

Abstract
The global motions and exchange kinetics of a model protein, ubiquitin, bound to the surface of negatively charged lipid-based nanoparticles (liposomes) are derived from combined analysis of exchange lifetime broadening arising from binding to nanoparticles of differing size. The relative contributions of residence time and rotational tumbling to the total effective correlation time of the bound protein are modulated by nanoparticle size, thereby permitting the various motional and exchange parameters to be determined. The residence time of ubiquitin bound to the surface of both large and small unilamellar liposomes is ∼20 μs. Bound ubiquitin undergoes internal rotation about an axis approximately perpendicular to the lipid surface on a low microsecond time scale (∼2 μs), while simultaneously wobbling in a cone of semiangle 30–55° centered about the internal rotation axis on the nanosecond time scale. The binding interface of ubiquitin with liposomes is mapped by intermolecular paramagnetic relaxation enhancement using Gd3+-tagged vesicles, to a predominantly positively charged surface orthogonal to the internal rotation axis.
Many biological processes involve transient interactions of proteins with very large macromolecular assemblies or cell surfaces. Recent NMR developments, involving the application of lifetime line broadening (ΔR2) and dark state exchange saturation transfer (DEST) spectroscopy, have allowed quantitative study of the dynamics of interaction of NMR visible molecules with very large (>1 MDa) NMR invisible species such as protofibrils and molecular machines at atomic resolution.1 In our previous studies,1 however, the transverse relaxation rates of the bound species were treated phenomenologically, i.e., not interpreted in the framework of a particular motional model for the bound (“dark”) state. Here, we show that global dynamics and exchange kinetics of a model protein on the surface of large nanoparticles can be quantitatively described by analysis of differential ΔR2 effects for particles of varying size.
The focus of the current work is the interaction of ubiquitin with lipid-based nanoparticles (liposomes). Liposomes have been extensively used as drug carriers and vehicles for studying biological membranes and their interactions with proteins.2 The interaction of ubiquitin with a wide variety of small-to-medium sized carbon, gold and silver nanoparticles has been the subject of recent investigations.3 However, no information is currently available on the dynamic behavior of ubiquitin on the surface of any nanoparticle.
Negatively charged and zwitterionic liposomes were made using 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocho-line (POPC), respectively (see SI). Liposomes were prepared as large (LUV) and small (SUV) unilamellar vesicles with mean diameters of 103 and 27 nm, respectively, and corresponding polydispersity indices of 0.1 and 0.2 (Figure S1A,B). The molecular tumbling correlation time (τR) of LUV and SUV nanoparticles, calculated using the Stokes–Einstein equation, is 100 and 1.8 μs, respectively.
The 15N–ΔR2 profiles (measured in the rotating frame to suppress chemical exchange line broadening; see SI) for U-[15N/2H]-ubiquitin in the presence of LUVs and SUVs are shown in Figure 1A,B, respectively. Substantial ΔR2 values are observed in the presence of negatively charged POPG vesicles but not zwitterionic POPC particles, indicating that ubiquitin interacts only with negatively charged liposomes. There are no observable exchange induced chemical shifts, and the ΔR2 effect can be entirely attributed to line broadening caused by rapid decay of transverse relaxation during the lifetime of the bound state that is transferred by exchange to the observable free state.1d The 15N–ΔR2 values are weakly dependent upon the static magnetic field (Figures 1 and S2): the average ratio of ΔR2 values at 800 to 500 MHz is ∼1.3 for LUVs (Figure 1C) and SUVs (Figure 1D), very close to the expected ratio of 1.3 from a relaxation mechanism based on the one-bond 1H–15N dipolar interaction and a −170 ppm 15N chemical shift anisotropy (for a slowly tumbling particle in the absence of exchange), and much smaller than any contribution from chemical exchange line broadening, which scales as the square of the magnetic field (8002/5002 = 2.56). Thus, one can conclude that the exchange rate constant , where koff and are the dissociation and apparent first order association rate constants, respectively, for the binding of ubiquitin to liposomes, is higher than for ubiquitin and that is larger than .1b,d
Figure 1.
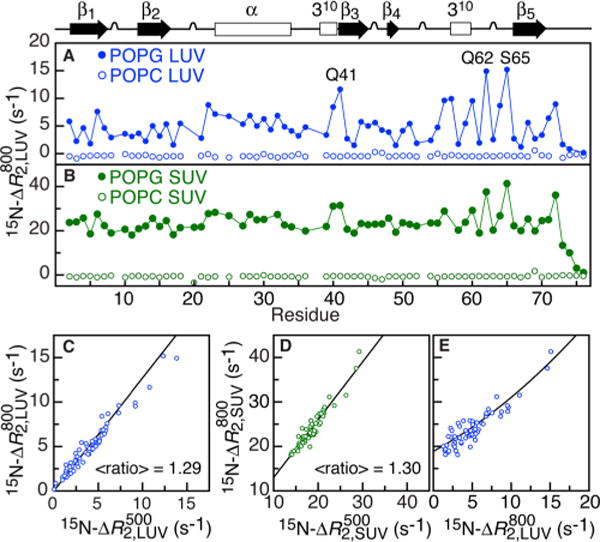
15N–ΔR2 profiles for 0.8 mM U-[15N/2H] ubiquitin (dissolved in H2O) in the presence of a 1:2 ratio (mol/mol on a lipid molecule basis) negatively charged (POPG, filled symbols) and zwitterionic (POPC, open symbols) liposomes of differing size: (A) LUVs (blue) and (B) SUVs (green). 15N–ΔR2 dependence on magnetic field strength for (C) LUVs and (D) SUVs, and (E) on nanoparticle size. The black lines in C–E represent simulated correlations derived from global best-fitting of all LUV and SUV 15N–ΔR2 data (see text, Figure 2 and SI). Experimental temperature = 25 °C.
The 15N–ΔR2 data for LUVs and SUVs (Figure 1A,B) reveal two unexpected features. First, the ΔR2 values for SUVs are 3–15-fold (average ∼4-fold) higher than those for LUVs (Figure 1E), even though the lipid monomer and ubiquitin concentrations are the same in both samples. This can be attributed to the larger concentration of SUV particles owing to their smaller size, hence the larger fraction of bound ubiquitin, and implies that the overall effective correlation time, τC = (1/τR+ kex)−1, of bound ubiquitin is dominated, at least in the case of LUVs, not by τR but by the lifetime (τex = 1/kex) of the complex. Note if τex >> τR (∼100 μs) for LUVs, would be limited by the static 1H–15N dipolar coupling of ∼20 kHz , and no sequence variation would be observed. Second, the ΔR2 values would be expected to be uniform for the binding of a rigid protein to a large particle, yet their range spans over a factor of ∼8 for LUVs (Figure 1A,C) and ∼2 for SUVs (Figure 1B,D) (excluding the disordered C-terminal tail comprising residues 73–76). In contrast, the 15N–R2 values for free ubiquitin, excluding the C-terminal tail and a small number of residues undergoing chemical exchange,4 are close to uniform with only minor variations owing to a very small degree of diffusion anisotropy.5 The experimental variation in 15N–ΔR2 values cannot therefore be ascribed to local motion at the individual N–H bond vector level but rather to a global motional phenomenon involving the whole ubiquitin molecule on the surface of the nanoparticles.
The values are related to molecular motion through the spectral density function J(ω) at a finite number of frequencies ω. Rotation of ubiquitin on the surface of the nanoparticle about an internal axis (Figure 2A) will result in a [P2(cos α)]2 = (3 cos2 α − 1)2/4 dependence6 of with a double-dip minimum at the magic angle (α = 54.74° and 180° − 54.74° = 125.26°) and a saddle point at α = 90°, where α is the angle between a given N–H bond vector (obtained from the molecular coordinates) and the axis of internal rotation. Internal rotation can therefore be described by an order parameter Sr2, given by the square of the second-order Legendre polynomial P2(cos α), and an internal rotation correlation time τr. can be further modulated by a faster motion described by an order parameter Sw2 and a correlation time τw. Assuming separation of time scales (τr > 5τw), J(ω) can be expressed, using the extended Lipari–Szabo formalism,7 as
| (1) |
where τr′ = (1/τC + 1/τr)−1 and τw′ = (1/τC + 1/τw)−1, and the overall order parameter S2 is given by Sw2Sr2 (see SI for further details). The time scale τr is expected to be intermediate between the nanosecond time scale applicable for free ubiquitin and the low-microsecond time scale for transmembrane proteins;9 the faster motion can be considered as wobbling in a cone of semiangle β0 (with Sw2 = [cos β0(1 + cos β0)]2/4),7 which is equivalent to restricted rotational diffusion centered about the internal axis of rotation, and is expected to take place on a nanosecond time scale.9
Figure 2.
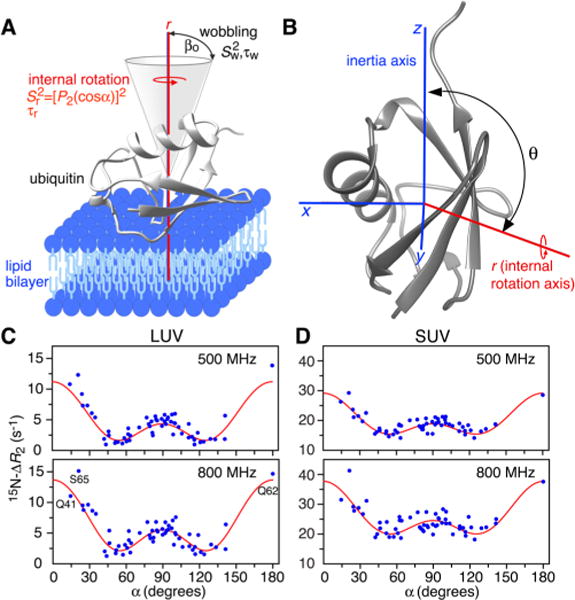
Global dynamics of ubiquitin on the surface of liposome nanoparticles. (A) Ubiquitin (gray ribbon, PDB 1UBQ8) rotates about an internal rotation axis (denoted as r and displayed in red), shown as perpendicular to the surface of the lipid bilayer (blue) while wobbling in a cone of semiangle β0. (B) Relationship between the axis r (red) and the x, y, and z axes of the inertia tensor (blue, used as an arbitrary internal reference frame): θ is the angle between the r and z axes; and φ the angle (not shown) between the x axis and the projection of the r axis on the x–y plane. Dependence of 15N–ΔR2 in the presence of (C) LUVs and (D) SUVs on the angle α between the N–H bond vectors and the internal rotation axis: experimental data are shown as blue circles; the best-fit curves, from global fitting to all LUV and SUV data, are in red (Sw2 = 0.5 and τw = 300 ns; see text and SI).
Binding of ubiquitin to liposomes is fast on the relaxation time scale and for LUVs τex < τR. Consequently, if the LUV and SUV 15N–ΔR2 data are treated independently, direct characterization of the binding equilibrium is impossible without some simplifying assumptions (see SI for details). Since τC is dramatically different for LUVs and SUVs (as manifested by the much smaller variation in 15N–ΔR2 for SUVs than LUVs; cf. Figure 1A,B) the solution to this problem lies in combined analysis of the LUV and SUV 15N–ΔR2 data with the reasonable assumption that the parameters describing the global dynamics of ubiquitin on the nanoparticle surface (i.e., τr, τw, Sw2, and the polar angles θ and φ defining the orientation of the internal rotation axis in the molecular frame of the inertia tensor; cf. Figure 2B) and the dissociation rate constant koff are independent of nanoparticle size.
Even if the LUV and SUV data are analyzed simultaneously, not all global dynamics parameters can be reliably extracted because only the product of Sw2 and τC enters into the first term of the spectral density (eq 1). Hence, only a range of possible values for the fast motional parameters, Sw2 and τw, can be determined. Numerical analyses of the LUV and SUV data separately on a grid of Sw2 and τw values for a set of fixed values of koff (Figure S3) define realistic bounds of 0.7 > Sw2 > 0.2, corresponding to wobbling within a cone of semiangle β0 ≈ 30° to 55°, and 0 < τw < 500 ns (see SI).
Simultaneous nonlinear least-squares fitting of the LUV and SUV 15N–ΔR2 data at multiple fields using the above assumptions, with Sw2 = 0.5 and τw = 300 ns, results in excellent fits to the ΔR2 data (Figures 2C,D and S2B) and yields values of koff = 51(±16) × 103 s−1, τr = 2.0 ± 0.4 μs, and (θ, φ) = (108 ± 1°, −4.5 ± 1°). and 1140 ± 410 s−1 for LUVs and SUVs, respectively, corresponding to liposome-bound ubiquitin populations of 0.13 and 2.2%. The τC values for LUVs and SUVs thus differ by an order of magnitude (16 versus 1.6 μs, respectively) with the former being dominated by 1/koff (∼20 μs) and the latter by the SUV molecular tumbing time τR (∼1.8 μs) The values of the various parameters are only slightly dependent on the choice of Sw2 and τw values, and remain within the reported uncertainties for τw < 500 ns. At 800 MHz, the calculated values range from ∼2000 to 14,000 s−1 for LUVs and ∼1000 to 1600 s−1 for SUVs (Figure S4). The dependence of 15N–ΔR2 on the angle α subtended by each N–H bond vector and the internal rotation axis is shown in Figure 2C,D (and Figure S2B). A similar dependence of the amide proton 1HN–ΔR2 values on α is also observed (Figure S5). Although 15N–ΔR2 and 1HN–ΔR2 values are correlated (Figure S5B), the 1HN–ΔR2 values are “contaminated” by dipolar interactions between spatially close labile protons in U-[15N/2H] ubiquitin dissolved in H2O and are therefore less suitable for quantitative analysis.
The lipid monomer concentration used in the NMR samples is 1.6 mM resulting in nanoparticle concentrations of 16 and 230 nM for LUVs and SUVs, respectively. The calculated bimolecular association rate constant for the binding of ubiquitin to both LUVs and SUVs is therefore ∼4 × 109 M−1 s−1, which compares reasonably well with predicted values of 1 to 5 × 1010 M−1 s−1 for a nonspecific diffusion limited process (see SI).
Analysis of the global dynamics of ubiquitin bound to liposomes defines the orientation of the internal rotation axis in the molecular frame (Figure 2B) but not relative to the lipid bilayer. The latter can be inferred using intermolecular transverse (Γ2) paramagnetic relaxation enhancement (PRE) measurements10 with Gd3+-paramagnetically tagged LUVs (see SI) to map the ubiquitin binding surface. The backbone amide and methyl proton (of Leu, Val, Ile-δ1) Γ2 PRE profiles are shown in Figure 3A. An approximately uniform 1H-Γ2 PRE background of ∼40 s−1 arises from diffusion of ubiquitin in the magnetic field generated by the Gd3+-tagged LUVs, an effect analogous to the blood-oxygen-level dependent (BOLD) effect in functional MRI.11 Superimposed on the background are regions with enhanced intermolecular PRE effects, which reach a maximal value of Γ2 ≈ 70 s−1 (with background subtracted) that is limited under the current exchange conditions by the value of . The intermolecular PRE effects are also manifested by wider paramagnetic 1H-DEST profiles (Figure S6).
Figure 3.
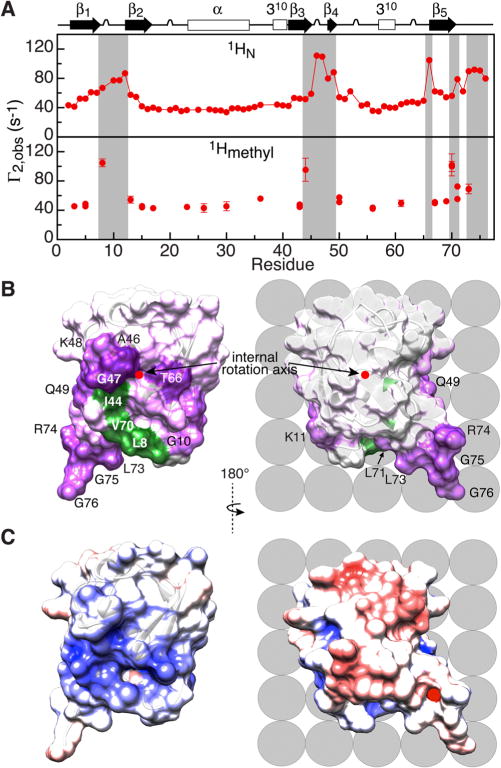
Proton intermolecular transverse PREs (Γ2) observed for U-[15N/2H]/[1Hmethyl/13Cmethyl-ILV] ubiquitin in the presence of negatively charged Gd3+-tagged POPG LUVs. (A) 1H-Γ2 profiles for backbone amide (top) and Ile/Leu/Val methyl (bottom) protons. (B) PRE mapping of the interaction surface of ubiquitin. The internal rotation axis (red dot) is perpendicular to the plane of the figure and orthogonal to the view shown in Figure 2A. 1HN-Γ2 PREs are color-coded from purple (110 s−1) to white (background = 40 s−1), and residues with 1HN-Γ2 ≥ 77 s−1 are labeled; residues with large (>80 s−1) 1Hmethyl-Γ2 values are colored in green. (C) Molecular surface of ubiquitin (same views as in panel B) color-coded according to electrostatic potential (±5 kT with blue, positive; white, neutral; and red, negative). Lipid molecules on the nanoparticle surface are shown schematically as gray spheres.
Although the complexity of the paramagnetic labeling pattern (multiple Gd3+ tags on the surface of LUVs separated by average distances that exceed protein dimensions by more than a factor of 2) and the dynamics of the bound state preclude quantitative interpretation of the intermolecular PRE data, it allows one to map the preferential mode of interaction of ubiquitin with POPG LUVs (Figure 3B). For comparison, the molecular surface of ubiquitin color-coded according to electrostatic potential is shown in Figure 3C. The predominant interaction of ubiquitin with the negatively charged surface of POPG LUVs involves only a single face of the protein, comprising a central hydrophobic region surrounded by a ring of positive charges, orthogonal to the internal axis of rotation (Figure 3B). This same face has also been implicated in the interaction of ubiquitin with negatively charged silver,3c Ln-doped SrF23e, and hydroxylated C60 fullerenol3d nanoparticles.
In summary, we have shown that it is possible to quantitatively characterize the global dynamics and exchange kinetics of an interaction occurring on the microsecond time scale between an NMR visible species (ubiquitin) and large lipid-based macromolecular assemblies (nanoparticles) with molecular tumbling times (τR) in the microsecond range. This was achieved by exploiting the different contributions of τR and lifetime (τex) of the complex to the total effective correlation time (τC) of ubiquitin bound to the surface of nanoparticles of drastically differing sizes (τex < τR for LUVs and >τR for SUVs). Ubiquitin rotates on the low microsecond time-scale around an internal axis approximately normal to the nanoparticle surface while undergoing faster rocking motions within a cone centered about the internal rotation axis (Figure 2A). In addition, the interaction surface on the protein can be delineated qualitatively from intermolecular PRE measurements using paramagnetically tagged liposomes (Figure 3B).
Supplementary Material
Acknowledgments
We thank Attila Szabo for useful discussions. This work was supported by the Intramural Program of NIDDK, NIH, and by the AIDS Targeted Antiviral Program of the Office of the Director of the NIH (to G.M.C. and A.B.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b02654.
Experimental details, theory, fitting procedures, and additional data (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.(a) Fawzi NL, Ying J, Ghirlando R, Torchia DA, Clore GM. Nature. 2011;480:268–272. doi: 10.1038/nature10577. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Libich DS, Fawzi NL, Ying J, Clore GM. Proc Natl Acad Sci U S A. 2013;110:11361–11366. doi: 10.1073/pnas.1305715110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Libich DS, Tugarinov V, Clore GM. Proc Natl Acad Sci U S A. 2015;112:8817–8823. doi: 10.1073/pnas.1510083112. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Anthis NJ, Clore GM. Q Rev Biophys. 2015;48:35–116. doi: 10.1017/S0033583514000122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Puri A, Loomis K, Smith B, Lee JH, Yavlovich A, Heldman E, Blumenthal R. Crit Rev Ther Drug Carrier Syst. 2009;26:523–580. doi: 10.1615/critrevtherdrugcarriersyst.v26.i6.10. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sapsford KE, Algar WR, Berti L, Gemmill KB, Casey BJ, Oh E, Stewart MH, Medintz IL. Chem Rev. 2013;113:1904–2074. doi: 10.1021/cr300143v. [DOI] [PubMed] [Google Scholar]; (c) Ceccon A, Lelli M, D’Onofrio M, Molinari H, Assfalg M. J Am Chem Soc. 2014;136:13158–13161. doi: 10.1021/ja507310m. [DOI] [PubMed] [Google Scholar]; (d) Ceccon A, Busato M, Perez Santero S, D’Onofrio M, Musiani F, Giorgetti A, Assfalg M. Chem Bio Chem. 2015;16:2633–2645. doi: 10.1002/cbic.201500451. [DOI] [PubMed] [Google Scholar]
- 3.(a) Calzolai L, Franchini F, Gilliland D, Rossi F. Nano Lett. 2010;10:3101–3105. doi: 10.1021/nl101746v. [DOI] [PubMed] [Google Scholar]; (b) Wang A, Vo T, Le V, Fitzkee NC. J Phys Chem B. 2014;118:14148–14156. doi: 10.1021/jp506506p. [DOI] [PubMed] [Google Scholar]; (c) Brahmkhatri VP, Chandra K, Dubey A, Atreya HS. Nanoscale. 2015;7:12921–12931. doi: 10.1039/c5nr03047a. [DOI] [PubMed] [Google Scholar]; (d) Zanzoni S, Ceccon A, Assfalg M, Singh RK, Fushman D, D’Onofrio M. Nanoscale. 2015;7:7197–7205. doi: 10.1039/c5nr00539f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zanzoni S, Pedroni M, D’Onofrio M, Speghini A, Assfalg M. J Am Chem Soc. 2016;138:72–75. doi: 10.1021/jacs.5b11582. [DOI] [PubMed] [Google Scholar]
- 4.Massi F, Grey MJ, Palmer AG. Protein Sci. 2005;14:735–742. doi: 10.1110/ps.041139505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tjandra N, Feller SE, Pastor RW, Bax A. J Am Chem Soc. 1995;117:12562–12566. [Google Scholar]
- 6.Wittebort RJ, Szabo A. J Chem Phys. 1978;69:1722–1736. [Google Scholar]
- 7.Clore GM, Szabo A, Bax A, Kay LE, Driscoll PC, Gronenborn AM. J Am Chem Soc. 1990;112:4989–4991. [Google Scholar]
- 8.Vijay-Kumar S, Bugg CE, Cook WJ. J Mol Biol. 1987;194:531–544. doi: 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]
- 9.Thomas DD. In: Techniques for the Analysis of Membrane Proteins. Reagan CI, Cherry RJ, editors. Chapman and Hall; London: 1986. pp. 376–431. [Google Scholar]
- 10.Clore GM, Iwahara J. Chem Rev. 2009;109:4108–4139. doi: 10.1021/cr900033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ogawa S, Lee TM, Kay AR, Tank DW. Proc Natl Acad Sci U S A. 1990;87:9868–9872. doi: 10.1073/pnas.87.24.9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.