

Abstract
Polyketide synthase (PKS) enzymes continue to hold great promise as synthetic biology platforms for the production of novel therapeutic agents, biofuels and commodity chemicals. Dehydratase (DH) catalytic domains play an important role during polyketide biosynthesis through the dehydration of the nascent polyketide intermediate to provide olefins. Our understanding of the detailed mechanistic and structural underpinning of DH domains that control substrate specificity and selectivity remains limited, thus hindering our efforts to rationally re-engineer PKSs. The curacin pathway houses a rare plurality of possible double bond permutations containing conjugated olefins as well as both cis- and trans-olefins, providing an unrivaled model system for polyketide dehydration. All four DH domains implicated in curacin biosynthesis were characterized in vitro using synthetic substrates and activity was measured by LC-MS/MS analysis. These studies resulted in complete kinetic characterization of the all trans trienoate-forming CurK dehydratase, whose kcat of 72 s−1 is more than three-orders of magnitude greater than any previously reported PKS DH domain. A novel stereospecific mechanism for diene formation involving a vinylogous enolate intermediate is proposed for the CurJ and CurH dehydratases based on incubation studies with truncated substrates. A synthetic substrate was co-crystallized with a catalytically inactive Phe substitution in the His-Asp catalytic dyad of CurJ DH to elucidate substrate-enzyme interactions. The resulting complex suggested the structural basis for dienoate formation and provided the first glimpse into the enzyme-substrate interactions essential for the formation of olefins in polyketide natural products. This examination of both canonical and non-canonical dehydration mechanisms reveals hidden catalytic activity inherent in some DH domains that may be leveraged for future applications in synthetic biology.
Keywords: curacin, polyketide dehydratase, vinylogous syn elimination, biosynthesis, dehydration kinetics, co-crystallization, PKS module stuttering
TOC image
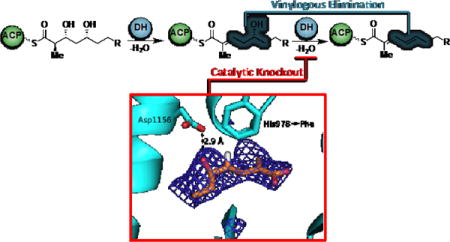
INTRODUCTION
Polyketide secondary metabolites are an exquisite example of Nature’s rich diversity, both in terms of molecular complexity and functionality. Small molecules from this natural product family cover a wide range of marketed medicinal agents, providing utility as hypolipodemic (lovastatin), antimicrobial (erythromycin), antineoplastic (ixabepilone), antifungal (amphotericin B), and immunosuppressive (FK-506) therapeutics. The variety in polyketide biological activity is largely attributed to their structural diversity arising from their assembly-line construction by polyketide synthases (PKSs). Type I PKSs consist of modular cassettes of megaenzymes with catalytic domains for extending, editing and transferring polyketide chains. The minimal components necessary for elongation of a polyketide intermediate include an acyl carrier protein (ACP), acyltransferase (AT), and ketosynthase (KS) domains. Each module may additionally house processing domains that alter the substituents and oxidation states at the α- and β-centers. These include C-methyltransferase (CMT), ketoreductase (KR), O-methyltransferase (OMT), dehydratase (DH), and enoyl reductase (ER) domains affording α-alkylated, β-hydroxy, β-methoxy, α,β-unsaturated or α,β-saturated products. Many of these transformations result in stereoselective formation of an optically enriched product. Subsequent elongation and processing steps by the downstream modules lead to the mature polyketide chain. Chain termination by a thioesterase (TE) domain results in lactonization or hydrolysis to a macrolactone or carboxylic acid, respectively. The released polyketide is often subjected to tailoring events catalyzed by post-PKS tailoring enzymes that further diversify the natural product scaffold.
Curacin A, a mixed polyketide-nonribosomal peptide natural product isolated from the cyanobacteria Moorea producens (formerly Lyngbya majuscula) is a potent antiproliferative agent that arrests mitosis through the inhibition of tubulin polymerization.1 Curacin A is biosynthesized via a mixed nonribosomal peptide synthase-polyketide synthase pathway incorporating three malonyl-CoA units in a cyclopropyl moiety, one L-cysteine in a thiazolidine-forming cyclization reaction, and seven units of malonyl-CoA through KS-catalyzed Claisen condensations. Both C-methyl and O-methyl substituents arise from two equivalents of S-adenosylmethionine (SAM) during PKS processing steps. The curacin (Cur) biosynthetic pathway has provided a wealth of information about non-canonical enzymatic processes in polyketide biosynthesis including a GCN5-related N-acetyltransferase (GNAT)-like strategy for polyketide chain initiation, cyclopropane synthesis through β-branching and cryptic halogenation, as well as polyketide release via off-loading of a terminal alkene.2–4 Moreover, the Cur pathway has yielded tremendous structural insight into this unprecedented PKS chemistry and three-dimensional structures of fourteen proteins in the Cur pathway have been published.5–13
We selected the curacin biosynthesis as a platform for studying dehydratase activity in PKSs. The pathway uniquely produces nearly every possible type of olefin substitution pattern (mono-, di-, and trisubstituted) as well as both cis and trans geometric isomers (Figure 1). Additionally, curacin B and C constitute geometric isomers of curacin A and suggest a degree of pathway flexibility in regards to olefin formation (Figure 2).14 Bioinformatic analysis of the curacin gene cluster shows that PKS modules CurG and CurI are missing DH domains while CurF contains an extraneous DH domain.10,15 Neighboring curacin modules could potentially accommodate for the missing DHs. This process, termed ‘domain stuttering’, involves direct transfer of an ACP-bound intermediate to the ACP of the next domain.16 This product could be β-processed (dehydrated by a DH, in this case) then transferred back to the KS domain of the same module for a typical round of extension and β-processing. We hypothesized these non-canonical dehydration events may occur through a simpler and more efficient process by an alternative mechanism. The Smith lab previously disclosed the structural characterization of all four excised DH domains from CurF, CurH, CurJ and CurK providing a solid structural foundation and a unique opportunity to capture a bound substrate.10 The conserved, catalytic His-Asp dyad residues necessary for syn-dehydration and the active site channels are surprisingly quite similar giving few clues on the identity of their native substrates.17,18 This set the stage for our present study using synthetic substrates to probe the substrate specificity and dehydration mechanism.
Figure 1.
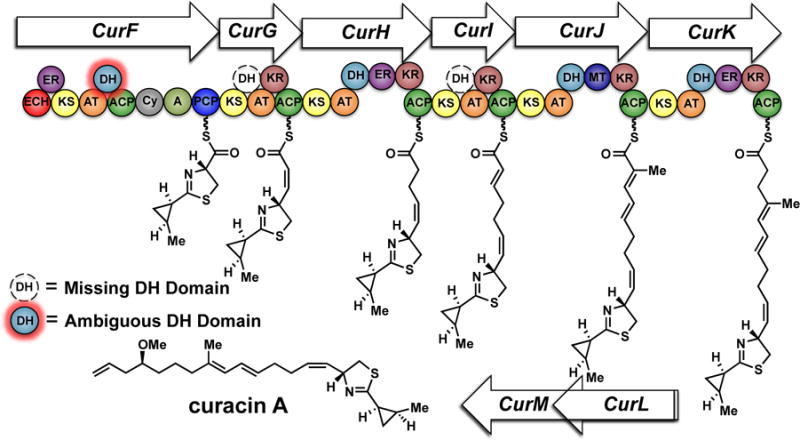
The curacin A mixed NRPS-PKS biosynthetic pathway. The extraneous CurF-DH with no predicted function is highlighted in red. The absent dehydratases in CurG and CurI are shown with dashed lines in black and white.
Figure 2.
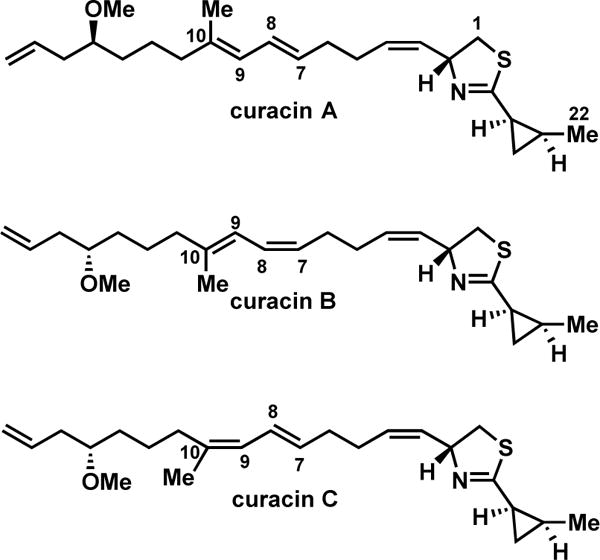
The natural geometric isomers of curacin. Both the Δ7,8 and Δ9,10 cis-isomers of curacin A have been isolated from Moorea producens. The biosynthetic origin of these isomerizations remains unknown.
RESULTS
Substrate Design and Synthesis
We set out to synthesize potential substrates to profile the dehydratase activity of the four DH domains in the pathway (Figure 3). The distal thiazoline and cyclopropyl moieties were eliminated to reduce molecular complexity and enhance solubility of the substrates. Synthesis of the truncated CurK-DH tetraketide 1 began with a Horner-Wadsworth-Emmons reaction between trans-2-pentenaldehyde (9) and triethyl 2-phosphonopropionate (10) to afford the trans-trans-dienoate 11 in quantitative yield (Scheme 1A). The dienoate was reduced with diisobutylaluminum hydride (DIBAL-H) and oxidized to aldehyde 13 employing MnO2. Asymmetric aldol reaction of 13 with Nagao’s D-valine-derived thiazolidinethione 14 provided alcohol 15 as a single diastereomer.19 Facile displacement of the chiral auxiliary with N-acetylcysteamine (NAC) furnished 1. The enantiomeric tetraketide 2 was prepared in an analogous fashion using the L-valine-derived thiazolidinethione (Supporting Information). The CurK product 7 was obtained chemoenzymatically from 1 employing CurK-DH.
Figure 3.
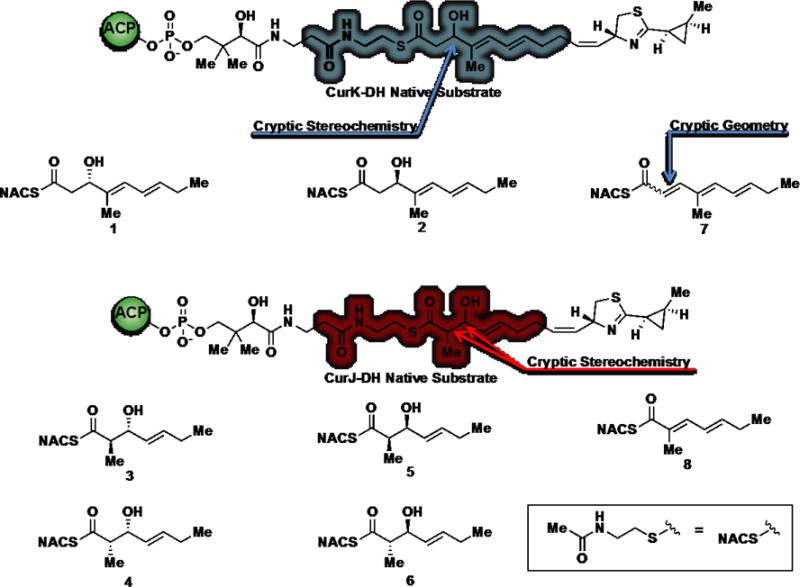
CurJ and CurK truncated substrates and products.
Scheme 1.
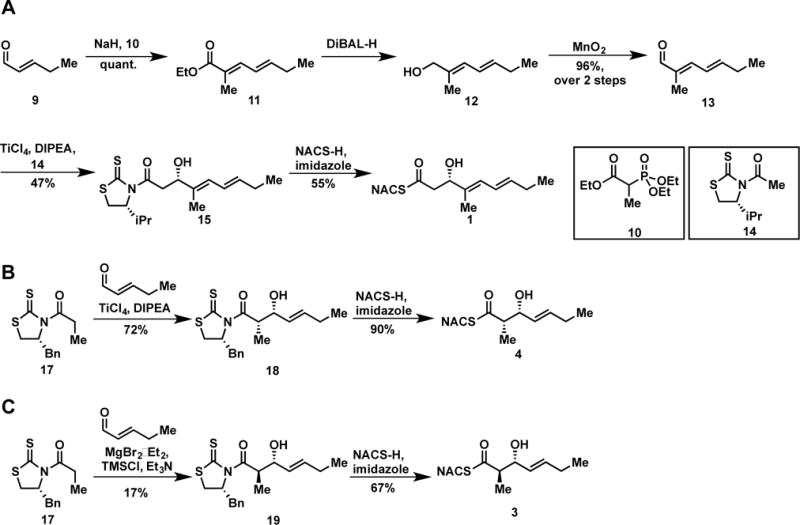
Synthesis of CurK-DH substrate 1 and the CurJ substrates 3 and 4.
The truncated CurJ triketide substrates were synthesized using Crimmins’ and Evans’ aldol chemistry that allows access to all four possible stereoisomers from common intermediate 9 and the D-phenylalanine-derived thiazolidinethione 17 or its enantiomer by varying the Lewis acid and amine base (Scheme 1).20,21 The syn-aldol adduct 18 was obtained in a respectable 72% yield and converted to the required NAC thioester 4 by treatment with N-acetylcysteamine (Scheme 1B). The anti-aldol adduct 19 was isolated in 17% yield and analogously elaborated to the NAC thioester 3 (Scheme 1C). We attribute the consistently poor yield of the anti-aldol transformation to a limitation of the Evans’ methodology when using aliphatic enals with an exchangeable γ-proton that may polymerize under the reaction conditions. The enantiomers of 3 and 4 were synthesized in an analogous way utilizing L-phenylalanine-derived thiazolidinethione auxiliary to afford 6 and 5, respectively (Supporting Information). Additionally, the truncated CurJ product 8 was synthesized through a Horner-Wadsworth Emmons reaction (Supporting Information).22,23
Biochemical Characterization of Cur DHs with 1 and 2
The four recombinant dehydratase domains (CurK-DH, CurJ-DH, CurH-DH, and CurF-DH) were individually incubated overnight at 37 °C with NAC esters 1 and 2 and analyzed via LC-MS/MS. CurK-DH dehydrated its truncated substrate in a completely stereospecific fashion, selectively acting only on the D-alcohol 1 with no turnover for L-alcohol 2, whereas CurH-DH was unable to process either 1 or 2 (Figure 4, Panels A and B). This matches the predicted stereospecificity of the CurK-KR containing the signature LDD motif for B-type ketoreductases.24 Curiously, CurF-DH and CurJ-DH also turned over 1, but not 2, in a stereospecific fashion. While CurF-DH maintained comparable total conversions with that of CurK-DH (55% of CurK-DH), the CurJ-DH was drastically deficient in its ability to catalyze the same transformation (7% of CurK-DH) (Figure 4, Panel C). A single reaction product from 1 and CurK-DH was observed, which was isolated in 25% overall yield from the overnight reaction mixture. The dehydrated substrate was analyzed via 1H and 13C NMR and confirmed as the all trans-trienoate 7. This result is consistent with the empirically based prediction that the enzymatic dehydration of D-alcohols affords E-olefins.25,26 A time course study of the dehydration of 1 (1 mM) by CurK-DH (5 μM) revealed that the enzyme rapidly dehydrates nearly all of the substrate, reaching completion within 20 min (Figure 4, Panel D). Steady-state kinetic analysis of the CurK-DH catalyzed dehydration of 1 to 7 was quantified by LC-MS/MS under initial velocity (v0) conditions (Materials and Methods). The resulting saturation curve (v0 versus [1]) was fit to the Michaelis-Menten equation to provide an apparent KM of 12.0 ± 4.7 mM and kcat of 72 ± 21 s−1 resulting in a specificity constant (kcat/KM) of (6.00 ± 1.41) × 103 M−1 s−1. While the KM value is similar to other NAC thioester substrates with their cognate excised PKS DH domains, the kcat value is nearly three orders of magnitude greater than any previously characterized PKS DH domain.27,28
Figure 4.
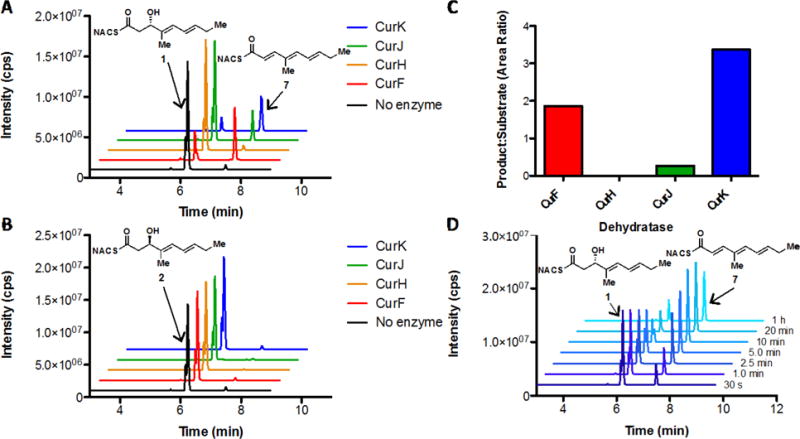
Incubation studies of synthetic CurK-DH substrates 1 and 2. Panels A and B show the extracted chromatograms of 14 h incubations of all curacin DH domains with substrates 1 and 2, respectively. CurF-DH, CurJ-DH and CurK-DH all display stereospecificity for D-alcohol (1) over the L-alcohol (2). Panel C displays the ratio of peak areas (7:1) for each curacin dehydratase enzyme subtracting negative control (no enzyme). CurF-DH and CurK-DH efficiently produce the triene 7, with a lesser amount produced by CurJ-DH. The time course extracted chromatograms of CurK-DH (5 μM) with substrate 1 (1 mM) are displayed in panel D. The enzyme reaches equilibrium by 20 min. The transition m/z 268→121 was used to monitor both residual substrates (1 and 2) as well as product (7) formation for all extracted chromatograms due to spontaneous dehydration of the substrate under the MS ionization conditions. The recurring shoulder on compounds 1 and 2 is believed to be minor trans, cis-isomers arising from isomerization during assay conditions.
To probe the reaction mechanism and facilitate co-crystallization with substrates we prepared catalytically inactive point mutants His996Phe and Asp1169Asn of the His-Asp catalytic dyad in CurK-DH based on sequence alignment and the reported CurK-DH structure.10 The His996Phe mutant was devoid of activity while the Asp1169Asn mutant retained small, but measurable activity showing 2–3% conversion of 1 to 7 after 12 hours. Crystal structures of both His996Phe and Asp1169Asn CurK-DH mutants were determined at 1.4 and 1.65 Å resolution, respectively (Table 1).10 The structures of the mutant proteins were virtually identical to the wild type CurK-DH (RMSD of 0.11 Å for 509 Cα atoms) with no significant conformational changes in the active site. Co-crystallization trials of substrate 1 and either mutant of wild type CurK-DH resulted in no crystal growth under conditions that readily yielded crystals in absence of substrate. Crystal soaking experiments with 1 resulted in a rapid dissolution of the crystals, suggesting that substrate binding may induce a conformational change and destabilize the crystal form.
Table 1.
Crystallographic Information
| CurJ H978F | CurK H996F | CurK D1169N | |
|---|---|---|---|
|
| |||
| Data | |||
| Space group | P212121 | P212121 | P212121 |
| Unit cell a, b, c (Å) | 47.55, 70.58, 176.02 | 38.11, 94.57, 152.00 | 38.22, 94.51, 151.70 |
| Wavelength (Å) | 1.000 | 1.000 | 1.000 |
| dmin (Å) | 2.4 (2.486 – 2.4) | 1.428 (1.479 – 1.428) | 1.648 (1.707 – 1.648) |
| Observations (#) | 302,198 (30,140) | 1,300,944 (96,298) | 860, 172 (75,239) |
| Unique reflections (#) | 23,985 (2,331) | 100,090 (8,843) | 67,404 (6,535) |
| Mean I/σI | 18.08 (2.07) | 28.15 (2.74) | 25.41 (2.07) |
| Rmerge | 0.09 (1.44) | 0.04 (0.72) | 0.05 (1.10) |
| CC1/2 | 0.99 (0.77) | 1.00 (0.83) | 1.00 (0.70) |
| CC* | 1.00 (0.93) | 1.00 (0.95) | 1.00 (0.91) |
| Completeness (%) | 100 (100) | 97 (87) | 100 (99) |
| Wilson B (Å2) | 62.4 | 19.4 | 26.5 |
|
| |||
| Refinement | |||
| Reflections (#) | 23,944 (2304) | 100,088 (8,843) | 67,401 (6,535) |
| Rwork | 0.188 (0.349) | 0.20 (0.25) | 0.20 (0.28) |
| Rfree | 0.242 (0.376) | 0.22 (0.29) | 0.23 (0.31) |
| RMSD bonds (Å) | 0.009 | 0.013 | 0.01 |
| RMSD angles (Å) | 1.06 | 1.32 | 1.21 |
| Atoms (#) | |||
| Protein | 4,587 | 4,444 | 4,436 |
| Solvent | 22 | 168 | 46 |
| Ligand | 11 | 0 | 0 |
| Avg B-factors (Å2) | |||
| Protein | 111 | 27.6 | 36.2 |
| Solvent | 99.8 | 30 | 30.4 |
| Ligand | 62.6 | n/a | n/a |
| Ramachandran | |||
| Favored (%) | 97.3 | 98 | 97.4 |
| Allowed (%) | 2.2 | 1.6 | 2.6 |
| Outliers (%) | 0.5 | 0.4 | 0.0 |
CurK-DH and CurF-DH Outcompete CurJ-DH in the Dehydration of its Predicted Substrate
We next evaluated the diastereomeric triketide substrates 3–6 with all four Cur DHs (Figure 3). Interestingly, only CurK-DH and CurF-DH were able to turn over the predicted CurJ-DH substrate 3, arising from the B1-type CurJ-KR, affording the conjugated trans-trans-dienoate 8 (Figure 5, Panel A).29 Neither of the syn-triketide substrates 4 and 5 nor the anti-triketide 6 were processed by any of the Cur DHs (Figure 5, Panels B, C and D). This reaffirms the substrate specificity of CurF-DH and CurK-DH for D-configured alcohols at the β-position. The trans-olefin can only be formed through a syn-elimination of the α-proton and β-hydroxy group in 3 and thus provides the first confirmation for this mechanism for a PKS substrate lacking an α-substituent. From a biosynthetic perspective, the inability of CurJ-DH to process any of the truncated triketide substrates was unexpected, especially in light of the high activity of CurK-DH with its predicted truncated tetraketide substrate 1. This is in stark contrast to CurJ-DH’s ability to accommodate substrate 1, a D-alcohol lacking an α-substituent, albeit with marginal catalytic efficacy. Furthermore, these results rule out the classic “stuttered” dehydration pathway because the same substrate would be used for the second elimination. As both CurH and CurJ immediately follow modules without DH domains (i.e. CurG and CurI) we sought an alternative hypothesis for dehydration.10
Figure 5.
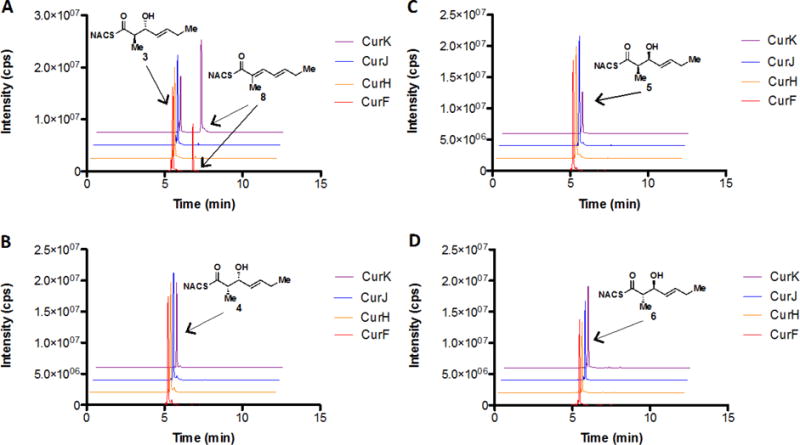
Overnight incubations of the putative triketide CurJ-DH substrates with all curacin PKS dehydratases. Panels A, B, C, and D show the LC-MS/MS extracted ion chromatograms with diastereomers 3, 4, 5 and 6, respectively. In each panel, the LC-MS/MS extracted ion chromatograms are stacked with the traces for each enzyme color-coded: red for CurF, orange for CurH, blue for CurJ, and purple for CurK. The transition m/z 242→95 was used to monitor both remaining substrate as well as product formation due to spontaneous dehydration under the MS ionization conditions. The anti-triketide 3 (panel A) is the only substrate that showed turnover by the appearance of a peak at 6.8 min corresponding to the dehydration product (this was only observed with CurK-DH and CurF-DH).
CurJ-DH and CurH-DH Harbor Vinylogous Dehydration Activity
The simplest proposal is that the KR products from modules lacking dehydratases are carried through to the next module. After extension and reduction, the resulting β,δ-dihydroxy thioester could first be eliminated in the normal fashion (α, β) followed by a vinylogous elimination (γ, δ) yielding the diene (Figure 6). Truncated δ-hydroxy-α,β -unsaturated thioester substrates 20 and 21 were synthesized to test this hypothesis (Supporting Information and Figure 6). No dehydration products were observed upon incubation with the L-alcohol substrate 20 (Figure 7, Panel A). Overnight incubation with all four DH domains revealed that only CurH-DH and CurJ-DH were able to stereospecifically process the D-alcohol substrate 21 to afford the trans-trans-dienoate product 8 (Figure 7, Panel B). Since the reaction is theoretically reversible, we next investigated the hydration reaction through incubation of trans-trans-dienoate 8 with each Cur DH domain. Consistent with the previous results, only CurH-DH and CurJ-DH were able to catalyze the reverse reaction, namely the regiospecific and stereospecific conversion of trans-trans-dienoate 8 exclusively to 21 when compared to controls, albeit at a low total conversion (<1%) (Figure 7, Panel C). Both the inability of the enzyme to reach a stable equilibrium and markedly slow reaction rate compared to the α,β-dehydration catalyzed by CurK indicate that the truncations of substrate 21 severely attenuate enzyme reactivity. The mechanism of this novel elimination was then probed through site-directed mutagenesis of the His-Asp catalytic dyad in both CurH-DH and CurJ-DH. In this case, the catalytic histidine was mutated to alanine and phenylalanine to afford CurH(H971A), CurH(H971F), CurJ(H978A), and CurJ(H978F) mutants. Similarly, the catalytic aspartate was mutated to asparagine yielding CurH(D1136N) and CurJ(D1156N). All mutants were completely devoid of activity towards 21 highlighting their critical role in this novel elimination reaction (Figure 7, Panel D).
Figure 6.
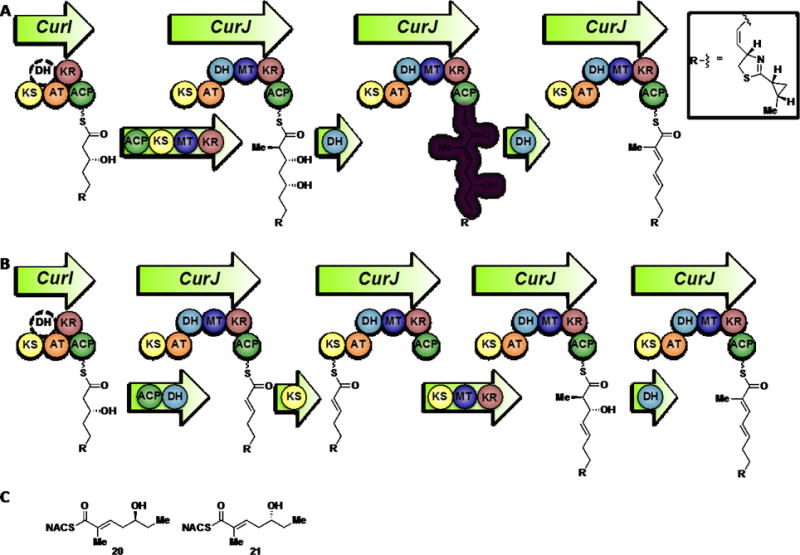
Proposed routes for elimination in the case of missing dehydratases. Panel A displays our proposed vinylogous elimination route in module J. The polyketide intermediate from CurI is loaded onto CurJ for normal extension, methylation and ketoreduction. The CurJ-DH eliminates the β-alcohol followed by a subsequent elimination of the δ-alcohol. Panel B shows a classical stuttering mechanism based on predictions in other biosynthetic pathways. The polyketide intermediate from CurI is transferred directly onto the CurJ-ACP and dehydrated by the CurJ-DH. The ACP bound intermediate is then transferred to the CurJ-KS for normal extension, methylation and ketoreduction. A final dehydration by CurJ-DH furnishes the putative CurJ product. Panel C presents the enantiomeric vinylogous CurJ-DH substrates 20 and 21. The design of the vinylogous substrates is based on the putative native substrate shown highlighted in violet in Panel A.
Figure 7.
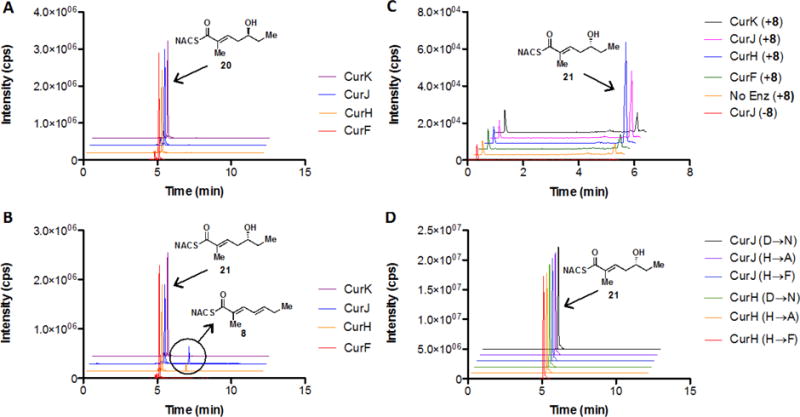
The vinylogous elimination of δ-hydroxy-α,β-unsaturated thioester substrates by curacin DH domains. Panels A and B display the LC-MS/MS extracted ion chromatograms obtained after overnight incubations of substrates 20 and 21 with each Cur DH monitoring the transition at m/z 242→95 for product dienoate 8 (peak at 6.8 minutes) and m/z 260→141 (peak at 5.1 minutes) for residual substrates 20 and 21. In each panel, the LC-MS/MS extracted ion chromatograms are stacked with the traces for each enzyme color-coded: red for CurF-DH, orange for CurH-DH, blue for CurJ-DH, and purple for CurK-DH. The L-alcohol substrate 20 shown in Panel A is not a substrate for any of the Cur DHs whereas the D-alcohol substrate 21 is converted to the corresponding dienoate 8 only by CurJ-DH and CurH-DH. Panel C shows the hydration of dienoate 8. Compared to negative control (without 8 or without CurJ-DH), only CurH-DH and CurJ-DH form 21 in the presence of 8. Panel D shows overnight incubation of the six CurJ-DH and CurH-DH catalytic dyad point mutants scanning for the same transitions as previous panels. All mutants are devoid of activity as seen by the absence of a peak at 6.8 min.
To visualize the binding of a vinylogous substrate to a DH, 21 was co-crystallized with the catalytically inactive H978F variant of the CurJ DH. Crystals grew more slowly in presence of compound (30 days compared to 2 days without substrate). In the 2.4-Å structure of the DH dimer, new density appeared in the active site of one monomer, adjacent to Asp1156 and within the hydrophobic substrate tunnel (Figure 8). Strong density was present for only the acyl portion of 21 and we assume that the polar N-acetylcysteamine was hydrolyzed during the long crystal growth period. The substitution of Phe for the catalytic His978 resulted in no change to the protein backbone conformation. The best fit of hydrolyzed 21 to the new density placed the δ-OH group within hydrogen bonding distance of Asp1156, in the same position as a water molecule in the free-enzyme structure.10 The acid 21 was bound such that dehydration would proceed via the classic syn elimination mechanism of both PKS and FAS DHs.
Figure 8.
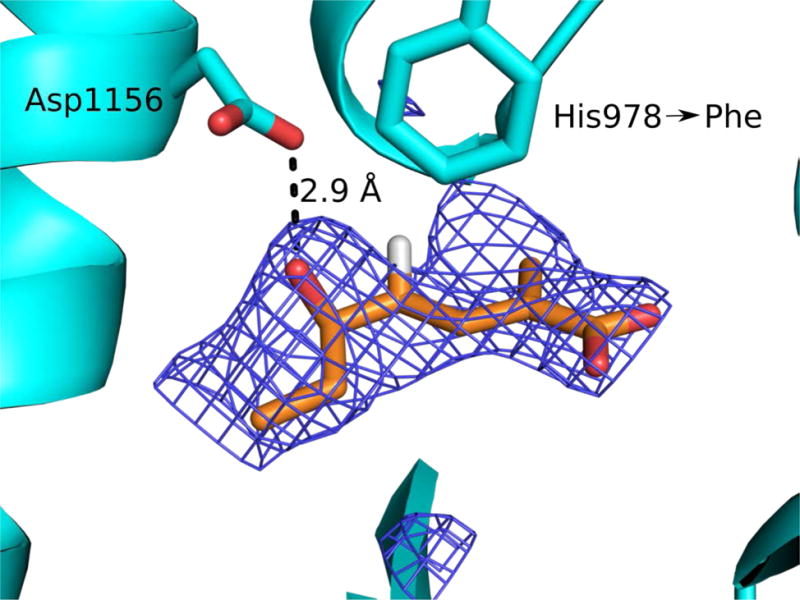
The CurJ DH H978F variant co-crystallized with the hydrolyzed form of compound 21. The δ-hydroxyl group of carboxylate 21 is within hydrogen bonding distance of Asp1156 (blue SA omit electron density contoured at 2.5σ). Hydrolyzed 21 (sticks with atom coloring: orange C, red O, white H; only the pro(R) H atom is shown) is within 4Å of the Phe substitution for His978 (sticks with cyan C).
DISCUSSION
Enigmatic Dehydration in Polyketide Synthases
Curiously, there are numerous examples in the literature of missing DH domains when diene final products or intermediates are predicted. Notable cases come from the biosynthesis of columbamides, stigmatellin, epothilones, leinamycin, thuggacin, and the myxalamides (Figure 9).30–34 Unexpectedly, many of these anomalies involve the formation of a cis-alkene during the distal dehydration. The classic interpretations of these cases invoke either a trans-acting dehydratase, separate from the PKS pathway, or a stuttered dehydration process.30,35 Vinylogous dehydration of δ-hydroxy-α,β-unsaturated thioester substrates as described here provides an alternative mechanism.
Figure 9.
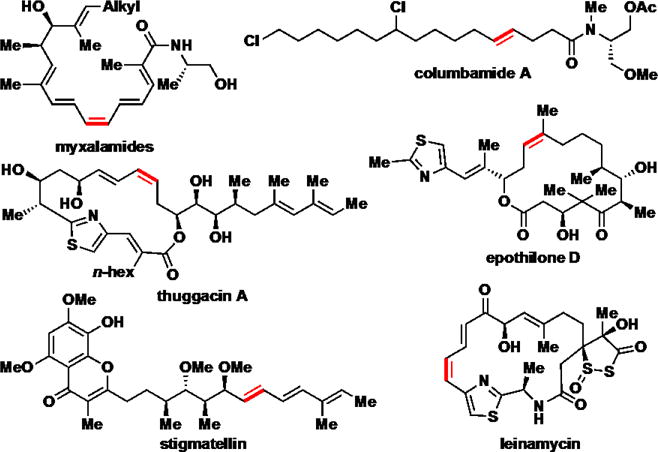
Select examples of sequential dehydration with a missing DH domain. Alkenes arising from modules lacking a dehydratase are highlighted in red. The subsequent module contains an active dehydratase, possibly capable of vinylogous dehydration.
Crystal Structures and Substrate Specificity
The crystallization behavior of both CurK DH and CurJ DH in the presence of NAC-linked substrates is suggestive of a conformational change in the protein at the entrance to the substrate tunnel. For both DHs, crystals either did not grow in the presence of substrate or grew only after thioester hydrolysis. Similarly, pre-grown DH crystals dissolved when presented with substrate. The structure of CurJ DH H978F with the hydrolyzed 21 reveals an ideal orientation for hydrogen bonding of the catalytic Asp1156 carboxylate with the substrate hydroxyl. The site is occupied by a water molecule in structures of the DH free enzyme.10 We found that the best fit of hydrolyzed 21 to the experimental density was in a conformation that would yield a trans-cis dienoate. As CurJ DH produced the trans-trans dienoate from 21 (Figure 7, Panel B), we assume that hydrolysis of the NAC thioester accounts for the predominant bound conformation.
Mechanistic Analogy to Canonical Eliminations in DH Domains
Vinylogous dehydration by DH domains closely parallels canonical dehydrations. Stereospecificity appears to be maintained between the two events in the module. Moreover, the empirical trend that D-alcohols form E-substituted olefins holds true in this distal elimination. The catalytic His-Asp dyad is essential for this novel, second transformation by the dehydratase domain as the respective point mutants are catalytically incompetent. Given these results, we predict the reaction is a net syn-elimination as previously established in α,β-dehydrations in PKS and FAS systems.36,37 The proposed mechanism shares similarities to three reported biosynthetic enzymes: FabA, an isomerizing DH involved in corallopyronin A biosynthesis, and a recently characterized isomerase (Figure 10).18,38,39 The first step involves deprotonation of the γ–proton, pro-(R) in the case of FabA, by a general base (likely the catalytic His) to afford a vinylogous enolate.40 Until this point, the reaction mechanism mirrors that of a classic isomerase. Instead of suprafacial protonation at the α-position, the subsequent elimination occurs with simultaneous protonation of the δ-hydroxyl group by the catalytic aspartic acid residue, forming the γ,δ-olefin. The dual activities would require that the substrate shift in the active site tunnel first to place the α-position by the catalytic His and the β-hydroxy by the catalytic Asp for the canonical dehydration, and then the γ-position by the catalytic His and δ-hydroxy by the catalytic Asp for the vinylogous dehydration.
Figure 10.
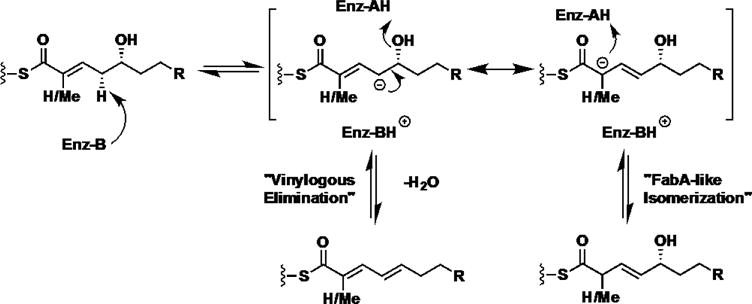
FabA-isomerization mechanism and proposed mechanism of vinylogous dehydration by curacin dehydratases J and H. The two events share a common intermediary vinylogous enolate and only differ only in the final re-protonation or elimination. Site-directed mutagenesis of the catalytic dyad indicates that both the canonical histidine and aspartic residues are the general base and acid, respectively. FabA (Escherichia coli β-hydroxydecanoyl thiol ester dehydrase) catalyzes elimination and bond isomerization in fatty acid biosynthesis.
In the present study, we found that CurF-DH activity unexpectedly closely mirrors that of the CurK-DH in its stereospecific processing of 1 over 2. This seemingly nonsensical mimicry of dehydration activity is puzzling due to the fact that CurK-DH is predicted to process the longest dehydratase substrate in the Cur pathway, and CurF DH has no known function. By analyzing CurF polyketide cassette (ER-KS-AT-DH) against other possible pathways in the producing organism, we found that JamJ from the jamaicamide pathway has high homology (>72% identical) and the predicted action in its pathway matches that of the extra modules in CurF (i.e. a complete reductive processing sequence to form an alkane intermediate). The majority of the modules needed for jamaicamide synthesis appear to be vestigial in CurF, and vice versa. For example, the ER domain is responsible for cyclopropane formation in curacin biosynthesis, whereas the vestigial JamJ ER domain has no activity with the natural jamaicamide β,γ-olefin intermediate but has robust reductive activity with α,β-olefin substrates.2 Additionally, there is an NRPS module set (PCP-A-Cy) for thiazoline production appended to the end of the vestigial CurF PKS module. Genetic analysis of several PKS biosynthetic clusters has recently revealed that analogous instances of surplus DH domains present in a pathway compared to a predicted activity is a more common occurrence than missing DH domains. This often arises due to two unique evolutionary pressures: The necessity to retain a hydroxyl group in the pathway for the final natural product or the duplication and insertion of a full or partial module cassette to fulfill another pathway role. In the former case, the DH domain is often catalytically inactivated by a knockout mutation in the catalytic region. Classic examples include rifamycin (DH2 and 5), amphotericin B (DH15, 17 and 18) and FK506 (DH3, 4 and 8).41–43 The latter case is often harder to interpret biosynthetically as the partial module harbors necessary activity for the natural product biosynthesis (i.e. CurF ER), but also a superfluous and active DH domain (i.e. CurF DH). Examples include rifamycin (DH6, 7 and 8), epothilone A and B (DH7 and 8) and FK506 (DH2).41,43,44,45 Our results indicate this may be diagnosed by both sequence comparison to other modules in the organism and substrate-dehydratase interrogation, looking for high sequence identity and unexpected substrate selectivity, respectively.
CONCLUSION
The curacin biosynthetic pathway continues to provide unique insight into non-canonical PKS enzymology. The present study focused on the mechanism and timing of DH-catalyzed processing in assembly of the polyketide segment introduced by the five Cur PKS monomodules CurG-CurK. Retrobiosynthetic analysis indicated five dehydrations are required; however, DH domains are missing in both CurG and CurI while an extraneous DH domain is present in CurF. To uncover the anomalous biosynthetic features of DH-catalysis in this segment of the polyketide, we employed diffusible small-molecule truncated NAC thioester substrates in conjunction with LC-MS/MS analysis, site-directed mutagenesis, and co-crystallization studies.
We initially focused on the dehydration event in CurK using a tetraketide substrate and showed that CurK processed its predicted D-alcohol substrate 1 to the all trans-trienoate 7 more than three-orders of magnitude greater than any previously reported DH with its native substrate,27,28 highlighting the high intrinsic activity of Cur DHs when presented with their native substrates. Therefore, the inability of the CurJ-DH, which is the DH domain in the preceding module, to turn over any of the four diastereomeric triketide substrates including 21, with the bioinformatically predicted 2R, 3S stereochemistry, suggested an alternative substrate. We hypothesized that CurJ may be responsible for a double dehydration of a β,δ-dihydroxy thioester substrate since the antecedent DH domain is absent in CurI. This would require a canonical dehydration to form a δ-hydroxy-α,β-unsaturated thioester intermediate, which would then undergo a second vinylogous elimination, at the same active site, to afford the trans-trans-dienoate product. Using a synthetic triketide δ-hydroxy-α,β-unsaturated thioester substrate, we unequivocally demonstrated that CurJ can catalyze this novel vinylogous elimination as well as the reverse hydration reaction in a completely stereospecific and regiospecific manner. The vinylogous elimination required the His-Asp catalytic dyad as point mutations to either of these residues completely abolished activity. CurH analogously lacks a DH domain in the preceding module and was also shown to catalyze a vinylogous elimination. By contrast, CurK does not catalyze the vinylogous elimination, but only the canonical dehydration as expected based on the collinear organization of CurJ and CurK, which both contain functional DH domains. Interestingly, the substrate specificity of CurF-DH mirrored that of CurK-DH, suggesting CurF-DH is vestigial and may have evolutionary arisen from an earlier gene duplication event.
To provide a structural framework for the novel vinylogous elimination, we co-crystallized a mutagenized CurJ(H978A) with δ-hydroxy-α,β-unsaturated thioester 21. The binding pose shows that the catalytic Asp1156 side chain is positioned to hydrogen bond with a hydroxyl group - or water molecule, as in the free-enzyme structures - and that this could be either a β-hydroxy or a δ-hydroxy.
The vinylogous elimination mechanism, catalyzed by both CurH-DH and CurJ-DH, compensates for the two modules lacking dehydratases (CurG and CurI) offering an alternative to the stuttered module hypothesis. The proposed mechanism closely resembles that of FabA in E. coli, which catalyzes α,β to γ,δ-isomerization of unsaturated fatty acids utilizing a His-Asp catalytic dyad. This discovery expands the growing number of known transformations carried out by the hotdog-fold family of enzymes. More broadly this new mechanism may be operative in other PKS systems missing DH domains.
EXPERIMENTAL SECTION
General Chemistry Procedures
All chemical reagents were used as provided unless indicated otherwise. Freshly distilled reagents were purified as reported.46 Tetrahydrofuran (THF) and dichloromethane (CH2Cl2) were purified via passage through neutral alumina columns. All reactions were performed under an argon atmosphere using oven-dried (150 °C) glassware. Compounds were purified by flash chromatography using silica gel (300–400 mesh) in the indicated solvent system. TLC was performed using 250 μm, F254 silica gel plates and visualized by UV or through staining with para-anisaldehyde. Optical rotations were acquired on a polarimeter at the indicated temperature using the sodium D line (λ = 589 nm) unless otherwise specified and reported as follows: = rotation (c g/100 mL, solvent). 1H and 13C NMR spectra were recorded on a 400 MHz NMR spectrometer. Chemical shifts are reported in ppm based on an internal standard of residual CHCl3 (7.26 ppm in 1H NMR and 77.16 in 13C NMR). Proton chemical data are reported in the following format: chemical shift (ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, sept = septet, m = multiplet, br = broad peak), coupling constant(s), and integration. High-resolution mass spectra (HRMS) were obtained on a time-of-flight (TOF) mass spectrometer using either PEG or PPG standards to calibrate the instrument.
Ethyl (2E,4E)-2-methylhepta-2,4-dienoate (11)
To a solution of triethyl 2-phosphonopropionate (2.81 mL, 13.1 mmol, 1.10 equiv) in THF (36 mL) at 0 °C was added sodium hydride (60% in oil, 0.595 g, 14.9 mmol, 1.25 equiv) and the reaction stirred for 2 h. Freshly distilled 2-pentenaldehyde 945 (1.16 mL, 11.9 mmol, 1.00 equiv) was then added dropwise over 20 min. After stirring at 0 °C for 5.5 h, the reaction was quenched with H2O (20 mL). The biphasic solution was separated and the aqueous layer was extracted with Et2O (4 × 20 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by silica gel flash chromatography (95:5 pentane–Et2O) afforded the title compound (2.00 g, quant.) as a transparent light yellow oil: Rf = 0.22 (98:2 pentane–Et2O); 1H NMR (CDCl3, 400 MHz) δ 7.16 (d, J = 11.2 Hz, 1H), 6.33 (dd, J = 14.8, 11.2 Hz, 1H), 6.11 (dt, J = 15.2, 6.4 Hz, 1H), 4.20 (q, J = 7.2 Hz, 2H), 2.21 (quint, J = 7.2 Hz, 2H), 1.93 (s, 3H), 1.30 (t, J = 7.2 Hz, 3H), 1.05 (t, J = 7.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 168.8, 144.6, 138.7, 125.3, 125.2, 60.6, 26.5, 14.5, 13.3, 12.7; HRMS (ESI+) m/z calcd for C10H16O2Na+ [M + Na]+ 191.1043, found 191.1039 (error 2.1 ppm).
(2E,4E)-2-Methylhepta-2,4-dienal (13)
To a solution of 11 (486 mg, 2.89 mmol, 1.00 equiv) in CH2Cl2 (13.0 mL) at 0 °C was slowly added diisobutylaluminum hydride (1.0 M in hexanes, 6.93 mL, 6.93 mmol, 2.40 equiv). After 40 min the reaction was quenched via successive addition of MeOH (3.5 mL) and saturated aqueous sodium potassium tartrate (10.0 mL). The resulting biphasic mixture was stirred vigorously at 23 °C for 14 h. The reaction mixture was partitioned between H2O (10.0 mL) and CH2Cl2 (10 mL). The biphasic mixture was separated and the aqueous layer was extracted with CH2Cl2 (3 × 12 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure to afford the allylic alcohol 12, which was carried onto the next reaction without further purification: Rf = 0.58 (8:2 hexanes–EtOAc).
To a solution of the crude alcohol 12 prepared above in CH2Cl2 (15.0 mL) were sequentially added anhydrous MgSO4 (1.04 g, 8.67 mmol, 3.00 equiv) and MnO2 (88% activated, 1.76 g, 20.2 mmol, 7.00 equiv). The resulting gray solution was vigorously stirred at 23 °C for 24 h. The reaction mixture was filtered through a pad of Celite (2.00 cm) and the filtrate concentrated under reduced pressure. Purification by silica gel flash chromatography (98:2 pentane–Et2O) afforded the title compound (0.345 g, 96% over 2 steps) as a colorless oil: Rf = 0.43 (9:1 pentane–Et2O); 1H NMR (CDCl3, 400 MHz) δ 9.42 (s, 1H), 6.83 (d, J = 11.2 Hz, 1H), 6.52 (ddt, J = 14.8, 11.2, 1.6 Hz, 1H), 6.28 (dt, J = 14.8, 6.4 Hz, 1H), 2.32–2.23 (m, 2H), 1.84 (t, J = 0.8 Hz, 3H), 1.10 (t, J = 7.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 195.3, 149.5, 147.3, 136.2, 125.1, 26.7, 13.1, 9.5; HRMS (ESI+) m/z calcd for C8H12ONa+ [M + Na]+ 147.0780, found 147.0794 (error 9.5 ppm).
(4R)-3-[(3S,4E,6E)-3-Hydroxy-4-methylnona-4,6-dienoyl]-4-isopropylthiazolidine-2-thione (15)
To a solution of 14 (0.484 g, 2.38 mmol, 1.60 equiv) in CH2Cl2 (12.0 mL) at −40 °C was added TiCl4 (0.277 mL, 2.53 mmol, 1.70 equiv) whereupon the solution changed from bright yellow to orange-red. The solution was stirred for 30 min at −40 °C, then freshly distilled iPr2NEt (0.441 mL, 2.53 mmol, 1.70 equiv) was added dropwise. After 2.25 h the blood-red solution was cooled to −78 °C and aldehyde 13 (185.0 mg, 1.49 mmol, 1.00 equiv, dried overnight over 3 Å molecular sieves) was added over one min. The aldol reaction was stirred at −78 °C for 3.5 h and quenched with saturated aqueous NH4Cl (6.0 mL). The biphasic solution was separated and the aqueous layer was extracted with CH2Cl2 (4 × 10 mL). The combined organics were dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by silica gel flash chromatography (7:3 hexane–EtOAc) afforded the title compound (0.230 g, 47%) as a viscous bright yellow oil: Rf = 0.41 (7:3 hexane–EtOAc); = −246 (c 1.00, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 6.24 (dd, J = 14.8, 10.8 Hz, 1H), 6.09 (d, J = 10.8 Hz, 1H), 5.76 (dt, J = 14.8, 6.8 Hz, 1H), 5.14 (t, J = 6.8 Hz, 1H), 4.61 (dd, J = 8.8, 0.8 Hz, 1H), 3.59–3.48 (m, 2H), 3.42 (dd, J = 17.2, 9.2 Hz, 1H), 3.03 (d, J = 11.6 Hz, 1H), 2.65 (br s, 1H), 2.37 (sext, J = 6.4 Hz, 1H), 2.13 (pent, J = 7.2 Hz, 2H), 1.77 (s, 3H), 1.06 (d, J = 6.8, 3H), 1.02 (d, J = 7.4, 3H), 0.99 (t, J = 8.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 203.2, 172.9, 137.5, 135.2, 125.9, 124.9, 73.0, 71.7, 44.1, 31.0, 30.8, 26.2, 19.2, 18.0, 13.8, 13.0; HRMS (ESI+) m/z calcd for C16H25NO2S2Na+ [M + Na]+ 350.1219, found 350.1207 (error 3.4 ppm).
S-(2-Acetamidoethyl) (S,4E,6E)-3-hydroxy-4-methylnona-4,6-dienethioate (1)
A solution of aldol adduct 15 (15.0 mg, 0.0458 mmol, 1.00 equiv) in CH2Cl2 (3.00 mL) was treated with N-acetylcysteamine (5.0 μL, 0.050 mmol, 1.1 equiv) and imidazole (9.4 mg, 0.14 mmol, 3.0 equiv) and stirred at 23 °C for 22 h. The reaction mixture was partitioned between H2O (5 mL) and EtOAc (5 mL) and the layers were separated. The aqueous layer was extracted with EtOAc (4 × 10 mL) and the combined organic fractions were dried (Na2SO4), filtered and concentrated under reduced pressure. The crude residue was purified by flash chromatography (95:5 CH2Cl2–MeOH) to afford the title compound (7.2 mg, 55%) as a colorless oil: Rf = 0.39 (95:5 CH2Cl2–MeOH); = −21 (c 0.52, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 6.22 (dd, J = 15.2, 10.4 Hz, 1H), 6.07 (d, J = 10.8 Hz, 1H), 5.83 (br s, 1H), 5.76 (dt, J = 15.2, 6.4 Hz, 1H), 4.53 (dd, J = 9.2, 3.6 Hz, 1H), 3.48–3.40 (m, 2H), 3.05 (app. sext, J = 8.0 Hz, 2H), 2.84 (dd, J = 15.2, 8.8 Hz, 1H), 2.76 (dd, J = 14.8, 4.0 Hz, 1H), 2.48 (br s, 1H), 2.13 (app. quint, J = 7.2 Hz, 2H), 1.96 (s, 3H), 1.75 (s, 3H), 1.01 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 199.0, 170.6, 138.0, 134.9, 126.3, 124.7, 74.0, 49.6, 39.5, 29.0, 26.1, 23.4, 13.8, 12.6; HRMS (ESI+) m/z calcd for C14H23NO3SNa+ [M + Na]+ 308.1291, found 308.1275 (error 5.2 ppm).
(4R)-4-Benzyl-3-[(2S,3R,4E)-3-hydroxy-2-methylhept-4-enoyl]thiazolidine-2-thione (18)
To a solution of thiazolidinethione 17 (0.250 g, 0.940 mmol, 1.00 equiv) in CH2Cl2 (4.00 mL) at 0 °C was added TiCl4 (0.110 mL, 0.990 mmol, 1.05 equiv). The resulting bright orange, opaque solution was stirred for 9 min. Next, freshly distilled iPr2EtN (0.183 mL, 1.05 mmol, 1.12 equiv) was added and the reaction was further stirred for 40 min at 0 °C. To the dark red mixture was slowly added freshly distilled 2-pentenaldehyde (0.138 mL, 1.41 mmol, 1.50 equiv) causing a color change to dark brown. After 2 h, the reaction was quenched with saturated aqueous NH4Cl (5 mL). The biphasic mixture was warmed to 23 °C and the layers separated. The aqueous layer was extracted with CH2Cl2 (3 × 10 mL) and the combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography (8:2 hexanes–EtOAc) afforded the title compound (0.252 g, 72 %) as a viscous, yellow oil: Rf = 0.20 (8:2 hexanes–EtOAc); = −191.6 (c 1.00, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 7.37–7.27 (m, 5H), 5.82 (ddt, J = 15.6, 6.4, 1.2 Hz, 1H), 5.50 (ddt, J = 15.2, 6.0, 1.2 Hz, 1H), 5.39 (ddd, J = 10.6, 6.8, 4 Hz, 1H), 4.79 (dq, J = 6.8, 3.2 Hz, 1H), 4.59–4.54 (m, 1H), 3.37 (dd, J = 11.6, 6.8 Hz, 1H), 3.24 (dd, J = 13.2, 4.0 Hz, 1H), 3.04 (dd, J = 13.2, 10.4 Hz, 1H), 2.88 (d, J = 11.6 Hz, 1H), 2.74 (d, J = 2.8 Hz, 1H), 2.08 (app. quint, J = 7.2, 2H), 1.19 (d, J = 6.8 Hz, 3H), 1.01 (t, J = 7.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 201.8, 177.8, 136.6, 135.0, 129.6, 129.1, 127.9, 127.4, 72.5, 69.1, 43.5, 37.1, 31.9, 25.5, 13.6, 11.6; HRMS (ESI+) m/z calcd for C18H23NO2S2Na+ [M + Na]+ 372.1062, found 372.1087 (error 6.7 ppm).
(4R)-4-Benzyl-3-[(2R,3R,4E)-3-hydroxy-2-methylhept-4-enoyl]thiazolidine-2-thione (19)
To a solution of 17 (0.150 g, 0.566 mmol, 1.00 equiv) in anhydrous EtOAc (1.4 mL) were sequentially added anhydrous MgBr2•OEt2 (14.6 mg, 0.0566 mmol, 0.100 equiv), 2-pentenaldehyde (60.9 μL, 0.623 mmol, 1.10 equiv), Et3N (0.158 mmol, 1.13 mmol, 2.00 equiv) and Me3SiCl (0.108 mL, 0.849 mmol, 1.50 equiv), which all had been freshly distilled. The reaction mixture was stirred at 23 °C for 26 h, then filtered through a silica gel plug (2.00 cm), washing with Et2O (20 mL), and the filtrate was concentrated under reduced pressure. The crude residue was dissolved in a biphasic mixture of THF (10 mL) and aqueous hydrochloric acid (1 N, 2 mL) and vigorously stirred at 23 °C for 2 h. The reaction was then quenched with saturated aqueous sodium bicarbonate (10 mL) and the biphasic solution was separated. The aqueous layer was extracted with EtOAc (3 × 15 mL) and the combined organic fractions were dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by silica gel flash chromatography (8:2 hexane–EtOAc) afforded the title compound (35.8 mg, 17%) as a viscous, yellow oil: Rf = 0.28 (8:2 hexane–EtOAc); = −297.1 (c 1.00, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 7.37–7.27 (m, 5H), 5.79 (dt, J = 15.2, 5.6 Hz, 1H), 5.42 (ddt, J = 15.2, 7.2, 1.6 Hz, 1H), 5.25 (ddd, J = 10.8, 6.8, 3.6 Hz, 1H), 4.34 (dq, J = 7.6, 6.8 Hz, 1H), 4.26 (t, J = 7.6 Hz, 1H), 3.39 (dd, J = 11.2, 6.8 Hz, 1H), 3.26 (dd, J = 13.2, 3.6 Hz, 1H), 3.06 (dd, J = 12.8, 10.8 Hz, 1H), 2.90 (d, J = 11.6 Hz, 1H), 2.16 (br, 1H), 2.06 (app. quint, J = 6.4, 2H), 1.22 (d, J = 6.8 Hz, 3H), 1.00 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 201.5, 178.2, 136.7, 136.3, 129.6, 129.3, 129.0, 127.3, 76.7, 69.2, 45.2, 36.8, 32.8, 25.4, 14.9, 13.5; HRMS (ESI+) m/z calcd for C18H23NO2S2Na+ [M + Na]+ 372.1062, found 372.1049 (error 3.5 ppm).
S-(2-Acetamidoethyl) (2R,3R,4E)-3-hydroxy-2-methylhept-4-enethioate (3)
To a solution of 19 (31.1 mg, 0.0890 mmol, 1.00 equiv) in CH2Cl2 (4.00 mL) was added imidazole (18.2 mg, 0.267 mmol, 3.00 equiv) and N-acetylcysteamine (10.4 μL, 0.0980 mmol, 1.10 equiv). The reaction mixture was vigorously stirred at 23 °C for 21 h. The crude reaction mixture was concentrated under reduced pressure and directly purified by flash chromatography (95:5 CH2Cl2–MeOH) to afford the title compound (16.8 mg, 67%) as a light yellow, viscous oil: Rf = 0.30 (95:5 CH2Cl2–MeOH); = −34.5 (c 0.29, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 6.00 (br s, 1H), 5.76 (dt, J = 15.6, 6.0 Hz, 1H), 5.40 (ddt, J = 15.6, 6.4, 1.2 Hz, 1H), 4.19 (t, J = 7.6 Hz, 1H), 3.55–3.37 (m, 2H), 3.13–2.98 (m, 2H), 2.76 (app. quint, J = 7.2 Hz, 1H), 2.42 (br s, 1H), 2.06 (app. quint, J = 7.6 Hz, 2H), 1.94 (s, 3H), 1.11 (d, J = 6.8 Hz, 3H), 1.00 (t, J = 7.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 203.6, 170.6, 136.6, 128.8, 75.5, 54.5, 39.5, 28.7, 25.4, 23.3, 15.1, 13.5; HRMS (ESI+) m/z calcd for C12H21NO3SNa+ [M + Na]+ 282.1134, found 282.1153 (error 6.7 ppm).
General biology procedures
All chemical reagents were purchased from Sigma-Aldrich and were used directly without further purification. E. coli BL21(DE3) cells were from New England BioLabs. IPTG was acquired through Gold Biotechnology. His60 Ni Superflow resin was purchased from Clontech Laboratories, Inc. OD600 was measured on an Eppendorf BioPhotometer. Sonication was carried out with a Branson Sonifier 450. Gel filtration purification was performed on HiLoad 16/600 Superdex 200 pg column (GE Healthcare). The protein mass spectra data was obtained on a Bruker BioTOF II mass spectrometer. LC–MS/MS was conducted with an AB Sciex QTRAP 5500 mass spectrometer and Shimadzu LC system.
Protein purification and crystallization
DHs were purified as described previously.10 For co-crystallization of substrates with inactive CurJ DH variants, the protein was pre-incubated 2 hr on ice in 5 mM substrate, and then crystallized at 20°C by sitting-drop vapor diffusion from a 1:1 mixture of protein stock (15 mg/mL DH, 5 mM substrate, 50 mM Tris pH 7.5, 150 mM NaCl, 10% glycerol) with well solution (18% polyethylene glycol (PEG) 3350, 5% 1,4-butanediol, 250 mM NaCl, 100 mM Bis-Tris propane pH 6.5). Crystals were harvested directly from drops into liquid N2.
Data collection and structure determination
Data were collected at the Advanced Photon Source (APS, Argonne National Laboratory) on GM/CA beamline 23ID-D. CurJ H978F crystals diffracted to 2.4 Å in the presence of 5 mM substrate. The crystals were nearly isomorphous with the previously reported crystals of wild-type CurJ DH.10 Data were processed using XDS.47 The structure was solved by molecular replacement in PHENIX48 using the wild-type CurJ (PDB 3KG8) as a search model. Refinement was accomplished in PHENIX and model building with coot49. Ligand restraint files were built using eLBOW50 in PHENIX. The structure was validated with MolProbity51; figures were generated with PyMOL.52
Steady-State Kinetic Studies of CurK-DH
Incubation and sample preparation
Substrate 1 (0.25–6.0 mM) was incubated with recombinant CurK-DH (20 nM) and reaction buffer (50 mM Tris, 150 mM NaCl, pH 8.0) in a total volume of 50 μL at 23 °C. The reaction was quenched at 4 min by transfer of 5 μL of the reaction mixture into 495 μL of quench solution (1:1 MeCN–H2O). The quenched mixture was briefly vortexed and centrifuged (21,000 × g, 5 min). A portion of the centrifuged supernatant (60 μL) was transferred to an HPLC vial containing the internal standard (10 μL of a 320 nM solution of diene 8), mixed, injected and analyzed by LC–tandem MS.
Instrumentation
Reverse-phase liquid chromatography was conducted on a Shimadzu UFLC XR, SIL-20AC autosampler and Prominence CTO-20A column oven. A SCIEX QTRAP 5500 (Framingham, MA) was used for mass detection with an electrospray ionization source. All instrumentation was run using Analyst Software (1.5.2, AB SCIEX)
Chromatography
Reverse phase HPLC was conducted using a Kinetix reverse-phase C18 column (50 mm × 2.1 mm, 2.6 μm, Phenomenex®, Torrance, CA) operated at 0.4 mL min−1 with a column temperature of 35 °C. The mobile phase A (0.1% formic acid in H2O) and mobile phase B (acetonitrile) were run at the following gradient program: 0 min, 5% B; 2 min, 5% B; 7 min, 55% B; 8 min, 70% B; 9 min, 70% B; 10.5 min, 5% B; 12 min, 5% B, STOP. The injection volume was 10 μL.
Mass Spectrometry
Analytes were analyzed by MS in positive ionization mode by Multiple Reaction Monitoring (MRM). Optimal MRM settings were determined by direct infusion of the analyte solution (10 nM, 1:1 H2O–MeCN with 0.1% formic acid) onto the MS by a syringe pump at a flow of 10 μL/min. The transitions and retention times are displayed in Table 2.
Table 2.
LC-MS/MS transitions, parameters and retention times for 1–8, 20 and 21.
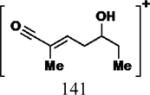
| Cmpd | Precursor Ion (m/z) | Product Ion (m/z) | DPa (V) | CEa (V) | CXPa (V) | Retention Time (min) |
|---|---|---|---|---|---|---|
| 1, 2 |
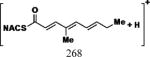
|
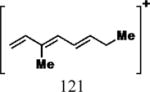
|
35 | 25 | 15 | 6.17 |
| 7 |
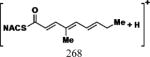
|
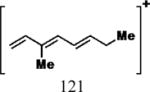
|
35 | 25 | 15 | 7.44 |
| 3, 4, 5, 6 |
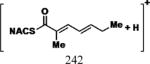
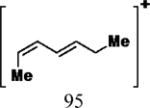
|
|
35 | 30 | 15 | 5.52 (3 and 6) 5.24 (4 and 5) |
| 8 |
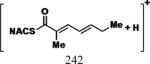
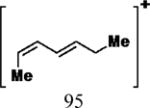
|
|
35 | 30 | 15 | 6.80 |
| 20, 21 |
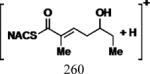
|
|
35 | 17 | 15 | 5.11 |
Mass spectrometry parameter abberviations: DP = de-clustering potential, CE = collision energy, CXP = collision cell exit potential.
Standard Curve and Data Analysis
A standard curve for the triene product 7 was generated by injecting the authentic standard at varying concentrations with a fixed concentration of internal standard 8. Transitions and retention times are given in Table S1. Analyte and internals standard peak areas were calculated using MultiQuant software (version 2.0.2). Each analyte peak area was normalized to internal standard (8) peak area and converted to an analyte concentration using the standard curve equation. Each reaction was performed in duplicate. The calculated initial velocity at each substrate concentration was used to generate a Michaelis-Menten curve (eq. 1, Figure S3) utilizing Prism 5 software (Version 5.0b) (Figure S2):
| (eq. 1) |
where v is the reaction rate, Vmax is the maximum enzymatic reaction rate, [S] is the concentration of substrate and KM is the Michaelis-Menten constant.
Overnight Incubation of CurDHs with Synthetic NAC Substrates
Incubations were carried out following a similar procedure to that of the kinetic analysis of CurK-DH. Briefly, substrates 1–6, 20–21, or product 8 (1 mM) were individually incubated with enzyme (5 μM) (CurF-DH, CurH-DH, CurJ-DH and CurK-DH) in reaction buffer (50 mM Tris, 150 mM NaCl, pH 8.0) at 23 °C for 14 h (or a set time period in the case of the CurK-DH time course experiment) in a total volume of 100 μL. Samples (5 μL) were quenched with 495 μL quench solution (1:1 MeCN–H2O). The mixture was centrifuged and the supernatant was directly analyzed by LC-MS/MS using the method described above. Chromatograms were generated by extraction of raw the LC-MS/MS data with Analyst software and plotted with Prism software.
Chemoenzymatic Synthesis of 1 by CurK-DH. S-(2-Acetamidoethyl) (2E,4E,6E)-4-methylnona-2,4,6-trienethioate (7)
A solution sterile H2O (3.82 mL) and a small amount of 10× TRIS buffer solution (1.50 M NaCl, 0.500M TRIS-HCl, pH 8.0) (0.600 mL) was briefly vortexed. To the buffered solution was added a DMSO– H2O (1:1) stock solution (50 mM) of the β-hydroxy thioester substrate 1 (0.700 mL, 10.0 mg, 35.0 μmol). The mixture was vortexed to mix and the CurK dehydratase (8.19 mg/mL, 0.684 mL, 5.60 mg, 0.160 μmol) was added. The mixture was briefly inverted and covered with aluminum foil to shield from light. The reaction mixture was shaken (200 rpm) at 23 °C in the dark for 14 h. The reaction mixture was extracted with EtOAc (4 × 10.0 mL) and the combined extracts were dried (Na2SO4), filtered and concentrated under reduced pressure. The crude extract was purified by flash chromatography (95:5 CH2Cl2–MeOH) affording the title compound (2.52 mg, 25%) as a transparent light yellow oil: Rf = 0.35 (95:5 CH2Cl2–MeOH); 1H NMR (CDCl3, 400 MHz) δ 7.30 (d, J = 15.6 Hz, 1H), 6.46 (d, J = 11.2 Hz, 1H), 6.40 (app. t, J = 13.6 Hz, 1H), 6.15 (d, J = 15.2 Hz, 1H), 6.05 (dt, J = 13.6, 6.4 Hz, 1H), 5.98 (br, 1H), 3.48 (q, J = 6.0 Hz, 2H), 3.12 (t, J = 6.4 Hz, 2H), 2.22 (app. quint, J = 6.8 Hz, 2H), 1.97 (s, 3H), 1.87 (s, 3H), 1.06 (t, J = 7.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 190.5, 170.6, 146.5, 143.6, 141.6, 131.2, 125.8, 122.8, 40.3, 28.5, 26.6, 23.4, 13.4, 12.5; HRMS (ESI+) m/z calcd for C14H21NO2SNa+ [M + Na]+ 290.1185, found 290.1208 (error 7.9 ppm).
Supplementary Material
Acknowledgments
This work was supported by a grant (GM081544 to J.L.S.) from the National Institutes of Health. Mass spectrometry was carried out with the assistance of Dr. Bruce A. Witthuhn in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, University of Minnesota, funded in part by Cancer Center Support Grant CA-77598. We also gratefully acknowledge financial support from the Department of Medicinal Chemistry, University of Minnesota (to W.D.F).
Abbreviations
- ACP
acyl carrier protein
- AT
acyltransferase domain
- CMT
C-methyltransferase domain
- CoA
coenzyme-A
- Cps
counts per second
- Cur
curacin
- DIBAL-H
diisobutylaluminum hydride
- DIPEA
diisopropylethylamine
- DH
dehydratase domain
- ER
enoyl reductase domain
- FAS
fatty acid synthase
- GNAT
GCN5(yeast histone)-related N-acetyltransferase
- IPTG
isopropyl β-D-1-thiogalctopyranoside
- KR
ketoreductase
- KS
ketosynthase domain
- LC–MS/MS
liquid chromatography-tandem mass spectrometry
- MRM
multiple reaction monitoring
- NAC
N-acetylcysteamine
- OMT
O-methyltransferase
- PKS
polyketide synthase
- SAM
S-adenosylmethionine
- TE
thioesterase domain
- TOF
time-of-flight
Footnotes
Supporting Information. Full 1H and 13C NMR spectra of all described compounds, procedures for the synthesis of compounds 8, 20, and 21 as well as LC–MS/MS data are included as Figures S1–2 and Table S1–2. A figure depicting ligand omit difference density is included as Figure S3. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
“All authors contributed to writing the manuscript and approve the submitted version. All organic syntheses, chemoenzymatic syntheses, compound identification, incubation studies, LC-MS/MS analyses and kinetic characterizations were carried out by W.D.F. The dehydratase mutagenesis and the crystallographic experiments and analysis were completed by G.J.D.”
References
- 1.Gerwick WH, Proteau PJ, Nagle DG, Hamel E, Blokhin A, Slate DL. J Org Chem. 1994;59:1243. [Google Scholar]
- 2.Gu L, Wang B, Kulkarni A, Geders TW, Grindberg RV, Gerwick L, Hakansson K, Wipf P, Smith JL, Gerwick WH, Sherman DH. Nature. 2009;459:731. doi: 10.1038/nature07870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gu L, Wang B, Kulkarni A, Gehret JJ, Lloyd KR, Gerwick L, Gerwick WH, Wipf P, Håkansson K, Smith JL, Sherman DH. J Am Chem Soc. 2009;131:16033. doi: 10.1021/ja9071578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gu L, Eisman EB, Dutta S, Franzmann TM, Walter S, Gerwick WH, Skiniotis G, Sherman DH. Angew Chem, Int Ed. 2011;50:2795. doi: 10.1002/anie.201005280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCarthy JG, Eisman EB, Kulkarni S, Gerwick L, Gerwick WH, Wipf P, Sherman DH, Smith JL. ACS Chem Biol. 2012;7:1994. doi: 10.1021/cb300385m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khare D, Wang B, Gu L, Razelun J, Sherman DH, Gerwick WH, Håkansson K, Smith JL. Proc Natl Acad Sci U S A. 2010;107:14099. doi: 10.1073/pnas.1006738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khare D, Hale Wendi A, Tripathi A, Gu L, Sherman David H, Gerwick William H, Håkansson K, Smith Janet L. Structure. 2015;23:2213. doi: 10.1016/j.str.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu L, Geders TW, Wang B, Gerwick WH, Håkansson K, Smith JL, Sherman DH. Science. 2007;318:970. doi: 10.1126/science.1148790. [DOI] [PubMed] [Google Scholar]
- 9.Whicher JR, Smaga SS, Hansen DA, Brown WC, Gerwick WH, Sherman DH, Smith JL. Chem Biol. 2013;20:1340. doi: 10.1016/j.chembiol.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akey DL, Razelun JR, Tehranisa J, Sherman DH, Gerwick WH, Smith JL. Structure. 2010;18:94. doi: 10.1016/j.str.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gehret JJ, Gu L, Gerwick WH, Wipf P, Sherman DH, Smith JL. J Biol Chem. 2011;286:14445. doi: 10.1074/jbc.M110.214635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geders TW, Gu L, Mowers JC, Liu H, Gerwick WH, Håkansson K, Sherman DH, Smith JL. J Biol Chem. 2007;282:35954. doi: 10.1074/jbc.M703921200. [DOI] [PubMed] [Google Scholar]
- 13.Akey DL, Gehret JJ, Khare D, Smith JL. Nat Prod Rep. 2012;29:1038. doi: 10.1039/c2np20016c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoo H-D, Gerwick WH. J Nat Prod. 1995;58:1961. [Google Scholar]
- 15.Chang Z, Sitachitta N, Rossi JV, Roberts MA, Flatt PM, Jia J, Sherman DH, Gerwick WH. J Nat Prod. 2004;67:1356. doi: 10.1021/np0499261. [DOI] [PubMed] [Google Scholar]
- 16.Moss SJ, Martin CJ, Wilkinson B. J Nat Prod. 2004;21:575. doi: 10.1039/b315020h. [DOI] [PubMed] [Google Scholar]
- 17.Helmkamp GM, Bloch K. J Biol Chem. 1969;244:6014. [PubMed] [Google Scholar]
- 18.Leesong M, Henderson BS, Gillig JR, Schwab JM, Smith JL. Structure. 1996;4:253. doi: 10.1016/s0969-2126(96)00030-5. [DOI] [PubMed] [Google Scholar]
- 19.Nagao Y, Hagiwara Y, Kumagai T, Ochiai M, Inoue T, Hashimoto K, Fujita E. J Org Chem. 1986;51:2391. [Google Scholar]
- 20.Crimmins MT, Chaudhary K. Org Lett. 2000;2:775. doi: 10.1021/ol9913901. [DOI] [PubMed] [Google Scholar]
- 21.Evans DA, Downey CW, Shaw JT, Tedrow JS. Org Lett. 2002;4:1127. doi: 10.1021/ol025553o. [DOI] [PubMed] [Google Scholar]
- 22.Wadsworth WS, Emmons WD. J Am Chem Soc. 1961;83:1733. [Google Scholar]
- 23.Rathke MW, Nowak M. J Org Chem. 1985;50:2624. [Google Scholar]
- 24.Zheng J, Taylor CA, Piasecki SK, Keatinge-Clay AT. Structure. 2010;18:913. doi: 10.1016/j.str.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 25.Caffrey P. ChemBioChem. 2003;4:654. doi: 10.1002/cbic.200300581. [DOI] [PubMed] [Google Scholar]
- 26.Reid R, Piagentini M, Rodriguez E, Ashley G, Viswanathan N, Carney J, Santi DV, Hutchinson CR, McDaniel R. Biochemistry. 2003;42:72. doi: 10.1021/bi0268706. [DOI] [PubMed] [Google Scholar]
- 27.Fiers WD, Dodge GJ, Li Y, Smith JL, Fecik RA, Aldrich CC. Chem Sci. 2015;6:5027. doi: 10.1039/c5sc01505g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Dodge GJ, Fiers WD, Fecik RA, Smith JL, Aldrich CC. J Am Chem Soc. 2015;137:7003. doi: 10.1021/jacs.5b02325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keatinge-Clay AT. Chem Biol. 2007;14:898. doi: 10.1016/j.chembiol.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 30.Tang L, Ward S, Chung L, Carney JR, Li Y, Reid R, Katz L. J Am Chem Soc. 2004;126:46. doi: 10.1021/ja030503f. [DOI] [PubMed] [Google Scholar]
- 31.Tang G-L, Cheng Y-Q, Shen B. Chem Biol. 2004;11:33. doi: 10.1016/j.chembiol.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 32.Buntin K, Irschik H, Weissman KJ, Luxenburger E, Blöcker H, Müller R. Chem Biol. 2010;17:342. doi: 10.1016/j.chembiol.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 33.Gaitatzis N, Silakowski B, Kunze B, Nordsiek G, Blöcker H, Höfle G, Müller R. J Biol Chem. 2002;277:13082. doi: 10.1074/jbc.M111738200. [DOI] [PubMed] [Google Scholar]
- 34.Kleigrewe K, Almaliti J, Tian IY, Kinnel RB, Korobeynikov A, Monroe EA, Duggan BM, Di Marzo V, Sherman DH, Dorrestein PC, Gerwick L, Gerwick WH. J Nat Prod. 2015;78:1671. doi: 10.1021/acs.jnatprod.5b00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dickschat JS, Vergnolle O, Hong H, Garner S, Bidgood SR, Dooley HC, Deng Z, Leadlay PF, Sun Y. ChemBioChem. 2011;12:2408. doi: 10.1002/cbic.201100474. [DOI] [PubMed] [Google Scholar]
- 36.Castonguay R, Valenzano CR, Chen AY, Keatinge-Clay A, Khosla C, Cane DE. J Am Chem Soc. 2008;130:11598. doi: 10.1021/ja804453p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sedgwick B, Morris C, French SJ. J Chem Soc. 1978;193 [Google Scholar]
- 38.Gay DC, Spear PJ, Keatinge-Clay AT. ACS Chem Biol. 2014;9:2374. doi: 10.1021/cb500459b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lohr F, Jenniches I, Frizler M, Meehan MJ, Sylvester M, Schmitz A, Gutschow M, Dorrestein PC, Konig GM, Schaberle TF. Chem Sci. 2013;4:4175. [Google Scholar]
- 40.Schwab JM, Klassen JB. J Am Chem Soc. 1984;106:7217. [Google Scholar]
- 41.Schupp T, Toupet C, Engel N, Goff S. FEMS Microbiol Lett. 1998;159:201. doi: 10.1111/j.1574-6968.1998.tb12861.x. [DOI] [PubMed] [Google Scholar]
- 42.Caffrey P, Lynch S, Flood E, Finnan S, Oliynyk M. Chem Biol. 2001;8:713. doi: 10.1016/s1074-5521(01)00046-1. [DOI] [PubMed] [Google Scholar]
- 43.Motamedi H, Shafiee A. Eur J Biochem. 1998;256:528. doi: 10.1046/j.1432-1327.1998.2560528.x. [DOI] [PubMed] [Google Scholar]
- 44.Molnár I, Schupp T, Ono M, Zirkle RE, Milnamow M, Nowak-Thompson B, Engel N, Toupet C, Stratmann A, Cyr DD, Gorlach J, Mayo JM, Hu A, Goff S, Schmid J, Ligon JM. Chem Biol. 2000;7:97. doi: 10.1016/s1074-5521(00)00075-2. [DOI] [PubMed] [Google Scholar]
- 45.In curacin biosynthetic nomenclature, proteins and modules are only designated by an equivalent, alphabetical identity. The CurF ER domain is considered part of the hydroxymethyl-glutaryl-CoA synthase cassette for beta-branching and is appended onto the first PKS module (module F). To draw an analogy between traditional PKS pathways, this has been simplified to include the ER in the Cur F PKS module.
- 46.Armarego WLF, Perrin DD. Purification of laboratory chemicals. 4th. Butterworth Heinemann; Oxford; Boston: 1996. [Google Scholar]
- 47.Kabsch W. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P, Lohkamp B, Scott WG, Cowtan K. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moriarty NW, Grosse-Kunstleve RW, Adams PD. Acta Crystallogr Sect D Biol Crystallogr. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen VB, Arendall WB, III, Head JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.The PyMol Molecular Graphics System, Version 1.3–1.4, Schrodinger, LLC. at < http://www.pymol.org/>.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.