altered to phenylalanine. Arrowhead indicates point of gene fusion; asterisks indicate point mutations within the kinase domain. (J) Western blot of developing whole wing discs (765>KIF5B-RET) expressing different variants of KIF5B-RET. (K) Summary table comparing three RET-fusion isoforms with WT-RET and their ability to activate the four markers and induce filopodia/invadopodia.
In these models we evaluated the state of RET activation (pRET), canonical RET signaling (pSRC), and emergent KIF5B-RET signaling (pEGFR). While ptc>KIF5B-RET showed strong activation of these three markers (Fig. 3B–B″), removing the motor domain (ptc>KIF5B-RET[ΔMD]) led to complete loss of detectable pEGFR signal; (Fig. 3C′). Deletion of the motor domain also resulted in loss of pERK levels as assessed by western blot analysis (Fig. 3J). This indicated that the kinesin motor domain is primarily required for KIF5B-RET to activate EGFR and downstream MAPK signaling. ptc>KIF5B-RET[ΔMD] wing epithelia still retained significant but reduced pRET levels (Fig. 3C), suggesting that the high levels of pRET and pSRC activity observed in KIF5B-RET cells depends on recruitment and activation of EGFR through the motor domain.
Inactivating the kinase domain (ptc>KIF5B-RET[3Y-3F]; Fig. 3E–E″) or removing the coiled-coil domain (ptc>KIF5B-RET[ΔCC]; Fig. 3D–D″) led to loss of all three markers, indicating that dimerization through the coiled-coil domain and an active RET kinase domain were essential for full KIF5B-RET activity (also see Fig. 3J). Activation of other downstream pathways as measured by pERK, pJNK, and RAC1 levels were also downregulated in both KIF5B-RET[3Y-3F] and KIF5B-RET[ΔCC] variants (Fig. 3J).
We next tested if other RTK’s were also recruited to this complex. Indeed KIF5B-RET upregulated activated FGFR (pFGFR) levels (ptc>KIF5B-RET; Fig. 3B‴ and 3J) as well as total levels of Drosophila PDGFR/VEGFR ortholog (Pvr; western blot, Suppl. Fig 2A). We restricted our analysis to RTK’s for which working phospho-specific antibodies in Drosophila were available, i.e., pEGFR and pFGFR (Gibson et al., 2012). Activation of pFGFR was also dependent on the kinesin portion of the fusion protein (Fig. 3C‴ and D‴) as well as the kinase domain of RET (Fig. 3E‴). The strong activation and localization of multiple RTK’s (pEGFR and pFGFR) by KIF5B-RET was unique, as two RET fusions implicated in different cancers did not show similar upregulation of these pathways. CCDC6-RET activated pRET, pSRC, and pFGFR weakly and did not activate pEGFR (Fig. 3G–G‴; Fig. 3K) while NCoA4-RET activated all three markers moderately (Fig. 3H–H‴; Fig. 3K). Neither of these other RET-fusion proteins provoked formation of filopodia/invadopodia processes (Fig. 3G–H and Fig. 3K).
The failure of ptc>KIF5B-RET[3Y-3F] to activate EGFR, SRC, and FGFR is especially notable, indicating that RET kinase domain activity is required to establish a multi-kinase RET-SRC-EGFR-FGFR signaling hub. That is, the RET kinase domain acts with EGFR, FGFR, and SRC to mediate the full range of KIF5B-RET signaling. The requirement of pEGFR and pFGFR for an intact motor domain also suggested a novel multi-RTK dependent signaling mechanism dependent on the kinesin domain, a possibility we further explored.
KIF5B-RET regulates pEGFR and pFGFR through RAB GTPases
Kinesin motor domain proteins transport cargo to distant cellular sites including signaling effectors such as GRB2 and RAB vesicles (Hirokawa et al., 2009). RAB vesicles regulate receptor tyrosine kinase recycling and internalization; kinesin motors help deliver these kinase/vesicle complexes to specific sites for localized signaling (Hirokawa et al., 2009). We identified a panel of 16 Kinesin cargo adaptor molecules including eight RAB proteins and by targeted knockdown assessed if they are required to recruit and activate pEGFR and pFGFR (Fig. 4A, Suppl. Fig. 2B; (Hirokawa et al., 2009).
We assessed requirement of these genes for KIF5B-RET function using a previously developed quantitative Drosophila viability assay (Dar et al., 2012). Expression of oncogenic KIF5B-RET in multiple developing tissues (ptc>KIF5B-RET) led to highly penetrant pupal lethality: only 2.3% of developing animals eclosed as adults. We found that RNA interference (RNAi)-mediated knockdown of components of the Src-invadopodia-complex and of individual rab genes increased the number of ptc>KIF5B-RET animals reaching adult stages (Fig. 4A); this indicated that RAB proteins normally function to promote KIF5B-RET activity. Knockdown of RAB vesicles could in principle traffic different RTK’s and indeed knockdown of Ret, Pvr, FGFR, as well as InR, all increased adult eclosion rates (Fig. 4A).
Focusing on the key rab gene rab9, knockdown (ptc>KIF5B-RET, rab9RNAi) led to strong reduction of KIF5B-RET-mediated pEGFR and pFGFR activation, indicating that the RAB machinery is involved in the recruitment and activation of pEGFR and pFGFR (Fig. 4B–C″ and Suppl. Fig. 2C). Knockdown of rab9 significantly reduced pSRC levels but knockdown EGFR, FGFR, and the other tested RTK’s had mild effects on pSRC (Suppl. Fig. 2D) and pRET levels (Suppl. Fig. 2E). This suggested that removal of individual RTK’s bound to KIF5B-RET had little effect on pSRC activation, but simultaneous removal of multiple RTK’s (pEGFR, pFGFR, etc), e.g., through rab9 knockdown, compromised pSRC activation. This data together with our SAR analysis indicated that KIF5B-RET molecule had a bipartite function: the RET kinase domain function (pSrc activation) could be uncoupled from the kinesin domain function (pEGFR and pFGFR activation) but there was mutual dependence of these kinase molecules to establish full activity of KIF5B-RET.
Knockdown of another Kinesin adaptor protein, Glutamate Receptor Interacting Protein 1 (GRIP1; (Setou et al., 2002) also increased survival of ptc>KIF5B-RET flies to adulthood (Fig. 4A). GRIP1 knockdown also resulted in loss of pEGFR activation by KIF5B-RET(Fig. 4F, J); notably, levels of pSRC and pRET were not affected, uncoupling EGFR and SRC activation (Fig. 4D, E, H, I). GRIP1 knockdown did not affect pFGFR activation indicating that recruitment of pFGFR relies on an yet unknown adaptor protein (Fig. 4G, K). GRIP1 knockdown also suppressed aspects of EMT including cell polarity as assessed by restoration of proper E-cadherin localization, providing further evidence that the GRIP1-EGFR axis is functionally required for KIF5B-RET-mediated transformation (Fig. 4L, M). GRIP1 has been previously implicated in modulating EGFR function in human cells (Yokomaku et al., 2005), and our findings extend these observations to KIF5B-RET.
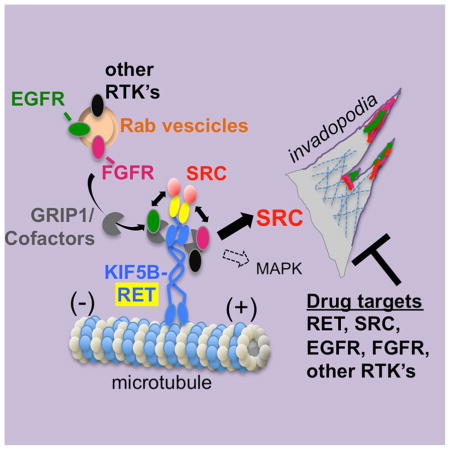
Taken together our fly studies support a model in which the KIF5B-RET fusion protein recruits a multi-protein ‘signaling hub’ through its KIF5B kinesin domain plus its RET kinase domain. This ‘signaling hub’ includes cofactors/adaptors such as RAB proteins and GRIP1 that, through their association with KIF5B-RET’s motor domain, serve as specific activators of multiple RTK’s like EGFR and FGFR.
HBEC3[KIF5B-RET] lung cells exhibit multiple aspects of transformation
We developed a lung cancer cell line model to determine which aspects of this ‘signaling hub’ observed in Drosophila were also relevant to KIF5B-RET-transformed human cells. To model lung adenocarcinoma (LADC) we used HBEC3-KT cells, normal human bronchial epithelial cells immortalized with CDK4 and hTERT (Sato et al., 2006). Using the pLenti6 vector system (Invitrogen), we generated multiple independent stable HBEC3 transformants that expressed KIF5B-RET.
Independent HBEC3[KIF5B-RET] lines exhibited significant differences with the parental line: most parental HBEC3 cells died a few days after reaching confluence, whereas HBEC3[KIF5B-RET] cells survived for several weeks (Fig. 5A, B); transformants showed robust growth in the absence of serum in contrast to parental cells (Fig. 5C). Analysis of cell biological features indicated that HBEC3[KIF5B-RET] cells extended striking filopodia-like processes (Fig. 5E) as well as large numbers of Actin-rich puncta compared to parental cells (Fig. 5J). Actin-rich puncta are characteristic of invadopodia and are used to measure the relative frequency of invadopodia (Hoshino et al., 2013). HBEC3[KIF5B-RET] cells displayed a 7-fold increase in invadopodia-like structures (Suppl. Fig 3B), mirroring results in our Drosophila KIF5B-RET model.
Figure 5. HBEC3[KIF5B-RET] lung cells exhibit multiple aspects of transformation.

(A–B) Phase contrast images of parental HBEC3 and stably transfected KIF5B-RET cells. Parental cells started dying after 7 days while KIF5B-RET cells maintained cell-cell contacts and survived beyond 21 days. (C) Incucyte based live imaging (every 6 hours) and confluency analysis. Parental HBEC3 cells (top panel) grew poorly in media lacking growth factor EGF, while KIF5B-RET cells (bottom panel) grew rapidly to reach confluency in 4 days. (D–E) Phase contrast images of KIF5B-RET cells extending long filopodia-like processes (E; asterisk). (F) Cell Signaling Pathscan RTK Antibody Array images of protein lysates from parental HBEC3 and KIF5B-RET cells. Examples of kinases deregulated in KIF5B-RET cells provided as a list matched to colored boxes. Array list in Suppl. Fig. 3A. (G) Western blot of protein lysates from parental HBEC3 and two independent KIF5B-RET transfected cell lines. KIF5B-RET cells showed upregulation of EMT markers Slug and N-Cadherin, and the protumorigenic stem cell marker Sox2. dpERK levels were reduced in KIF5B-RET cells. (H) Western blot demonstrating that KIF5B-RET transfected cell lines upregulate KIF5B and pRET[Y1062] levels within physiological levels. In addition, Rab9 and Rab7 were moderately upregulated while Rab5 was more weakly upregulated. Regarding EGFR, pEGFR[Y845] was upregulated while pEGFR[Y1068] was downregulated. (I–J) Actin cytoskeleton of KIF5B-RET transfected and parental cells visualized by Phalloidin-Rhodamine staining. KIF5B-RET cells contained a large number Actin-rich puncta (arrows) characteristic of invadopodia. There was a 7-fold increase (see Suppl. Fig. 3B) in Actin-rich puncta in KIF5B-RET cells (median 15.5/cell) compared to parental HBEC3 cells (median 2.5/cell). (K–L) KIF5B-RET cells showed strong upregulation of tumorigenic stem cell transcription factor Sox2 in the nuclei. Parental cells showed low levels of Sox2 that was mostly excluded from nuclei.
Western blot analysis of HBEC3[KIF5B-RET] cells indicated upregulation of EMT markers N-cadherin and Slug (Fig. 5G), phenocopying important aspects of our Drosophila model. Stem cell fate factor SOX2 controls genetic programs that drive tumorigenesis and cancer cell motility including lung cancers (Boumahdi et al., 2014; Siegle et al., 2014). Some studies, including ours, have also shown that SOX2 levels are often upregulated in cancer cells after therapeutic treatment (Rothenberg et al., 2015); Das et al, unpublished), suggesting a role for stem cell fate effectors in promoting tumorigenesis as well as resistance to therapy. We found that SOX2 levels were strongly upregulated in multiple HBEC3[KIF5B-RET] cell lines compared to HBEC3 parental cells when grown to confluency (Fig. 5G, K, L). We conclude that KIF5B-RET promotes key aspects of cellular transformation in human bronchial epithelial cells.
HBEC3[KIF5B-RET] cells signal through multiple cancer-related axes
To explore the overall state of the HBEC3[KIF5B-RET] kinase network we performed a phospho-kinase array analysis (Cell Signaling RTK Pathscan Array). HBEC3[KIF5B-RET] cells showed broader activation of kinases compared to parental cells (Fig. 5F). Similar to our Drosophila KIF5B-RET models, pSRC levels were strongly upregulated. Phosphorylation of RTKs such as HER2, HER3, FGFR1, FGFR3, and FGFR4 were also upregulated, as was the PI3K pathway effector AKT at two positions (471, 304). Interestingly, we observed a strong downregulation of the RAS pathway effector pERK1/2 (see also Fig. 5G).
Using western blot analysis we confirmed modest increases in levels of the N-terminal region of KIF5B and phosphorylation of RET at position 1062 (pRET[Y1062]; Fig. 5H). RAB proteins RAB5, RAB7, and RAB9 were moderately upregulated within physiological levels (Fig 5H). We observed a significant increase in a SRC-dependent tyrosine phosphorylation in EGFR (pEGFR[Y845]); interestingly, we observed a significant decrease in MAPK-dependent EGFR phosphorylation, pEGFR[Y1068]. This switch from ERK-dominant to SRC-dominant EGFR phosphorylation mirrors the overall changes in activity of these two cytoplasmic kinases (Fig. 5F, G). These results raise an interesting mechanistic question: what components of the KIF5B-RET ‘signaling hub’ regulate this ERK-to-SRC switch in signaling specificity?
HBEC3[KIF5B-RET] cells showed cofactor-dependent pEGFR signal switching
In situ immunofluorescence staining confirmed that HBEC3[KIF5B-RET] cells grown to confluence exhibited increased pEGFR[Y845] and decreased pEGFR[Y1068]; conversely, parental cells exhibited low pEGFR[Y845] and high pEGFR[Y1068] levels (Fig. 6A, B, E, F). This further emphasizes a KIF5B-RET switch from ERK-dominant to SRC-dominant signaling. Removal of EGF from the growth media resulted in complete loss of the high basal pEGFR[Y1068] signal of parental cells (Suppl. Fig. 4D, E), indicating these phosphorylation sites are indeed bona fide predictors of EGFR signaling.
Figure 6. HBEC3[KIF5B-RET] cells show cofactor-dependent pEGFR signal switching.

(A–D) Immunofluorescence images showing upregulation of pEGFR-Y845 levels in KIF5B-RET cells (B) compared to parental cells (A). siRNA-mediated knockdown of GRIP1 (C) and SRC (D) suppressed the increase of pEGFR-Y845 levels in KIF5B-RET cells. DAPI labels nuclei and Phalloidin-Rhodamine labels Actin cytoskeleton. (E–H) Immunofluorescence images showing strong downregulation of pEGFR-Y1068 levels in KIF5B-RET cells (F) compared to parental cells (E). siRNA mediated knockdown of GRIP1 (G) and SRC (H) suppressed the effect of KIF5B-RET by moderately increasing levels of pEGFR-Y1068. (I) Western blot of KIF5B-RET cells with knockdown of indicated genes. Numbers indicate final concentration of siRNA (nM) in media. While KIF5B-RET cells showed low levels of dpERK (lane 1; also see Fig. 5F, G), knockdown of SRC, RAB9A, or GRIP1, led to increased dpERK levels. RAB9A knockdown led to reduction of pSRC levels similar to the fly experiments. RAB9A and SRC blot panels confirm that siRNA knockdown led to strong reduction in RAB9A and SRC levels.
We next performed siRNA mediated knockdown of components of the KIF5B-RET ‘signaling hub’ to establish their functional requirement. Using siRNA-directed knockdown on confluent HBEC3[KIF5B-RET] cells, reduction of GRIP1, SRC, or RAB9A all led to strong suppression of the increased pEGFR[Y845] signal induced by KIF5B-RET (Fig. 6C, D and Suppl. Fig. 4C). In contrast, knockdown of these components moderately increased the pEGFR[Y1068] signal in HBEC3[KIF5B-RET] cells (Fig. 6G, H and Suppl. Fig. 4G). Knockdown of GRIP1, SRC, or RAB9A in KIF5B-RET cells also led to re-establishment of high pERK levels (Fig 6I). Together, this data indicates that SRC, GRIP1, and RAB9A normally mediate KIF5B-RET-mediated switching of EGFR from predominantly phospho-Y1068 to predominantly phospho-Y845, that is, switching from an ERK-associated phosphorylation event to a SRC-associated event.
This switch in preferred EGFR phosphorylation sites distinguishes activating point mutant RET from KIF5B-RET. We explored ways to incorporate these findings to identify therapeutic approaches better tailored for suppressing KIFB-RET-mediated transformation.
Inhibiting EGFR signaling improves therapeutic targeting of the KIF5B-RET network
Using our fly viability assay we screened a panel of 66 FDA approved drugs for their ability to rescue KIF5B-RET induced pupal lethality (ptc>KIF5B-RET). The list (Suppl. Fig. 6) included the polypharmacological lead AD80, a kinase inhibitor previously demonstrated to inhibit RET and downstream signaling components (Dar et al., 2012). In this assay, AD80 showed the best efficacy profile, rescuing pupal viability from ~2% (controls) to ~70% viability (Fig. 7A). Other RET pathway inhibitors regorafenib and sorafenib and the MEK inhibitor trametinib were less effective, while FDA approved drugs for RET-driven tumors including vandetanib and cabozantinib provided only marginal improvement in fly viability. HBEC3[KIF5B-RET] cells were also poorly responsive to vandetanib or cabozantinib (Fig. 7C, Suppl. Fig. 7A, B), further indicating that canonical RET inhibitors are not effective in reducing KIF5B-RET-mediated transformation. Interestingly, neither was as effective as the EGFR inhibitor erlotinib in confluent HBEC3[KIF5B-RET] cells (Fig. 7C), again emphasizing the importance of EGFR in KIF5B-RET-mediated transformation.
Figure 7. Inhibiting EGFR signaling is optimal for therapeutics targeting KIF5B-RET network.

(A) Screening a panel of 66 FDA-approved and investigational drugs (See list Suppl. Fig. 6) in a ptc>KIF5B-RET fly viability assay. Mean shown as column graphs and SEM as error bars. Ratio represents the mean of the number of adults/pupae per vial; see experimental procedures. Asterisks indicate a significance of P<0.05 for each genotype compared to DMSO control (mean=2.1%). RET inhibitors regorafenib (=13.7%), sorafenib (=8.6%), AD80(=70.3%) and MEK inhibitor trametinib (=16.7%) showed ability to improve pupae to adult survival. (B) Drugs and combinations that suppressed pupal lethality in ptc>KIF5B-RET flies. 59.2% of AD80 treated (n=203) and 30.6% of sorafenib treated (400 μM, n=369) flies eclosed as adults vs. only 3.4% of DMSO control (n=509). Combined sorafenib/gefitinib treatment (n=253) led to 60% eclosure as adults. Sorafenib (200 μM, n=330) in combination with paclitaxel (0.5 μM) significantly increased viability (n=435) to 20%. (C) Effect of various inhibitors on growth rate of human HBEC3[KIF5B-RET] cells. Dose response curve fitted to non-linear regression model using PRISM software; IC50’s in brackets. Left panel: confluent cells showed weak sensitivity to clinically approved RET inhibitors vandetanib or cabozantinib (IC50s >10 μM) but were sensitive to both sorafenib and AD80 (IC50s = ~0.5 μM). EGFR inhibitor erlotinib showed intermediate effect with (IC50 = ~10 μM). Right panel: Combination of sorafenib/erlotinib or sorafenib/paclitaxel potently inhibited (IC50 = ~0.1 μM) growth of HBEC3[KIF5B-RET] cells. Doses of paclitaxel (3 nM) and erlotinib (0.1 μM) used in the combinations had little effect on growth of HBEC3[KIF5B-RET] cells by themselves (left panel). (D) Summary table of KIF5B network relevant targets for the drugs AD80, sorafenib, and vandetanib. (E) Model: KIF5B-RET fusion protein recruits pEGFR, pFGFR, and other RTK’s from RAB-vesicles via GRIP1 and other cofactors. pSRC activated by RET kinase domain cooperates with the recruited RTK’s for optimal signaling. EGFR signaling in lung cells is MAPK dominant (pEGFR[Y1068]) but active SRC switches EGFR signaling to a SRC dominant (pEGFR[Y845]) form.
To further assess the importance of inhibiting both RET and EGFR activity, we used fly wing discs as an in vivo assay: ptc>KIF5B-RET larvae were fed drugs orally and larval wing epithelia examined for pEGFR activity. Efficacy in flies and cell lines tracked with activity against EGFR: AD80 proved the most potent compound for inhibiting both pRET and pEGFR activation (Suppl. Fig. 5A, B). The FDA drug sorafenib—currently in clinical trials for RET-based cancers (Lam et al., 2010)—also showed significant inhibition of pEGFR in vivo though less than AD80 (Suppl. Fig. 5C). Fly tissues treated with AD80 still retained some pRET function (Suppl. Fig. 5B‴) in vivo, highlighting the need for targeting other relevant pathways for optimal therapeutics.
Our Drosophila structure/function studies demonstrated a key, previously undescribed, requirement for KIF5B-RET function: kinesin domain dependent EGFR activation. Kinesin motors depend on microtubules for cargo transport. We therefore tested clinically relevant microtubule inhibitors for their ability to inhibit EGFR activation. Microtubule inhibitors vincristine and paclitaxel moderately inhibited pEGFR activation (Suppl. Fig. 5D, E).
Addressing multiple pathways improves efficacy against KIF5B-RET transformation
Polypharmacological lead compound AD80’s targets include RET, SRC, and multiple EGFR targets including BRAF and S6K. In addition AD80 also inhibits FGFR, PDGFR, VEGFR, and InR (Supplement-(Dar et al., 2012) targets that were genetically required for KIFB-RET function in fly assays. AD80 provided the strongest rescue in our KF5B-RET Drosophila survival assay (Fig. 7A, B). The multikinase inhibitors AD80 and sorafenib also showed strong activity in confluent human HBEC3[KIF5B-RET] cells: both showed strongly reduced IC50 in HBEC3[KIF5B-RET] cells compared to the parental line—especially in confluent cultures—indicating that KIF5B-RET has conferred a dependence on RET kinase signaling (Fig. 7C, Suppl. Fig. 7A, B, C). AD80 was the most potent on low confluency cells (Suppl. Fig. 7B).
One key difference between the experimental compound AD80 and the FDA-approved drug sorafenib is that sorafenib is a poor inhibitor of SRC (Apsel et al., 2008), the primary signaling axis activated by EGFR in the context of KIF5B-RET. One prediction is that sorafenib’s efficacy would be enhanced by adding EGFR inhibitors such as gefitinib or erlotinib or by adding microtubule inhibitors like paclitaxel that inhibited pEGFR recruitment by KIF5B-RET. Indeed rescue of sorafenib-treated ptc>KIF5B-RET flies was improved when treatment included gefitinib, matching levels of AD80 rescue; sorafenib also performed better in the presence of paclitaxel (Fig. 7B). HBEC3[KIF5B-RET] cells showed strongly increased sensitivity when sorafenib was combined with low dose (0.1 μM) erlotinib or low dose paclitaxel (3 nM); AD80 showed little improvement when combined with erlotinib (Fig. 7C; Suppl. Fig. 7A, B; data not shown). While our manuscript was in submission a study, consistent with our findings, showed that RET and EGFR inhibitor combinations worked well against some RET-fusions (Vaishnavi et al., 2017).
Finally the BRAF-inhibitor vemurafenib was less effective on HBEC3[KIF5B-RET] cells than parental lines (Suppl. Fig. 7C), further highlighting the shift away from pERK1/2 activation. In summary our therapeutic studies indicate that combining available RET inhibitors with EGFR inhibitors or microtubule regulators can be a potent therapeutic strategy that better accounts for the unusual signaling networks activated by KIF5B-RET.
Discussion
Gene fusions represent one of the earliest described genetic aberrations linked to cancer (Mertens et al., 2015). Targeting the kinase portion of the fusion protein has proven a useful strategy: for example, treating patients with a BCR-ABL fusion using Abl inhibitors provided the first example of a successful treatment with targeted therapy (Cohen et al., 2002). Currently the therapeutic paradigm has focused primarily on targeting the kinase domain of the kinase fusion protein with tyrosine kinase inhibitors (TKIs). Model studies or patient trials in ALK, ROS1, BRAF, and RET- fusion containing cancers have focused on driver-specific TKIs as a primary therapeutic strategy (Galetta et al., 2012); TKIs have recently been approved for PDGFRB- and ALK- fusion containing epithelial tumors (Forde and Rudin, 2012; Wright and Petersen, 2007).
Our current findings challenge this paradigm of therapeutics that solely target the kinase function of fusion oncogenes. Our studies have shown that: a) the N-terminal of KIF5B-RET fusion protein recruits multiple RTK’s, b) RET kinase domain depends on components, e.g., SRC, GRIP1, RAB9A, for oncogenic signaling c) the assembled kinases form a multi-protein signaling hub, where they reinforce mutual phosphorylation, d) optimal therapy against KIF5B-RET requires inhibition of RET, SRC, EGFR, FGFR (Fig. 7D, E). Recent clinical trials of patients with RET-fusions showed KIF5B-RET patients to be the least responsive to RET inhibitor vandetanib(Yoh et al., 2017) compared to CCDC6-RET patients. In other trials of RET-fusion patients - using using various RET inhibitors (Drilon et al., 2016) (Gautschi et al., 2017) - objective response of KIF5B-RET patients was consistently low suggesting failure of single agent RET inhibitor therapy. These clinical findings support our studies. In these trials, KIF5B-RET fusions represented the most common RET-rearranged events in LADC patients, suggesting a potentially significant impact of our therapeutic findings for treatment.
Our studies have also uncovered a mechanism whereby KIF5B-RET protein switches intracellular signaling from being pEGFR-Y1068/MAPK dominant to being pEGFR-Y845/SRC dominant (Fig. 7E). Strong upregulation of pSRC and pEGFR in KIF5B-RET cells could likely increase the binding and affinity of EGFR to adaptors to potentiate downstream signaling (Begley et al., 2015), or promote invasive behavior in transformed cells (Mader et al., 2011). KIF5B-RET studies in mammalian cells have shown similar strong upregulation of the SRC pathway (Lin et al., 2016). In addition analysis a cohort of patients with KIF5B-RET rearrangement (Sarfaty et al., 2016) shows frequent visceral metastases, which is consistent with our findings of increased SRC-dependent local invasion through invadopodia formation.
With respect to the ‘signaling hub’ promoting oncogenic signaling by KIF5B-RET: we propose that the identity of RTK’s recruited by the RAB-vesicle would depend on the cell type and KIF5B-RET fusions occurring in other cancers could potentially assemble a different palate of RTK’s, with different signaling outcomes, thus requiring different therapeutics. This could explain why, in contrast to published KIF5B-RET data, our studies using human lung cells identified this multi-RTK containing signaling hub.
Finally, we have shown that KIF5B-RET signaling can be therapeutically inhibited by targeting RET, EGFR, FGFR, as well as SRC. Additional components of this hub like the RAB and kinesin machinery could be potentially targeted opening additional therapeutic windows into treating KIF5B-RET tumors. This would be especially important as RABs and Kinesins could be recruiting other, yet unidentified, signaling components into the KIF5B-RET signaling hub. Future studies can focus on whether additional components are recruited to this multi-protein signaling hub and the best therapeutic options for inhibiting them.
Experimental Procedures
Antibodies and Histology
Third instar wing discs were staged and fixed in 4% paraformaldehyde. Immuno-fluorescence was performed as described(Das et al., 2013). Antibodies used: anti-pRET[Y905], anti-pJnk, anti-pAkt, anti-SOX2, anti-slug, anti-NCadherin, anti-pEGFR[Y845], anti-pEGFR[Y1068], anti-pFGFR[Y653/654], anti-Rab5, anti-Rab7, anti-Rab9(Cell Signaling), anti-pSRC[Y418] (Invitrogen), anti-dpERK (SIGMA), plus anti-Actin,, anti-E-cadherin, anti-α-Catenin, anti-Rho1, anti-Syntaxin, anti-β-tubulin (Developmental Studies Hybridoma Bank), anti-Actin, anti-GAPDH antibodies (Santa Cruz Biotechnology), anti-Rac1 antibody (BD biosciences), anti-KIF5B(Abcam), anti-EGFR (Julia Cordero), anti-Arp3(William Theurkauf).
Comprehensive Statistical Analysis
For viability pupal analysis in Figure 4 & 7 mean and standard error of the mean (SEM) were calculated and 4–5 vials/experiments, biological replicates, per dose were analyzed and repeated at least 2 times. Each vial had between 20–80 developing embryos and total (n) indicated in legend represents total number of embryos analyzed. For the large 66 drug library screen 8 vials per drug, biological replicates, with approximately 20 pupae per vial were analyzed; ratio of eclosed adults/pupae was plotted. To assess statistical significance of difference between means, t-Test with Welch’s correction was performed using PRISM software. The correction was used to account for samples with unequal variances and unequal sample sizes. For MTT on cancer cancer cells each dose was performed in quadruplicates and mean signal and SEM analyzed.
Fly Stocks, Genetics, and Subcloning
Fly stocks: Bloomington and VDRC Drosophila stock centers. UAS-KIF5B-RET flies were generated by cloning a human cDNA obtained from Dr Ohno. For structure function analysis of hKIF5B-RET variants were generated using standard molecular biology subcloning techniques and clone into pUAST-attB vector. RET-WT DNA was obtained from Dr. Plaza-Menacho. Injection and creation of attp40 transgenics was done by BESTGENE INC.
Inhibitor Studies in Flies
Drugs were obtained from LC laboratories or Selleck Chemicals and were dissolved in DMSO as stock solutions ranging from 1–200mM. Drugs (500–1000 μl) were diluted in molten (~50–60°C) enriched fly food, aliquoted into 5 ml vials. 30–60 embryos of each genotype were raised on drug-containing food until they matured as third-instar larvae (wing disc western assay assay) or allowed to proceed to adulthood (viability assay and wing vein quantitation assay).
MTT Assay
Cancer cell lines were cultured in Airway Epithelial Basal Media supplemented with Bronchial Epithelial growth kit from ATCC. Cells were grown in 75 cm2 sterile polystyrene culture flasks to 80% confluency, trypsinized, and re-seeded in equal aliquots into 96-well plates. After 2 days and ~50% confluency, media was removed and replaced with DMSO or drug containing media. Cells were allowed to grow another 4 days (all other fast growing cancer lines) after which MTT assay was performed. Spectrophotometric readings at 590 nm and 630 nm using a 96-well plate reader were used to establish growth and viability of cells. Each drug dose was tested in quadruplicates and experiments repeated twice.
Phospho-protein Array Analysis
PathScan RTK Signaling Antibody Array Kit (Cat # 7982) was used to assess kinase activity of human cancer cell lines. Briefly, human cancer cells were plated at 50–60% confluency in 100 cm2 tissue culture plates in respective media and allowed to grow for 4, 5 days. Cells were washed with cold 1x PBS, scraped off into 1x Lysis buffer from kit, and lysates extracted. BIORAD protein assay was used to assess protein concentration of lysate. Antibody array was incubated with lysates at 0.5 mg/ml total protein concentration, as recommended by manufacturer, and developed according to manufacturer protocols.
Tissue Culture Studies
Cells were grown in Airway Epithelial Basal Media supplemented with Bronchial Epithelial growth kit from ATCC. Cells were transfected with Lipofectamine or HIPERFECT for siRNA experiments and cell lysis performed 24 and 48 hrs after transfection. For imaging studies, cells were grown to 50 or 100% confluency in Lab-TekII 8-chamber slides (Fisher), transfected, and growth for further 24–48 hrs in media.
Western Blot of Fly Wing Discs
30 third-instar discs of each genotype (765>UAS-transgene) were dissolved in Lysis Buffer (50 mM Tris, 150 mM NaCl, 1% Triton-X100, 1 mM EDTA) supplemented with protease inhibitor cocktail (Sigma) and phosphatase inhibitor cocktail (Sigma). Total protein in each sample was quantitated using BIORAD protein assay. Samples resolved on Invitrogen NU-PAGE gradient SDS-page and transferred by standard protocols. Membranes were stripped with SIGMA Restore stripping buffer and reprobed with other antibodies to assess signal under exactly the same loading conditions.
Western Blot of Cancer Cell Lines
HBEC3 cell lines were grown in 100cm2 well plates in Bronchial Airway Epithelial Media (ATCC) media each supplemented with pen/strep antibiotics. After required growth cells were washed twice with cold PBS, and then lysed in RIPA buffer (25 mM Tris pH 7.6; 150 mM NaCl; 1% NP-40; 0.1% SDS) containing protease and phosphatase inhibitors and sonicated (Roche). Lysate protein concentration was assessed using BIORAD protein assay. 5 or 10ug of total cell lysate were separated by SDS-PAGE, transferred to PVDF membrane and blotted for the indicated proteins using commercial antibodies. Membranes were stripped and probed as described above.
Whole Mount Imaging of Fly Wings
For adult wing vein analysis, wings were dissected and kept in 100% ethanol overnight, mounted on slides in 80% glycerol in phosphate buffered saline solution, and imaged by regular light microscopy using Leica DM5500 Q microscope.
Supplementary Material
Highlights.
KIF5B-RET kinesin domain activates various RTKs via RAB vesicles
The RET kinase domain activates canonical pathways including SRC
This multi-kinase hub shows enhanced signaling activity and promotes invadopodia
Optimal drugs against KIF5B-RET should target RET, EGFR, FGFR, PDGFR, and SRC
Acknowledgments
We thank members of the Cagan laboratory members, including Vanessa Barcessat, for technical assistance and for helpful discussions. We thank the Bloomington Drosophila Stock Center for Drosophila reagents. Microscopy was performed in part at the Microscopy Shared Resource Facility at the Icahn School of Medicine at Mount Sinai. This research was supported by National Institutes of Health grants R01-CA170495, R01-CA170495 and R01-CA109730, Department of Defense grant W81XWH-15-1-0111 and American Cancer Society grants 120886-PFM-11-137-01-DDC (T. D.) and 120616-RSGM-11-018-01-CDD (R. C.).
Footnotes
Author Contributions
TD and RC conceived and designed the project. TD performed all experiments. TD and RC acquired research funds. TD and RC wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM, Knight ZA. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol. 2008;4:691–699. doi: 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley MJ, Yun CH, Gewinner CA, Asara JM, Johnson JL, Coyle AJ, Eck MJ, Apostolou I, Cantley LC. EGF-receptor specificity for phosphotyrosine-primed substrates provides signal integration with Src. Nat Struct Mol Biol. 2015;22:983–990. doi: 10.1038/nsmb.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beldjord C, Desclaux-Arramond F, Raffin-Sanson M, Corvol JC, De Keyzer Y, Luton JP, Plouin PF, Bertagna X. The RET protooncogene in sporadic pheochromocytomas: frequent MEN 2-like mutations and new molecular defects. J Clin Endocrinol Metab. 1995;80:2063–2068. doi: 10.1210/jcem.80.7.7608256. [DOI] [PubMed] [Google Scholar]
- Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, Delatte B, Caauwe A, Lenglez S, Nkusi E, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014;511:246–250. doi: 10.1038/nature13305. [DOI] [PubMed] [Google Scholar]
- Buszczak M, Paterno S, Lighthouse D, Bachman J, Planck J, Owen S, Skora AD, Nystul TG, Ohlstein B, Allen A, et al. The carnegie protein trap library: a versatile tool for Drosophila developmental studies. Genetics. 2007;175:1505–1531. doi: 10.1534/genetics.106.065961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark ES, Whigham AS, Yarbrough WG, Weaver AM. Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia. Cancer Res. 2007;67:4227–4235. doi: 10.1158/0008-5472.CAN-06-3928. [DOI] [PubMed] [Google Scholar]
- Cohen MH, Williams G, Johnson JR, Duan J, Gobburu J, Rahman A, Benson K, Leighton J, Kim SK, Wood R, et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin Cancer Res. 2002;8:935–942. [PubMed] [Google Scholar]
- Dar AC, Das TK, Shokat KM, Cagan RL. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature. 2012;486:80–84. doi: 10.1038/nature11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das TK, Sangodkar J, Negre N, Narla G, Cagan RL. Sin3a acts through a multi-gene module to regulate invasion in Drosophila and human tumors. Oncogene. 2013;32:3184–3197. doi: 10.1038/onc.2012.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Rekhtman N, Arcila M, Wang L, Ni A, Albano M, Van Voorthuysen M, Somwar R, Smith RS, Montecalvo J, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol. 2016;17:1653–1660. doi: 10.1016/S1470-2045(16)30562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forde PM, Rudin CM. Crizotinib in the treatment of non-small-cell lung cancer. Expert Opin Pharmacother. 2012;13:1195–1201. doi: 10.1517/14656566.2012.688029. [DOI] [PubMed] [Google Scholar]
- Galetta D, Rossi A, Pisconti S, Colucci G. The emerging role of ALK inhibitors in the treatment of advanced non-small cell lung cancer. Expert Opin Ther Targets. 2012;16(Suppl 2):S45–S54. doi: 10.1517/14728222.2011.642372. [DOI] [PubMed] [Google Scholar]
- Gautschi O, Milia J, Filleron T, Wolf J, Carbone DP, Owen D, Camidge R, Narayanan V, Doebele RC, Besse B, et al. Targeting RET in Patients With RET-Rearranged Lung Cancers: Results From the Global, Multicenter RET Registry. J Clin Oncol. 2017;35:1403–1410. doi: 10.1200/JCO.2016.70.9352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson NJ, Tolbert LP, Oland LA. Activation of glial FGFRs is essential in glial migration, proliferation, and survival and in glia-neuron signaling during olfactory system development. PLoS One. 2012;7:e33828. doi: 10.1371/journal.pone.0033828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth AC, Fish M, Nusse R, Calos MP. Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics. 2004;166:1775–1782. doi: 10.1534/genetics.166.4.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19:1893–1907. doi: 10.1158/1055-9965.EPI-10-0437. [DOI] [PubMed] [Google Scholar]
- Ju YS, Lee WC, Shin JY, Lee S, Bleazard T, Won JK, Kim YT, Kim JI, Kang JH, Seo JS. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2012;22:436–445. doi: 10.1101/gr.133645.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim FD, Rubin GM. Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development. 1998;125:1–9. doi: 10.1242/dev.125.1.1. [DOI] [PubMed] [Google Scholar]
- Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, Sakamoto H, Tsuta K, Furuta K, Shimada Y, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18:375–377. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam ET, Ringel MD, Kloos RT, Prior TW, Knopp MV, Liang J, Sammet S, Hall NC, Wakely PE, Jr, Vasko VV, et al. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J Clin Oncol. 2010;28:2323–2330. doi: 10.1200/JCO.2009.25.0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson S, Cagan RL. Drosophila Cancer Models Identify Functional Differences between Ret Fusions. Cell Rep. 2016;16:3052–3061. doi: 10.1016/j.celrep.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Wang S, Xie W, Zheng R, Gan Y, Chang J. Apatinib inhibits cellular invasion and migration by fusion kinase KIF5B-RET via suppressing RET/Src signaling pathway. Oncotarget. 2016;7:59236–59244. doi: 10.18632/oncotarget.10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, Curran JA, Balasubramanian S, Bloom T, Brennan KW, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382–384. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Falola J, Zhu X, Gu Y, Kim LT, Sarosi GA, Anthony T, Nwariaku FE. Antiproliferative effects of Src inhibition on medullary thyroid cancer. J Clin Endocrinol Metab. 2004;89:3503–3509. doi: 10.1210/jc.2003-031917. [DOI] [PubMed] [Google Scholar]
- Mader CC, Oser M, Magalhaes MAO, Bravo-Cordero JJ, Condeelis J, Koleske AJ, Gil-Henn H. An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 2011;71:1730–1741. doi: 10.1158/0008-5472.CAN-10-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Blanco E, Roch F, Noll E, Baonza A, Duffy JB, Perrimon N. A temporal switch in DER signaling controls the specification and differentiation of veins and interveins in the Drosophila wing. Development. 1999;126:5739–5747. doi: 10.1242/dev.126.24.5739. [DOI] [PubMed] [Google Scholar]
- McCoach CE, Doebele RC. The minority report: targeting the rare oncogenes in NSCLC. Curr Treat Options Oncol. 2014;15:644–657. doi: 10.1007/s11864-014-0310-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens F, Johansson B, Fioretos T, Mitelman F. The emerging complexity of gene fusions in cancer. Nat Rev Cancer. 2015;15:371–381. doi: 10.1038/nrc3947. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaza-Menacho I, Barnouin K, Goodman K, Martínez-Torres RJ, Borg A, Murray-Rust J, Mouilleron S, Knowles P, McDonald NQ. Oncogenic RET kinase domain mutations perturb the autophosphorylation trajectory by enhancing substrate presentation in trans. Mol Cell. 2014;53:738–751. doi: 10.1016/j.molcel.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read RD, Goodfellow PJ, Mardis ER, Novak N, Armstrong JR, Cagan RL. A Drosophila model of multiple endocrine neoplasia type 2. Genetics. 2005;171:1057–1081. doi: 10.1534/genetics.104.038018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg SM, Concannon K, Cullen S, Boulay G, Turke AB, Faber AC, Lockerman EL, Rivera MN, Engelman JA, Maheswaran S, et al. Inhibition of mutant EGFR in lung cancer cells triggers SOX2-FOXO6-dependent survival pathways. Elife. 2015;4 doi: 10.7554/eLife.06132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarfaty M, Moore A, Neiman V, Dudnik E, Ilouze M, Gottfried M, Katznelson R, Nechushtan H, Sorotsky HG, Paz K, et al. RET Fusion Lung Carcinoma: Response to Therapy and Clinical Features in a Case Series of 14 Patients. Clin Lung Cancer. 2016 doi: 10.1016/j.cllc.2016.09.003. [DOI] [PubMed] [Google Scholar]
- Sato M, Vaughan MB, Girard L, Peyton M, Lee W, Shames DS, Ramirez RD, Sunaga N, Gazdar AF, Shay JW, et al. Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res. 2006;66:2116–2128. doi: 10.1158/0008-5472.CAN-05-2521. [DOI] [PubMed] [Google Scholar]
- Setou M, Seog DH, Tanaka Y, Kanai Y, Takei Y, Kawagishi M, Hirokawa N. Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature. 2002;417:83–87. doi: 10.1038/nature743. [DOI] [PubMed] [Google Scholar]
- Siegle JM, Basin A, Sastre-Perona A, Yonekubo Y, Brown J, Sennett R, Rendl M, Tsirigos A, Carucci JA, Schober M. SOX2 is a cancer-specific regulator of tumour initiating potential in cutaneous squamous cell carcinoma. Nat Commun. 2014;5:4511. doi: 10.1038/ncomms5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, Asaka R, Hamanaka W, Ninomiya H, Uehara H, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:378–381. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- Vaishnavi A, Schubert L, Rix U, Marek LA, Le AT, Keysar SB, Glogowska MJ, Smith MA, Kako S, Sumi NJ, et al. EGFR Mediates Responses to Small-Molecule Drugs Targeting Oncogenic Fusion Kinases. Cancer Res. 2017 doi: 10.1158/0008-5472.CAN-17-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Larson DE, Cagan RL. Csk-deficient boundary cells are eliminated from normal Drosophila epithelia by exclusion, migration, and apoptosis. Dev Cell. 2006;10:33–44. doi: 10.1016/j.devcel.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Wright TI, Petersen JE. Treatment of recurrent dermatofibrosarcoma protuberans with imatinib mesylate, followed by Mohs micrographic surgery. Dermatol Surg. 2007;33:741–744. doi: 10.1111/j.1524-4725.2007.33154.x. [DOI] [PubMed] [Google Scholar]
- Yoh K, Seto T, Satouchi M, Nishio M, Yamamoto N, Murakami H, Nogami N, Matsumoto S, Kohno T, Tsuta K, et al. Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): an open-label, multicentre phase 2 trial. Lancet Respir Med. 2017;5:42–50. doi: 10.1016/S2213-2600(16)30322-8. [DOI] [PubMed] [Google Scholar]
- Yokomaku D, Jourdi H, Kakita A, Nagano T, Takahashi H, Takei N, Nawa H. ErbB1 receptor ligands attenuate the expression of synaptic scaffolding proteins, GRIP1 and SAP97, in developing neocortex. Neuroscience. 2005;136:1037–1047. doi: 10.1016/j.neuroscience.2005.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.