

Abstract
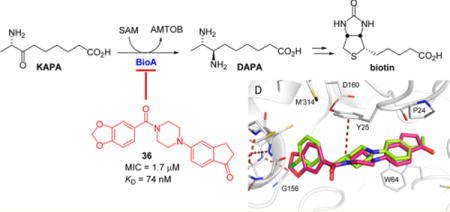
The pyridoxal 5′-phosphate (PLP)-dependent transaminase BioA catalyzes the second step in the biosynthesis of biotin in Mycobacterium tuberculosis (Mtb) and is an essential enzyme for bacterial survival and persistence in vivo. A promising BioA inhibitor 6 containing an N-aryl, N′-benzoylpiperazine scaffold was previously identified by target-based whole-cell screening. Here, we explore the structure–activity relationships (SAR) through the design, synthesis, and biological evaluation of a systematic series of analogues of the original hit using a structure-based drug design strategy, which was enabled by cocrystallization of several analogues with BioA. To confirm target engagement and discern analogues with off-target activity, each compound was evaluated against wild-type (WT) Mtb in biotin-free and -containing medium as well as BioA under- and overexpressing Mtb strains. Conformationally constrained derivative 36 emerged as the most potent analogue with a KD of 76 nM against BioA and a minimum inhibitory concentration of 1.7 μM (0.6 μg/mL) against Mtb in biotin-free medium.
Graphical abstract
INTRODUCTION
Tuberculosis (TB) is both an ancient scourge and a current global health crisis.1 The common ancestor of Mycobacterium tuberculosis (Mtb) and other members of the Mtb complex is thought to have evolved in Africa approximately 70,000 years ago,2 and evidence of Mtb infection has been found in New Kingdom Egyptian3 as well as pre-Columbian Peruvian4 mummies. Today, TB is one of the top 10 causes of death worldwide.5 In 2015, there were approximately 1.8 million deaths from TB, including 400,000 deaths of those also infected with HIV according to the World Health Organization (WHO).5 On the basis of the Joint United Nations Programme on HIV/AIDS (UNAIDS) reports, TB is the leading cause of death in HIV-positive populations,6 accounting for more than 35% of all deaths from AIDS-associated diseases.5,6 Although the worldwide prevalence of TB is decreasing, it remains a threat, particularly to the HIV-positive population.7
The current first-line therapy recommendation is for 2 months of HRZE (isoniazid, rifiampicin, pyrazinamide, and ethambutol, respectively) followed by a 4 month continuation of HR therapy.8 Although treatment-resistant TB has existed since the introduction of modern TB therapeutics,9 drug-resistant TB has now emerged as a global challenge, making the standard drugs isoniazid and rifampicin ineffective toward combating some cases of Mtb.10 The World Health Organization estimates that nearly half a million new cases of multidrug resistant tuberculosis (MDR-TB) will occur each year.11 MDR-TB is particularly prevalent in China, India, and Russia; these three countries account for more than 50% of all MDR-TB cases.10 Moreover, extensively drug-resistant TB (XDR-TB) is more challenging to treat, resulting in an extremely high mortality rate. On its own, XDR-TB is a burgeoning major global health problem.12,13 Treatment success rates for TB, MDR-TB, and XDR-TB are 83, 52, and 28%, respectively.5 Control of the TB epidemic is further complicated by latent infections, and it is estimated that one in three people worldwide are infected with Mtb.14,15 Consequently, there is an urgent need to develop drugs that act on novel targets.14,16–20
One approach to designing new TB therapies is to target metabolic processes essential in both active and latent stages of Mtb. Biotin (vitamin H, vitamin B7) biosynthesis is one such pathway.21 Biotin 5 (Scheme 1) is an essential cofactor utilized by carboxylases in fatty acid metabolism and gluconeogenesis pathways.22 Humans obtain biotin from intestinal microflora, dietary sources, and recycling.23 However, Mtb synthesizes biotin de novo24 because it cannot obtain sufficient levels of biotin to support growth in either mouse or man,28 which makes the biotin biosynthesis pathway an attractive target for new drug discovery.
Scheme 1. Late Steps of the Biotin Biosynthesis Pathway in Mtba.
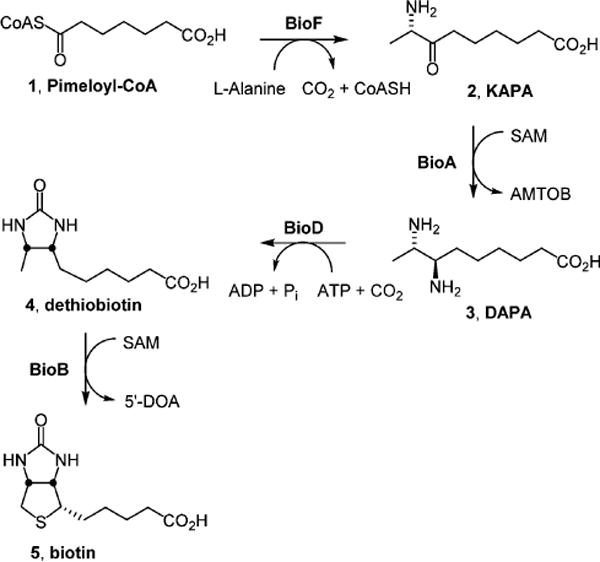
aBioF catalyzes the decarboxylative condensation of pimeloyl-CoA 1 with alanine to furnish 8-aminopelaragonic acid (KAPA, 2). The PLP-dependent aminotransferase BioA performs the reductive amination of KAPA 2 to 7,8-diaminopelargonic acid (DAPA, 3). BioD-mediated carboxylation of DAPA 3 provides dethiobiotin 4, which is converted to biotin 5 by the radical SAM enzyme BioB.
Biotin biosynthesis in Mtb involves the conversion of pimeloyl-CoA 1 to biotin 5 (Scheme 1) using the reactions catalyzed by enzymes 7-keto-8-aminopelargonic acid (KAPA) synthase BioF, 7,8-diaminopelargonic acid (DAPA) synthase BioA, dethiobiotin (DTB) synthetase (BioD), and biotin synthase (BioB). BioA, the target enzyme of the present work, is the second enzyme in this pathway. A pyridoxal 5′-phosphate (PLP)-dependent enzyme, BioA catalyzes the transamination of KAPA 2 to DAPA 3 using S-adenosylmethionine (SAM) as an amino donor.25–27 BioA has been shown to be essential for bacterial persistence in a murine model of TB, making it a promising target for novel TB therapy.28 BioA has been structurally characterized29 and is amenable to biochemical characterization.27 As such, it has been a target of our drug discovery efforts, and we have identified several small molecule inhibitors of BioA, including reversible covalently modifying hydrazines,30 an irreversible covalent inhibitor developed from amiclenomycin31–33 and compounds identified through conventional in vitro screening approaches.34
Most recently, a number of potent hits have been identified by a target-based whole-cell screening approach including N-aryl, N′-benzoylpiperazine 6, which was identified as the most promising hit based on its combination of potency and selectivity (Figure 1A).35 The compound shows good inhibition of BioA with an IC50 of 155 nM and reasonable on-target activity against whole-cell Mtb in biotin-free medium with a minimum inhibitor concentration of 26 μM and no observed activity in biotin-supplemented medium.35,36 It has a structural core comprised of a piperazine ring flanked by two aryl groups each attached to a piperazine nitrogen: a 4-acetylphenyl ring on one side and a methylene-3,4-dioxybenzoyl group on the other. To guide the structure-based optimization of 6, we cocrystallized 6 in complex with BioA and subsequently solved the X-ray crystal structure at 1.6 Å resolution (Table S1; PDB ID: 4XJP).36 The complex shows that inhibitor 6 extends across the length of a partially induced binding groove at the BioA monomer–monomer interface and atop the PLP cofactor. However, comparison of the shape of the BioA pocket to the shape of the ligand in the complex led us to believe that opportunities for further inhibitor optimization exist.
Figure 1.
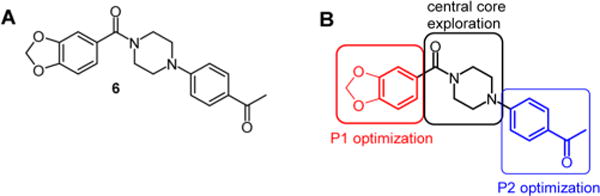
(A) Structure of initial hit N-aryl, N′-benzoylpiperazine 6. (B) Summary of SAR analysis.
Guided by molecular structures, we have prepared three series of analogues of 6 to explore the structure–activity relationship (SAR) of this scaffold (Figure 1B). The first series investigated the replacement of the benzodioxole group with a number of aromatic systems, a process that we call P1 (pocket 1) optimization. The second series explored modification of the 4-acetylphenyl ring with various aromatic systems while keeping the best aryl moiety at the P1 optimization site, which we call P2 optimization. The third series replaced the central piperazine core with other isosteres. Combining some of the best features from these series has led to the discovery of inhibitors more potent than the original hits.
RESULTS AND DISCUSSION
Structure–Activity Relationship (SAR) Studies
Assessment of the antitubercular activity of all compounds was performed using the protocol described by Park et al.35 The minimum inhibitory concentrations (MICs) were determined with wild-type (WT) Mtb H37Rv in biotin-free and -containing medium to identify compounds with biotin-dependent activity. To provide further support for the mechanism of action, we also evaluated the activity of every compound against BioA under- and overexpressing Mtb strains (BioA-UE and BioA-OE, respectively). BioA-UE expresses approximately 20% of BioA relative to WT Mtb and is therefore more sensitive to BioA inhibitors, whereas BioA-OE expresses approximately 1200% of BioA relative to WT Mtb and thus should be more resistant to BioA inhibitors.
P1 Optimization
The benzodioxole group of 6 binds into a pocket (P1) at one end of the ligand binding groove. Unlike P2, which is in part comprised of conformationally malleable amino acid side chains, P1 is lined largely by backbone atoms that appear to be more rigid (Figure 2A). In P1, the benzodioxole group is bound by Gly156-Tyr157 and a helical turn formed by residues Cys168-Gly172, and it stacks directly alongside the peptide joining Gly172 to Gly173. Most of the protein heteroatoms lining this pocket are already engaged in hydrogen bonding with other protein atoms. Gly156 O, for example, is H-bonded to Asp169 NH. Consequently, the subsite is polar, but it does not afford many opportunities for additional H-bonding. Such a site is well-suited to accommodate the benzodioxole moiety of 6.
Figure 2.
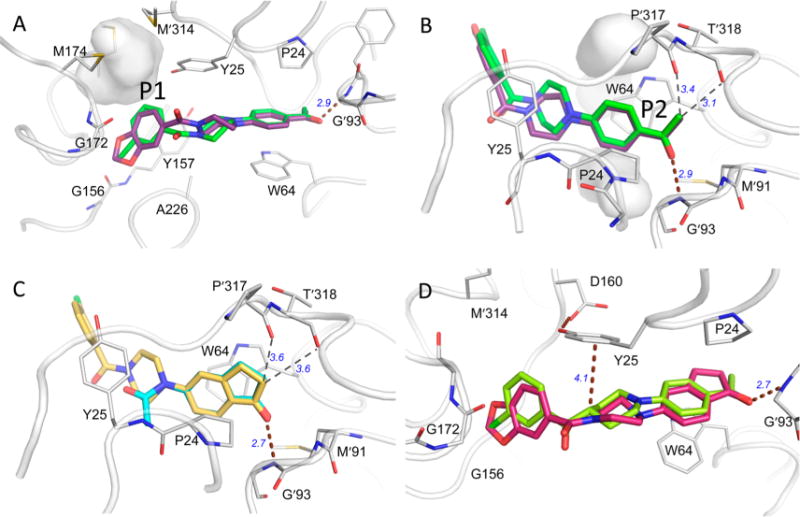
(A) Comparison of 6 (purple, PDB ID: 4XJP) and 12 (green, PDB ID: 4W1X) in the binding pocket with residues that make substantial contact with the ligands. H-bonds are shown with brown dashed lines. Weaker nonbonded close contacts are illustrated with thin dashed lines. (A) The P1 subsite: the gray surface surrounds a cavity between Met165, Met 174, and Met′314 left unoccupied by ligand or protein atoms following ligand binding. (B) The P2 subsite: unoccupied cavities are also found both above and below the acetylphenyl ring in complexes with 6 and 12. (C) Position of inden-1-one-containing fragment38 (cyan, PDB ID: 4WYF) compared to that of compound 26 (yellow, PDB ID: 4XJO) in the same orientation as panel B. (D) Crystal structures with compounds 36 (red, PDB ID: 5KGS) and 39 (lime, PDB ID: 4KGT).
There is, however, poor shape complementarity between the benzodioxole group and the pocket surrounding it, suggesting that better substituents to fill this pocket might be found. A series of alternatives to the benzodioxole group were explored (Table 1). It seemed that the 1,3-benzodioxole moiety of 6 was inadequate to completely fill the subsite, so larger benzodioxoles, i.e., 1,4-benzodioxole 7 and 1,5-benzodioxole 8, were explored. The dimension of the P1 pocket was further tested with smaller 5-membered heterocycles 15 and 16. All of these modifications drastically reduced potency under all conditions. In retrospect, the larger aliphatic bridges of compounds 7 and 8 likely cannot be accommodated alongside the Gly172 peptide bond as in 6. If the ring were to flip 180° around the aryl-amide bond, a steric clash would arise with Tyr25.
Table 1.
SAR of the Methylene-3,4-dioxybenzoyl Moiety
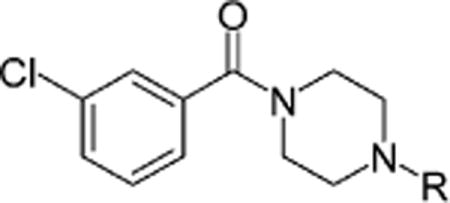
| |||||
|---|---|---|---|---|---|
| MIC50 (μM)
|
|||||
| Compound | R | BioA-UEa | BioA-OEb | WT-biotinc | WT+biotind |
| 6 |
|
0.4 (1.7)e | 31.1 (>50)e | 5.6 (23.4)e | >50 (>50)e |
| 7 |
|
10.4 | >50 | >50 | >50 |
| 8 |
|
21.1 | >50 | >50 | >50 |
| 9 |
|
1.9 | 17.7 | 10.2 | 10.7 |
| 10 |
|
>50 | >50 | >50 | >50 |
| 11 |
|
18.3 | >50 | 28.6 | 39.1 |
| 12 |
|
0.5 | 7.5 | 5.1 | 30.1 |
| 13 |
|
>50 | >50 | >50 | >50 |
| 14 |
|
14.6 | >50 | >50 | >50 |
| 15 |
|
>50 | >50 | >50 | >50 |
| 16 |
|
11.3 | >50 | >50 | >50 |
| 17 |
|
>50 | >50 | >50 | >50 |
| 18 |
|
>50 | >50 | >50 | >50 |
| 19 |
|
10.6 | 47.6 | 8.7 | 6.2 |
| 20 |
|
>50 | >50 | >50 | >50 |
Minimum inhibitory concentration (MIC) that results in 50% inhibition against the Mtb BioA underexpression strain.
MIC that results in 50% inhibition against the Mtb BioA overexpression strain.
MIC that results in 50% inhibition against Mtb H37Rv without supplemental biotin.
MIC that results in 50% inhibition against Mtb H37Rv with 1 μM supplemental biotin.
MIC that results in 90% inhibition.
On the other hand, the 3,4-dichlorophenyl analogue 9, which is a close conformational mimic of the 1,3-benzodioxole moiety, was reasonably well-tolerated with a 2-fold loss in potency against WT Mtb in biotin-free medium. The findings that this compound had equal susceptibility against WT Mtb in biotin-containing medium indicates undesirable off-target activity. Removing the 4-chloro group produced the 3-chlorophenyl analogue 12, which showed slightly improved activity against WT Mtb over the original hit 6 and, unlike 9, maintained on-target activity as judged by its activity profile. Comparison of the crystal structure of the complex of BioA and 12 (PDB ID: 4W1X) with 6 (PDB ID: 4XJP) shows that these two compounds bind very similarly.35,36
There also appeared to be a penetrable gap between Tyr25 and Cys168 that might be filled with substituents extending from the 5-position of the 3-chorophenyl ring. This led to the synthesis of analogues 10 and 11 but neither produced improvements in potency over 6. One explanation for the inactivity of select compounds 10 and 11 may be that any atom directly connected to the 5-position of the chlorobenzene ring must pass very close to the OH of Tyr25, which is already donating the hydrogen to Asp160 and might therefore repel the large chlorine (10) or electronegative oxygen (11). Replacement of the phenyl ring with a pyridyl ring in chloropyridyl 13 and pyridyl 14 obliterated activity, which together with the lack of activity of the aforementioned thiazole 15 and pyrazole 16 derivatives demonstrates that the presence of a phenyl ring at the P1 position is highly favorable. To complete our exploration of the P1 subsite, we prepared a series of aliphatic analogues (17–20) that were devoid of activity except for 19, which was nearly equipotent to 6 against WT Mtb. However, the MIC of 19 was insensitive to exogenous biotin or BioA underexpression suggesting the activity of this compound is largely due to off-target effects.
P2 Optimization
Compounds 6 and 12 share a common orientation for the 4-acetylphenyl group in a hydrophobic pocket (P2) bound by the side chains of Pro24 and Trp64 and loops Met′91-Gly′93 and Pro′317-Thr′318 (Figure 2A). (Here, and throughout this discussion, a prime in the residue number identifies residues in the alternate monomer of the BioA homodimer). Trp64 is induced into a side chain conformation that enables π–π stacking extending across the length of the acetylphenyl group, affirming the importance of an aromatic substituent in P2. The acetyl carbonyl oxygen accepts a hydrogen bond from the backbone amide of Gly′93, but backbone carbonyl groups from residues Pro′317 and Thr′318 that are directed into the subsite make only weakly stabilizing polar interactions with the acetyl methyl group (e.g., C–H···O hydrogen bonds). The next priority in exploring analogue SAR was to modify the acetylphenyl group of 12 to explore more diverse functional groups (Table 2).
Table 2.
SAR of the Acetylphenyl Moiety
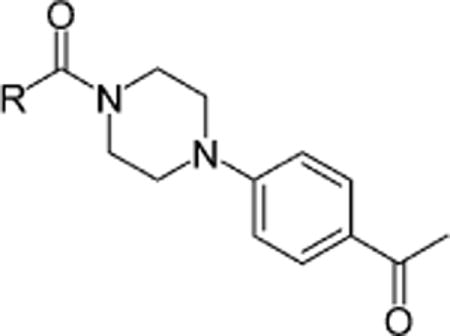
| |||||
|---|---|---|---|---|---|
| MIC50 (μM)
|
|||||
| Compound | R | BioA-UE | BioA-OE | WT-biotin | WT+biotin |
| 12 |
|
0.5 | 7.5 | 5.1 | 30.1 |
| 21 |
|
>50 | >50 | >50 | >50 |
| 22 |
|
24.4 | >50 | 22.0 | >50 |
| 23 |
|
>50 | >50 | >50 | >50 |
| 24 |
|
>50 | >50 | >50 | >50 |
| 25 |
|
0.4 | 38.9 | 6.7 | >50 |
| 26 |
|
0.3 | 8.1 | 4.4 | 47.0 |
| 27 |
|
1.8 | >50 | 35.0 | >50 |
| 28 |
|
1.2 | >50 | 15.2 | >50 |
| 29 |
|
12.8 | >50 | 40.8 | 48.0 |
| 30 |
|
26.6 | >50 | >50 | 47.1 |
| 31 |
|
>50 | >50 | >50 | >50 |
| 32 |
|
44.8 | 45.9 | 18.5 | 22.7 |
| 33 |
|
4.7 | 20.9 | 15.4 | 11.8 |
| 34 |
|
1.1 | 7.6 | 9.9 | 19.0 |
| 35 |
|
15.1 | >50 | 20.4 | 39.1 |
The dimensions of P2 were tested with biphenyl moieties (e.g., 21 and 22) that are privileged structures in drug discovery37 as well as smaller heterocyclic biphenyls (23–25). Of the compounds bearing a biphenyl-like group, only oxazole 25 was as effective as the original hit. Most other functional group substitutions at the 4-position of the phenyl ring were inactive. The introduction of a methyl group in 23 and 24 obliterated activity. The loss of activity by several of these can be attributed in retrospect to the apparent inflexibility of the backbone of residues ′317–′318, which, left unmoved, forms a stout wall of H-bond acceptors inside the pocket that must resist interactions with H-bond acceptors of 31–33. The position of the acetyl group is critical; meta-acetyl analogue 30 lost nearly 10–50-fold in activity compared to the para-acetyl 12 against WT Mtb and Bio-UE Mtb strains. The thiophene of 34 likely serves as a respectable isostere of 12 and retains comparable (but not superior) activity. The replacement of the keto group with a ketoxime group of 35 resulted in inferior activity. Several 3,4-bicyclic analogues (26–29) were inspired by the overlay of the acetylphenyl group of 6 or 12 with an inden-1-one moiety found and characterized in fragment screening (Figure 2B,C).38 These compounds generally retained some biological activity, but only compound 26 with the identical inden-1-one of the original fragment hit improved upon the activity of 12. A crystal structure of the complex with 26 (Table S1; PDB ID: 4XJO36) was determined. This structure confirms the predicted binding mode and affirms the position of the inden-1-one with an H-bond to Gly′93 NH (2.8 Å) as well as tight packing against carbonyl oxygen atoms of Pro′317 and Thr′318 by carbon atoms in the five-membered carbocycle (3.5 and 3.6 Å, respectively).
Central Core Exploration
Several heterocyclic alternatives to the piperazinyl carbonyl linker in the central core were explored (Table 3). Smaller inflexible triazole linkers (37 and 38) were inactive. Interestingly, the corresponding piperidinyl analogues of 12 and 6 (39 and 40, respectively) showed divergent activity. The chlorophenyl analogue 39 delivered modest antitubercular activity, although much of that activity may be off-target. The benzodioxole analogue 40 was completely inactive. We obtained a crystal structure of the complex with 39 (Table S1, Figure 2D, PDB ID: 5KGT). Both 6 and 12 derive some potency from planar stacking between the amide bond of the piperazinyl carbonyl and Tyr25.38 The amide plane is absent in piperidinyl analogues, but in the complex with 39, the ring pucker permits a perfectly positioned C–H on C4 of the piperidine to donate a H-bond to the Tyr25 π-system, whereas the position of the chlorophenyl ring is consistent with the positioning of the same moiety in the complex with 12. To rationalize the complete lack of activity for 40, the analogous crystal structure of 6 was examined. This structure shows that the piperazine in 6 must twist significantly to accommodate the benzodioxole in P1; a comparable twist applied to the piperidine of 40 likely forces C4 out of proper alignment for a good interaction with Tyr25, resulting in lower affinity and inactivity.
Table 3.
SAR of the Piperazine Linker
| Compound | Structure | MIC50 (μM)
|
|||
|---|---|---|---|---|---|
| BioA-UE | BioA-OE | WT-biotin | WT+biotin | ||
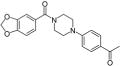
| 6 |
|
0.4 | 31.1 | 5.6 | >50 |
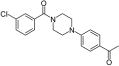
| 12 |
|
0.5 | 7.5 | 5.1 | 30.1 |
| 36 |
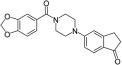
|
0.3 | 2.8 | 1.7 | >50 |
| 37 |
|
24.4 | >50 | 22.0 | >50 |
| 38 |
|
>50 | >50 | >50 | >50 |
| 39 |
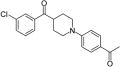
|
6.8 | 9.2 | 4.1 | 3.3 |
| 40 |
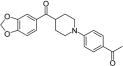
|
>50 | >50 | >50 | >50 |
Combining the best of the P1 substituents with the best P2 group and flexible piperazinyl linker resulted in compound 36, which has potent antitubercular activity with an improved MIC of 1.7 μM against WT Mtb in biotin-free medium (Table 3). Moreover, 36 displays biotin and BioA-dependent activity, consistent with the desired mechanism of action (Table 3 and Figure 3). We obtained the crystal structure of BioA with 36 (Table S1, Figure 2D, PDB ID: 5KGS), which showed conserved binding modes for each of its distinct substituents. The binding mode of the inden-1-one P2 substituent is highly similar to that of 26 in structure 4XJO36 (Table S1). A slight difference in planar orientation can be attributed to a twist in the piperazinyl carbonyl core that appears to coincide with the P1 substituent identity; one position is observed with the benzodioxole P1 group (6, 36), whereas the other position is observed in compounds with 3-chlorophenyl P1 groups (12 and 26). To yield the conserved piperazinyl carbonyl core twist, the benzodioxole group of 36 is bound in the same fashion as the benzodioxole group of 6.
Figure 3.
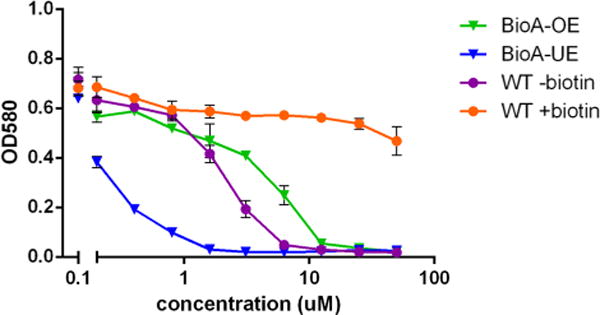
Inhibition of Mtb growth by compound 36. A representative concentration–response plot for 36 showing Mtb growth on the y-axis as measured by the optical density at 580 nm versus the log10 of the compound concentration on the x-axis. WT Mtb in biotin-free medium (purple), WT Mtb in biotin-containing medium (orange), Bio-UE (blue), and Bio-OE (green) Mtb strains in biotin-free medium.
Overall, the SAR studies have shown that compounds 6, 12, 25–28, and 36 are effective inhibitors of both BioA and Mtb with MIC values from 1.7 (36) to 35 μM (27). Other compounds (9, 11, 19, 22, and 32–35) inhibit WT Mtb with MIC values from 9.9 (34) to 40.8 μM (29), but these compounds do not show biotin-dependent activity nor the predicted monotonic behavior with BioA expression levels from the BioA-UE, BioA-OE, WT-biotin, and WT+biotin inhibition experiments.
Binding Affinity by Isothermal Titration Calorimetry (ITC)
BioA binding affinity (KD) and thermodynamic parameters (ΔH, ΔS) of selected compounds were determined by isothermal titration calorimetry (ITC) as shown in Table 4 with representative ITC data illustrated in Figure 4. The KD values correlate well with the corresponding MIC values. The most potent compound, 36, possessed a BioA KD value of 74 nM. The tested compounds have a range of potencies spanning approximately 1 order of magnitude from 74 to (36) to 886 nM (28).
Table 4.
ITC Data of Selected Compounds
| Compound | Structure |
KD (nM) |
ΔH (kcal/mol) |
−TΔS (kcal/mol) |
|---|---|---|---|---|
| 635 |
|
108 ± 1 | −11.8 | 2.3 |
| 12 |
|
157 ± 6 | −4.4 | −4.9 |
| 26 |
|
132 ± 74 | −7.3 | −2.1 |
| 27 |
|
685 ± 14 | −3.2 | −5.2 |
| 28 |
|
866 ± 19 | −3.5 | −4.7 |
| 34 |
|
322 ± 3 | −7.0 | −1.8 |
| 36 |
|
74 ± 1 | −11.1 | 1.4 |
Figure 4.

Representative ITC binding isotherms (top) and integrated enthalpy curves (bottom) for 12, 26, and 36 (100 μM) with BioA (10 μM).
Changes in the P1 group produce a consistent energetic profile. Compounds incorporating the benzodioxole (compounds 6 or 36) reflect a significant enthalpic benefit upon binding but at moderate entropic cost. This is a pattern expected to be found when good specific interactions that restrict binding conformation are made. In the case of the benzoxazole, the excellent geometry of weak CH···O interactions with Gly156 O and Cys168 CβH, and Gly227 CαH can explain this stabilization pattern. Compounds that incorporate the chlorophenyl in P1 consistently show a more balanced contribution to binding that are favorable in both enthalpy and entropy. The chlorine of 12 lies centered between these same protein structural elements but does not impose the same directional restraints as C···H interactions made by the methylene bridge of the dioxolane.
We anticipated that cyclizing the acetylphenyl P2 group of 6 or 12 (resulting in 36 or 26, respectively) might result in a smaller entropic binding penalty, but no consistent trend confirming this prediction emerges from the ITC data. Alternate P2 closed-ring systems (27 and 28) were also explored using ITC. However, neither of these compounds were especially potent or had significantly different thermodynamic parameters (Table 4).
In addition to potency, the thermodynamic parameters of binding can be key elements in determining the best clinical candidate. Enthalpy-driven hits are often preferred because they may be more amenable to future optimization.39–41 In our series, the most potent inhibitor (36) is driven by enthalpic stabilization but has an entropic binding cost, which may provide an opportunity for future improvement.
Synthetic Chemistry
For the preparation of inhibitors in the P1 optimization series that involved modification of the 1,3-benzodioxole ring with various other aromatic systems, the synthesis started from condensation of piperazine 41 with 4′-fluoroacetophenone to afford monoaroylated intermediate 42 (Scheme 2). Subsequent coupling with various carboxylic acids using standard EDC-mediated conditions provided compounds 6–11 and 13–20. Similarly, compound 36 was synthesized by the condensation of 41 with 5-fluoro-1-indanone to furnish intermediate 43, which after EDC coupling with piperonylic acid afforded 36 (Scheme 2).
Scheme 2.
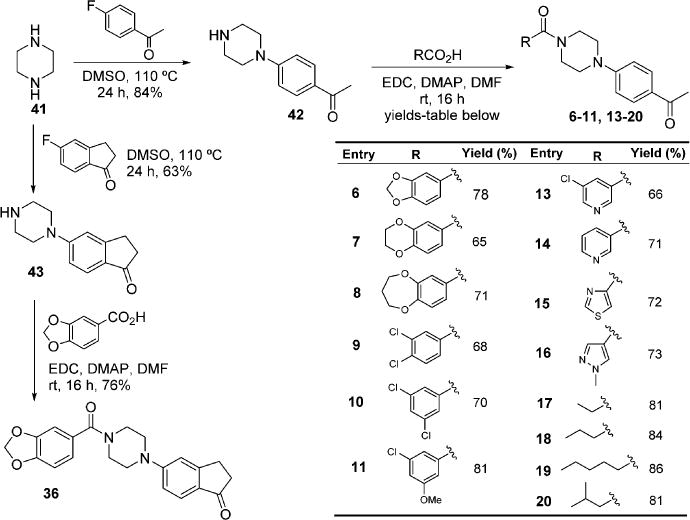
Modification of the Benzodioxoloyl Ring
The synthesis of inhibitors in the P2 optimization series with modification of the acetylphenyl ring started with monoaroylation of piperazine 41 with 3-chlorobenzoyl chloride generated N-(3-chlorobenzoyl)piperazine 44 using a previously reported procedure (Scheme 3).42 Palladium-catalyzed amination43 in the presence of Cs2CO3 with an aryl bromide afforded compounds 12, 21–24, and 26–34. However, preparation of the remaining two compounds in the series involved the functional group interconversions of methyl ketone 12 to isoxazole 25 and oxime 35. Condensation of 12 with N,N-dimethylformamide dimethyl acetal in o-xylene provided the keto-enamine intermediate, which when treated with hydroxylamine hydrochloride in EtOH, underwent subsequent condensation to afford 25 (Scheme 3). Ketoxime 35 was prepared by the reaction of ketone 12 with hydroxylamine hydrochloride in EtOH.
Scheme 3.
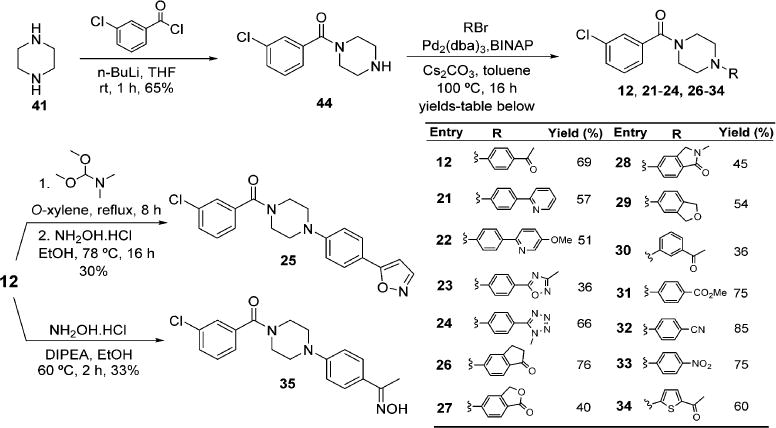
Modification of the Acetophenyl Ring
For linker optimization, two different linker moieties were employed for the replacement of piperazine, 1,2,3-triazoles, and piperidine. Synthesis of inhibitors with the 1,2,3-triazoles linker began with substitution of 3-chlorobenzyl chloride 45 with sodium azide to provide intermediate 46.44 Subjection of azide 46 and the corresponding terminal alkynes to standard click chemistry conditions afforded 37 and 38 (Scheme 4). For the synthesis of inhibitors with a piperidine linker, the amino group of isonipecotic acid 47 was Boc protected followed by the conversion of the carboxylic acid to Weinreb amide 48 (Scheme 5).45 Addition of an aryllithium species to 48 afforded compounds 49 and 50. Deprotection of the Boc group with TFA followed by palladium-catalyzed amination with 4-bromoacetophenone furnished analogues 39 and 40.
Scheme 4.
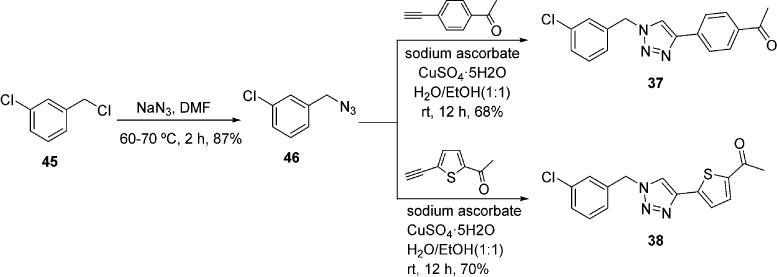
Modification of Linker with 1,2,3-Triazoles
Scheme 5.
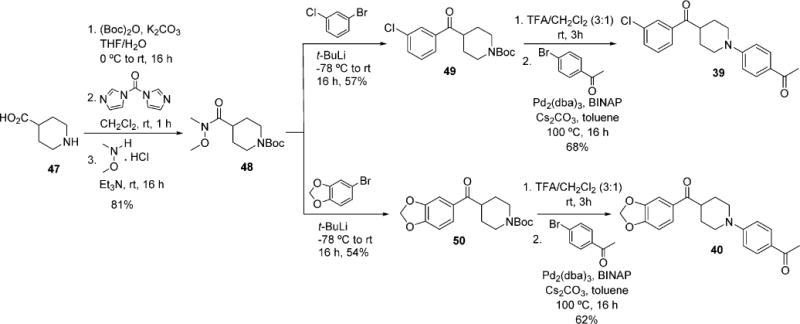
Modification of Linker with Piperidine
CONCLUSIONS
The SAR of N-aryl, N′-benzoylpiperazine 635 was explored through the preparation of 34 analogues that comprehensively examined the P1 group, P2 group, and piperazinyl carbonyl core. The synthetic modifications were informed by multiple cocrystal structures of analogues in the series. After investigating smaller and larger cyclic groups as well as aliphatic groups, the best P1 groups were found to be benzodioxole and 3-chlorophenyl. In the P2 group, only modification to an inden-1-one group resulted in analogues with increased antitubercular activity. Explorations of the central core led to the conclusion that the piperazinyl carbonyl core geometry is necessary for optimal P1 group binding. Although many of the analogues displayed the expected BioA and biotin-dependent whole-cell activity, we also observed several compounds that did not and therefore act through alternate mechanisms, which helped clarify interpretation of the SAR. For example, compound 19 containing a pentyl side chain at P1 and compound 32 with a 4-cyanophenyl group at P2 each displayed nearly equal activity in biotin-free and -containing media. Moreover, the activity did not vary significantly with BioA expression levels. Given the relatively simple nature of the initial N-aryl, N′-benzoylpiperazine scaffold, it is not altogether surprising that some analogues could engage alternate targets because N-arylpiperazines have many reported bioactivities. However, our analysis rapidly identified these off-targets hits and allowed us to remain focused on compounds that operate through the desired mechanism of action. Compound 26 containing the optimal P1 and P2 groups maintained the excellent bioactivity of the initial hit, but successfully removed the methylenedioxy group, a known toxicophore, and one rotatable bond through cyclization of the methyl ketone back onto the phenyl ring. Compound 36 emerged as the most potent analogue with a minimum inhibitory concentration of 1.7 μM against Mtb in biotin-free medium. Structures of BioA with both 26 and 36 indicate that the binding modes are nearly identical to 6, and thermodynamic data show that the binding is strongly enthalpy driven. Optimization of the thermodynamic parameters of compounds like 36 to achieve additional entropic stabilization may present a future avenue for the development of even more potent BioA inhibitors.
EXPERIMENTAL SECTION
BioA Protein Expression and Purification
N-terminally His-tagged BioA was overexpressed in Escherichia coli and purified using Ni affinity and size exclusion chromatography as described previously.34,46 Full saturation of the active site by the PLP cofactor was ensured by adding 1 mM PLP to pooled protein as eluted from the SEC column and confirmed using differential scanning fluorimetry as previously described.34,46
Crystallography
Crystals of BioA were obtained by hanging drop vapor diffusion at 20 °C and a microseeding technique under conditions of 100 mM HEPES pH 7.5 (Sigma Life Science), 100 mM MgCl2 (EMD Chemicals), and 10–14% PEG 8000 (Acros Organics); drops consisted of 2 μL of protein solution (13 mg/mL) in buffer from purification, 1.5 μL of well solution, and 0.5 μL of seed solution as previously reported.30,35,38 Complexes were obtained by soaking using a 5 mM final concentration of the ligand. Co-crystals may also be obtained using the drop setup as above supplemented with 0.15 μL of ligand solution (50 mM). Crystals were cryoprotected using well solution supplemented with 15% PEG 400 (Sigma Life Science), followed by flash vitrification in liquid nitrogen.
Data for structures 4XJO, 4XJP, and 5KGS were collected at APS beamline 17-ID (IMCA-CAT) equipped with a Dectris Pilatus 6 M detector at 100 K. Data for structure 5KGT were collected at ALS beamline 4.2.2 equipped with an RDI 8 M detector at 100 K. The data were processed using AutoProc47 or XDS48 and solved with Phaser,49 using the coordinates from structure 4W1X or 3TFT.31,35 Refinement was carried out in Phenix,50,51 and visualization was performed in Coot.52 Ligand restraints were calculated using JLigand.53
Isothermal Titration Calorimetry (ITC)
ITC was conducted on a MicroCal Auto-iTC200 microcalorimeter (Malvern Instruments Ltd., UK) with a cell volume of 200 μL and a syringe volume of 40 μL. All experiments were performed at 25 °C in ITC buffer (25 mM HEPES [pH 7.5] and 50 mM NaCl). BioA was exchanged into this buffer using an Amicon Ultra concentrator, and the final enzyme concentration was determined using a NanoDrop instrument with the calculated extinction coefficient ε280 (ε280 = 64900 M−1 cm−1). The concentration of BioA was 10 μM. The ligand concentration was 100 μM, which was prepared by diluting a 20 mM DMSO stock with the ITC buffer. DMSO was added to the corresponding BioA protein solution to equalize the DMSO concentrations. All titrations were performed with a stirring speed of 750 rpm and a 150 s interval between 2 μL injections. The initial injection was not used for data fitting. Titrations were run past the point of enzyme saturation to determine and correct the heats of dilution. Data were fit to a theoretical titration curve using the Origin software package (version 7.0) provided with the instrument to obtain KA (the association constant in M−1) and ΔH (enthalpy) of binding. The thermodynamic parameters (ΔG and −TΔS) are calculated using eq 1
| (1) |
where ΔG, ΔH, and ΔS are the changes in free energy, enthalpy, and entropy of binding, respectively, R = 1.98 cal mol−1 K−1, and T is the absolute temperature. The affinity of the ligands for BioA is provided as the dissociation constant (KD = 1/KA). Average thermodynamic parameters and standard errors (Table 4) were computed from triplicate experiments.
Mtb Whole-Cell Growth Assay (MIC Determination)
The Mtb whole-cell assay for the MIC determination was performed using a method previously described.35 Briefly, Mtb WT, BioA-UE, and BioA-OE were grown in Sauton’s medium (0.5 g KH2PO4, 0.5 g of MgSO4·7H2O, 2.0 g of citric acid, 0.05 g of ferric ammonium citrate, 60 mL of glycerol, 4.0 g of asparagine, 0.1 mL of 1% ZnSO4, and 0.02% tyloxapol in 1 L) containing 1 μM biotin to an OD580 nm between 1.0 and 1.2. The cells were subsequently harvested by centrifugation, washed twice with biotin-free Sauton’s medium, and diluted in 96-well plates to an OD580 nm of 0.03. To set up the Mtb WT with or without biotin plates, 1 μM biotin or no biotin was added to the plates, respectively. To set up the Mtb BioA-UE or BioA-OE plates, 200 ng/mL anhydrotetracycline was added to the plates containing BioA-UE or BioA-OE stain, respectively, to achieve the desired level of BioA expression.
The compounds were added to final concentrations between 0.2 and 50 μM. Wells containing no compound were used as controls. Plates were incubated at 37 °C, and optical density was measured after 9 days. All growth assays were performed in triplicate with two biological replicates. MIC values were calculated using Prism (version 5.01).
Synthesis
General
Chemicals and solvents were purchased from Acros Organics, Alfa Aesar, Sigma-Aldrich, and TCI America and were used as received. An anhydrous solvent dispensing system using two packed columns of neutral alumina was used for drying THF and CH2Cl2, and the solvents were dispensed under argon gas (Ar). EtOAc and hexanes were purchased from Fisher Scientific. TLC analyses were performed on TLC silica gel plates 60 F254 from EMD Millipore Inc. and were visualized with UV light. Purification by flash chromatography was performed using a medium-pressure flash chromatography system equipped with flash column silica cartridges with the indicated solvent system. 1H and 13C spectra were recorded on a 400 MHz Varian NMR spectrometer. Proton chemical shifts are reported in ppm from an internal standard of residual chloroform (7.26), methanol (3.31), dimethyl sulfoxide (2.50), or monodeuterated water (HDO, 4.79); carbon chemical shifts are reported in ppm from an internal standard of residual chloroform (77.2), methanol (49.1), or dimethyl sulfoxide (39.5). Proton chemical data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, dt = doublet of triplets, t = triplet, q = quartet, pent = pentet, m = multiplet, ap = apparent, br = broad), coupling constant(s), integration. High-resolution mass spectra (HRMS) were obtained on a Bruker BioTOF II instrument. Purity of the final compounds was determined by 1H NMR and reverse-phase HPLC and was ≥95%.
General Procedure A: EDC Coupling
To a solution of 42 (1.2 mmol, 1.20 equiv) and the corresponding carboxylic acid (1 mmol, 1.00 equiv) in DMF (7 mL) was added EDC (1 mmol, 1.00 equiv) followed by DMAP (1 mmol, 1.00 equiv) at 23 °C. The reaction was stirred for 16 h at 23 °C and quenched with water (25 mL). The mixture was extracted with EtOAc (3 × 50 mL). The combined organic layers were washed by saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography on silica gel (1:1 hexanes/EtOAc) afforded the coupled product.
1-(4-(4-(Benzo[d][1,3]dioxole-5-carbonyl)piperazin-1-yl)phenyl)-ethanone 6
Compound 42 (98 mg, 0.48 mmol) was coupled with piperonylic acid (66 mg, 0.40 mmol) using general procedure A for EDC coupling. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (110 mg, 78%) as a white solid: mp 140–141 °C; Rf = 0.23 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 8.5 Hz, 2H), 6.94–6.91 (m, 2H), 6.85–6.79 (m, 3H), 5.97 (s, 2H), 3.73 (s, 4H), 3.35–3.33 (m, 4H), 2.48 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.30, 169.8, 153.6, 148.9, 147.6, 130.2, 128.7, 128.2, 121.6, 113.8, 108.1, 107.9, 101.4, 47.5, 26.0; HRMS (ESI+) calcd for C20H20N2NaO4+ 375.1315, found 375.1324 (error 2.4 ppm).
1-(4-(4-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carbonyl)piperazin-1-yl)phenyl)ethanone 7
Compound 42 (98 mg, 0.48 mmol) was coupled with 1,4-benzodioxane-6-carboxylic acid (72 mg, 0.40 mmol) using general procedure A for EDC coupling. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (95 mg, 65%) as a white solid: mp 173–174 °C; Rf = 0.14 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.86 (d, J = 8.5 Hz, 2H), 6.98–6.91 (m, 2H), 6.88–6.84 (m, 3H), 4.26 (s, 4H), 3.75 (s, 4H), 3.35 (s, 4H), 2.50 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.4, 169.9, 153.7, 145.1, 143.3, 130.3, 128.23, 128.16, 120.7, 117.2, 116.7, 113.8, 64.4, 64.2, 47.6, 26.1; HRMS (ESI+) calcd for C21H22N2NaO4+ 389.1472, found 389.1462 (error 2.6 ppm).
1-(4-(4-(3,4-Dihydro-2H-benzo[b][1,4]dioxepine-7-carbonyl)-piperazin-1-yl)phenyl)ethanone 8
Compound 42 (98 mg, 0.48 mmol) was coupled with 3,4-dihydro-2H-1,5-benzodioxepine-7-carboxylic acid (78 mg, 0.40 mmol) using general procedure A for EDC coupling. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (108 mg, 71%) as a white solid: mp 183–185 °C; Rf = 0.14 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 8.4 Hz, 2H), 7.05–6.97 (m, 3H), 6.83 (d, J = 8.5 Hz, 2H), 4.26–4.18 (m, 4H), 3.73 (s, 4H), 3.34 (s, 4H), 2.48 (s, 3H), 2.3–1.96 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 196.3, 169.6, 153.6, 152.4, 150.7, 10.2, 130.0, 128.2, 122.3, 121.5, 120.8, 113.8, 70.4, 70.3, 47.5, 41.9, 31.2, 26.0; HRMS (ESI+) calcd for C22H24N2NaO4+ 403.1628, found 403.1640 (error 3.0 ppm).
1-(4-(4-(3,4-Dichlorobenzoyl)piperazin-1-yl)phenyl)ethanone 9
Compound 42 (43 mg, 0.21 mmol) was coupled with 3,4-dichlorobenzoic acid (34 mg, 0.18 mmol) using general procedure A for EDC coupling. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (46 mg, 68%) as a white solid: mp 123–124 °C; Rf = 0.33 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.89–7.86 (m, 2H), 7.54–7.49 (m, 2H), 7.27 (dd, J = 8.3, 2.3 Hz, 1H), 6.88–6.85 (m, 2H), 3.86 (s, 2H), 3.63 (s, 2H), 3.37 (s, 4H), 2.52 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.4, 167.9, 153.5, 135.0, 134.4, 133.1, 130.7, 130.3, 129.3, 128.6, 126.4, 114.1, 47.6, 42.2, 26.1; HRMS (ESI+) calcd for C19H18Cl2N2NaO2+ 399.0638, found 399.0630 (error 2.0 ppm).
1-(4-(4-(3,5-Dichlorobenzoyl)piperazin-1-yl)phenyl)ethanone 10
Compound 42 (98 mg, 0.48 mmol) was coupled with 3,5-dichlorobenzoic acid (76 mg, 0.40 mmol) using general procedure A for EDC coupling. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (105 mg, 70%) as a white solid: mp 194–195 °C; Rf = 0.41 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.87 (d, J = 8.5 Hz, 2H), 7.42 (d, J = 2.3 Hz, 1H), 7.31–7.30 (m, 2H), 6.86 (d, J = 8.5 Hz, 2H), 3.88 (s, 2H), 3.59 (s, 2H), 3.39 (s, 4H), 3.33 (s, 1H), 2.51 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.4, 167.3, 153.5, 138.0, 135.5, 130.3, 130.0, 128.6, 125.5, 114.1, 47.9, 41.8, 26.1; HRMS (ESI+) calcd for C19H18Cl2N2NaO2+ 399.0638, found 399.0628 (error 2.5 ppm).
1-(4-(4-(3-Chloro-5-methoxybenzoyl)piperazin-1-yl)phenyl)-ethanone 11
Compound 42 (98 mg, 0.48 mmol) was coupled with 3-chloro-5-methoxybenzoic acid (75 mg, 0.40 mmol) using general procedure A for EDC coupling. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (121 mg, 81%) as a white solid: mp 196–197 °C; Rf = 0.22 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 8.5 Hz, 2H), 6.95–6.92 (m, 2H), 6.84–6.82 (m, 3H), 3.85 (s, 2H), 3.85–3.78 (m, 5H), 3.56 (s, 2H), 3.35 (s, 4H), 2.48 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.3, 168.5, 160.3, 153.5, 137.6, 135.1, 130.2, 128.3, 119.0, 115.6, 113.8, 111.2, 55, 47.6, 41.6, 26.0; HRMS (ESI+) calcd for C20H21ClN2NaO3+ 395.1133, found 395.1143 (error 2.5 ppm).
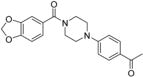
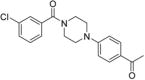
1-(4-(4-(3-Chlorobenzoyl)piperazin-1-yl)phenyl)ethanone 12
Compound 44 (0.20 g, 0.90 mmol) was coupled with 4′-bromoacetophenone (0.15 g, 0.75 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (0.18 g, 69%) as a white solid: mp 142–143 °C; Rf = 0.65 (EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.89–7.86 (m, 2H), 7.43–7.29 (m, 4H), 6.88–6.84 (m, 2H), 3.89 (s, 2H), 3.60 (s, 2H), 3.37 (s, 4H), 2.51 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.4, 168.8, 153.6, 137.0, 134.7, 130.3, 130.1, 130.0, 128.5, 127.3, 125.1, 114.0, 47.7 (br), 41.8 (br), 26.1; HRMS (ESI+) calcd for C19H19ClN2NaO2+ 365.1027, found 365.1029 (error 0.5 ppm).
1-(4-(4-(5-Chloronicotinoyl)piperazin-1-yl)phenyl)ethanone 13
Compound 42 (98 mg, 0.48 mmol) was coupled with 5-chloronicotinic acid (63 mg, 0.40 mmol) using general procedure A for EDC coupling. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (90 mg, 66%) as a white solid: mp 161–162 °C; Rf = 0.13 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.64–8.58 (m, 1H), 8.53 (s, 1H), 7.85 (d, J = 8.5 Hz, 2H), 7.76 (s, 1H), 6.84 (d, J = 8.5 Hz, 2H), 3.89 (s, 2H), 3.62–3.57 (m, 2H), 3.43–3.31 (m, 4H), 2.48 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.3, 166.1, 153.4, 149.9, 145.5, 134.7, 132.2, 131.9, 130.2, 128.6, 114.0, 47.1, 41.8, 26.1; HRMS (ESI+) calcd for C18H18ClN3NaO2+ 366.0980, found 366.0967 (error 3.6 ppm).
1-(4-(4-Nicotinoylpiperazin-1-yl)phenyl)ethanone 14
Compound 42 (74 mg, 0.36 mmol) was coupled with nicotinic acid (37 mg, 0.30 mmol) using general procedure A for EDC coupling. Purification by flash chromatography (2:1 DCM/CH3CN) afforded the title compound (66 mg, 71%) as a white solid: mp 142–143 °C; Rf = 0.24 (2:1 DCM−CH3CN); 1H NMR (400 MHz, CDCl3) δ 8.68 (d, J = 5.2 Hz, 2H), 7.88 (d, J = 8.5 Hz, 2H), 7.78 (dt, J = 7.9, 1.9 Hz, 1H), 7.38 (dd, J = 7.9, 5.0 Hz, 1H), 6.87 (d, J = 8.6 Hz, 2H), 3.92 (s, 2H), 3.62 (s, 2H), 3.39 (s, 4H), 2.51 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.4, 167.8, 153.5, 151.0, 147.9, 135.1, 131.1, 130.3, 128.6, 123.5, 114.0, 47.6, 42.0, 26.1; HRMS (ESI+) calcd for C18H19N3NaO2+ 332.1369, found 332.1360 (error 2.7 ppm).
1-(4-(4-(Thiazole-4-carbonyl)piperazin-1-yl)phenyl)ethanone 15
Compound 42 (74 mg, 0.36 mmol) was coupled with 4-thiazolecarboxylic acid (39 mg, 0.30 mmol) using general procedure A for EDC coupling. Purification by flash chromatography (EtOAc) afforded the title compound (68 mg, 72%) as a white solid: mp 118–119 °C; Rf = 0.24 (EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.79 (d, J = 2.1 Hz, 1H), 8.05 (d, J = 2.2 Hz, 1H), 7.87–7.85 (m, 2H), 6.85 (d, J = 8.9 Hz, 2H), 4.10 (s, 2H), 3.92 (s, 2H), 3.41 (s, 4H), 2.49 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.4, 162.3, 153.7, 151.9, 130.3, 128.1, 125.3, 113.6, 47.8, 47.1, 46.4, 42.3, 26.0; HRMS (ESI+) calcd for C16H17N3NaO2S+ 338.0934, found 338.0928 (error 1.8 ppm).
1-(4-(4-(1-Methyl-1H-pyrazole-4-carbonyl)piperazin-1-yl)phenyl)-ethanone 16
Compound 42 (74 mg, 0.36 mmol) was coupled with 1-methyl-1H-pyrazole-4-carboxylic acid (38 mg, 0.30 mmol) using general procedure A for EDC coupling. Purification by flash chromatography (1:4 DCM/CH3CN) afforded the title compound (68 mg, 73%) as a white solid: mp 170–171 °C; Rf = 0.18 (1:4 DCM/CH3CN); 1H NMR (400 MHz, CDCl3) δ 7.86 (d, J = 8.0, 2H), 7.72 (s, 1H), 7.62 (s, 1H), 6.84 (d, J = 8.0, 2H), 3.89 (s, 3H), 3.85 (t, J = 4.0 Hz, 4H), 3.38 (t, J = 4.0 Hz, 4H), 2.48 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.4, 167.8, 153.5, 151.0, 147.9, 135.1, 131.1, 130.3, 128.6, 123.5, 114.0, 47.7, 41.8, 26.1; HRMS (ESI+) calcd for C17H20N4NaO2+ 335.1478, found 335.1477 (error 0.3 ppm).
1-(4-(4-Acetylphenyl)piperazin-1-yl)propan-1-one 17
Compound 42 (49 mg, 0.24 mmol) was coupled with propionic acid (0.015 mL, 0.20 mmol) using general procedure A for EDC coupling. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (42 mg, 81%) as a white solid: mp 75–76 °C; Rf = 0.07 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.6 (d, J = 8 Hz, 2H), 6.5 (d, J = 8 Hz, 2H), 3.76 (d, J = 5.6 Hz, 2H), 3.61 (d, J = 5.4 Hz, 2H), 3.34 (dt, J = 12.1, 5.0 Hz, 4H), 2.50 (d, J = 2.2 Hz, 3H), 2.37 (qd, J = 7.5, 2.1 Hz, 2H), 1.16 (td, J = 7.4, 2.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 196.4, 172.3, 153.6, 130.3, 128.2, 113.7, 47.5, 47.3, 44.7, 40.9, 26.4, 26.1, 9.3; HRMS (ESI+) calcd for C15H20N2NaO2+ 283.1417, found 283.1423 (error 2.1 ppm).
1-(4-(4-Acetylphenyl)piperazin-1-yl)butan-1-one 18
Compound 42 (49 mg, 0.24 mmol) was coupled with butyric acid (0.018 mL, 0.20 mmol) using general procedure A for EDC coupling. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (46 mg, 84%) as a white solid: mp 78–79 °C; Rf = 0.19 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 8.9 Hz, 2H), 3.75 (t, J = 5.3 Hz, 2H), 3.62 (t, J = 5.2 Hz, 2H), 3.44–3.21 (m, 4H), 2.49 (s, 3H), 2.32 (t, J = 7.6 Hz, 2H), 1.63–1.69 (m, 2H), 0.96 (t, J = 8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 196.3, 171.5, 153.6, 130.3, 128.1, 113.6, 47.58, 47.3, 44.9, 40.8, 35.0, 26.0, 18.6, 13.9; HRMS (ESI+) calcd for C16H22N2NaO2+ 297.1573, found 297.1580 (error 2.4 ppm).
1-(4-(4-Acetylphenyl)piperazin-1-yl)hexan-1-one 19
Compound 42 (49 mg, 0.24 mmol) was coupled with hexanoic acid (0.025 mL, 0.20 mmol) using general procedure A for EDC coupling. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (52 mg, 86%) as a white solid: mp 73–74 °C; Rf = 0.20 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 8.4 Hz, 2H), 6.83 (d, J = 8.5 Hz, 2H), 3.74 (t, J = 5.2 Hz, 2H), 3.61 (t, J = 5.1 Hz, 2H), 3.32 (dt, J = 11.6, 5.1 Hz, 4H), 2.48 (s, 3H), 2.32 (t, J = 7.7 Hz, 2H), 1.62 (t, J = 7.4 Hz, 2H), 1.31 (d, J = 6.4 Hz, 4H), 0.87 (d, J = 6.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 196.3, 171.7, 153.6, 130.3, 128.1, 113.6, 47.5, 47.3, 44.9, 40.9, 33.1, 31.5, 26.1, 24.9, 22.4, 13.9; HRMS (ESI+) calcd for C18H26N2NaO2+ 325.1886, found 325.1901 (error 4.6 ppm).
1-(4-(4-Acetylphenyl)piperazin-1-yl)-3-methylbutan-1-one 20
Compound 42 (49 mg, 0.24 mmol) was coupled with isovaleric acid (0.022 mL, 0.20 mmol) using general procedure A for EDC coupling. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (47 mg, 81%) as a white solid: mp 57–58 °C; Rf = 0.21 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 8 Hz, 2H), 6.84 (d, J = 8 Hz, 2H), 3.76 (t, J = 5.5 Hz, 2H), 3.63 (t, J = 4.0 Hz, 2H), 3.49–3.16 (m, 4H), 2.49 (s, 3H), 2.23 (dd, J = 7.0, 2.4 Hz, 2H), 2.12 (t, J = 6.5 Hz, 1H), 0.97 (dd, J = 6.6, 2.5 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 196.3, 171.0, 153.6, 130.3, 128.1, 113.7, 47.6, 47.3, 45.1, 41.9, 40.9, 26.1, 25.7, 22.6; HRMS (ESI+) calcd for C17H24N2NaO2+ 311.1730, found 311.1738 (error 2.6 ppm).
(3-Chlorophenyl)(4-(4-(pyridin-2-yl)phenyl)piperazin-1-yl)-methanone 21
Compound 44 (92 mg, 0.41 mmol) was coupled with 2-(4-bromophenyl)pyridine (80 mg, 0.34 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (73 mg, 57%) as a light brown solid: mp 158–159 °C; Rf = 0.19 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.64 (dt, J = 4.9, 1.4 Hz, 1H), 7.99–7.90 (m, 2H), 7.75–7.63 (m, 2H), 7.47–7.28 (m, 3H), 7.16 (ddd, J = 6.7, 4.8, 1.5 Hz, 1H), 7.05–6.96 (m, 3H), 3.94 (s, 2H), 3.61 (s, 2H), 3.34 (s, 2H), 3.25 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 168.7, 156.8, 151.1, 149.4, 137.2, 136.6, 134.6, 131.1, 129.91, 129.87, 127.7, 127.2, 125.1, 121.2, 119.5, 116.1, 48.9 (br), 47.3, 42.0; HRMS (ESI+) calcd for C22H20ClN3NaO+ 400.1187, found 400.1176 (error 2.7 ppm).
(3-Chlorophenyl)(4-(5-methoxypyridin-2-yl)piperazin-1-yl)-methanone 22
Compound 44 (92 mg, 0.41 mmol) was coupled with 2-(4-bromophenyl)-5-methoxypyridine (90 mg, 0.34 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (71 mg, 51%) as a light brown solid: mp 165–166 °C; Rf = 0.70 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.86 (d, J = 8.3 Hz, 2H), 7.59 (d, J = 8.7 Hz, 1H), 7.46–7.28 (m, 5H), 6.98 (d, J = 8.4 Hz, 2H), 4.05–3.88 (m, 5H), 3.59 (s, 2H), 3.30 (s, 2H), 3.21 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 168.7, 154.3, 150.6, 149.8, 137.2, 136.8, 134.6, 131.3, 130.0, 129.9, 127.3, 127.2, 125.1, 121.4, 119.9, 116.4, 55.6, 49.4, 47.4, 42.1; HRMS (ESI+) calcd for C23H22ClN3NaO2+ 430.1293, found 430.1283 (error 2.3 ppm).
(3-Chlorophenyl)(4-(4-(3-methyl-1,2,4-oxadiazol-5-yl)phenyl)-piperazin-1-yl)methanone 23
Compound 44 (88 mg, 0.39 mmol) was coupled with 5-(4-bromophenyl)-3-methyl-1,2,4-oxadiazole (76 mg, 0.32 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (44 mg, 36%) as a light brown solid: mp 195–196 °C; Rf = 0.21 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.07–7.96 (m, 2H), 7.48–7.26 (m, 4H), 7.00–6.91 (m, 2H), 3.92 (s, 2H), 3.63 (s, 2H), 3.39 (s, 4H), 2.44 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 175.3, 168.9, 167.5, 153.3, 137.0, 134.7, 130.2, 130.0, 129.6, 127.3, 125.2, 114.9, 114.8, 47.7, 42.0; HRMS (ESI+) calcd for C20H19ClN4NaO2+ 405.1089, found 405.1102 (error 3.2 ppm).
(3-Chlorophenyl)(4-(4-(1-methyl-1H-tetrazol-5-yl)phenyl)-piperazin-1-yl)methanone 24
Compound 44 (0.12 g, 0.55 mmol) was coupled with 5-(4-bromophenyl)-1-methyl-1H-tetrazole (0.11 g, 0.46 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (0.12 g, 66%) as a light brown solid: mp 202–203 °C; Rf = 0.21 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 8.4 Hz, 2H), 7.42–7.27 (m, 4H), 6.96 (d, J = 8.4 Hz, 2H), 4.33 (s, 3H), 3.90 (s, 2H), 3.57 (s, 2H), 3.32 (s, 2H), 3.23 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 168.7, 165.0, 151.9, 137.1, 134.6, 130.0, 129.9, 127.8, 127.2, 125.1, 118.8, 115.8, 48.7, 48.5, 47.1, 41.9, 39.3; HRMS (ESI+) calcd for C19H19ClN6NaO+ 405.1201, found 405.1216 (error 3.7 ppm).
(3-Chlorophenyl)(4-(4-(isoxazol-5-yl)phenyl)piperazin-1-yl)-methanone 25
The solution of 12 (50 mg, 0.15 mmol) and N,N-dimethylformamide dimethyl acetal (0.022 mL, 0.15 mmol) in O-xylene (2 mL) was refluxed for 8 h. After removing the solvent under reduced pressure, hydroxylamine hydrochloride (10 mg, 0.14 mmol) was added followed by EtOH (2 mL). The mixture was heated at 78 °C for 10 h. After cooling to 23 °C, H2O (5 mL) was added. The mixture was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed by saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography on silica gel (1:1 hexanes/EtOAc) afforded the title compound (16 mg, 30%) as a light brown solid: mp 108–109 °C; Rf = 0.21 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.24 (d, J = 1.9 Hz, 1H), 7.74–7.68 (m, 2H), 7.46–7.35 (m, 3H), 7.32 (dt, J = 7.4, 1.5 Hz, 1H), 6.99–6.94 (m, 2H), 6.38 (d, J = 1.9 Hz, 1H), 3.92 (s, 2H), 3.60 (s, 2H), 3.32 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 169.3, 168.8, 151.6, 150.8, 137.1, 134.7, 130.1, 130.0, 127.3, 127.2, 125.2, 119.2, 115.9, 97.1, 48.5, 47.4, 41.8; HRMS (ESI+) calcd for C20H18ClN3NaO2+ 390.0980, found 390.0992 (error 3.1 ppm).
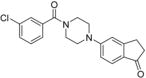
5-(4-(3-Chlorobenzoyl)piperazin-1-yl)-2,3-dihydro-1H-inden-1-one 26
Compound 44 (31 mg, 0.14 mmol) was coupled with 5-bromo-1-indanone (25 mg, 0.12 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (32 mg, 76%) as a light brown oil: Rf = 0.21 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 8.6 Hz, 1H), 7.47–7.30 (m, 4H), 6.87 (dd, J = 8.7, 2.2 Hz, 1H), 6.82 (d, J = 2.1 Hz, 1H), 3.89 (s, 2H), 3.63 (s, 2H), 3.42 (s, 4H), 3.06–3.03 (m, 2H), 2.66–2.63 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 205.0, 168.9, 157.8, 155.3, 136.9, 134.7, 130.2, 130.0, 128.8, 127.3, 125.2, 114.9, 110.6, 77.2, 47.9, 42.0, 36.4, 25.9; HRMS (ESI+) calcd for C20H19ClN2NaO2+ 377.1027, found 377.1017 (error 2.7 ppm).
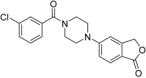
5-(4-(3-Chlorobenzoyl)piperazin-1-yl)isobenzofuran-1(3H)-one 27
Compound 44 (154 mg, 0.68 mmol) was coupled with 5-bromophthalide (121 mg, 0.57 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (81 mg, 40%) as a light brown solid: mp 154–155 °C; Rf = 0.11 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 8.6 Hz, 1H), 7.47–7.33 (m, 3H), 7.35–7.24 (m, 1H), 6.99 (dd, J = 8.7, 2.1 Hz, 1H), 6.82 (d, J = 2.1 Hz, 1H), 5.20 (s, 2H), 3.92 (s, 2H), 3.64 (s, 2H), 3.41 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 168.8, 154.9, 149.2, 136.8, 134.7, 130.2, 130.0, 127.2, 126.8, 125.1, 116.2, 116.0, 106.4, 69.1, 47.97, 47.0, 41.8; HRMS (ESI+) calcd for C19H17ClN2NaO3+ 379.0820, found 379.0804 (error 4.2 ppm).
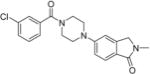
5-(4-(3-Chlorobenzoyl)piperazin-1-yl)-2-methylisoindolin-1-one 28
Compound 44 (72 mg, 0.32 mmol) was coupled with 5- bromo-2-methylisoindolin-1-one (60 mg, 0.26 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (43 g, 45%) as a light brown solid: mp 155–156 °C; Rf = 0.11 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.69 (d, J = 8.4 Hz, 1H), 7.43–7.26 (m, 4H), 6.97 (dd, J = 8.5, 2.2 Hz, 1H), 6.89 (d, J = 2.1 Hz, 1H), 4.29 (s, 2H), 3.92 (s, 2H), 3.60 (s, 2H), 3.30 (s, 4H), 3.14 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.8, 168.6, 153.4, 143.0, 137.1, 134.7, 130.1, 130.0, 127.3, 125.2, 124.8, 124.5, 116.1, 109.3, 51.9, 49.1, 47.5, 42.1, 29.4; HRMS (ESI+) calcd for C20H20ClN3NaO2+ 392.1136, found 392.1146 (error 2.6 ppm).
(3-Chlorophenyl)(4-(1,3-dihydroisobenzofuran-5-yl)piperazin-1-yl)methanone 29
Compound 44 (112 mg, 0.50 mmol) was coupled with 5-bromo-1,3-dihydroisobenzofuran (84 mg, 0.42 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (78 mg, 54%) as a light brown oil: Rf = 0.32 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.42–7.29 (m, 4H), 7.13 (d, J = 8.2 Hz, 1H), 6.85 (dd, J = 8.2, 2.2 Hz, 1H), 6.80 (d, J = 2.2 Hz, 1H), 5.05 (q, J = 2.4 Hz, 4H), 3.92 (s, 2H), 3.58 (s, 2H), 3.20 (s, 3H), 3.12 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 168.7, 150.8, 140.5, 137.2, 134.6, 131.5, 129.93, 129.89, 127.2, 125.1, 121.5, 116.6, 109.5, 73.5, 73.2, 50.3, 47.6, 42.1; HRMS (ESI+) calcd for C19H19ClN2NaO2+ 365.1027, found 365.1014 (error 3.6 ppm).
1-(3-(4-(3-Chlorobenzoyl)piperazin-1-yl)phenyl)ethanone 30
Compound 44 (121 mg, 0.54 mmol) was coupled with 3′-bromoacetophenone (0.06 mL, 0.45 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (56 mg, 36%) as a light brown oil: Rf = 0.34 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.51–7.29 (m, 7H), 7.11 (dd, J = 8.3, 2.5 Hz, 1H), 3.91 (s, 2H), 3.58 (S, 2H), 3.28 (s, 2H), 3.20 (s, 2H), 2.57 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 198.2, 168.7, 151.0, 138.0, 137.1, 134.6, 130.0, 129.9, 129.4, 127.2, 125.1, 121.3, 120.8, 115.3, 49.4, 47.3 (br), 42.0, 26.7; HRMS (ESI+) calcd for C19H19ClN2NaO2+ 365.1027, found 365.1038 (error 3.0 ppm).
Methyl 4-(4-(3-Chlorobenzoyl)piperazin-1-yl)benzoate 31
Compound 44 (202 mg, 0.90 mmol) was coupled with methyl 4-bromobenzoate (161 mg, 0.75 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (201 mg, 75%) as a light brown solid: mp 165–165 °C; Rf = 0.37 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.93–7.89 (m, 2H), 7.41–7.26 (m, 4H), 6.86–6.83 (m, 2H), 3.83 (s, 5H), 3.56 (s, 2H), 3.33 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 168.7, 166.8, 153.5, 137.0, 134.5, 131.1, 130.0, 129.9, 127.2, 125.1, 120.6, 114.1, 51.6, 47.8, 47.1, 41.9; HRMS (ESI+) calcd for C19H19ClN2NaO3+ 381.0976, found 381.0961 (error 3.9 ppm).
4-(4-(3-Chlorobenzoyl)piperazin-1-yl)benzonitrile 32
Compound 44 (154 mg, 0.68 mmol) was coupled with 4- bromobenzonitrile (104 mg, 0.57 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (157 mg, 85%) as a light brown oil: Rf = 0.35 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.49–7.46 (m, 2H), 7.41–7.33 (m, 3H), 7.30–7.27 (m, 1H), 6.86–6.83 (m, 2H), 3.86 (s, 2H), 3.58 (s, 2H), 3.34 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 168.6, 152.7, 136.7, 134.5, 133.3, 130.0, 129.8, 127.1, 125.0, 119.5, 114.5, 101.0, 47.2, 41.5; HRMS (ESI+) calcd for C18H16ClN3NaO+ 348.0874, found 348.0892 (error 5.2 ppm).
(3-Chlorophenyl)(4-(4-nitrophenyl)piperazin-1-yl)methanone 33
Compound 44 (124 mg, 0.55 mmol) was coupled with 1-bromo-4-nitrobenzene (92 mg, 0.46 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (119 mg, 75%) as a yellow solid: mp 102–103 °C; Rf = 0.40 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 8 Hz, 2H), 7.42–7.32 (m, 4H), 6.83 (d, J = 8 Hz, 2H), 3.89 (s, 2H), 3.63 (s, 2H), 3.45 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 168.7, 154.2, 138.8, 136.7, 134.5, 130.1, 129.9, 127.2, 125.7, 125.0, 112.9, 46.8, 41.6; HRMS (ESI+) calcd for C17H16ClN3NaO3+ 368.0772, found 368.0786 (error 3.8 ppm).
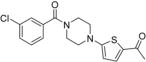
1-(5-(4-(3-Chlorobenzoyl)piperazin-1-yl)thiophen-2-yl)ethanone 34
Compound 44 (154 mg, 0.68 mmol) was coupled with 2- acetyl-5-bromothiophene (117 mg, 0.57 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (119 mg, 60%) as a light brown solid: mp 131–132 °C; Rf = 0.25 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.47–7.32 (m, 4H), 7.31–7.25 (m, 1H), 6.07 (dd, J = 4.3, 1.3 Hz, 1H), 3.86 (s, 2H), 3.62 (s, 2H), 3.32 (s, 4H), 2.41 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 189.1, 168.8, 165.8, 136.7, 134.73, 134.68, 130.2, 130.0, 129.8, 127.2, 125.1, 105.1, 49.7, 46.5, 41.3, 25.2; HRMS (ESI+) calcd for C17H17ClN2NaO2S+ 371.0591, found 371.0610 (error 5.1 ppm).
(3-Chlorophenyl)(4-(4-(1-(hydroxyimino)ethyl)phenyl)piperazin-1-yl)methanone 35
To a solution of 12 (177 mg, 0.51 mmol) in EtOH (2 mL) was added DIPEA (0.14 mL, 0.82 mmol) and hydroxylamine hydrochloride (57 mg, 0.82 mmol). The mixture was heated at 60 °C for 2 h. After cooling, EtOH was removed in vacuo, and then H2O (20 mL) was added. The mixture was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed by saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography on silica gel (EtOAc) afforded the title compound (60 mg, 33%) as a white solid: mp 180–181 °C; Rf = 0.74 (EtOAc); 1H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H), 7.58–7.55 (m, 2H), 7.44–7.30 (m, 4H), 6.91–6.88 (m, 2H), 3.93 (s, 2H), 3.59 (s, 2H), 3.29 (s, 2H), 3.20 (s, 2H), 2.25 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.8, 155.3, 151.2, 137.1, 130.0, 129.9, 128.4, 127.3, 127.0, 125.2, 115.9, 49.1, 47.3, 42.1, 11.9; HRMS (ESI+) calcd for C19H20ClN3NaO2+ 380.1136, found 380.1156 (error 5.3 ppm).
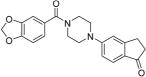
5-(4-(Benzo[d][1,3]dioxole-5-carbonyl)piperazin-1-yl)-2,3-dihydro-1H-inden-1-one 36
Compound 43 (212 mg, 0.98 mmol) was coupled with piperonylic acid (132 mg, 0.82 mmol) using general procedure A for EDC coupling. Purification by flash chromatography (EtOAc) afforded the title compound (218 mg, 73%) as a white solid: mp 158–159 °C; Rf = 0.43 (EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 8.0 Hz, 1H), 6.94–6.90 (m, 2H), 6.85–6.78 (m, 3H), 5.97 (s, 2H), 3.74 (s, 4H), 3.37 (s, 4H), 3.01–2.98 (m, 2H), 2.60–2.58 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 204.8, 169.8, 157.7, 155.3, 149.0, 147.6, 128.7, 128.4, 125.0, 121.6, 114.6, 110.3, 108.1, 108.0, 101.4, 47.7, 36.3, 25.8; HRMS (ESI+) calcd for C21H20N2NaO4+ 387.1315, found 387.1326 (error 2.8 ppm).
1-(4-(1-(3-Chlorobenzyl)-1H-1,2,3-triazol-4-yl)phenyl)ethanone 37
To a solution of 4644 (84.0 mg, 0.50 mmol, 1.00 equiv) and 1-(4-ethynylphenyl)ethanone (72 mg, 0.50 mmol) in EtOH/H2O (1:1, 6 mL) were added CuSO4·5H2O (12.0 mg, 0.05 mmol, 0.10 equiv) in one portion followed by sodium ascorbate (30.0 mg, 0.15 mmol, 0.30 equiv) in one portion at 23 °C. The resulting yellowish cloudy suspension was stirred for 12 h at 23 °C; 10 mL of cold 1:1 hexanes/EtOAc was added for trituration, and the suspension was filtered to afford the title compound (105 mg, 68%) as a light brown solid: mp 164–165 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.68 (s, 1H), 8.14 (t, J = 1.9 Hz, 1H), 8.08–8.02 (m, 5H), 7.84 (dt, J = 7.6, 1.4 Hz, 1H), 7.67 (t, J = 7.9 Hz, 1H), 6.32 (s, 2H), 2.61 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 191.3, 145.4, 135.94, 135.88, 135.0, 134.0, 133.9, 131.0, 129.0, 128.0, 126.9, 125.1, 124.2, 56.2, 26.7; HRMS (ESI+) calcd for C17H14ClN3NaO+ 334.0718, found 334.0722 (error 1.2 ppm).
1-(5-(1-(3-Chlorobenzyl)-1H-1,2,3-triazol-4-yl)thiophen-2-yl)-ethanone 38
Using the procedure for the synthesis of 37, compound 46 (97 mg, 0.58 mmol) was reacted with 1-(5-ethynylthiophen-2-yl)ethanone (88 mg, 0.58 mmol). Purification by adding 10 mL of cold 1:1 hexanes/EtOAc for trituration followed by filtration afforded the title compound (128 mg, 70%) as a light brown solid: mp 174–175 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.62 (s, 1H), 8.12 (t, J = 1.9 Hz, 1H), 8.04 (d, J = 7.8 Hz, 1H), 7.96 (d, J = 3.9 Hz, 1H), 7.84–7.79 (m, 1H), 7.67 (t, J = 7.9 Hz, 1H), 7.60 (d, J = 3.9 Hz, 1H), 6.32 (s, 2H), 2.56 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 191.2, 190.7, 142.5, 140.9, 135.8, 134.9, 134.0, 133.9, 131.0, 128.0, 126.9, 125.3, 123.9, 56.3, 26.5; HRMS (ESI+) calcd for C15H12ClN3NaOS+ 340.0282, found 340.0291 (error 2.6 ppm).
1-(4-(4-(3-Chlorobenzoyl)piperidin-1-yl)phenyl)ethanone 39
The mixture of 49 (61 mg, 0.19 mmol) in 4 mL of 1:3 TFA/CH2Cl2 was stirred at rt for 3 h. After removing the solvent under reduced pressure, the residue was coupled with 1-(4-bromophenyl)ethanone (24 mg, 0.12 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (28 mg, 68%) as a white solid: mp 121–122 °C; Rf = 0.33 (2:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.92–7.82 (m, 4H), 7.56 (d, J = 8.0 Hz, 1H), 7.45 (t, J = 8.0 Hz, 1H), 6.89 (d, J = 8.0 Hz, 2H), 3.96 (d, J = 9.0 Hz, 2H), 3.46–3.41 (m, 1H), 3.10–3.03 (m, 2H), 2.51 (s, 3H), 2.04–1.84 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 200.6, 196.4, 153.9, 137.4, 135.1, 133.1, 130.4, 130.1, 128.3, 127.4, 126.3, 113.6, 47.3, 43.3, 27.8, 26.1; HRMS (ESI+) calcd for C20H20ClNNaO2+ 364.1075, found 364.1069 (error 1.6 ppm).
1-(4-(4-(Benzo[d][1,3]dioxole-5-carbonyl)piperidin-1-yl)phenyl)-ethanone 40
The mixture of 50 (63 mg, 0.19 mmol) in 4 mL of 1:3 TFA/CH2Cl2 was stirred at rt for 3 h. After removing the solvent under reduced pressure, the residue was coupled with 1-(4-bromophenyl)ethanone (24 mg, 0.12 mmol) using general procedure B. Purification by flash chromatography (1:1 hexanes/EtOAc) afforded the title compound (26 mg, 62%) as a white solid: mp 170–171 °C; Rf = 0.30 (2:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 8.0 Hz, 2H), 7.59 (dd, J = 8.0, 4.0 Hz, 1H), 7.44 (d, J = 4.0 Hz, 1H), 6.89 (dd, J = 8.0, 4.0 Hz, 3H), 6.05 (s, 2H), 3.98 (dt, J = 12.0, 4.0 Hz, 2H), 3.42–3.37 (m, 1H), 3.07 (td, J = 12.0, 4.0 Hz, 2H), 2.52 (s, 3H), 1.98–1.88 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 199.9, 196.4, 154.0, 151.8, 148.4, 130.5, 130.4, 124.3, 113.6, 108.1, 108.0, 101.9, 47.4, 43.0, 28.2, 26.1; HRMS (ESI+) calcd for C21H21NNaO4+ 374.1363, found 374.1370 (error 1.9 ppm).
1-(4-(Piperazin-1-yl)phenyl)ethanone 42
To a solution of piperazine 41 (775 mg, 9.00 mmol, 3.00 equiv) in DMSO (12 mL) was added 4′-fluoroacetophenone (0.36 mL, 3.00 mmol, 1.00 equiv), and the mixture was heated at 110 °C for 24 h. After cooling to 23 °C, the reaction mixture was quenched with water (50 mL). The mixture was extracted with EtOAc (3 × 30 mL). The combined organic layers were washed by saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography on silica gel (130:25:4 CHCl3/MeOH/NH4OH) afforded the title compound (531 mg, 84%) as a light yellow solid: mp 102–103 °C; Rf = 0.30 (130:25:4 CHCl3/MeOH/NH4OH); 1H NMR (400 MHz, CDCl3) δ 7.86 (d, J = 8.9 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 3.33–3.30 (m, 4H), 3.04–3.01 (m, 4H), 2.51 (s, 3H), 2.03 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 196.5, 154.5, 130.3, 127.6, 113.4, 48.4, 45.8, 26.1.
(3-Chlorophenyl)(piperazin-1-yl)methanone 44
To a solution of piperazine 41 (1.00 g, 11.6 mmol, 1.05 equiv) in THF (50 mL) was added n-BuLi (2.5 M in hexanes, 10.2 mL, 25.5 mmol, 2.32 equiv) at 23 °C. The reaction was stirred for 1 h at 23 °C, and then 3-chlorobenzoyl chloride (1.41 mL, 11.0 mmol, 1.00 equiv) was added. After 10 min, the reaction mixture was quenched with MeOH (20 mL) and concentrated under reduced pressure. The residue was partitioned between EtOAc (50 mL) and saturated aqueous NaHCO3 (50 mL). The aqueous layer was extracted with EtOAc (2 × 30 mL). The combined organic layers were washed by saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography on silica gel (130:25:4 CHCl3/MeOH/NH4OH) afforded the title compound (1.60 g, 65%) as a light brown solid: mp 86–87 °C; Rf = 0.43 (130:25:4 CHCl3/MeOH/NH4OH); 1H NMR (400 MHz, CDCl3) δ 7.43–7.30 (m, 3H), 7.31–7.23 (m, 1H), 3.74 (br s, 2H), 3.37 (br s, 2H), 2.93 (br s, 2H), 2.82 (br s, 2H), 1.71 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 167.9, 137.1, 133.8, 129.3, 129.0, 126.5, 124.4, 48.3, 45.7, 45.2, 42.6; HRMS (ESI+) calcd for C11H13ClN2NaO+ 247.0609, found 247.0615 (error 2.4 ppm).
General Procedure B: Palladium-Catalyzed Coupling
A mixture of 44 (1 mmol, 1.20 equiv), aryl bromide (0.82 mmol, 1.00 equiv), Cs2CO3 (1.64 mmol, 2.00 equiv), Pd2(dba)3 (0.033 mmol, 0.04 equiv), and BINAP (0.066 mmol, 0.08 equiv) in toluene (6 mL) was purged with argon for 3 min. The reaction mixture was heated at 100 °C for 16 h. After cooling to 23 °C, H2O (20 mL) was added. The mixture was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed by saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography on silica gel (1:1 hexanes/EtOAc) afforded the coupled product.
tert-Butyl 4-(3-Chlorobenzoyl)piperidine-1-carboxylate 49
To the solution of 1-bromo-3-chlorobenzene (0.12 mL, 1 mmol) in THF (5 mL) at −78 °C was added 2.5 M t-BuLi (0.44 mL, 1.1 mmol). The mixture was stirred at −78 °C for 30 min, and 4845 (245 mg, 0.9 mmol) was added. The mixture was slowly warmed to rt, stirred for 16 h, and quenched by adding H2O (10 mL). The mixture was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed by saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography on silica gel (5:1 hexanes/EtOAc) afforded the title compound (183 mg, 57%) as a white solid: Rf = 0.23 (5:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.89 (s, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.40–7.44 (m, 1H), 4.15 (s, 2H), 3.38 (tt, J = 12.0, 4.0 Hz, 1H), 2.93 (t, J = 12.0 Hz, 2H), 1.84–1.81 (m, 2H), 1.73–1.64 (m, 2H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 200.7, 171.9, 154.6, 137.4, 135.1, 133.0, 130.1, 128.3, 126.3, 79.7, 43.6, 43.3, 28.4.
tert-Butyl 4-(Benzo[d][1,3]dioxole-5-carbonyl)piperidine-1-carboxylate 50
To the solution of 5-bromobenzo[d][1,3]dioxole (0.12 mL, 1 mmol) in THF (5 mL) at −78 °C was added 1.7 M t-BuLi (1.3 mL, 2.2 mmol). The mixture was stirred at −78 °C for 30 min, and 4830 (245 mg, 0.9 mmol) was added. The mixture was warmed to rt slowly and stirred for 16 h. The reaction was quenched by the addition of H2O (10 mL). The mixture was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed by saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography on silica gel (5:1 hexanes/EtOAc) afforded the title compound (161 mg, 54%) as a white solid: Rf = 0.16 (5:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.47 (d, J = 8.0 Hz, 1H), 7.32 (s, 1H), 6.77 (d, J = 8.0 Hz, 1H), 5.95 (s, 2H), 4.09–4.06 (m, 2H), 3.27–3.21 (m, 1H), 2.83–2.77 (m, 2H), 1.74–1.71 (m, 2H), 1.64–1.58 (m, 2H), 1.38 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 199.8, 154.4, 151.6, 148.1, 130.3, 124.1, 107.8, 107.7, 101.7, 79.3, 53.2, 43.0, 28.4, 28.2.
Supplementary Material
Acknowledgments
This work was supported by NIH Grant AI091790. Isothermal titration calorimetry was carried out using an ITC-200 microcalorimeter funded in part by the NIH Shared Instrumentation Grant S10-OD017982. This research used resources at the Industrial Macromolecular Crystallography Association Collaborative Access Team (IMCA-CAT) beam-line 17-ID supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. This research also used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The authors also acknowledge the use of beamline 4.2.2 (Molecular Biology Consortium) at the Advanced Light Source and thank the beamline director, Jay Nix. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231.
ABBREVIATIONS USED
- AMTOB
S-adenosyl-2-oxo-4-methylthiobutyric acid
- BINAP
2,2′-bis(diphenylphosphino)-1,1′-binaphthyl
- Boc
N-tert-butoxycarbonyl
- DAPA
7,8-diaminopelargonic acid
- DIPEA
N,N-diisopropylethylamine
- DMAP
4-dimethylaminopyridine
- DOA
5′-deoxyadenosine
- EDC
1-ethyl-3-(3-(dimethylamino)-propyl)carbodiimide
- KAPA
7-keto-8-aminopelargonic acid
- ITC
isothermal titration calorimetry
- Mtb
Mycobacterium tuberculosis
- PLP
pyridoxal 5′-phosphate
- SAM
S-adenosylmethionine
- TB
tuberculosis
- TFA
trifluoroacetic acid
- WT
wild-type
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b00189.
X-ray crystallographic data and copies of 1H and 13C NMR spectra (PDF)
Molecular formula strings (CSV)
ORCID
Courtney C. Aldrich: 0000-0001-9261-594X
Notes
The authors declare no competing financial interest.
References
- 1.Daniel TM. The History of Tuberculosis. Respir Med. 2006;100(11):1862–1870. doi: 10.1016/j.rmed.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M, Parkhill J, Malla B, Berg S, Thwaites G, Yeboah-Manu D, Bothamley G, Mei J, Wei L, Bentley S, Harris SR, Niemann S, Diel R, Aseffa A, Gao Q, Young D, Gagneux S. Out-of-Africa Migration and Neolithic Coexpansion of Mycobacterium Tuberculosis with Modern Humans. Nat Genet. 2013;45(10):1176–1182. doi: 10.1038/ng.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nerlich AG, Haas CJ, Zink A, Szeimies U, Hagedorn HG. Molecular Evidence for Tuberculosis in an Ancient Egyptian Mummy. Lancet. 1997;350(9088):1404. doi: 10.1016/S0140-6736(05)65185-9. [DOI] [PubMed] [Google Scholar]
- 4.Salo WL, Aufderheide AC, Buikstra J, Holcomb TA. Identification of Mycobacterium Tuberculosis DNA in a Pre-Columbian Peruvian Mummy. Proc Natl Acad Sci USA. 1994;91(6):2091–2094. doi: 10.1073/pnas.91.6.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization. Global Tuberculosis Report 2016. Geneva, Switzerland: 2016. [Google Scholar]
- 6.UNAIDS. UNAIDS Fact Sheet November 2016. Geneva, Switzerland: 2016. Joint United Nations Programme on HIV/AIDS. [Google Scholar]
- 7.Uplekar M, Weil D, Lonnroth K, Jaramillo E, Lienhardt C, Dias HM, Falzon D, Floyd K, Gargioni G, Getahun H, Gilpin C, Glaziou P, Grzemska M, Mirzayev F, Nakatani H, Raviglione M. WHO’s New End TB Strategy. Lancet. 2015;385(9979):1799–1801. doi: 10.1016/S0140-6736(15)60570-0. [DOI] [PubMed] [Google Scholar]
- 8.World Health Organization. Treatment of Tuberculosis Guidelines. 4th. Geneva, Switzerland: 2010. [PubMed] [Google Scholar]
- 9.Espinal MA. The Global Situation of MDR-TB. Tuberculosis. 2003;83(1–3):44–51. doi: 10.1016/s1472-9792(02)00058-6. [DOI] [PubMed] [Google Scholar]
- 10.World Health Organization. Drug-Resistant TB: Surveillance and Response: Supplement to Global Tuberculosis Report 2014. Geneva, Switzerland: 2014. [Google Scholar]
- 11.World Health Organization. Multidrug and Extensively Drug-Resistant TB (M/XDR-TB): 2010 Global Report on Surveillance and Response. World Health Organization; Geneva, Switzerland: 2010. [Google Scholar]
- 12.Dheda K, Migliori GB. The Global Rise of Extensively Drug-Resistant Tuberculosis: Is the Time to Bring Back Sanatoria Now Overdue? Lancet. 2012;379(9817):773–775. doi: 10.1016/S0140-6736(11)61062-3. [DOI] [PubMed] [Google Scholar]
- 13.Migliori GB, Besozzi G, Girardi E, Kliiman K, Lange C, Toungoussova OS, Ferrara G, Cirillo DM, Gori A, Matteelli A, Spanevello A, Codecasa LR, Raviglione MC, SMIRA/TBNET Study Group Clinical and Operational Value of the Extensively Drug-Resistant Tuberculosis Definition. Eur Respir J. 2007;30(4):623–626. doi: 10.1183/09031936.00077307. [DOI] [PubMed] [Google Scholar]
- 14.Barry CE, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, Schnappinger D, Wilkinson RJ, Young D. The Spectrum of Latent Tuberculosis: Rethinking the Biology and Intervention Strategies. Nat Rev Microbiol. 2009;7:845–855. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dye C. Global Burden of Tuberculosis: Estimated Incidence, Prevalence, and Mortality by Country. JAMA J Am Med Assoc. 1999;282(7):677. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- 16.Zumla A, Nahid P, Cole ST. Advances in the Development of New Tuberculosis Drugs and Treatment Regimens. Nat Rev Drug Discovery. 2013;12(5):388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- 17.Mdluli K, Kaneko T, Upton A. The Tuberculosis Drug Discovery and Development Pipeline and Emerging Drug Targets. Cold Spring Harbor Perspect Med. 2015;5(6):a021154–a021154. doi: 10.1101/cshperspect.a021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cole ST, Riccardi G. New Tuberculosis Drugs on the Horizon. Curr Opin Microbiol. 2011;14(5):570–576. doi: 10.1016/j.mib.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 19.Evangelopoulos D, McHugh TD. Improving the Tuberculosis Drug Development Pipeline. Chem Biol Drug Des. 2015;86(5):951–960. doi: 10.1111/cbdd.12549. [DOI] [PubMed] [Google Scholar]
- 20.Koul A, Arnoult E, Lounis N, Guillemont J, Andries K. The Challenge of New Drug Discovery for Tuberculosis. Nature. 2011;469(7331):483–490. doi: 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- 21.Salaemae W, Azhar A, Booker GW, Polyak SW. Biotin Biosynthesis in Mycobacterium Tuberculosis: Physiology, Biochemistry and Molecular Intervention. Protein Cell. 2011;2(9):691–695. doi: 10.1007/s13238-011-1100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marquet A, Bui BT, Florentin D. Biosynthesis of Biotin and Lipoic Acid. Vitam Horm (London, UK) 2001;61:51–101. doi: 10.1016/s0083-6729(01)61002-1. [DOI] [PubMed] [Google Scholar]
- 23.Said HM. Cell and Molecular Aspects of Human Intestinal Biotin Absorption. J Nutr. 2008;139(1):158–162. doi: 10.3945/jn.108.092023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cronan JE, Lin S. Synthesis of the A,ω-Dicarboxylic Acid Precursor of Biotin by the Canonical Fatty Acid Biosynthetic Pathway. Curr Opin Chem Biol. 2011;15(3):407–413. doi: 10.1016/j.cbpa.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stoner GL, Eisenberg MA. Purification and Properties of 7, 8-Diaminopelargonic Acid Aminotransferase. J Biol Chem. 1975;250(11):4029–4036. [PubMed] [Google Scholar]
- 26.Stoner GL, Eisenberg MA. Biosynthesis of 7, 8-Diaminopelargonic Acid from 7-Keto-8-Aminopelargonic Acid and S-Adenosyl-L-Methionine. The Kinetics of the Reaction. J Biol Chem. 1975;250(11):4037–4043. [PubMed] [Google Scholar]
- 27.Mann S, Ploux O. 7,8-Diaminoperlargonic Acid Aminotransferase from Mycobacterium Tuberculosis, a Potential Therapeutic Target: Characterization and Inhibition Studies. FEBS J. 2006;273(20):4778–4789. doi: 10.1111/j.1742-4658.2006.05479.x. [DOI] [PubMed] [Google Scholar]
- 28.Park SW, Klotzsche M, Wilson DJ, Boshoff HI, Eoh H, Manjunatha U, Blumenthal A, Rhee K, Barry CE, Aldrich CC, Ehrt S, Schnappinger D. Evaluating the Sensitivity of Mycobacterium Tuberculosis to Biotin Deprivation Using Regulated Gene Expression. PLoS Pathog. 2011;7(9):e1002264. doi: 10.1371/journal.ppat.1002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dey S, Lane JM, Lee RE, Rubin EJ, Sacchettini JC. Structural Characterization of the Mycobacterium Tuberculosis Biotin Biosynthesis Enzymes 7,8-Diaminopelargonic Acid Synthase and Dethiobiotin Synthetase. Biochemistry. 2010;49(31):6746–6760. doi: 10.1021/bi902097j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dai R, Wilson DJ, Geders TW, Aldrich CC, Finzel BC. Inhibition of Mycobacterium Tuberculosis Transaminase BioA by Aryl Hydrazines and Hydrazides. ChemBioChem. 2014;15(4):575–586. doi: 10.1002/cbic.201300748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi C, Geders TW, Park SW, Wilson DJ, Boshoff HI, Abayomi O, Barry CE, Schnappinger D, Finzel BC, Aldrich CC. Mechanism-Based Inactivation by Aromatization of the Transaminase BioA Involved in Biotin Biosynthesis in Mycobaterium Tuberculosis. J Am Chem Soc. 2011;133(45):18194–18201. doi: 10.1021/ja204036t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kitahara T, Hotta K, Yoshida M, Okami Y. Biological Studies on Amiclenomycin. J Antibiot. 1975;28(3):215–221. doi: 10.7164/antibiotics.28.215. [DOI] [PubMed] [Google Scholar]
- 33.Okami Y, Kitahara T, Hamada M, Naganawa H, Kondo S, Maeda K, Takeuchi T, Umezawa H. Studies on a New Amino Acid Antibiotic, Amiclenomycin. J Antibiot. 1974;27(9):656–664. doi: 10.7164/antibiotics.27.656. [DOI] [PubMed] [Google Scholar]
- 34.Wilson DJ, Shi C, Duckworth BP, Muretta JM, Manjunatha U, Sham YY, Thomas DD, Aldrich CC. A Continuous Fluorescence Displacement Assay for BioA: An Enzyme Involved in Biotin Biosynthesis. Anal Biochem. 2011;416(1):27–38. doi: 10.1016/j.ab.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park SW, Casalena DE, Wilson DJ, Dai R, Nag PP, Liu F, Boyce JP, Bittker JA, Schreiber SL, Finzel BC, Schnappinger D, Aldrich CC. Target-Based Identification of Whole-Cell Active Inhibitors of Biotin Biosynthesis in Mycobacterium Tuberculosis. Chem Biol (Oxford, UK) 2015;22(1):76–86. doi: 10.1016/j.chembiol.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dai R. Fragment Based Inhibitor Design of Mycobacterium Tuberculosis BioA Dissertation. University of Minnesota; Minneapolis, MN: 2015. [Google Scholar]
- 37.Costantino L, Barlocco D. Privileged Structures as Leads in Medicinal Chemistry. Curr Med Chem. 2006;13(1):65–85. [PubMed] [Google Scholar]
- 38.Dai R, Geders TW, Liu F, Park SW, Schnappinger D, Aldrich CC, Finzel BC. Fragment-Based Exploration of Binding Site Flexibility in Mycobacterium Tuberculosis BioA. J Med Chem. 2015;58(13):5208–5217. doi: 10.1021/acs.jmedchem.5b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaires JB. Calorimetry and Thermodynamics in Drug Design. Annu Rev Biophys. 2008;37(1):135–151. doi: 10.1146/annurev.biophys.36.040306.132812. [DOI] [PubMed] [Google Scholar]
- 40.Freire E. Do Enthalpy and Entropy Distinguish First in Class from Best in Class? Drug Discovery Today. 2008;13(19–20):869–874. doi: 10.1016/j.drudis2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruben AJ, Kiso Y, Freire E. Overcoming Roadblocks in Lead Optimization: A Thermodynamic Perspective. Chem Biol Drug Des. 2006;67(1):2–4. doi: 10.1111/j.1747-0285.2005.00314.x. [DOI] [PubMed] [Google Scholar]
- 42.Wang T, Zhang Z, Meanwell NA. Benzoylation of Dianions: Preparation of Monobenzoylated Derivatives of Symmetrical Secondary Diamines. J Org Chem. 1999;64(20):7661–7662. [Google Scholar]
- 43.Wolfe JP, Wagaw S, Buchwald SL. An Improved Catalyst System for Aromatic Carbon–Nitrogen Bond Formation: The Possible Involvement of Bis(Phosphine) Palladium Complexes as Key Intermediates. J Am Chem Soc. 1996;118(30):7215–7216. [Google Scholar]
- 44.Chakraborty A, Dey S, Sawoo S, Adarsh NN, Sarkar A. Regioselective 1,3-Dipolar Cycloaddition Reaction of Azides with Alkoxy Alkynyl Fischer Carbene Complexes. Organometallics. 2010;29(23):6619–6622. [Google Scholar]
- 45.Li J, Zhang X, Zhang Z, Padakanti PK, Jin H, Cui J, Li A, Zeng D, Rath NP, Flores H, Perlmutter JS, Parsons SM, Tu Z. Heteroaromatic and Aniline Derivatives of Piperidines As Potent Ligands for Vesicular Acetylcholine Transporter. J Med Chem. 2013;56(15):6216–6233. doi: 10.1021/jm400664x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Geders TW, Gustafson K, Finzel BC. Use of Differential Scanning Fluorimetry to Optimize the Purification and Crystallization of PLP-Dependent Enzymes. Acta Crystallogr, Sect F: Struct Biol Cryst Commun. 2012;68(5):596–600. doi: 10.1107/S1744309112012912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, Womack T, Bricogne G. Data Processing and Analysis with the autoPROC Toolbox. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67(4):293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kabsch W. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66(2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser Crystallographic Software. J Appl Crystallogr. 2007;40(4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66(2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards Automated Crystallographic Structure Refinement with Phenix.refine. Acta Crystallogr, Sect D: Biol Crystallogr. 2012;68(4):352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emsley P, Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60(12):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 53.Lebedev AA, Young P, Isupov MN, Moroz OV, Vagin AA, Murshudov GN. JLigand: A Graphical Tool for the CCP 4 Template-Restraint Library. Acta Crystallogr, Sect D: Biol Crystallogr. 2012;68(4):431–440. doi: 10.1107/S090744491200251X. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.