

Abstract
Purpose
The most common cause of Fuchs' endothelial corneal dystrophy (FECD) is an intronic CTG repeat expansion in TCF4. Expanded CUG repeat RNA colocalize with splicing factor, muscleblind-like 1 (MBNL1), in nuclear foci in endothelium as a molecular hallmark. Myotonic dystrophy type 1 (DM1) is a neuromuscular disorder caused by a CTG repeat expansion in the 3′-untranslated region (UTR) of DMPK. In this study, we examine for RNA-MBNL1 foci in endothelial cells of FECD subjects with DM1, test the hypothesis that DM1 patients are at risk for FECD, and determine prevalence of TCF4 and DMPK expansions in a FECD cohort.
Methods
Using FISH, we examined for nuclear RNA-MBNL1 foci in endothelial cells from FECD subjects with DM1. We examined 13 consecutive unrelated DM1 patients for FECD using slit-lamp and specular microscopy. We genotyped TCF4 and DMPK repeat polymorphisms in a FECD cohort of 317 probands using short-tandem repeat and triplet repeat-primed PCR assays.
Results
We detected abundant nuclear RNA foci colocalizing with MBNL1 in endothelial cells of FECD subjects with DM1. Six of thirteen DM1 patients (46%) had slit-lamp and specular microscopic findings of FECD, compared to 4% disease prevalence (P = 5.5 × 10-6 ). As expected, 222 out of 317 (70%) FECD probands harbored TCF4 expansion, while one subject harbored DMPK expansion without prior diagnosis of DM1.
Conclusions
Our work suggests that DM1 patients are at risk for FECD. DMPK mutations contribute to the genetic burden of FECD but are uncommon. We establish a connection between two repeat expansion disorders converging upon RNA-MBNL1 foci and FECD.
Keywords: Fuchs' endothelial corneal dystrophy, myotonic dystrophy, triplet repeat expansion, DMPK, nuclear RNA foci
Fuchs' endothelial corneal dystrophy (FECD, Mendelian Inheritance in Man [MIM] 136800) is an age-related degenerative disorder of the endothelium resulting in corneal edema and loss of vision. FECD affects 4% of whites over the age of 40 in the United States1 and is the leading indication for corneal transplantation.2 The corneal endothelium, the inner postmitotic hexagonal monolayer of cells responsible for maintenance of stromal dehydration, is prone to oxidative damage, apoptosis, and premature senescence in FECD.3–9 The basement membrane of the endothelium, Descemet's membrane, becomes diffusely thickened and develops focal excrescences termed guttae that are visible with slit-lamp and specular biomicroscopy.10 FECD is a clinical diagnosis based on the presence of confluent central guttae on slit-lamp microscopy. Progressive loss of central endothelial cell density results in corneal edema, scarring, and loss of vision.
FECD can be inherited as an autosomal dominant trait with genetic heterogeneity.11 Rare heterozygous mutations in collagen, type VIII, alpha 2 gene (COL8A2, MIM 120252) cause an early-onset corneal endothelial dystrophy.12 Other genes including solute carrier family 4, sodium borate transporter, member 11 (SLC4A11, MIM 610206), transcription factor 8 (TCF8, MIM 189909), lipoxygenase homology domains 1 (LOXHD1, MIM 613267), and ATP/GTP binding protein-like 1 (AGBL1, MIM 615523) collectively account for a small fraction of adult-onset FECD cases.13–20 Genome-wide association studies of adult-onset FECD have implicated transcription factor 4 (TCF4, MIM 602272) and more recently KN motif– and ankyrin repeat domain–containing protein 4 (KANK4, MIM 614612), laminin gamma-1 (LAMC1, MIM150290), and Na+, K+ transporting ATPase, beta-1 polypeptide (ATP1B1, MIM 182330), with the TCF4 locus noted to have a predominant effect.21,22
CTG triplet repeat expansions in the third intron of TCF4 (CTG18.1 locus) are the most common genetic cause of adult-onset FECD cases in the United States23,24 TCF4 is a conserved class I basic helix-loop-helix (bHLH) transcription factor that binds to the canonical E-box promoter sequences of target genes.25,26 The CTG18.1 locus was discovered in 1997 by the repeat expansion detection assay, with expanded alleles of greater than 37 CTG repeats found to be unstable and present in 3% of subjects in Caucasian pedigrees.27 TCF4 expansions of greater than 40 CTG repeats confer significant risk for the development of FECD with an odds ratio (OR) of 32.3 in whites.24 The expanded allele was shown to cosegregate with complete penetrance in 52% of 29 white FECD families and with incomplete penetrance in an additional 10% of these families.24 Transethnic studies have been performed in Singapore-Chinese, Indian, and Japanese documenting the association of the triplet repeat expansion with FECD in nonwhite populations.28–30
Myotonic dystrophy type 1 (DM1) is a paradigm for genetic disorders caused by CTG expansions. In DM1, the expansion is within the 3′-untranslated region (UTR) of the dystrophia myotonia protein kinase gene.31,32 The expanded DM1 repeat RNA associates with the splicing factor muscleblind-like 1 (MBNL1) in nuclear foci that can be visualized by fluorescent in situ hybridization (FISH) and that are a molecular hallmark for disease.33,34 Association of MBNL1 with mutant RNA affects the cellular pool of free MBNL1 and triggers missplicing of some MBNL1 target genes in affected brain, muscle, and heart tissues.34 Accumulation of expanded CUG repeat RNA nuclear foci35 with colocalization with MBNL1 and missplicing of target genes36 has been recently reported in endothelial cells of FECD subjects with the TCF4 repeat expansion.
Gattey et al.37 reported FECD in four DM1 subjects including a mother–daughter pair. No molecular studies were performed and because these are both common disorders, it can be concluded that additional studies were warranted. In this study, we explored the association between DM1 and FECD. We detected the presence of nuclear RNA-MBNL1 foci in endothelial cells from an organ donor whose corneas were found to be unsuitable for transplantation for the findings of FECD. Surprised that the donor did not harbor a TCF4 expansion, we hypothesized correctly that the subject harbored a CTG repeat expansion in the 3′ UTR of the DMPK gene and subsequently confirmed a clinical diagnosis of DM1. Additionally, we tested the hypothesis that DM1 patients are at risk for FECD and determined prevalence of TCF4 and DMPK triplet repeat expansions in a University of Texas Southwestern (UTSW) FECD cohort.
Methods
Subjects
The study was approved by the UTSW Institutional Review Board (IRB) and conducted in adherence to the tenets of the Declaration of Helsinki.
We obtained corneas from a 54-year-old white male organ donor with “muscular dystrophy” who had succumbed to a cardiac arrest from the eye bank at UT Transplant Services. Certified eye bank technicians had examined the corneas using Cellchek EB-10 specular microscopy (Konan Medical, Irvine, CA, USA) and detected FECD findings of confluent endothelial guttae and decreased endothelial cell density, and therefore found them to be unsuitable for transplantation. Additional control tissues were also obtained from the eye bank.
To test the hypothesis that patients with DM1 are at risk for FECD, we examined 13 consecutive unrelated patients with an established diagnosis of DM1 over the age of 40 (mean = 54.8, standard deviation [SD] = 10.3) from the UTSW Neuromuscular Cardiomyopathy Clinic (Table 1). Clinical genetic testing results for DM1 were obtained where available. All DM1 subjects were white. All subjects underwent an eye examination including slit-lamp microscopy by a cornea fellowship-trained ophthalmologist (VVM). Inclusion criterion for FECD was the presence of slit-lamp examination findings of grade 2 or higher on the modified Krachmer FECD grading scale: grade 0: no central guttae; grade 1: up to 12 scattered central guttae; grade 2: ≥12 scattered central guttae; grade 3: 1- to 2-mm confluent central guttae; grade 4: 2–5 mm of confluent central guttae; grade 5: >5-mm confluent central guttae without stromal edema; grade 6: >5-mm confluent central guttae with stromal edema.13 Specular microscopy of the corneal endothelium was performed by certified ophthalmic technicians using a Konan SL Specular Microscope (Konan Medical). The endothelial cell density and morphology parameters were calculated by the center method using the microscope's automated software. We obtained central corneal thickness (CCT) measurements using a Corneo-gage Plus ultrasonic pachymeter (Sonomed, New Hyde Park, NY, USA). The average of three separate measurements was used as the CCT.
Table 1.
Demographic Information and Microscopy Results of DM1 Patients
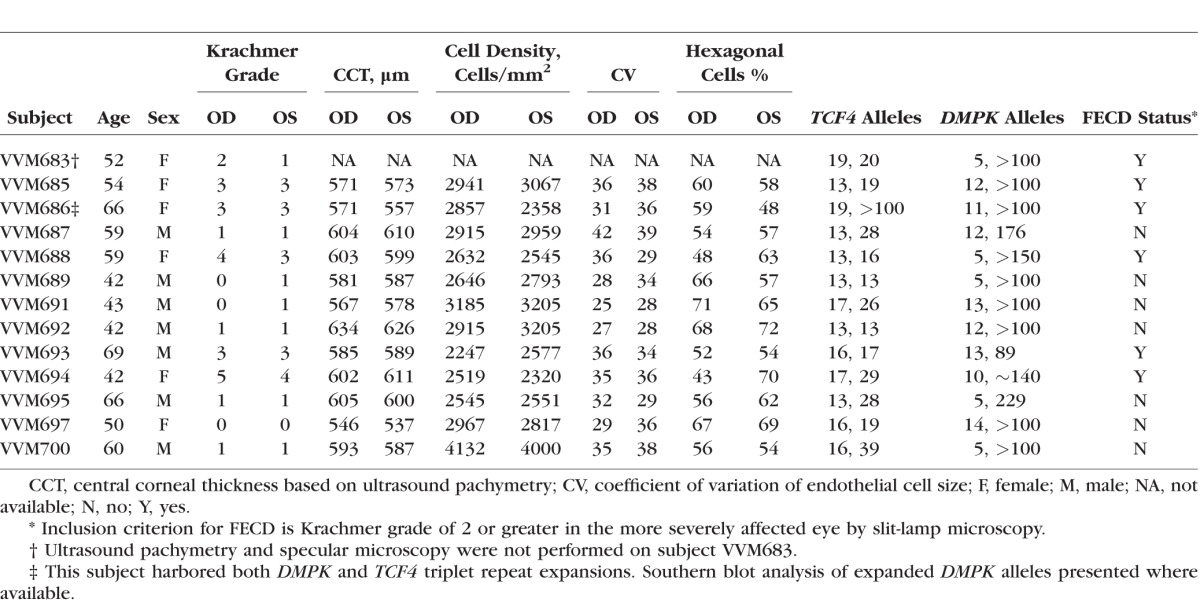
Then, we screened a cohort of 317 FECD probands recruited at a cornea referral practice at UTSW for the prevalence of the DMPK and TCF4 triplet repeat expansions. All subjects had undergone an eye examination including slit-lamp biomicroscopy by a cornea fellowship-trained ophthalmologist (VVM) and were found to have slit-lamp examination findings of grade 2 or higher on the modified Krachmer FECD grading scale. The triplet repeat expansions in DMPK and TCF4 were genotyped using a combination of short-tandem repeat (STR) and triplet repeat-primed polymerase chain reaction (TP-PCR) assays.
FISH
Corneal endothelial cells from an organ donor with FECD were examined for the presence for expanded CUG repeat RNA foci. FISH with chemically synthesized (CAG)6CA-5′ Texas red-labeled 2-O-methyl RNA 20-mers probe (8 μL at 20 ng/μL) (Integrated DNA Technologies, Coralville, IA, USA) and staining with 4′,6-diamidino-2-phenylindole, H-1500 DAPI (Vector Labs, Burlingame, CA, USA) of endothelial cells from this subject and controls were performed as we previously reported.35 Cells were imaged at ×60 magnification using a Widefield Deltavision pDV fluorescence microscope (GE Healthcare, Chicago, IL, USA). Images were processed using ImageJ software (https://imagej.nih.gov/ij/). Fifteen representative images were analyzed to derive percentage of cells with RNA foci. After performing the FISH assay, we stained the cells with anti-MBNL1 antibody as previously described.22
Genotyping of DMPK and TCF4 Triplet Repeat Polymorphisms
DNA from the organ donor corneal tissue was extracted with Trizol reagent (Life Technologies, Carlsbad, CA, USA) per the manufacturer's protocol. Genomic DNA was extracted from leukocytes of peripheral blood samples from each study subject using Autogen Flexigene (Qiagen, Valencia, CA, USA).
We genotyped the TCF4 CTG18.1 triplet repeat polymorphism using a combination of STR analysis and TP-PCR assay and examined the amplicons with the ABI 3730XL DNA analyzer (Applied Biosystems, Foster City, CA, USA) as previously reported.24 We genotyped the CTG triplet repeat locus at the 3′ UTR of DMPK with STR analysis and TP-PCR using locus-specific primers (Supplementary Table 1).
Results
Abundant discrete, punctate nuclear RNA foci were identified in 85% of the endothelial cells examined from the subject (16-1348) with FECD and muscular dystrophy (Fig. 1A). Nuclear RNA foci were detected in 61% of the endothelial cells from the subject (16-3407) with FECD and TCF4 triplet repeat expansion included as a positive control (Fig. 1A). Additionally, we demonstrated colocalization of the splicing factor MBNL1 with the nuclear RNA foci (Fig. 1B) in the subject (16-1348) with FECD and muscular dystrophy. Genotyping results indicated that the subject (16-1348) did not have a TCF4 triplet expansion but rather had homozygous alleles with 12 CTG repeats at the CTG18.1 locus (Supplementary Fig. S1). Then, we hypothesized that the subject (16-1348) harbored a DMPK triplet repeat expansion. STR analysis detected one allele at the DMPK locus with 10 CTG repeats, and the TP-PCR assay detected an expansion at the second allele (Supplementary Fig. S1). A certified eye bank technician contacted the family of the organ donor for additional past medical history and learned that a clinical diagnosis of DM1 for the subject (16-1348) had been made 12 years prior at the age of 42. The subject's medical diagnosis had been confirmed with clinical genetic testing which showed evidence of a trinucleotide repeat expansion in the myotonic dystrophy alleles using Southern blot analysis. The CTG repeat numbers of the two DMPK alleles were 10 and approximately 300, respectively.
Figure 1.

Nuclear RNA foci accumulate and colocalize with MBNL1 in corneal endothelial cells with triplet repeat expansion in DMPK gene. (A) FISH with a (CAG)6CA-5′ Texas red-labeled 2-O-methyl RNA 20-mers probe (Integrated DNA Technologies) on endothelial cells of FECD/DM1 subject (16-1348) with an expanded DMPK allele with 300 CTG repeats revealed punctate, nuclear RNA foci (red). RNA foci were present in endothelial cells from a FECD subject (16-3407) with an expanded TCF4 allele with 71 CTG repeats and absent in cells from unaffected subject (16-0729) without the DMPK and TCF4 repeat expansions. DNA was stained with DAPI (blue). The scale bar represents 25 μm. (B) Colocalization of MBNL1 with the expanded CUG repeat nuclear foci. After hybridization with RNA probe (red), cells were stained with anti-MBNL1 antibody (green) and DAPI (blue). The scale bar represents 10 μm.
Then, we examined 13 consecutive unrelated patients with an established diagnosis of DM1 for findings of FECD. Our genotyping results confirmed that all DM1 patients had DMPK triplet repeat expansions; one DM1 subject harbored both DMPK and TCF4 triplet repeat expansions (Table 1). We observed that 6 out of 13 (46%) of the DM1 subjects had slit-lamp examination findings of grade 2 or higher on the modified Krachmer FECD grading scale, which is significantly higher than the 4% prevalence of FECD in the US population over the age 40 (P value = 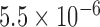 ). The female DM1 subjects were more likely to be affected with FECD than their male counterparts (P value =
). The female DM1 subjects were more likely to be affected with FECD than their male counterparts (P value = 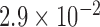 ; Table 2), compatible with the known female bias for FECD.38,39 There was no significant difference between the two groups in terms of age and CCT (Table 2). Specular microscopy confirmed the presence of guttae in all FECD subjects diagnosed by slit-lamp examination (Fig. 2). Based on specular microscopy, the eyes of FECD subjects had a lower endothelial cell density (P value =
; Table 2), compatible with the known female bias for FECD.38,39 There was no significant difference between the two groups in terms of age and CCT (Table 2). Specular microscopy confirmed the presence of guttae in all FECD subjects diagnosed by slit-lamp examination (Fig. 2). Based on specular microscopy, the eyes of FECD subjects had a lower endothelial cell density (P value = 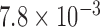 ) and lower percentage of hexagonal cells (P value =
) and lower percentage of hexagonal cells (P value = 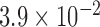 ) compared to the eyes of non-FECD subjects, which is compatible with the increased cellular senescence seen in FECD.
) compared to the eyes of non-FECD subjects, which is compatible with the increased cellular senescence seen in FECD.
Table 2.
A Comparison* of FECD and Non-FECD Subjects Among DM1 Patients†
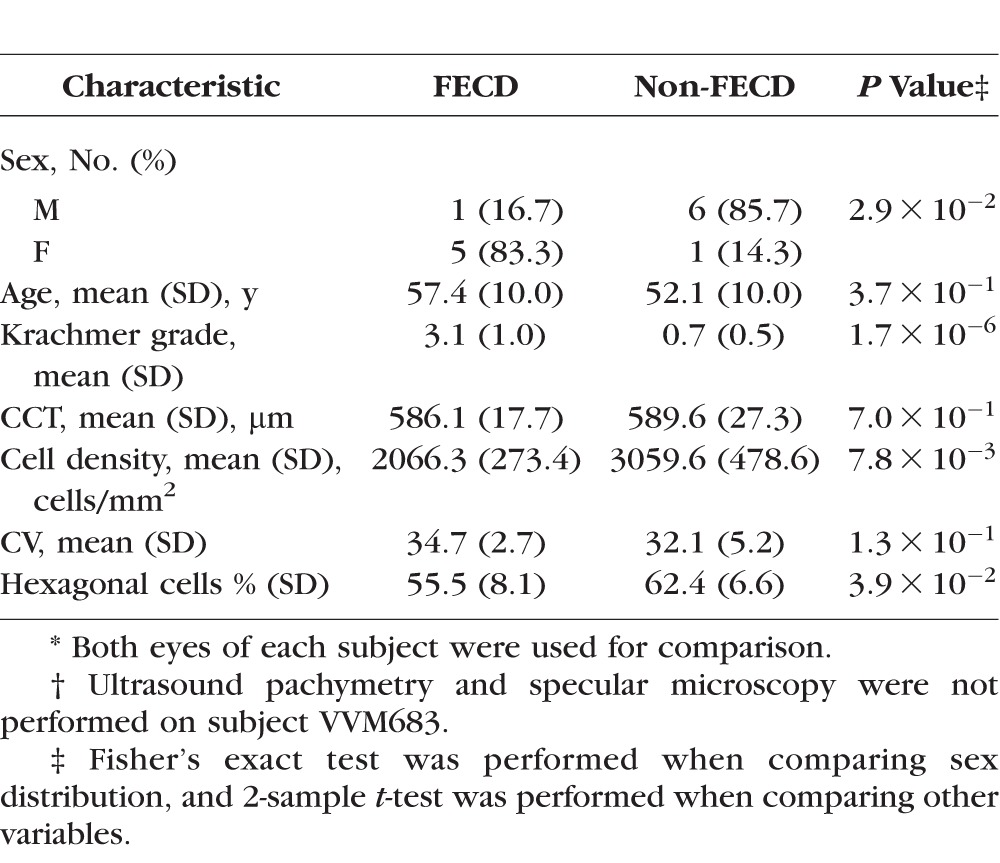
Figure 2.
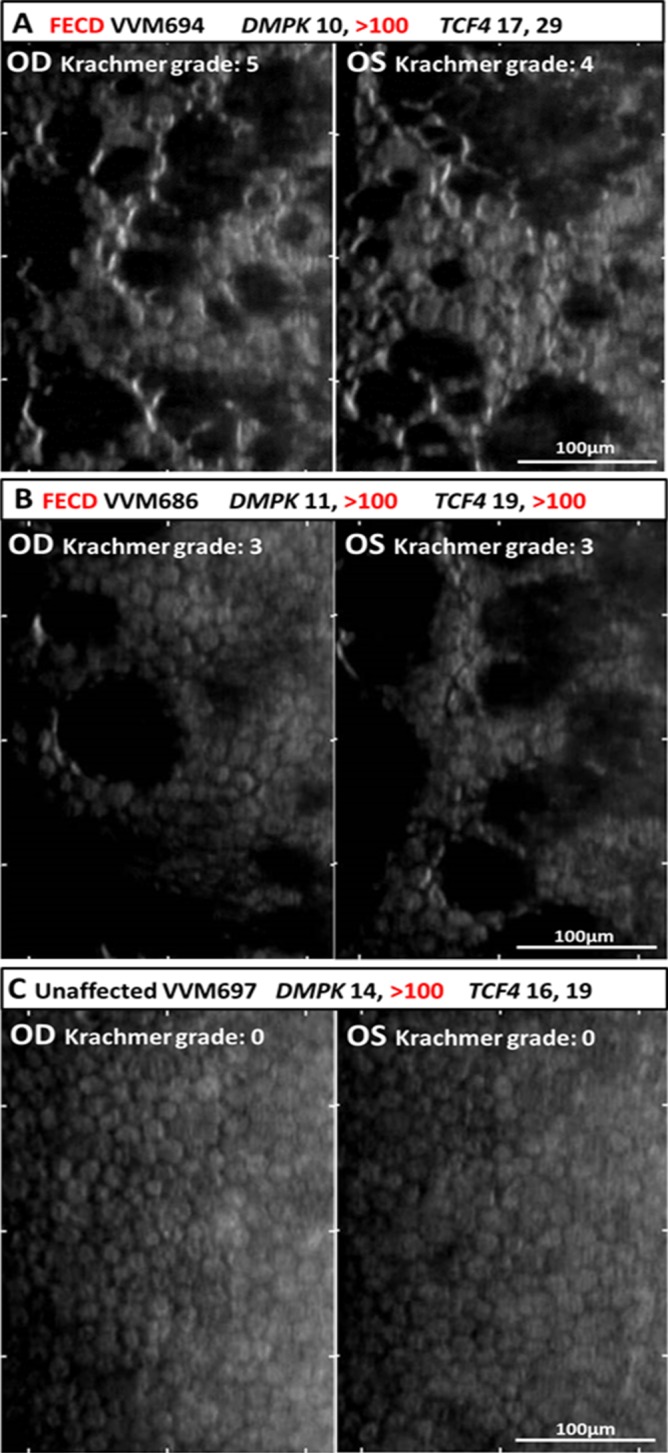
Specular microscopy of corneal endothelium in myotonic dystrophy type 1 patients. (A) Specular microscopy of the central endothelium of DM1 subject with FECD Krachmer grade of 5 OD (oculus dexter, right eye) and 4 OS (oculus sinister, left eye) revealed typical FECD findings including numerous focal dark spots corresponding to corneal guttae, increased cellular polymorphism (loss of normal hexagonal pattern), and polymegathism (variation in cell size). Photomicrograph images represent a surface area of 400 by 220 μm. Bar represents 100 μm. (B) Specular imaging of endothelium of DM1 subject harboring both DMPK1 and TCF4 triplet repeat expansions with FECD Krachmer grade of 3 in each eye. Numerous focal dark spots corresponding to corneal guttae are shown in OD. Large dark areas corresponding to confluence of the corneal guttae and marked loss of endothelial cell density and grotesque morphology of remaining cells are shown in OS. (C) Specular images of DM1 subject without FECD showing absence of guttae and normal endothelial cell density and morphology in both eyes.
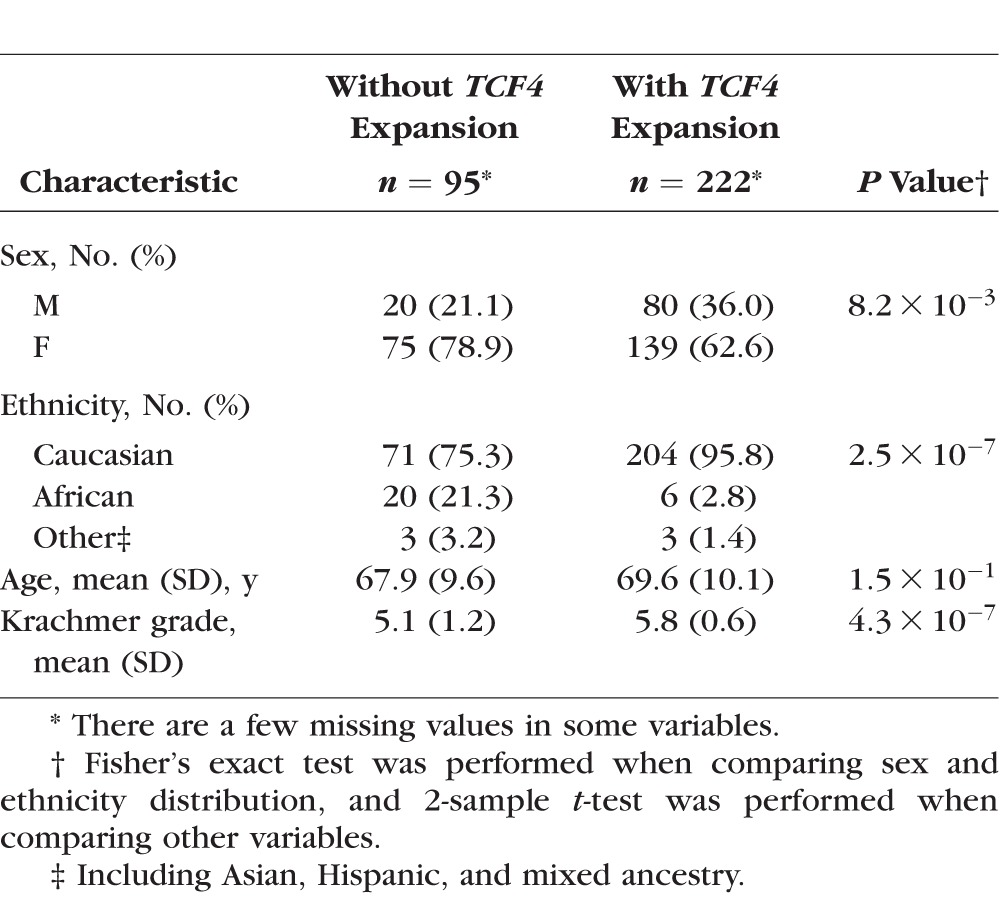
Next, we examined prevalence of the TCF4 and DMPK triplet repeat polymorphisms in the UTSW FECD cohort and found that 222 of 317 (70%) probands harbored TCF4 expansions. As we had previously reported, the subjects with the TCF4 triplet repeat expansion had a greater clinical severity of disease in comparison to their counterparts without the expansion (Table 3).40 Out of 95 FECD subjects who did not harbor an expansion in TCF4, only 1 subject was identified with a DMPK triplet repeat expansion with alleles of 15 and 71 CTG repeats (Supplementary Fig. S2). She had undergone cataract surgery and corneal transplantation in both eyes for Krachmer grade 6 severity of FECD. Review of her past medical history revealed no prior clinical diagnosis of myotonic dystrophy.
Table 3.
Demographic Information of FECD Cohort
Discussion
Ocular findings frequently associated with DM1 include ptosis, cataracts, reticular macular dystrophy, and peripheral pigmentary retinopathy.41 Our results indicate that FECD may also be a common ocular finding, with 46% of our DM1 patients affected by FECD. A previous clinical study of DM1 subjects with a mean age of 38 (SD = 13.3) years found no abnormalities in corneal endothelial cell density or morphology using specular morphology.42 We intentionally screened DM1 subjects over the age of 40 years because FECD is a disease of middle age. Additional studies on larger DM1 cohorts are warranted to validate our findings on the penetrance of the FECD trait with expansions in the DMPK triplet repeat polymorphism and to determine any sex bias. Further studies are also warranted to assess FECD clinical severity and any positive correlation to CTG repeat number as previously reported with TCF4 triplet repeat expansions.40 Nearly all DM1 subjects develop a cataract.41 Patients with comorbid FECD should be counseled that they are at increased risk of corneal edema that may require corneal transplantation at the time of or after their cataract surgery.
We found a subject in our UTSW FECD cohort with a DMPK expansion without a prior clinical diagnosis of DM1. Individuals with small DMPK expansions have a mild DM1 phenotype. They may be asymptomatic except for cataracts and lead active lives with normal life spans.43
Our observations confirm that TCF4 triplet repeat expansions are the predominant cause of FECD. DMPK1 triplet repeat expansions, however, can also contribute to the overall genetic burden of this disease and provide a different molecular and clinical perspective on the pathogenesis of FECD. Several genetic diseases are caused by CTG expansions, and the link between molecular mechanism and disease is best characterized for DM1. As a result, DM1 offers insights that may prove valuable for FECD, where we are in the early stages of understanding mechanism and therapeutic development.
DM1 and FECD, however, are not identical diseases even though they both originate from noncoding CTG expansions. The DMPK expansion in DM1 results in a multiorgan disease that involves various tissues in the eye including lens, retina, and corneal endothelium. In contrast, the TCF4 repeat expansion appears to affect the corneal endothelium without any clinically apparent sequela to other ocular tissues or bodily organs. We speculate that differences in triplet repeat length and/or tissue specific factors define the phenotypic spectrum of these two triplet repeat expansions.
We report here that mutant expansions in DMPK and TCF4 share important similarities, including (1) nuclear foci that contain expanded CUG repeats, (2) association of foci with MBNL1 protein, and (3) an ability to cause FECD. These results suggest that the triplet expansions in both DMPK and TCF4 may cause the same corneal endothelial tissue phenotype of FECD through shared molecular mechanisms that are triggered by toxic gain-of-function RNA. These findings provide a new window on the molecular pathogenesis of FECD and suggest that the DM1 paradigm can be used to facilitate therapeutic development.
Supplementary Material
Acknowledgments
The authors thank the patients for their participation in this study and also thank Aimee Tilley and Bryan Gallerson for study coordination efforts. We thank eye bank technician Christina Megedyuk and eye bank director Donna Drurry at Transplant Services at the University of Texas Southwestern for their efforts. We thank the collaborating corneal specialists Wayne R. Bowman, James P. McCulley, Steven Verity, Sandhya Iyer, Brad Bowman, and Walter Beebe. We thank David R. Corey, PhD, for helpful discussions on the project and manuscript.
Supported by National Institutes of Health Grants R01EY022161 (VVM) and P30EY020799, and an unrestricted grant from Research to Prevent Blindness, New York, New York, USA. Support also came from National Institutes of Health Grants U54AR068791 (PPM) and UL1TR001105 (CX).
Disclosure: V.V. Mootha, None; B. Hansen, None; Z. Rong, None; P.P. Mammen, None; Z. Zhou, None; C. Xing, None; X. Gong, None
References
- 1. Lorenzetti DW, Uotila MH, Parikh N, Kaufman HE. . Central cornea guttata. Incidence in the general population. Am J Ophthalmol. 1967; 64: 1155– 1158. [PubMed] [Google Scholar]
- 2. 2015 Eye Banking Statisical Report. Washington, DC: Eye Bank Association of America; 2016. [Google Scholar]
- 3. Chi HH, Teng CC, Katzin HM. . Histopathology of primary endothelial-epithelial dystrophy of the cornea. Am J Ophthalmol. 1958; 45: 518– 535. [DOI] [PubMed] [Google Scholar]
- 4. Laing RA, Leibowitz HM, Oak SS, Chang R, Berrospi AR, Theodore J. . Endothelial mosaic in Fuchs' dystrophy. A qualitative evaluation with the specular microscope. Arch Ophthalmol. 1981; 99: 80– 83. [DOI] [PubMed] [Google Scholar]
- 5. Bigar F. . Specular microscopy of the corneal endothelium. Optical solutions and clinical results. Dev Ophthalmol. 1982; 6: 1– 94. [PubMed] [Google Scholar]
- 6. Borderie VM, Baudrimont M, Vallee A, Ereau TL, Gray F, Laroche L. . Corneal endothelial cell apoptosis in patients with Fuchs' dystrophy. Invest Ophthalmol Vis Sci. 2000; 41: 2501– 2505. [PubMed] [Google Scholar]
- 7. Li QJ, Ashraf MF, Shen DF,et al. . The role of apoptosis in the pathogenesis of Fuchs endothelial dystrophy of the cornea. Arch Ophthalmol. 2001; 119: 1597– 1604. [DOI] [PubMed] [Google Scholar]
- 8. Matthaei M, Zhu AY, Kallay L, Eberhart CG, Cursiefen C, Jun AS. . Transcript profile of cellular senescence-related genes in Fuchs endothelial corneal dystrophy. Exp Eye Res. 2014; 129: 13– 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jurkunas UV, Bitar MS, Funaki T, Azizi B. . Evidence of oxidative stress in the pathogenesis of fuchs endothelial corneal dystrophy. Am J Pathol. 2010; 177: 2278– 2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hogan MJ, Wood I, Fine M. . Fuchs' endothelial dystrophy of the cornea. 29th Sanford Gifford Memorial lecture. Am J Ophthalmol. 1974; 78: 363– 383. [DOI] [PubMed] [Google Scholar]
- 11. Magovern M, Beauchamp GR, McTigue JW, Fine BS, Baumiller RC. . Inheritance of Fuchs' combined dystrophy. Ophthalmology. 1979; 86: 1897– 1923. [DOI] [PubMed] [Google Scholar]
- 12. Biswas S, Munier FL, Yardley J,et al. . Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet. 2001; 10: 2415– 2423. [DOI] [PubMed] [Google Scholar]
- 13. Zhang C, Bell WR, Sundin OH,et al. . Immunohistochemistry and electron microscopy of early-onset fuchs corneal dystrophy in three cases with the same L450W COL8A2 mutation. Trans Am Ophthalmol Soc. 2006; 104: 85– 97. [PMC free article] [PubMed] [Google Scholar]
- 14. Vithana EN, Morgan P, Sundaresan P,et al. . Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2). Nat Genet. 2006; 38: 755– 757. [DOI] [PubMed] [Google Scholar]
- 15. Vithana EN, Morgan PE, Ramprasad V,et al. . SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Hum Mol Genet. 2008; 17: 656– 666. [DOI] [PubMed] [Google Scholar]
- 16. Riazuddin SA, Zaghloul NA, Al-Saif A,et al. . Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am J Hum Genet. 2010; 86: 45– 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Krafchak CM, Pawar H, Moroi SE,et al. . Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am J Hum Genet. 2005; 77: 694– 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mehta JS, Vithana EN, Tan DT,et al. . Analysis of the posterior polymorphous corneal dystrophy 3 gene, TCF8, in late-onset Fuchs endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2008; 49: 184– 188. [DOI] [PubMed] [Google Scholar]
- 19. Riazuddin SA, Parker DS, McGlumphy EJ,et al. . Mutations in LOXHD1, a recessive-deafness locus, cause dominant late-onset Fuchs corneal dystrophy. Am J Hum Genet. 2012; 90: 533– 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Riazuddin SA, Vasanth S, Katsanis N, Gottsch JD. . Mutations in AGBL1 cause dominant late-onset Fuchs corneal dystrophy and alter protein-protein interaction with TCF4. Am J Hum Genet. 2013; 93: 758– 764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baratz KH, Tosakulwong N, Ryu E,et al. . E2-2 protein and Fuchs's corneal dystrophy. N Engl J Med. 2010; 363: 1016– 1024. [DOI] [PubMed] [Google Scholar]
- 22. Afshari NA, Igo RP Jr, Morris NJ,et al. . Genome-wide association study identifies three novel loci in Fuchs endothelial corneal dystrophy. Nat Commun. 2017; 8: 14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wieben ED, Aleff RA, Tosakulwong N,et al. . A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS One. 2012; 7: e49083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mootha VV, Gong X, Ku HC, Xing C. . Association and familial segregation of CTG18.1 trinucleotide repeat expansion of TCF4 gene in Fuchs' endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2014; 55: 33– 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murre C, Bain G, van Dijk MA,et al. . Structure and function of helix-loop-helix proteins. Biochim Biophys Acta. 1994; 1218: 129– 135. [DOI] [PubMed] [Google Scholar]
- 26. Ephrussi A, Church GM, Tonegawa S, Gilbert W. . B lineage--specific interactions of an immunoglobulin enhancer with cellular factors in vivo. Science. 1985; 227: 134– 140. [DOI] [PubMed] [Google Scholar]
- 27. Breschel TS, McInnis MG, Margolis RL,et al. . A novel, heritable, expanding CTG repeat in an intron of the SEF2-1 gene on chromosome 18q21.1. Hum Mol Genet. 1997; 6: 1855– 1863. [DOI] [PubMed] [Google Scholar]
- 28. Xing C, Gong X, Hussain I,et al. . Transethnic replication of association of CTG18.1 repeat expansion of TCF4 gene with Fuchs' corneal dystrophy in Chinese implies common causal variant. Invest Ophthalmol Vis Sci. 2014; 55: 7073– 7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nanda GG, Padhy B, Samal S, Das S, Alone DP. . Genetic association of TCF4 intronic polymorphisms, CTG18.1 and rs17089887 with Fuchs' endothelial corneal dystrophy in Indian population. Invest Ophthalmol Vis Sci. 2014; 55: 7674– 7680. [DOI] [PubMed] [Google Scholar]
- 30. Nakano M, Okumura N, Nakagawa H,et al. . Trinucleotide repeat expansion in the TCF4 gene in Fuchs' endothelial corneal dystrophy in Japanese. Invest Ophthalmol Vis Sci. 2015; 56: 4865– 4869. [DOI] [PubMed] [Google Scholar]
- 31. Brook JD, McCurrach ME, Harley HG,et al. . Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992; 68: 799– 808. [DOI] [PubMed] [Google Scholar]
- 32. Mahadevan M, Tsilfidis C, Sabourin L,et al. . Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992; 255: 1253– 1255. [DOI] [PubMed] [Google Scholar]
- 33. Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE. . Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci U S A. 1997; 94: 7388– 7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. . Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. 2004; 13: 3079– 3088. [DOI] [PubMed] [Google Scholar]
- 35. Mootha VV, Hussain I, Cunnusamy K,et al. . TCF4 triplet repeat expansion and nuclear RNA foci in Fuchs' endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2015; 56: 2003– 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Du J, Aleff RA, Soragni E,et al. . RNA toxicity and missplicing in the common eye disease Fuchs endothelial corneal dystrophy. J Biol Chem. 2015; 290: 5979– 5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gattey D, Zhu AY, Stagner A, Terry MA, Jun AS. . Fuchs endothelial corneal dystrophy in patients with myotonic dystrophy: a case series. Cornea. 2014; 33: 96– 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Musch DC, Niziol LM, Stein JD, Kamyar RM, Sugar A. . Prevalence of corneal dystrophies in the United States: estimates from claims data. Invest Ophthalmol Vis Sci. 2011; 52: 6959– 6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Afshari NA, Pittard AB, Siddiqui A, Klintworth GK. . Clinical study of Fuchs corneal endothelial dystrophy leading to penetrating keratoplasty: a 30-year experience. Arch Ophthalmol. 2006; 124: 777– 780. [DOI] [PubMed] [Google Scholar]
- 40. Soliman AZ, Xing C, Radwan SH, Gong X, Mootha VV. . Correlation of severity of Fuchs endothelial corneal dystrophy with triplet repeat expansion in TCF4. JAMA Ophthalmol. 2015; 1– 6. [DOI] [PubMed]
- 41. Vos TA. . 25 years dystrophia myotonica (D.M.). Ophthalmologica. 1961; 141: 37– 45. [DOI] [PubMed] [Google Scholar]
- 42. Rosa N, Lanza M, Borrelli M,et al. . Corneal thickness and endothelial cell characteristics in patients with myotonic dystrophy. Ophthalmology. 2010; 117: 223– 225. [DOI] [PubMed] [Google Scholar]
- 43. Arsenault ME, Prevost C, Lescault A, Laberge C, Puymirat J, Mathieu J. . Clinical characteristics of myotonic dystrophy type 1 patients with small CTG expansions. Neurology. 2006; 66: 1248– 1250. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.