

An ultrathin graphene artificial interphase stabilizes active material and conductive carbon in aqueous energy storage systems.
Abstract
Aqueous lithium energy storage systems address environmental sustainability and safety issues. However, significant capacity fading after repeated cycles of charge-discharge and during float charge limit their practical application compared to their nonaqueous counterparts. We introduce an artificial solid electrolyte interphase (SEI) to the aqueous systems and report the use of graphene films as an artificial SEI (G-SEI) that substantially enhance the overall performance of an aqueous lithium battery and a supercapacitor. The thickness (1 to 50 nm) and the surface area (1 cm2 to 1 m2) of the G-SEI are precisely controlled on the LiMn2O4-based cathode using the Langmuir trough–based techniques. The aqueous battery with a 10-nm-thick G-SEI exhibits a discharge capacity as high as 104 mA·hour g−1 after 600 cycles and a float charge current density as low as 1.03 mA g−1 after 1 day, 26% higher (74 mA·hour g−1) and 54% lower (1.88 mA g−1) than the battery without the G-SEI, respectively. We propose that the G-SEI on the cathode surface simultaneously suppress the structural distortion of the LiMn2O4 (the Jahn-Teller distortion) and the oxidation of conductive carbon through controlled diffusion of Li+ and restricted permeation of gases (O2 and COx), respectively. The G-SEI on both small (~1 cm2 in 1.15 mA·hour cell) and large (~9 cm2 in 7 mA·hour cell) cathodes exhibit similar property enhancement, demonstrating excellent potential for scale-up and manufacturing.
INTRODUCTION
The global demand for safe and environmentally sustainable electrochemical energy storage has vastly increased in the recent years. Aqueous lithium-ion energy storage systems (ALESS), such as aqueous Li-ion batteries and supercapacitors, are designed to address safety and sustainability concerns (1, 2). However, significant capacity fading after repeated cycles of charge-discharge or during float charge has been one of the main problems that have made the ALESS less competitive than their nonaqueous counterparts (3, 4). Although the capacity of lithium manganese oxide (LiMn2O4) (LMO) is related to the bulk material properties, a complex surface chemistry is also important in determining the electrochemical performance of the electrode material (5). More specifically, structural instability of Li transition metal oxides (for example, LiNiO2, LiMn2O4, and LiMnPO4) and electrochemical oxidation of conductive carbon in the cathode are two main causes for the capacity fading of the ALESS (6–8).
The diffusion rate of Li+ inside the aqueous electrolytes is much higher than that of the bulk of cathode materials during the discharge process (6). As a result, the Li+ accumulate on the surface of the Li transition metal oxides, and a partial crystalline structural transformation of these particles may occur, which ultimately leads to the so-called Jahn-Teller distortion (9). This structural distortion undoubtedly affects the three-dimensional (3D) Li+ diffusion pathways, which severely reduces the capacity retention after repeated cycles of charge-discharge (10, 11). In addition, the electrochemical oxidation followed by the consumption of the carbon-based conductive particles is responsible for the irreversible capacity loss of the ALESS. This process is induced by contacting water at low potentials and/or by oxygen evolved from the water oxidation at high potentials (12, 13). The final products of these reactions are oxidized carbon and O2, CO, and CO2 gases that not only affect the physical contact between the electrolyte and the cathode but also decrease the electrical conductivity (electron mobility) across the cathode/electrolyte interface (14). All these factors may block Li+ diffusions (15), which limits the capacity of LMO under high C rates. There have been several approaches previously proposed to solve these issues in the cathodes. They are mainly based on doping the individual Li metal oxide particles with foreign atoms (Ni, Co, etc.), modification of the structure of conductive carbon, and the addition of organic/inorganic compounds into the cathode and/or electrolyte formulations (16–22). In particular, Langmuir-Blodgett (L-B) processes have recently been adapted to fabricate multilayered structures of nanomaterials on the separator of the lithium-sulfur systems for regulating mass and charge transport (23, 24). However, these approaches are often not precisely controlled at the nanoscale level and involve complicated chemistry and high-temperature processes (>800°C) with added polarization effects and limited scale-up potentials. Incorporation of a well-defined and stable interphase into the spacing between the cathode and the aqueous electrolyte without affecting the bulk of the cathode could be a more efficient way to simultaneously maintain the stability of the active materials and the conductive carbon (25).
Solid electrolyte interphase (SEI) in the nonaqueous Li storage systems forms in situ from the reactions between the electrode surface and the organic compounds in the electrolytes and can significantly alleviate irreversible side reactions (26). In general, the key features of an ideal SEI are as follows: (i) compatibility with the electrode surface, (ii) ionically conductive, (iii) impermeable to electrolyte molecules/gases, (iv) mechanically and chemically stable, and (v) thin and compact (27–29). These features could contribute to the suppression of structural distortion of the Li transition metal oxides and the electrochemical oxidation of the conductive carbon particles in the cathodes of the ALESS. However, the SEI cannot be formed spontaneously in the ALESS with aqueous electrolytes due to multiple reasons. Unlike the SEI in the nonaqueous systems, there are usually no polymerizable components in the aqueous electrolytes to integrate the possible SEI on the electrode surface. The inorganic hydroxides and oxides at the electrode/electrolyte interface would also not have sufficient ionic conductivity and act more like a passivation layer. To the authors’ knowledge, there has been no report on the formation of the in situ and/or ex situ SEI in the ALESS. Graphene films, made of 2D sp2 carbon nanosheets, would have the unique features to function as an ideal SEI on the cathode surface in the ALESS. Some of these features include excellent electrical conductivity and electrochemical stability with controlled permeation of Li+ and restricted permeation of the large cations (>4.5 Å) and gas molecules (22, 23).
Here, single- to multilayer graphene films as an artificial SEI (G-SEI) are fabricated and transferred onto the surface of the LMO-based cathode of a rechargeable hybrid aqueous lithium battery (ReHAB) (30–33) and a supercapacitor. In our scalable Langmuir trough–based techniques, under ambient conditions, the thickness (1 to 50 nm) and the surface area (1 cm2 to 1 m2) of the G-SEI can be fully controlled to obtain the most optimal electrochemical performance. The G-SEI with a thickness of 10 nm can contribute a reversible capacity of 35 mA·hour g−1, and the ReHAB maintains more than 87 and 88% of the initial capacity after 600 cycles of charge-discharge and during 1-day float charge, respectively. The ReHAB with the G-SEI on the cathode also shows only 1.1 mV hour−1 open-circuit voltage (OCV) decay rate after 72 hours of self-discharge compared to 3.2 mV hour−1 for the cathode without the G-SEI. The ReHAB also offers an excellent rate capability that is superior to most of the reported aqueous Li-ion batteries. We hypothesize that the G-SEI could simultaneously suppress the Jahn-Teller distortion and water-induced electrochemical oxidation of conductive carbon on the cathode/electrolyte interface through a controlled diffusion of Li+ and restricted permeation of gases (O2 and COx), respectively. Extensive microscopy and ex situ and in situ spectroscopy characterizations are used to understand the key functions of the G-SEI.
RESULTS
Fabrication and transfer of the G-SEI onto the cathode
The G-SEI in this study were fabricated from graphene oxide (GO) followed by in situ reduction on the cathode surface. GO was produced via an improved synthesis method (27, 34) with slight modifications in the procedure to obtain nanosheets with lower defects and larger lateral sizes for better stability and coverage on the cathode surface (Materials and Methods). Figure S1 shows the basic structural evaluations of the GO using spectroscopy and microscopy techniques. There is a very sharp (001) x-ray diffraction (XRD) peak at 9.1° corresponding to a d spacing of 9.7 Å in the GO that indicates a successful exfoliation of a graphitic (002) structure at 26.2° with a d spacing of 3.4 Å (fig. S1A). However, reduced GO (RGO) by hydrazine monohydrate shows a broader diffraction peak at 22.5° most likely due to the restacking of the exfoliated layers after reduction. Raman spectra in fig. S1B show a slight blueshifting of the G band (1580 to 1595 cm−1) and a significant contribution of the D band (ID/IG = 0.92) in GO compared to the graphite precursor (ID/IG = 0.34), mainly due to the incorporation of oxygen functionalities into the graphene network. In agreement with previous reports (35, 36), a higher contribution of the D band (ID/IG = 0.99) was detected in the RGO (fig. S1B) due to an increase in the number of aromatic domains responsible for the D band, but not necessarily their overall size, which is responsible for the G band. The high-resolution transmission electron microscopy (HRTEM) of a single GO layer with some wrinkles is shown in fig. S1C.
The GO films, made of millions of embedded GO nanosheets, were fabricated and transferred onto the cathode surface via the Langmuir trough–based techniques: L-B isotherm and dipping methods and the Langmuir-Schaefer (L-S) method (Materials and Methods). There are many variables that determine the quality of the GO films on the cathode before the in situ reduction. The key variables that were investigated in this study are surface pressure (mN m−1), coating angle, coating method, concentration of GO, and the films’ thickness. Figure 1A shows a typical surface pressure (mN m−1) change versus GO film area (cm2) upon compression in the L-B isotherm technique (Fig. 1A, inset). Considering GO as an amphiphilic nanoparticle (hydrophobic in the basal plane and hydrophilic on the edges) (37), three different regions can be distinguished in the surface pressure curve. The GO nanosheets are initially by far isolated similar to the gas molecules (phase I), and then they start meeting each other upon compression to form a compact film (phase II). Further compression of the film causes a collapse in the 2D GO film and its conversion into a 3D structure, where no significant changes in the surface pressure were observed (phase III). The atomic force microscopy (AFM) of a single-layer GO film (Fig. 1B) on highly ordered pyrolytic graphite (HOPG) substrate shows wrinkles inside the nanosheets across the film with an average lateral size of 10 μm. These wrinkles become more and more highlighted by increasing the number of GO layers to 5 and 10 layers, as shown in Fig. 1 (C and D, respectively). The selected-area electron diffraction (SAED) of hexagonal graphene structure is shown in Fig. 1A as inset. The AFM observations of the GO films on the cathodes confirmed that the best film coverage was obtained at surface pressures of 12 to 13 mN m−1, very close to the end of phase II (Fig. 1A), and right before the GO film collapses entering phase III. To find out the optimal electrochemical performance, GO films with four different thicknesses were fabricated and deposited onto the cathode surface: 1-layer (~1 nm), 5-layer (~5 nm), 10-layer (~10 nm), and 50-layer (~50 nm). The AFM and scanning electron microscopy (SEM) images from the surfaces of the Blank cathode with no GO film and 5-layer and 10-layer GO films are shown in Fig. 1 (E to G and H to J, respectively). Note that the 50-layer GO film is very similar to the 10-layer film. Obviously, by increasing the thickness of GO films (the number of layers), the cathode features (for example, LMO crystals and graphite particles in Fig. 1, E and H) underneath the films become less evident, and the wrinkles from the embedded GO nanosheets become more apparent (Fig. 1, F, G, I, and J). We noted that the AFM and SEM imaging techniques were not useful to detect the 1-layer GO film on the cathodes due to the roughness of the surfaces. One major advantage of the Langmuir trough–based techniques is that it can be applied onto any water-compatible electrode surface, where only a small amount of GO needs to be used. For example, only 2.5 mg of 1-nm-thick GO is required to cover a cathode with 1-m2 surface area. An in situ chemical reduction method using hydrazine monohydrate was used to convert the GO films sitting on the cathodes to the RGO films (Materials and Methods). Note that no obvious structural changes of the cathode materials (LMO and graphite) were observed after treating the Blank cathode with hydrazine (fig. S2). Contact angle measurements of the hydrazine aqueous solution on the 10GO cathode were conducted and compared with the Blank cathode (fig. S3). It is evident that the 10GO-coated cathode has a smaller contact angle (95°), mainly due to the oxygen functionalities, than the Blank cathode (145°). The adhesion force between the final RGO film (G-SEI) and the cathode surface is mainly through van der Waals (π-π) interactions. These RGO films preserved their morphological features after the reduction process and will function as the G-SEI onto the cathode surface (fig. S4). According to a previous review (38), the chemical reduction by hydrazine solution may only partially reduce the GO nanomaterials in comparison with the high-temperature annealing (>1000°C) at inert atmospheres (Ar and N2). Although the reported conductivities for the high-temperature treatment are one to two orders of magnitude higher than the hydrazine treatment, the reduction method cannot be used in our study due to degradation of the polymeric binder [for example, polyvinylidene difluoride (PVDF)] and the LMO particles at high temperatures. Other in situ reduction (chemical and nonchemical) processes can be further explored as a future work for this research. In addition, the in-plane (x-y direction) electrical conductivity of the G-SEI is reported to be much higher than the through-plane (z direction) conductivity (39). Despite the fact that the GO film on the cathode can be well wet by the hydrazine solution (fig. S3, contact angle of 95°), the upmost G-SEI surface and the edges and some voids between the cathode and the G-SEI might have been mainly reduced. To improve the homogeneity of the film at high thicknesses (Fig. 1J), the deposition process can be divided into multiple steps, that is, layer-by-layer deposition, instead of one-time deposition (the current method in this work). For example, deposition of the 10RGO film can be divided into 10 individual steps of 1RGO deposition, which would have limited scale-up potentials. Figure S4 shows a more homogeneous film after 10 times layer-by-layer deposition of 1RGO than the one-step deposition of 10RGO (Fig. 1J).
Fig. 1. Characterizations of G-SEI.
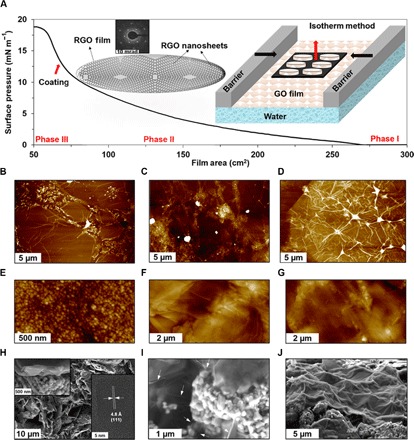
(A) Surface pressure versus trough area in the L-B method showing three different phases. Inset: SAED and schematics of an isotherm method and packed nanosheets in a GO film. (B to D) AFM images of 1-layer, 5-layer, and 10-layer GO films, respectively, on an HOPG substrate. AFM (E to G) and SEM (H to J) images of the Blank cathode and 5-layer and 10-layer GO films on the cathodes, respectively. Inset on the right side of (H) is the HRTEM of one LMO crystal. Arrows in (I) show the wrinkles of a GO film.
Battery, supercapacitor, and electrochemical testing
The cathodes with the RGO films were assembled in the ReHAB system and tested (see Materials and Methods). Cyclic voltammetry (CV) profiles of the batteries without the G-SEI and with 1-layer, 5-layer, 10-layer, and 50-layer G-SEI cathodes (denoted as Blank, 1RGO, 5RGO, 10RGO, and 50RGO, respectively) at a scan rate of 1 mV s−1 are shown in Fig. 2A. The mass loading of LMO was fixed at 7.8 ± 0.61 mg cm−2. There are two redox couples located at 1.82/1.78 V and 1.96/1.92 V versus Zn2+/Zn, corresponding to the two stages of extraction and insertion of Li+ from/into the host spinel structure of LMO in the aqueous electrolyte, respectively. These redox couples are LiMn2O4↔Li0.5Mn2O4 + 0.5Li+ + 0.5e− and Li0.5Mn2O4↔Mn2O4 + 0.5Li+ + 0.5e−, respectively (30). The symmetrical peaks in 1.76 to 1.90 V reveal that the Li+ extraction/insertion processes in all the cathodes are highly reversible. The G-SEI did not have any effect on the peak shape and the redox potentials, indicating that the charge-discharge process of the coated cathode has not been affected, which is analogous to the in situ formed SEI in organic electrolytes (15). The rate performances are shown in Fig. 2B, where 10RGO shows the best rate capability. To find out the effect of the chemical reduction, the rate capability results for the 10GO cathode are also shown in fig. S5, where worse discharge capacities at different C rates than the Blank cathode were obtained. A comparison of fig. S5 with Fig. 2B indicates that even in-plane (x-y direction) partial reduction of GO to RGO on the cathode surface provides sufficient conductive pathways for electron transport. When the rate increased to 10 C, a discharge capacity of 74 mA·hour g−1 was still maintained, 32% higher than the Blank cathode. A capacity of 114.53 mA·hour g−1 was still recoverable and sustainable up to 30 cycles without obvious loss when the current density returns to 1 C. Galvanostatic charge-discharge measurement was also carried out at a voltage range of 1.4 to 2.1 V at different C rates and shown in Fig. 2C. Consistent with the CV (Fig. 2A), the results show two plateaus in discharge curves, which are related to the two-stage Li+ extraction/insertion behavior.
Fig. 2. The ReHABs performance with the G-SEI (1RGO, 5RGO, 10RGO, and 50RGO) and without the G-SEI (Blank).
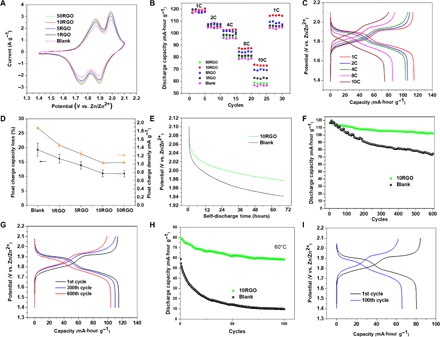
(A) CV profiles at a scan rate of 1 mV s−1. (B) Charge-discharge capacities as a function of cycle number at different C rates. (C) Charge-discharge curves of the ReHAB with the 10RGO cathode at different C rates. (D) Capacity loss (measured capacity after 1-day float charge versus initial capacity) and the corresponding float charge current density. (E) Voltage drop plateau in 72 hours of OCV tests. (F) Cyclic performance under a current density of 1 C at room temperature (25°C). (G) Corresponding charge-discharge curves at different cycles. (H) Cyclic performance of the ReHAB with the 10RGO cathode under a current density of 4 C at 60°C. (I) Corresponding charge-discharge curves.
For aqueous lithium batteries, high float charge capacity/current is one of the main drawbacks, which may decrease the cycle life under practical conditions. Thus, minimization of the float charge current is highly desirable yet quite challenging in designing highly stable aqueous batteries. The float charge current of the batteries was measured by maintaining the charged cells at 2.1 V for 1 day. The observed current drop and capacity loss are shown in Fig. 2D. The float charge capacity loss, the ratio of float charge capacity to the discharge capacity, was only 11.1% for the 10RGO cathode, much smaller than the Blank one (25.3%). Moreover, the float charge current density for the 10RGO cathode was as low as 1.03 mA g−1, 54% less than that with the Blank cathode (1.88 mA g−1). Further increases in the thickness of the G-SEI to 50-layer slightly increased the float charge capacity loss and the float charge current density. When the G-SEI are very thick (50RGO), they will behave like stacked graphite particles due to the strong π-π interaction between the layers, and unwanted polarization effects will occur, leading to higher float charge capacity loss (38, 40). The severe self-discharge is another major drawback of aqueous lithium batteries (41, 42). Here, the OCV of ReHABs was monitored to find out whether the G-SEI can suppress the self-discharge effect. It was observed that the OCV with the Blank cathode decays markedly in 72 hours (Fig. 2E) with a rate of 2.8 mV hour−1. This process was greatly inhibited by incorporation of the G-SEI, where the OCV remained at 1.97 V after 72 hours with a decay rate of only 1.1 mV hour−1, almost one-third of the Blank cathode. This result is much better than previously reported aqueous lithium batteries and lead acid systems (43, 44). Therefore, the G-SEI layer provides a great potential for practical applications, where the capacity and potential stability during float charge and self-discharge are major challenges (45).
The cyclic stability at 1 C for the ReHABs with the 10RGO and Blank cathodes is compared in Fig. 2F. After 600 cycles at room temperature, a discharge capacity of about 104 mA·hour g−1 (87% of initial capacity) was retained for the cell with the 10RGO cathode, whereas the cell with the Blank cathode had only 74 mA·hour g−1. Figure 2G shows the corresponding charge-discharge curves of the 10RGO cathode after different cycles. The discharge plateaus around 1.9 and 1.7 V agree well with the CV results. After 600 cycles, the plateau maintained its original shape, suggesting a superior stability of both the anode and the cathode in the cell. The cyclic performance at 1 C at 60°C was also tested (Fig. 2H). Whereas the Blank cathode showed a very low specific discharge capacity of 57 mA·hour g−1 at the first cycle leading to almost zero at the 100th cycle, the 10RGO cathode retained 73% after 100 cycles with a capacity of 61 mA·hour g−1. No obvious changes of plateaus can be seen from the corresponding charge-discharge curves (Fig. 2I), indicating good reversibility of the 10RGO cathode. Table 1 shows a comparison of the cycling stability of the 10RGO cathode in this study with the previous literature data for the LMO-based aqueous lithium batteries (19, 32, 46–56). Safety evaluation of batteries was determined from the solvent used in the electrolyte, as described in a previous literature (57). Under a relatively low C rate (0.2 C; fig. S6), the first charge capacity is higher than the discharge capacity, which is caused by the consumption of Li+ by surface reactions when they are extracted from the spinel LMO during the first charge (58). Previous reports also showed the same behavior as fig. S6, and it seems typical for LMO-based electrode materials (59–61). When the C rate was increased from 0.2 C (fig. S6) to 1 C (Fig. 2, C and G), a more symmetrical voltage profile could be obtained. This observation implies that the irreversible process only occurs under low extraction/insertion rate (0.2 C) in the batteries.
Table 1. A comparison of the cycling stability of various LMO-based rechargeable lithium battery systems.
CNT, carbon nanotubes; EC, ethylene carbonate; DMC, dimethyl carbonate; EMC, ethyl-methyl carbonate; Ppy, polypyrrole.
| Cathode | Anode | Electrolyte | Capacity retention | Cycles | Safety | Reference |
| LiMn2O4 | VO2 | Saturated LiNO3 (aqueous) | 83% | 42 | Good | (19) |
| LiMn2O4 | TiP2O7 | 5 M LiNO3 (aqueous) | 85% | 10 | Good | (46) |
| LiMn2O4 | LixV2O5/Ppy | 5 M LiNO3 (aqueous) | 82% | 60 | Good | (47) |
| LiMn2O4/CNT hybrids | Zn foil | 2 M Li2SO4 + 1M ZnSO4 (aqueous) | 68% | 300 | Good | (32) |
| LiMn2O4 | LiTi2(PO4)3 | 1 M Li2SO4 (aqueous) | 82% | 200 | Good | (48) |
| LiMn2O4 | LiV3O8 | 2 M Li2SO4 (aqueous) | 53% | 100 | Good | (49) |
| LiMn2O4 | V2O5 | Saturated LiNO3 (aqueous) | 89% | 100 | Good | (50) |
| LiMn2O4/CNT hybrids | Li foil | 1 M LiPF6 (EC/DMC/EMC) | 92% | 50 | Poor | (51) |
| LiMn2O4 | Na2V6O16·0.14H2O | Saturated Li2SO4 (aqueous) | 77% | 200 | Good | (52) |
| Porous LiMn2O4 spheres | Metallic lithium | 1 M LiPF6 (EC/DMC) | 73% | 1000 | Poor | (53) |
| AlF3-coated LiMn2O | Zn foil | 1 M Li2SO4 | 90% | 100 | Good | (54) |
| LiMn2O4 | Zn foil | Thixotropic gel | 61% | 1000 | Good | (55) |
| LiMn2O4 | Zn foil | Pb2+-containing gel | 75% | 300 | Good | (56) |
| LiMn2O4 electrode with RGO films as G-SEI | Zn foil | 1 M Li2SO4 + 2M ZnSO4 (aqueous) | 87% 73% (measured at 60°C) |
600 100 |
Good | This work |
To explore the versatility of the technique, we also used the dip-coating method to deposit GO films on the cathode. After reduction by hydrazine, the obtained 10RGO cathode also showed only 12.6% capacity loss after 1-day float charge (fig. S7), which is close to the cathode prepared by the isotherm technique. For comparison, the cathodes were also directly coated with pre-RGO using the same isotherm and dipping methods. There was no obvious improvement in the float charge effect for all the samples (fig. S8). The curling and agglomeration of RGO particles before coating resulted in the formation of a discontinuous layer on the cathode; thus, the ideal features of the G-SEI would not be obtained (27). The individual LMO particles were also coated with GO using the same L-B trough method and in situ reduced by hydrazine monohydrate. The rate capability was tested, and the results are shown in fig. S9. It is evident that coating the individual LMO particles with RGO is not as effective as the G-SEI on the entire cathode surface (compare fig. S9 with Fig. 2B). Ex situ thermally reduced GO powder at 900°C in Ar was used to form a higher conductive artificial SEI on the cathode’s surface using the same L-B trough method. The float charge and the rate capability of the thermally reduced 10RGO cathode are shown in fig. S10 and are compared with the Blank cathode. Evidently, minimal improvements in both float charge capacity (fig. S10A) and rate capability (fig. S10B) were obtained for the ex situ thermally reduced 10RGO compared with the in situ chemically reduced 10RGO (Fig. 2, B and D). Similar to the ex situ chemically reduced 10RGO by hydrazine (fig. S8), ex situ thermal reduction of GO most likely leads to curling and agglomeration of RGO particles on the cathode’s surface during the coating process. Therefore, a discontinuous coating on the cathode surface will be formed, and the ideal features of the G-SEI cannot be fully used. From these results, one can conclude that the uniform G-SEI with stable morphological preservation and sufficient electrical conductivity from in situ chemical reduction (Fig. 2, B and D) are much more effective than the nonuniform G-SEI from ex situ chemical/thermal reduction with higher electrical conductivity (figs. S8 and S10).
It is well known that the cathodes with large mass loading of active materials (>15 mg cm−2) are more favorable for scale-up purposes (62). The L-S method was used to fabricate the G-SEI (RGO films) on the larger-area (9 cm2 in 7 mA·hour cell) cathodes for potential applications in large-scale power sources such as UPS (uninterruptible power system), as shown in fig. S11. We have also adopted a water vapor reduction method that is a fully scalable and industrially safe approach for in situ reduction of the GO on these large-area cathodes to RGO. The corresponding float charge was measured for the 10RGO and Blank cathodes via the same way as conventional cathodes (fig. S12). It is evident that the 10RGO large-area cathode also showed the lowest float charge capacity loss (13.2%), 35% improvement compared to the Blank cathode (19.7%). These results showed the great scalability potentials of the Langmuir trough for artificial fabrication of the G-SEI. LMO-based supercapacitor is another important ALESS, where capacity fading after repeated cycles of charge-discharge remains an issue (63–65). Here, we have incorporated the G-SEI into the cathode of a Li-ion hybrid supercapacitor and assembled an asymmetrical supercapacitor according to a previous literature (63). Figure S13 shows a whole charge-discharge process of the supercapacitor (10RGO) at current densities from 1 to 16 A g−1. The charge-discharge process of the supercapacitor exhibits obvious voltage plateau, indicating the process of Li+ deintercalation/intercalation in LMO (63). The cycling stability of the supercapacitor with the G-SEI (10RGO) compared with the Blank cathode at 4 A g−1 is shown in fig. S14. The capacity retention of the 10RGO supercapacitor is 83% after 1000 cycles, 11% higher than that in the Blank cathode. This improvement in capacity stability is encouraging for further exploration of the G-SEI in other aqueous supercapacitor systems.
The electrochemical impedance spectroscopy (EIS) was used to find out the impedance of the RGO cathodes during cycling. Figure 3 (A and B) reveals the relationship between the impedance change during the cycling stability test for the Blank and 10RGO cathodes. The Nyquist plots consist of a depressed semicircle in the high-frequency region and a straight line in the low-frequency region. The semicircle at high-to-medium frequencies was assigned to the charge transfer resistance (Rct) inside the cathode, which reflects Li+ diffusion into the cathodes (66), whereas the real axis intercept is the equivalent series resistance (Rs). The evolution trends of Rct in the Blank and 10RGO cathodes were obtained for different cycles (Fig. 3C), and fitted by the relevant equivalent circuit models (fig. S15). It is obvious that the values of Rct of both cathodes all increase along with the cycle numbers but show totally different trends. The initial values (at the 5th and 10th cycles) of Rct in the 10RGO cathode are about 7% larger than those in the Blank cathode. However, in the following cycles, the Rct of the 10RGO cathode maintains the same, whereas these values in the Blank cathode increase significantly and exceed the Rct of 10RGO at the 20th cycle. The EIS measurements of the 10RGO and Blank cathodes were also conducted over time without cycling. Because there were no electrochemical charge-discharge processes to distort the LMO structure or to oxidize/consume carbon, the Rct did not have significant changes over time (fig. S16). On the other hand, the Rs of both the Blank and 10RGO cathodes increased either during repeated cycles of charge-discharge (fig. S17) or over time with cycling (fig. S18). It is known that Rs is closely related to the electronic resistance of the cathode and the ionic resistance of the liquid electrolyte (67, 68). Because the electrolyte properties remained unchanged over time, the increase in Rs may be due to the dissolution of Mn from the LMO particles and formation of a MnO2 layer around the LMO inside the bulk cathode (69, 70). These control experiments indicate that in aqueous lithium batteries, changes in Rct are highly related to the surface reactions during electrochemical charge-discharge, whereas the increase in Rs is a consequence of the inevitable degradation of LMO cathode materials.
Fig. 3. Impedance and Li+ diffusivity characterizations of 10RGO and Blank cathodes.
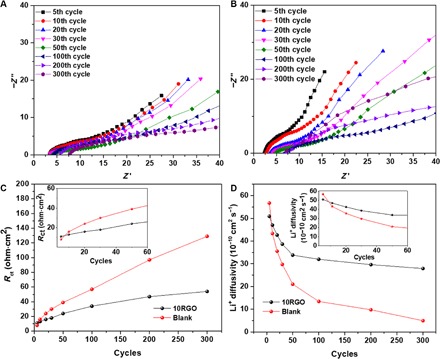
Nyquist plots of the (A) 10RGO and (B) Blank cathodes in different cycling stages. (C and D) Rct and Li+ diffusivity of the 10RGO and Blank cathodes as a function of cycle number, respectively.
Moreover, we have also fitted the Warburg element in the EIS spectra of the 10RGO and Blank cathodes during repeated cycles to quantify the diffusivity of Li+ in the cathode (DLi; Fig. 3D). Similar to Rct, the 10RGO sample has smaller DLi in the initial few cycles but shows much larger values than the Blank sample after 10 cycles. In the ALESS, the Jahn-Teller distortion of Li transition metal oxides and the carbon oxidation/consumption significantly affect the Li+ diffusion in electrode/electrolyte interfaces (71, 72). The results obtained here are also in agreement with the calculated DLi from the CV plots at different C rates. The calculated DLi (based on cathodic peaks; see the Supplementary Materials for calculations) before the cycling test was 6.19 × 10−9 and 7.32 × 10−9 cm2 s−1 for the 10RGO and Blank samples, respectively. After 300 cycles, the DLi of 10RGO can still be maintained at 3.64 × 10−9 cm2 s−1, whereas the DLi from the Blank cathode markedly decreased to 7.79 × 10−10 cm2 s−1. The stabilized Rct and DLi in 10RGO indicate the well-retained Li+ diffusion paths across the electrode/electrolyte interface during cycling, which is the main cause for the improved rate capability. In the following sections, detailed mechanisms of the effect of the RGO film, as the G-SEI, on these two unwanted phenomena are provided.
DISCUSSION
The G-SEI to suppress the oxidation/consumption of carbon
It is well known that the carbon-based conductive particles in the cathodes of the ALESS participate in the electrochemical reactions with water and oxygen during repeated cycles of charge-discharge. These particles will then be converted into oxidized carbon and, ultimately, COx gases that significantly affect the battery performance. The morphology, concentration of carbon, and oxidation state of the graphitic carbon particles on the surface and inside the bulk of the cathodes after the cycling stability test (300 cycles of charge-discharge) were evaluated using SEM, Raman, and x-ray photoelectron spectroscopy (XPS) techniques (Fig. 4). SEM images of the Blank and 10RGO cathodes after the cycling stability test are shown in Fig. 4 (A and B, respectively). A significant change in the surface morphologies was observed in the Blank cathode after the cycling stability test with only 58 atomic % (at %) carbon left, detected by energy-dispersive x-ray spectroscopy (EDX) (Fig. 4A). In addition, the surface is fully covered by the side products from the electrochemical reactions, such as graphite oxide and distorted LMO particles (compare Fig. 1H before the test with Fig. 4A after the test). However, the G-SEI with slight changes in morphology (compare Fig. 1J with Fig. 4B) are still evident in the SEM image of the 10RGO cathode, whereas 79 at % of carbon was retained in the cathode. Note that the approximate weight of RGO in a cathode with an area of 1 cm2 is only 0.005 mg, which is fully negligible compared to the weight of conductive graphite particles (0.45 mg). This indicates that the G-SEI significantly suppressed the carbon consumption of the cathode after the cycling stability test that leads to lower ohmic resistance (Rs), as shown in the EIS results (Fig. 3D). Figure 4C shows the Raman spectra of the Blank and 1RGO and 10RGO cathodes before and after the cycling stability test. The typical D and G bands of the graphitic structure were detected at 1325 and 1580 cm−1, respectively. A very low contribution of D and G bands was detected in the Blank cathode after the cycling test (Fig. 4C, Blank-After). These low graphitic properties are most likely due to the heavily oxidation/consumption of graphite particles that participated in the electrochemical oxidations. The ID/IG increases by increasing the thickness of the G-SEI (1RGO to 10RGO), very likely due to the already created defects inside the RGO films from the chemical oxidation/exfoliation process (fig. S1). Note that the G-SEI as a carbon allotrope itself might undergo the electrochemical oxidation similar to the graphitic carbon inside the cathode. However, the 2D unique gas and ion permeation properties of the G-SEI are the major advantages that simultaneously suppress the Jahn-Teller distortion and the carbon oxidation/consumption during the repeated cycles of charge-discharge. Chemical modification of the sp2 carbon in graphene via incorporation of more stable metal adatoms may improve the electrochemical stability of the G-SEI. In addition, the L-B trough technique in this study can be adapted in the future for other 2D nanomaterials (for example, WS2 and MoS2) to artificially create an SEI in both aqueous and nonaqueous lithium energy storage systems. High-resolution XPS analyses were conducted to systemically study the composition of carbon (C1s; Fig. 4, D and E) and oxygen (O1s; Fig. 4, F and G) species on the surface and in the bulk of the Blank cathodes before and after the cycling stability test. The deconvolution of C1s peaks, based on a Lorentz-Gaussian algorithm, indicated that the oxygen functional species on the surface of the Blank cathode after cycling appeared predominantly in the form of hydroxyl (C–OH; 286.2 eV) and carboxyl (O═C–OH; 290.1 eV) groups (compare Fig. 4, D and E). Note that all spectra were calibrated with a binding energy of 284.8 eV for the C═C bond. The characteristic peaks of the PVDF binder in the cathodes were also detected at 286.6 and 291.2 eV for the CH2 and CF2 groups, respectively. Note that the fluorine concentration was almost constant (~34 at %) for all cathodes with/without the G-SEI before and after the cycling test, indicating no change in the PVDF binder. Less oxidation of carbon was detected in the bulk of the Blank cathode after sputtering with Ar+ for 60 s (compare O1s peaks in Fig. 4, F and G). This observation confirms that the electrochemical oxidation reactions mostly occurred on the surface of the cathode in contact with the electrolyte. The C1s peaks in the cathodes with the G-SEI are similar to the Blank cathode before and after the cycling. However, the O1s peaks showed that both the surface and the bulk similarly contributed to the electrochemical reactions (fig. S19). That is, the G-SEI directed the electrochemical reactions from the surface to the bulk of the cathode.
Fig. 4. The morphology of cathode surface, concentration of carbon, and oxidation state of the graphitic carbon particles before and after the cycling stability test.
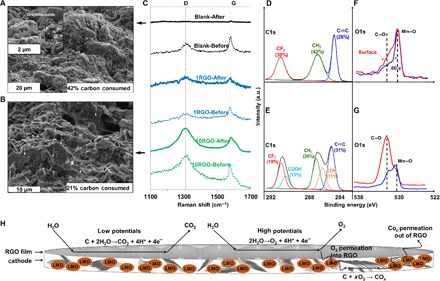
SEM images of the Blank (A) and 10RGO-coated (B) cathodes. (C) Raman spectra of the Blank and 1RGO and 10RGO cathodes after the cycling test. C1s (D and E) and O1s (F and G) high-resolution XPS spectra of the Blank cathode before and after the cycling test. a.u., arbitrary units. (H) Schematic of O2 and CO2 evolution and permeation on the surface and inside a RGO-coated cathode.
A gas permeation–based mechanism is proposed to explain the role of the G-SEI in the suppression of carbon oxidation/consumption after the cycling stability test. The typical electrochemical reactions among graphite conductive particles in the cathodes of the ReHAB and water (from aqueous electrolyte) at low potentials and oxygen (O2 from water oxidation) at high potentials are shown in Fig. 4H. It has been previously reported that the permeation of gases (for example, CO2 and O2) through the graphene films, such as GO or RGO, can be significantly decreased, up to two orders of magnitude, by increasing the thickness of the film (23, 73). Here, we propose that the permeation of O2 is restricted to the bulk of the cathode. The O2 is evolved on top of the G-SEI at high potentials. As a result, less graphite particles will be oxidized/consumed in the bulk through this reaction: C + xO2→COx, as confirmed by EDX and XPS. However, there is a possibility that O2 is also evolved in the small spacing between the G-SEI layer and the cathode surface due to the diffusion of water molecules into the G-SEI, which results in generating COx inside the bulk of the cathode (Fig. 4H). In this case, the G-SEI restrict the permeation of the COx from the bulk to the outer surface of the cathode. That is, the COx molecules will be trapped inside the bulk of the cathode that might slow down the kinetics of carbon oxidation by water at low potentials (Fig. 4H). In these two scenarios, the G-SEI will decrease the overall gas generation on the outer surface of the cathode in contact with the electrolyte due to the restricted permeation of gases from the bulk (COx) and from underneath the G-SEI (O2). It is noteworthy that in the aqueous electrolyte, the decomposition of aqueous electrolytes may inevitably occur because of the narrow electrochemical stability window of water (74). In addition, the delithiated spinel LMO also has the catalytic effect on water electrolysis (75). As a result, the generation of gases is inevitable in the aqueous electrolyte during long-term cycling, which causes a decrease of Li-ion diffusion or Li-ion conduction in the electrolyte and ultimately contributes to a tremendous impact on the reversible performance of batteries (76). Figure S20 shows the coulombic efficiencies of the 10RGO and Blank samples during 300 cycles at 4 C. Although the 10RGO sample shows relatively low coulombic efficiencies (around 96%) during 300 cycles at 4 C, the value is still higher than that of the Blank sample (around 92%) and is consistent with the previously reported coulombic efficiency for the LMO-based aqueous batteries (80 to 95%) (47, 77–79). The lower gas generation (bubble formation) on the outer surface of the G-SEI layer will maintain the effective contact area between the electrolyte and cathode during cycling, which increases the chance of transport of Li+ into the bulk of the cathode and leads to an improved coulombic efficiency.
The G-SEI to suppress the Jahn-Teller distortion
The G-SEI on the cathode also suppressed the Jahn-Teller distortion, which is responsible for capacity fading during repeated cycles of charge-discharge of aqueous Li+ batteries (16, 80) and supercapacitors (81, 82). Figure 5A shows the XRD peaks of the cathodes without (Blank) and with the G-SEI (1RGO, 5RGO, and 10RGO) before and after 300 cycles. Previous studies showed that when the Jahn-Teller distortion happens, Li+ are inserted into the empty 16c octahedral sites of the spinel, thus decreasing the intensity ratio of (311) to (400) peaks [I(311)/I(400)] (83). It is evident that the I(311)/I(400) for the cathodes with the G-SEI remained almost constant before and after the cycle life testing (0.9 to 1.1), indicating a greatly suppressed Jahn-Teller distortion. It is well known that manganese ions in the LMO structure are evenly distributed in two oxidation states, Mn3+ and Mn4+ (9). When the amount of Mn3+ on the surface of LMO particles is more than Mn4+, the spinel LMO is apt to exhibit the Jahn-Teller structural distortion and a crystalline transition from cubic to tetragonal occurs. That is, the more Mn3+ are on the LMO surface, the more serious is the Jahn-teller distortion. Therefore, the oxidation state of manganese reflects the extent of distortion in LMO. The characteristic Raman peaks of the LMO (Fig. 5B) were detected for all cathodes at 586 and 625 cm−1 for Mn3+ and Mn4+, respectively. Although a significant contribution of Mn3+ is evident in the Blank cathode after 300 cycles (Fig. 5B, Blank-After), all the cathodes with the G-SEI showed almost the same contributions of Mn3+ and Mn4+ before and after 300 cycles. This finding confirms that the LMO particles in the Blank cathode after repeated cycles were fully covered by a Mn3+-rich region, indicating a serious Jahn-Teller distortion. High-resolution Mn2p3/2 XPS peaks are also shown in Fig. 5C. The deconvoluted peaks of Mn2p3/2 at 641.5 and 643.5 eV are associated with Mn3+ and Mn4+, respectively. In agreement with the Raman results (Fig. 5B), the G-SEI layer maintained the equal contributions of Mn3+ and Mn4+ in the LMO structure after the cycling test (56% versus 44%), whereas the Blank cathode showed a significant increase in the Mn3+ contribution (compare the peaks in Fig. 5C). The high-resolution Li1s XPS peaks also showed a more significant shift to lower binding energies for the Blank cathode after repeated cycles than the RGO cathode (Fig. 5D). This shift is attributed to the loss of manganese that causes a change in the electronegativity of the LMO structure (18).
Fig. 5. Characterization of LMO cathodes before and after the cycling stability test.
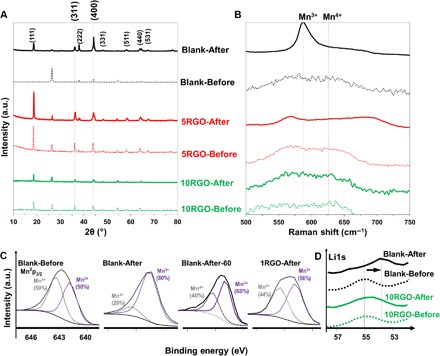
XRD (A), Raman (B), XPS Mn2p3/2 (C), and XPS Li1s (D) spectra for the Blank cathode before and after the 300 charge-discharge test in comparison with the cathodes with the G-SEI (RGO cathodes).
To further quantify the Jahn-Teller distortion in the Blank and 10RGO cathodes, in situ XRD patterns during discharge were recorded (fig. S21). It is evident that the Blank cathode has two distinct peaks during the discharge process from Li0.17Mn2O4 to Li0.32Mn2O4. These peaks indicated that there was a lattice transition from cubic to tetragonal, and a larger crystalline mismatch in the Blank cathode than the 10RGO cathode occurred. The changes in lattice parameters (Δa and Δc) and volumes (Δv) during discharge were also calculated from the in situ XRD patterns by Rietveld analysis using the Expo2013 software and are shown in Fig. 6 (A and B, respectively). Note that a cubic crystal is identified with one lattice constant (a), whereas a tetragonal crystal is identified with two constants (a and c). As a general trend, Δa, Δc, and Δv decreased by increasing the thickness of the G-SEI layer (Fig. 6, A and B, from 1RGO to 5RGO to 10RGO). For example, the 10RGO cathode showed a Δα of 0.153 Å, a Δc of 0.112 Å, and a Δv of 2.7 Å3, which correspond to 38, 61, and 73% decrease, respectively, in the lattices’ mismatch compared to the Blank cathode. The suppression of lattice changes from cubic to tetragonal will ultimately maintain the integrity of the LMO particles during the discharge and charge processes. As a result, the diffusion pathways of Li+ will remain the same, and the Rct (Fig. 3C) after 300 cycles does not significantly change, leading to greatly improved battery and supercapacitor performances (Fig. 2 and fig. S9).
Fig. 6. Quantifying the Jahn-Teller distortion in the Blank and 10RGO cathodes.
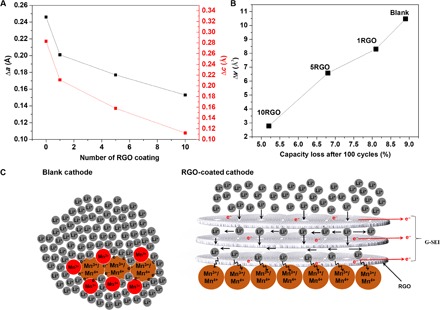
Lattice parameter (Δa and Δc) (A) and volume change (Δv) (B) during discharge process for the Blank and the RGO cathodes calculated from the in situ XRD patterns. (C) Schematics of the permeation-based mechanism for the Blank and RGO cathodes representing the Li+ diffusion and electron mobility.
It is well established that the onset of the Jahn-Teller distortion of LMO takes place when the Li+ accumulate on the surface of the cathode. It is speculated that under dynamic, nonequilibrium conditions during discharging, LMO crystals at the surface of the cathode are more lithiated than others in the bulk, thereby driving the composition of these LMO crystals’ surface into a Mn3+-rich region (Fig. 6C) and inducing the Jahn-Teller distortion (84). Therefore, one may expect that the nonequilibrium conditions could be avoided through a controlled diffusion of Li+ into the LMO particles. As a result, the accumulation of the Li+ and formation of Mn3+-rich regions around the LMO particles (responsible for the Jahn-Teller distortion) on the cathode surface will less likely happen. In addition, it has been previously reported that the Li+ can only diffuse perpendicularly into the graphene layers through the structural defects and will move parallel to the layers where no defects are found (85). Therefore, we propose a mechanism in which the G-SEI on the cathode surface can control the diffusion of Li+ and prevent their accumulation on the LMO particles while enhancing the electron mobility (Fig. 6C). During the discharge process of the RGO cathodes, the Li+ first diffuse into the G-SEI and then reach the cathode surface by passing through the defects. Contact angle measurements of the aqueous electrolyte on the surfaces of the 10RGO and Blank cathodes were conducted (fig. S22). The electrolyte was dispensed at a rate of 0.5 μl s−1, and the pictures were taken every 500 ms. It is evident that the 10RGO cathode has a slightly smaller contact angle (122°) than the Blank cathode (135°), most likely due to the oxygen functional residues from the GO film. A previous report (38) also confirmed that the in situ chemical treatment process using hydrazine monohydrate only partially reduces GO to RGO. These oxygen functional residues on the cathode surface facilitate the diffusion of the electrolyte into the bulk of the cathode. Because the density of defects is limited across the G-SEI (Fig. 6C), the Li+ have to diffuse more parallel to the film than perpendicular, which will ultimately elongate their diffusion process. As a result, the Li+ accumulate more on/inside the G-SEI than the surface of the cathode that will restrict the formation of Mn3+-rich regions, leading ultimately to the suppressed Jahn-Teller distortion. Obviously, the diffusion of Li+ will be further restricted by increasing the thickness of the G-SEI; thus, more suppression of the Jahn-Teller distortion will occur (Fig. 6, A and B).
On the other hand, the G-SEI may partially provide some capacity. This capacity contribution was provided at the working potential of the LMO active particles (between 1.2 and 4.2 V versus Li/Li+) (86) but not at the natural potential of graphite. To measure the pure capacity of the G-SEI due to the intercalation of Li+, we prepared the cathodes with the same formulation, but the LMO particles were substituted by the same amount of the inactive silica particles (10 μm in size). Figure S23 shows the capacity values (μA·hour cm−2) for the Blank and 10RGO silica cathodes after 10 cycles of charge-discharge at a fixed current density of 16 mA g−1 in a potential window of 1.4 to 2.1 V versus Zn/Zn+ (3.6 to 4.3 V versus Li/Li+). Whereas the capacity of the Blank silica cathode approaches zero after only five cycles, the 10RGO silica cathode showed a stable capacity of about 0.14 μA·hour cm−2. Considering the surface area of the cathode (1.13 cm2), the density of graphene (2 g cm−3), and the average thickness for 10RGO (20 nm with unavoidable wrinkling), the corresponding capacity will be approximately 35 mA·hour g−1. Note that the mass loading of RGO is extremely small (a graphene few layers); thus, the contributed capacity by RGO (0.14 μA·hour cm−2) to the total capacity of the cathode is negligible. The structural defects in the RGO film can provide abundant channels for Li+ diffusion (87–89). Therefore, the RGO film does not affect the electrochemical reactions between LMO and Li+. Figure S24 presents the discharge curve of the 10RGO/silica cathode at a current density of 16 mA g−1. On the basis of the discharge plateau, the voltage for Li intercalation into RGO is around 1.85 V, which is similar to the Li+ intercalation potential into LMO in the aqueous electrolyte. That is, a simultaneous intercalation of Li+ into both RGO and LMO is quite possible.
CONCLUSIONS
In summary, the Langmuir trough–based techniques were successfully developed to fabricate and transfer single- and multilayer G-SEI onto the surfaces of the cathodes of an aqueous lithium battery and a supercapacitor. The aqueous lithium battery maintained more than 88% of the initial capacity after 1-day float charge and 95% of the OCV after 72 hours of self-discharge. The G-SEI also offered an excellent rate capability (74 mA·hour g−1 at 10 C, 64% of the 0.2-C capacity) and cycling stability in both the aqueous battery (87% of initial capacity maintained after 600 cycles) and the supercapacitor (83% of initial capacity maintained after 1000 cycles). The G-SEI on the large cathodes (9 cm2 in 7 mA·hour cell) demonstrated similar property improvement to the small cathodes (1 cm2 in 1.15 mA·hour cell), confirming excellent scale-up potentials of the reported approach in the aqueous energy storage systems. On the basis of the in situ and ex situ characterizations, we proposed that the G-SEI layer simultaneously suppressed the accumulation of Li+ (leading to the Jahn-Teller distortion) and oxidation/consumption of conductive carbon on the cathode/electrolyte interface through a controlled diffusion of Li+ and restricted permeation of gases (O2 and COx) in/out of the cathode surface, respectively.
MATERIALS AND METHODS
Chemicals and materials
Sulfuric acid (H2SO4; 95%), potassium permanganate (KMnO4; 99%), hydrazine monohydrate (N2H4·H2O; 98%), diethyl ether (99%), hydrogen peroxide [H2O2; 30 weight % (wt %) in H2O], phosphoric acid (H3PO4; 85%), hydrochloric acid (HCl; 38%), 1,2-dichloroethane (anhydrous 99.8%), 1-methyl-2-pyrrolidinone (NMP; ≥99.5% purity), Li2SO4 (≥99% purity), ZnSO4 (≥99% purity), and ethanol (denatured, reagent grade) were all purchased from Sigma-Aldrich. Crystalline graphite powder and graphite foil were purchased from Alfa Aesar (325 mesh, 99%). PVDF (HSV 900) was purchased from Kynar. LMO and sphere-like graphite powders (KS-6) were purchased from MTI.
Synthesis of GO and RGO
GO was synthesized on the basis of the improved synthesis method by Zehtab Yazdi et al. (35) with slight modifications in the procedure to obtain GO sheets with lower defects and larger flake sizes. In a typical procedure, 3 g of graphite was stirred in 360 ml of concentrated H2SO4 for 1 hour. Then, 40 ml of H3PO4 was added, and the mixture was stirred for 15 min. KMnO4 (18 g) was then slowly added to the mixture and stirred for another 1 hour at room temperature, followed by 12 hours at 50°C. To stop the oxidation, we cooled down the mixture to room temperature and added 400 ml of cold distilled water containing 3 ml of H2O2 (30%). The mixture was centrifuged at 4000 rpm for 4 hours, and the supernatant was removed. The remaining GO paste was then purified with 200 ml of distilled water, 200 ml of 30% HCl, and 200 ml of ethanol, where the mixture was centrifuged after each stage at 4000 rpm for 4 hours and the supernatant was decanted away. The purified GO paste after this extensive purification process was dried at room temperature for 1 day under fume hood, and about 3 g of solid was obtained. To synthesize RGO, we reduced the GO product from the above procedure by either chemical treatment via hydrazine monohydrate (10 volume %) or hydrothermal method. In the chemical reduction method, 1 g of GO powder was dispersed in 1 liter of distilled water at room temperature for 4 hours to obtain a uniform and stable suspension. Hydrazine monohydrate (1 ml) was then added to the GO/water suspension and heated to 95°C for 2 hours. Finally, RGO was collected by vacuum filtration through a 5-μm polytetrafluoroethylene (PTFE) membrane. In the hydrothermal reduction treatment, a total of 25 ml of the GO aqueous solution was transferred to a Teflon-lined autoclave and heated at 180°C for 6 hours. The autoclave was then cooled to room temperature, and the product RGO was also collected through the same vacuum filtration procedure.
Coating procedure
To coat the graphene monolayers on the cathodes’ surfaces, we prepared an organic suspension of the nanoparticles. Graphene nanoparticles (GO or RGO) (1 mg) were fully dispersed in 3 ml of ethanol with magnetic stirring at room temperature for 1 hour, followed by 15-min bath sonication. Then, 37 ml of 1,2-dichloroethane was added to the mixture and stirred for 1 hour to obtain a uniform suspension. The L-B and L-S trough methods were used to coat the graphene monolayers on the cathodes. In a typical procedure, the trough (KSV minitrough with a maximum surface area of 273 cm2, made of PTFE with a dipping well of 37 mm) and the barriers were cleaned once by water and twice by ethanol and fully dried at room temperature under nitrogen gas. The trough was filled with 275 ml of pure water as the subphase. Then, the clean glass slides having the cathodes on one side were placed either at the bottom of the trough (for the L-B isotherm technique) or inside the well (for the L-B dipping technique). The barriers were then closed up to 160 mm from one side, and the air-water interface was cleaned by vacuum suction. This process was repeated at least three times until the surface pressure changes were less than 0.2 mN m−1 during compression and decompression, indicating that the air-water interface was clean. The organic suspension of graphene (GO or RGO) was then transferred to a clean 5-ml glass syringe and dripped onto the air-water interface of the trough via Teflon tubing with interconnects made of poly(ether ketone). The dripping was performed on the water interface, with a height of ~2 mm just above the interface, at a rate of 0.1 ml min−1 for a total of 1.5 ml that was controlled by a syringe pump. We waited for 30 min to make sure that the organic solvent was completely evaporated, and the graphene monolayer was uniformly distributed on the air-water interface. The barriers then compressed the graphene film at a rate of 30 mm min−1 until the surface pressure reached approximately 12 mN m−1. In the L-B isotherm technique, water subphase was sucked out with a KNF Laboport PTFE vacuum pump to slowly lower the surface of water and deposit compact graphene monolayer on the cathodes’ substrates. In the L-B dipping technique, the cathodes were pulled out of the well with a speed of 8 mm min−1, whereas the barriers also compressed the film with a speed of 30 mm min−1 to provide a continuous graphene coating on the cathodes. In the L-S method, the cathodes were moved down horizontally toward the graphene monolayer on the interface with a speed of 8 mm min−1 until they touched the water surface, and then they were immediately pulled out with the same speed. The L-S method was very useful for the cathodes with the high loading of active materials that could be disintegrated in the L-B isotherm and dipping techniques due to a slightly longer immersion time in water subphase. Coated cathodes were then removed from the trough and dried overnight at room temperature under the fume hood before the battery assembly. In situ reduction methods via hydrazine or water vapor were used when GO was used as the graphene precursor to coat the cathodes’ surfaces. The GO-coated cathodes were immersed in hydrazine monohydrate for 5 min and then washed with distilled water and dried at 50°C for 5 hours before the battery assembly. In a water vapor reduction method, the surfaces of GO-coated cathodes were exposed to continuous flow of water vapor for 5 hours and then cooled down to room temperature and dried.
Characterization techniques
XRD patterns of the samples were obtained using a Bruker D8 Discover x-ray diffractometer with Cu x-ray tube and a wavelength of 1.54 Å. The in situ XRD patterns were collected at beamline 1D using a Mar345 image plate detector with a wavelength of 1.0331 Å. For easy comparison, 2q of all XRD patterns was recalculated and converted to the corresponding angles for k = 1.54 Å (Cu Kα radiation). The SEM work was carried out in a Philips XL-30 ESEM, using 20-kV acceleration voltage. The AFM was carried out in a Bruker Dimension Icon machine using a ScanAsyst-Air probe on a silicon cantilever substrate. The samples of GO were prepared by drop-casting 40 μl of GO/water suspension (0.05 mg/ml) on an HOPG substrate, followed by drying at room temperature for 4 hours. The AFM images of the bare and coated cathodes were directly taken from the surface before assembling into the battery system. Careful adjustment of imaging parameters may be required for good qualities. The HRTEM was carried out in a Zeiss Libra 200 MC (Carl Zeiss Microscopy GmbH), at 200-kV acceleration voltage, with the standard single tilt holder. The images were captured by a TRS SharpEye (Troendle) at 2048 × 2048 pixels. A drop of this suspension was placed on the carbon side of a standard TEM grid that was covered with an approximately 40-nm-thin holey carbon film (EMS300-Cu) and placed on a filter paper to quickly dry. SAED patterns were taken using an aperture size of 30 μm and after calibrating with a gold polycrystalline sample. The structural defects of the samples were investigated using Raman spectroscopy. The Raman spectra were recorded by a Bruker Senterra Raman microscope with a laser radiation of 785 nm, an integration of 20 s, an objective of ×10, and a laser power of 20 mW. To measure the elemental composition, we used a Thermo VG Scientific ESCALAB 250 microprobe to record XPS spectra. The spectra were taken using a monochromatic aluminum source, at 1486.6 eV and 49.3 W, with a beam diameter of 200.0 μm. The samples were pressed on a piece of conductive carbon tape, and a double neutralization—a low-energy electron beam and low-energy Ar+ beam—was used during spectrum acquisition. The binding energies were reported relative to C1s at 284.8 eV. The chamber analysis pressure was 2.0 × 10−9 Pa during acquisition. The takeoff angle was 45°C. For each sample, a high-sensitivity mode spectrum was taken with a wide binding energy range of 0 to 1350 eV (survey) to determine the surface elemental composition of the samples. Then, a narrower binding energy window, with a pass energy of 23.50 eV, was used to obtain high-energy resolution spectra of the elements present in the sample to determine its chemical environment and quantification.
Fabrication of LMO-based cathodes
The LMO cathodes were prepared by casting a slurry of 86 wt % LMO, 7 wt % KS-6, and 7 wt % PVDF in NMP on graphite foil and vacuum drying at 60°C for 24 hours. Cathode discs of 12 mm diameter were cut (typical active material load of 5.8 mg cm−2) and soaked in the electrolyte solution under reduced pressure.
Assembly of ReHABs
Graphene-coated cathodes (diameter, 1.15 cm; thickness of the cathode film, 110 μm) were assembled with zinc discs (RotoMetals; diameter, 1.15 cm; thickness, 0.2 mm) and absorbed glass mat (NSG Corporation; diameter, 1.15 cm; thickness, 0.5 mm), which act as the anode and separator, respectively. The solution of 1 M Li2SO4 (≥99% purity; Sigma-Aldrich) and 2 M ZnSO4 (≥98% purity; Sigma-Aldrich) in deionized water was used as the electrolyte (pH 4).
Battery testing
CV measurement and EIS were performed on a Bio-Logic VMP3 electrochemical workstation, using Pt wire as the counter electrode, Ag/AgCl as the reference electrode, and 1 M Li2SO4/2 M ZnSO4 as the electrolyte. Swagelok-type cells were used for galvanostatic charge-discharge cycling at room temperature and 60°C, using a Neware battery tester at 4 C (1 C = 120 mA g−1) between 1.4 and 2.1 V. Rate capability test was performed under 1 to 10 C. Self-discharge tests of the ReHAB cells were performed at room temperature in Swagelok-type cells using a Neware battery tester (BTS, 5 V and 5 mA). The cells were galvanostatically charged and discharged for three cycles using 0.25-C equivalent rate (1 C = 130 mA g−1) between 1.4 and 2.1 V versus Zn/Zn2+, and charged to 2.1 V, and the OCV condition was left for 84 hours with continuous monitoring of the cell voltage. Float charge current tests were performed by charging the cell to 2.1 V at 0.2 C and holding it at that voltage for 1 day, during which time the current variation was recorded. The capacity loss (Closs) of float charge is calculated using Eq. 1
| (1) |
where Cf is the float charge capacity during 1-day float charge and Cdischarge is the capacity before 1-day float charge.
Supercapacitor testing
An asymmetrical supercapacitor device based on graphene-coated cathode (10RGO) was fabricated using activated carbon (AC) as the anode and 0.5 M aqueous solution of Li2SO4 as the electrolyte. The balancing weight ratio of AC to LMO (2:1) was calculated on the basis of their specific capacitances. The galvanostatic charge-discharge cycling test of the supercapacitor was performed in a CHI 760D electrochemistry workstation.
Supplementary Material
Acknowledgments
We thank M. Ahmed and M. Martz for their help in running SEM and contact angle measurements, respectively, and for the manuscript revision. Funding: This research was financially supported by Positec Inc., Natural Sciences and Engineering Research Council of Canada, Canadian Foundation for Innovation, the Canada Research Chairs program, and Mitacs (IT04444). Author contributions: J.Z. performed all electrochemical characterizations and battery testing, assisted in XRD, and wrote half of the manuscript. A.Z.Y. proposed, designed, and developed the Langmuir trough techniques; conducted detailed characterizations by XRD, XPS, Raman, HRTEM, AFM, and SEM; and wrote half of the manuscript. J.H. assisted in AFM imaging of the cathodes. G.V. assisted in using the trough to fabricate the G-SEI on the cathodes. P.C. oversaw all research phases, provided regular guidance to the research, and revised the manuscript. All authors discussed and commented on the manuscript. Competing interests: A.Z.Y., J.Z., and P.C. are authors on a pending patent application related to this work (CN 201710011769.7, provisional patent filed on 10 January 2017). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/9/e1701010/DC1
fig. S1. Characterizations of graphite, GO, and RGO.
fig. S2. XRD patterns of the Blank cathode treated with hydrazine.
fig. S3. Contact angle measurement results of the hydrazine aqueous solution on the GO-coated and Blank cathodes.
fig. S4. SEM image of the 10RGO cathode obtained by 10 times layer-by-layer deposition of 1RGO.
fig. S5. Rate capability results of the battery with the 10GO cathode (without hydrazine reduction) in comparison with the Blank cathode.
fig. S6. Typical charge-discharge curves of ReHAB with the 10RGO cathode at 0.2 C.
fig. S7. Capacity loss after 1-day float charge of the cathodes with different thicknesses of the G-SEI (1RGO, 5RGO, and 10RGO) and Blank cathodes (dip-coating GO and in situ reduction to RGO).
fig. S8. Capacity loss after 1-day float charge of the 1RGO, 5RGO, 10RGO, and Blank cathodes (directly dip-coating RGO).
fig. S9. Rate capability results of the pristine LMO and RGO-coated LMO particles in the cathode.
fig. S10. Capacity loss after 1-day float charge and rate capabilities of Blank and 10RGO (thermally reduced) cathodes.
fig. S11. The inside and outside of a large-scale battery cell (7 mA·hour) with 9-cm2 cathode surface area for potential applications in the UPS power sources.
fig. S12. Capacity loss after 1-day float charge of the 1RGO, 5RGO, 10RGO, and Blank cathodes in the large-scale cell (fig. S10) (mass loading of active materials, 25 to 28 mg cm−2; L-S method to coat GO followed by water vapor reduction).
fig. S13. Typical charge-discharge curves of the 10RGO-LMO/AC asymmetrical supercapacitor at different current densities.
fig. S14. Cycling stability of the 10RGO-LMO/AC asymmetrical supercapacitor at a current density of 4 A g−1.
fig. S15. Relevant equivalent circuit model for EIS data in Fig. 3.
fig. S16. Rct of the 10RGO and Blank cathodes as a function of time without repeated cycles.
fig. S17. Rs of the 10RGO and Blank cathodes as a function of repeated cycles of charge-discharge.
fig. S18. Rs of the 10RGO and Blank cathodes as a function of time without repeated cycles.
fig. S19. O1s peaks of the 10RGO cathode after the cycling test on the surface (red) and inside the bulk (blue).
fig. S20. Coulombic efficiencies of the 10RGO and Blank samples during 300 cycles at 4 C.
fig. S21. In situ XRD patterns of Blank and 10RGO cathode during discharge process (LixMn2O4, 0.11 < x < 1).
fig. S22. Contact angle results of the aqueous electrolytes on the Blank (left) and 10RGO (right) cathodes.
fig. S23. The cycling properties of the Blank and 10RGO cathodes that are made of inactive silica particles rather than the LMO.
fig. S24. Discharge curves of the 10RGO/silica cathode at a current density of 16 mA g−1.
fig. S25. Typical CVs of the 10RGO cathode at a scan rate of 0.1 to 0.7 mV s−1.
fig. S26. The peak current (ip) in the 10RGO cathode samples (peak 1 in this study) as a function of the square root of the scan rate (v).
Calculations of Li+ diffusivity based on EIS data
Calculations of Li+ diffusivity based on CV plot
Reference (90)
REFERENCES AND NOTES
- 1.Wu X.-L., Jiang L.-Y., Cao F.-F., Guo Y.-G., Guo L.-J., LiFePO4 nanoparticles embedded in a nanoporous carbon matrix: Superior cathode material for electrochemical energy-storage devices. Adv. Mater. 21, 2710–2714 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Suo L., Borodin O., Gao T., Olguin M., Ho J., Fan X., Luo C., Wang C., Xu K., “Water-in-salt” electrolyte enables high-voltage aqueous lithium-ion chemistries. Science 350, 938–943 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Tarascon J.-M., Tarascon M., Issues and challenges facing rechargeable lithium batteries. Nature 414, 359–367 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Ongaro F., Saggini S., Mattavelli P., Li-ion battery-supercapacitor hybrid storage system for a long lifetime, photovoltaic-based wireless sensor network. IEEE Trans. Power Electron. 27, 3944–3952 (2012). [Google Scholar]
- 5.Aurbach D., Gamolsky K., Markovsky B., Salitra G., Gofer Y., Heider U., Oesten R., Schmidt M., The study of surface phenomena related to electrochemical lithium intercalation into Lix MOy host materials (M = Ni, Mn). J. Electrochem. Soc. 147, 1322–1331 (2000). [Google Scholar]
- 6.Thackeray M. M., Structural considerations of layered and spinel lithiated oxides for lithium ion batteries. J. Electrochem. Soc. 142, 2558–2563 (1995). [Google Scholar]
- 7.Aydinol M., Kohan A., Ceder G., Ab initio calculation of the intercalation voltage of lithium-transition-metal oxide electrodes for rechargeable batteries. J. Power Sources 68, 664–668 (1997). [Google Scholar]
- 8.Chen H., Cong T. N., Yang W., Tan C., Li Y., Ding Y., Progress in electrical energy storage system: A critical review. Prog. Nat. Sci. 19, 291–312 (2009). [Google Scholar]
- 9.Li X., Xu Y., Wang C., Suppression of Jahn–Teller distortion of spinel LiMn2O4 cathode. J. Alloys Compd. 479, 310–313 (2009). [Google Scholar]
- 10.Pieczonka N. P. W., Liu Z., Lu P., Olson K. L., Moote J., Powell B. R., Kim J.-H., Understanding transition-metal dissolution behavior in LiNi0.5Mn1.5O4 high-voltage spinel for lithium ion batteries. J. Phys. Chem. C 117, 15947–15957 (2013). [Google Scholar]
- 11.Li B., Xing L., Xu M., Lin H., Li W., New solution to instability of spinel LiNi0.5Mn1.5O4 as cathode for lithium ion battery at elevated temperature. Electrochem. Commun. 34, 48–51 (2013). [Google Scholar]
- 12.Gallagher K. G., Fuller T. F., Kinetic model of the electrochemical oxidation of graphitic carbon in acidic environments. Phys. Chem. Chem. Phys. 11, 11557–11567 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Liu H., Xu Q., Yan C., Qiao Y., Corrosion behavior of a positive graphite electrode in vanadium redox flow battery. Electrochim. Acta 56, 8783–8790 (2011). [Google Scholar]
- 14.Y. Yu, Electrowinning of Zinc for the Anode of Rechargeable Hybrid Aqueous Batteries (University of Waterloo, 2015). [Google Scholar]
- 15.Edström K., Gustafsson T., Thomas J. O., The cathode–electrolyte interface in the Li-ion battery. Electrochim. Acta 50, 397–403 (2004). [Google Scholar]
- 16.Wang F. X., Xiao S. Y., Shi Y., Liu L. L., Zhu Y. S., Wu Y. P., Wang J. Z., Holze R., Spinel LiNixMn2−xO4 as cathode material for aqueous rechargeable lithium batteries. Electrochim. Acta 93, 301–306 (2013). [Google Scholar]
- 17.Guohua L., Guohua H., Uchida T., Wakihara M., The spinel phases LiMyMn2−yO4 (M = Co, Cr, Ni) as the cathode for rechargeable lithium batteries. J. Electrochem. Soc. 143, 178–182 (1996). [Google Scholar]
- 18.Pasta M., Wessells C. D., Huggins R. A., Cui Y., A high-rate and long cycle life aqueous electrolyte battery for grid-scale energy storage. Nat. Commun. 3, 1149 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Wang H., Huang K., Zeng Y., Yang S., Chen L., Electrochemical properties of TiP2O7 and LiTi2(PO4)3 as anode material for lithium ion battery with aqueous solution electrolyte. Electrochim. Acta 52, 3280–3285 (2007). [Google Scholar]
- 20.Zhu Q., Zheng S., Lu X., Wan Y., Chen Q., Yang J., Zhang L.-z., Lu Z., Improved cycle performance of LiMn2O4 cathode material for aqueous rechargeable lithium battery by LaF3 coating. J. Alloys Compd. 654, 384–391 (2016). [Google Scholar]
- 21.Stojković I. B., Cvjetićanin N. D., Mentus S. V., The improvement of the Li-ion insertion behaviour of Li1.05Cr0.10Mn1.85O4 in an aqueous medium upon addition of vinylene carbonate. Electrochem. Commun. 12, 371–373 (2010). [Google Scholar]
- 22.Wang F., Lin Y., Suo L., Fan X., Gao T., Yang C., Han F., Qi Y., Xu K., Wang C., Stabilizing high voltage LiCoO2 cathode in aqueous electrolyte with interphase-forming additive. Energy Environ. Sci. 9, 3666–3673 (2016). [Google Scholar]
- 23.Kim M. S., Ma L., Choudhury S., Moganty S. S., Wei S., Archer L. A., Fabricating multifunctional nanoparticle membranes by a fast layer-by-layer Langmuir–Blodgett process: Application in lithium–sulfur batteries. J. Mater. Chem. A 4, 14709–14719 (2016). [Google Scholar]
- 24.Kim M. S., Ma L., Choudhury S., Archer L. A., Multifunctional separator coatings for high-performance lithium–sulfur batteries. Adv. Mater. Interfaces 3, 1600450 (2016). [Google Scholar]
- 25.Wang Y., Yi J., Xia Y., Recent progress in aqueous lithium-ion batteries. Adv. Energy Mater. 2, 830–840 (2012). [Google Scholar]
- 26.Choi N.-S., Han J.-G., Ha S.-Y., Park I., Back C.-K., Recent advances in the electrolytes for interfacial stability of high-voltage cathodes in lithium-ion batteries. RSC Adv. 5, 2732–2748 (2015). [Google Scholar]
- 27.Nie M., Abraham D. P., Chen Y., Bose A., Lucht B. L., Silicon solid electrolyte interphase (SEI) of lithium ion battery characterized by microscopy and spectroscopy. J. Phys. Chem. C 117, 13403–13412 (2013). [Google Scholar]
- 28.Eom K., Jung J., Lee J. T., Lair V., Joshi T., Lee S. W., Lin Z., Fuller T. F., Improved stability of nano-Sn electrode with high-quality nano-SEI formation for lithium ion battery. Nano Energy 12, 314–321 (2015). [Google Scholar]
- 29.An S. J., Li J., Daniel C., Mohanty D., Nagpure S., Wood D. L. III, The state of understanding of the lithium-ion-battery graphite solid electrolyte interphase (SEI) and its relationship to formation cycling. Carbon 105, 52–76 (2016). [Google Scholar]
- 30.Yan J., Wang J., Liu H., Bakenov Z., Gosselink D., Chen P., Rechargeable hybrid aqueous batteries. J. Power Sources 216, 222–226 (2012). [Google Scholar]
- 31.Lu C., Hoang T. K. A., Doan T. N. L., Zhao H., Pan R., Yang L., Guan W., Chen P., Rechargeable hybrid aqueous batteries using silica nanoparticle doped aqueous electrolytes. Appl. Energy 170, 58–64 (2016). [Google Scholar]
- 32.Zhu X., Doan T. N. L., Yu Y., Tian Y., Sun K. E. K., Zhao H., Chen P., Enhancing rate performance of LiMn2O4 cathode in rechargeable hybrid aqueous battery by hierarchical carbon nanotube/acetylene black conductive pathways. Ionics 22, 71–76 (2016). [Google Scholar]
- 33.Lu C., Hoang T. K., Doan T. N. L., Acton M., Acton H., Guan W., Chen P., Influence of different silica gelling agents on the performance of aqueous gel electrolytes. J. Ind. Eng. Chem. 42, 101–106 (2016). [Google Scholar]
- 34.Marcano D. C., Kosynkin D. V., Berlin J. M., Sinitskii A., Sun Z., Slesarev A., Alemany L. B., Lu W., Tour J. M., Improved synthesis of graphene oxide. ACS Nano 4, 4806–4814 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Zehtab Yazdi A., Chizari K., Jalilov A. S., Tour J., Sundararaj U., Helical and dendritic unzipping of carbon nanotubes: A route to nitrogen-doped graphene nanoribbons. ACS Nano 9, 5833–5845 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Higginbotham A. L., Kosynkin D. V., Sinitskii A., Sun Z., Tour J. M., Lower-defect graphene oxide nanoribbons from multiwalled carbon nanotubes. ACS Nano 4, 2059–2069 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Dimiev A. M., Tour J. M., Mechanism of graphene oxide formation. ACS Nano 8, 3060–3068 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Pei S., Cheng H.-M., The reduction of graphene oxide. Carbon 2012, 50, 3210–3228 (2012). [Google Scholar]
- 39.P. Mukhopadhyay, R. K. Gupta, Graphite, Graphene, and Their Polymer Nanocomposites (CRC Press, 2012). [Google Scholar]
- 40.Lung-Hao Hu B., Wu F.-Y., Lin C.-T., Khlobystov A. N., Li L.-J., Graphene-modified LiFePO4 cathode for lithium ion battery beyond theoretical capacity. Nat. Commun. 4, 1687 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Konarov A., Gosselink D., Zhang Y., Tian Y., Askhatova D., Chen P., Self-discharge of rechargeable hybrid aqueous battery. ECS Electrochem. Lett. 4, A151–A154 (2015). [Google Scholar]
- 42.Rahmanifar M., Mousavi M., Shamsipur M., Heli H., A study on open circuit voltage reduction as a main drawback of Zn–polyaniline rechargeable batteries. Synth. Met. 155, 480–484 (2005). [Google Scholar]
- 43.Zhao H., Wu Q., Hu S., Xu H., Rasmussen C. N., Review of energy storage system for wind power integration support. Appl. Energy 137, 545–553 (2015). [Google Scholar]
- 44.Sinha S., Chandel S., Review of recent trends in optimization techniques for solar photovoltaic–wind based hybrid energy systems. Renew. Sustain. Energy Rev. 50, 755–769 (2015). [Google Scholar]
- 45.G. Pistoia, Electric and Hybrid Vehicles: Power Sources, Models, Sustainability, Infrastructure and the Market (Elsevier, 2010). [Google Scholar]
- 46.Zhao M., Zheng Q., Wang F., Dai W., Song X., Electrochemical performance of high specific capacity of lithium-ion cell LiV3O8//LiMn2O4 with LiNO3 aqueous solution electrolyte. Electrochim. Acta 56, 3781–3784 (2011). [Google Scholar]
- 47.Wang H., Zeng Y., Huang K., Liu S., Chen L., Improvement of cycle performance of lithium ion cell LiMn2O4/LixV2O5 with aqueous solution electrolyte by polypyrrole coating on anode. Electrochim. Acta 52, 5102–5107 (2007). [Google Scholar]
- 48.Luo J.-Y., Xia Y.-Y., Aqueous lithium-ion battery LiTi2(PO4)3/LiMn2O4 with high power and energy densities as well as superior cycling stability. Adv. Funct. Mater. 17, 3877–3884 (2007). [Google Scholar]
- 49.Wang G., Zhang H., Fu L., Wang B., Wu Y., Aqueous rechargeable lithium battery (ARLB) based on LiV3O8 and LiMn2O4 with good cycling performance. Electrochem. Commun. 9, 1873–1876 (2007). [Google Scholar]
- 50.Stojković I., Cvjetićanin N., Pašti I., Mitrić M., Mentus S., Electrochemical behaviour of V2O5 xerogel in aqueous LiNO3 solution. Electrochem. Commun. 11, 1512–1514 (2009). [Google Scholar]
- 51.Tang M., Yuan A., Xu J., Synthesis of highly crystalline LiMn2O4/multiwalled carbon nanotube composite material with high performance as lithium-ion battery cathode via an improved two-step approach. Electrochim. Acta 166, 244–252 (2015). [Google Scholar]
- 52.Zhou D., Liu S., Wang H., Yan G., Na2V6O16·0.14H2O nanowires as a novel anode material for aqueous rechargeable lithium battery with good cycling performance. J. Power Sources 227, 111–117 (2013). [Google Scholar]
- 53.Xi L. J., Wang H.-E., Lu Z. G., Yang S. L., Ma R. G., Deng J. Q., Chung C. Y., Facile synthesis of porous LiMn2O4 spheres as positive electrode for high-power lithium ion batteries. J. Power Sources 198, 251–257 (2012). [Google Scholar]
- 54.Tron A., Park Y. D., Mun J., AlF3-coated LiMn2O4 as cathode material for aqueous rechargeable lithium battery with improved cycling stability. J. Power Sources 325, 360–364 (2016). [Google Scholar]
- 55.Hoang T. K. A., Doan T. N. L., Lu C., Ghaznavi M., Zhao H., Chen P., Performance of thixotropic gel electrolytes in the rechargeable aqueous Zn/LiMn2O4 battery. ACS Sustainable Chem. Eng. 5, 1804–1811 (2017). [Google Scholar]
- 56.Hoang T. K., Acton M., Chen H. T., Huang Y., Doan T. N. L., Chen P., Sustainable gel electrolyte containing Pb2+ as corrosion inhibitor and dendrite suppressor for the zinc anode in the rechargeable hybrid aqueous battery. Mater. Today Energy 4, 34–40 (2017). [Google Scholar]
- 57.Doughty D., Roth E. P., A general discussion of Li ion battery safety. Electrochem. Soc. Interface 21, 37–44 (2012). [Google Scholar]
- 58.Guo Y.-G., Hu J.-S., Wan L.-J., Nanostructured materials for electrochemical energy conversion and storage devices. Adv. Mater. 20, 2878–2887 (2008). [Google Scholar]
- 59.Ding Y.-L., Xie J., Cao G.-S., Zhu T.-J., Yu H.-M., Zhao X.-B., Single-crystalline LiMn2O4 nanotubes synthesized via template-engaged reaction as cathodes for high-power lithium ion batteries. Adv. Funct. Mater. 21, 348–355 (2011). [Google Scholar]
- 60.Zhan D., Zhang Q., Hu X., Zhu G., Peng T., Single-crystalline LiMn2O4 nanorods as cathode material with enhanced performance for Li-ion battery synthesized via template-engaged reaction. Solid State Ion. 239, 8–14 (2013). [Google Scholar]
- 61.Xue P., Gao D., Chen S., Zhao S., Wang B., Li L., Improved high-temperature capacity retention of the LiMn2O4 cathode lithium-ion battery by ion exchange polymer coating. RSC Adv. 4, 52624–52628 (2014). [Google Scholar]
- 62.Choi N.-S., Yao Y., Cui Y., Cho J., One dimensional Si/Sn-based nanowires and nanotubes for lithium-ion energy storage materials. J. Mater. Chem. 21, 9825–9840 (2011). [Google Scholar]
- 63.Wang Y.-g., Xia Y.-y., A new concept hybrid electrochemical surpercapacitor: Carbon/LiMn2O4 aqueous system. Electrochem. Commun. 7, 1138–1142 (2005). [Google Scholar]
- 64.Wang F. X., Xiao S. Y., Zhu Y. S., Chang Z., Hu C. L., Wu Y. P., Holze R., Spinel LiMn2O4 nanohybrid as high capacitance positive electrode material for supercapacitors. J. Power Sources 246, 19–23 (2014). [Google Scholar]
- 65.Wang F. X., Xiao S. Y., Gao X. W., Zhu Y. S., Zhang H. P., Wu Y. P., Holze R., Nanoporous LiMn2O4 spinel prepared at low temperature as cathode material for aqueous supercapacitors. J. Power Sources 242, 560–565 (2013). [Google Scholar]
- 66.Liu L. L., Wang X. J., Zhu Y. S., Hu C. L., Wu Y. P., Holze R., Polypyrrole-coated LiV3O8-nanocomposites with good electrochemical performance as anode material for aqueous rechargeable lithium batteries. J. Power Sources 224, 290–294 (2013). [Google Scholar]
- 67.Lin W., Chen Y., Li P., He J., Zhao Y., Wang Z., Liu J., Qi F., Zheng B., Zhou J., Xu C., Fu F., Enhanced performance of lithium sulfur battery with a reduced graphene oxide coating separator. J. Electrochem. Soc. 162, A1624–A1629 (2015). [Google Scholar]
- 68.Yang X., Zhang L., Zhang F., Huang Y., Chen Y., Sulfur-infiltrated graphene-based layered porous carbon cathodes for high-performance lithium–sulfur batteries. ACS Nano 8, 5208–5215 (2014). [DOI] [PubMed] [Google Scholar]
- 69.Wu X., Wang Z., Li X., Guo H., Zhang Y., Xiao W., Effect of lithium difluoro (oxalato) borate and heptamethyldisilazane with different concentrations on cycling performance of LiMn2O4. J. Power Sources 204, 133–138 (2012). [Google Scholar]
- 70.Eddahech A., Briat O., Vinassa J.-M., Performance comparison of four lithium–ion battery technologies under calendar aging. Energy 84, 542–550 (2015). [Google Scholar]
- 71.Lesel B. K., Ko J. S., Dunn B., Tolbert S. H., Mesoporous LixMn2O4 thin film cathodes for lithium-ion pseudocapacitors. ACS Nano 10, 7572–7581 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Chang Z., Yang Y., Li M., Wang X., Wu Y., Green energy storage chemistries based on neutral aqueous electrolytes. J. Mater. Chem. A 2, 10739–10755 (2014). [Google Scholar]
- 73.Kim H. W., Yoon H. W., Yoon S.-M., Yoo B. M., Ahn B. K., Cho Y. H., Shin H. J., Yang H., Paik U., Kwon S., Choi J.-Y., Park H. B., Selective gas transport through few-layered graphene and graphene oxide membranes. Science 342, 91–95 (2013). [DOI] [PubMed] [Google Scholar]
- 74.Kim H., Hong J., Park K. Y., Kim H., Kim S. W., Kang K., Aqueous rechargeable Li and Na ion batteries. Chem. Rev. 114, 11788–11827 (2014). [DOI] [PubMed] [Google Scholar]
- 75.Robinson D. M., Go Y. B., Greenblatt M., Dismukes G. C., Water oxidation by λ-MnO2: Catalysis by the cubical Mn4O4 subcluster obtained by delithiation of spinel LiMn2O4. J. Am. Chem. Soc. 132, 11467–11469 (2010). [DOI] [PubMed] [Google Scholar]
- 76.Sun F., Markötter H., Manke I., Hilger A., Kardjilov N., Banhart J., Three-dimensional visualization of gas evolution and channel formation inside a lithium-ion battery. ACS Appl. Mater. Interfaces 8, 7156–7164 (2016). [DOI] [PubMed] [Google Scholar]
- 77.Tang W., Liu L., Zhu Y., Sun H., Wu Y., Zhu K., An aqueous rechargeable lithium battery of excellent rate capability based on a nanocomposite of MoO3 coated with PPy and LiMn2O4. Energy Environ. Sci. 5, 6909–6913 (2012). [Google Scholar]
- 78.Tang W., Tian S., Liu L. L., Li L., Zhang H. P., Yue Y. B., Bai Y., Wu Y. P., Zhu K., Nanochain LiMn2O4 as ultra-fast cathode material for aqueous rechargeable lithium batteries. Electrochem. Commun. 13, 205–208 (2011). [Google Scholar]
- 79.Wang G., Qu Q., Wang B., Shi Y., Tian S., Wu Y., An aqueous electrochemical energy storage system based on doping and intercalation: PPy//LiMn2O4. ChemPhysChem 9, 2299–2301 (2008). [DOI] [PubMed] [Google Scholar]
- 80.Wang X., Hou Y., Zhu Y., Wu Y., Holze R., An aqueous rechargeable lithium battery using coated Li metal as anode. Sci. Rep. 3, 1401 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Naveen A. N., Selladurai S., Investigation on physiochemical properties of Mn substituted spinel cobalt oxide for supercapacitor applications. Electrochim. Acta 125, 404–414 (2014). [Google Scholar]
- 82.Hao Y.-J., Wang Y.-Y., Lai Q.-Y., Zhao Y., Chen L.-M., Ji X.-Y., Study of capacitive properties for LT-Li4Mn5O12 in hybrid supercapacitor. J. Solid State Electrochem. 13, 905–912 (2009). [Google Scholar]
- 83.Chen R., Knapp M., Yavuz M., Heinzmann R., Wang D., Ren S., Trouillet V., Lebedkin S., Doyle S., Hahn H., Ehrenberg H., Indris S., Reversible Li+ storage in a LiMnTiO4 spinel and its structural transition mechanisms. J. Phys. Chem. C 118, 12608–12616 (2014). [Google Scholar]
- 84.Chung K. Y., Ryu C.-W., Kim K.-B., Onset mechanism of Jahn-Teller distortion in 4 V LiMn2O4 and its suppression by LiM0.05Mn1.95O4 (M = Co, Ni) coating. J. Electrochem. Soc. 152, A791–A795 (2005). [Google Scholar]
- 85.Yao F., Güneş F., Ta H. Q., Lee S. M., Chae S. J., Sheem K. Y., Cojocaru C. S., Xie S. S., Lee Y. H., Diffusion mechanism of lithium ion through basal plane of layered graphene. J. Am. Chem. Soc. 134, 8646–8654 (2012). [DOI] [PubMed] [Google Scholar]
- 86.Ai W., Zhou W., Du Z., Sun C., Yang J., Chen Y., Sun Z., Feng S., Zhao J., Dong X., Huang W., Yu T., Toward high energy organic cathodes for Li-ion batteries: A case study of vat dye/graphene composites. Adv. Funct. Mater. 27, 1603603 (2017). [Google Scholar]
- 87.Zhou L.-J., Hou Z. F., Wu L.-M., First-principles study of lithium adsorption and diffusion on graphene with point defects. J. Phys. Chem. C 116, 21780–21787 (2012). [Google Scholar]
- 88.Fan Z.-J., Yan J., Wei T., Ning G.-Q., Zhi L.-J., Liu J.-C., Cao D.-X., Wang G.-L., Wei F., Nanographene-constructed carbon nanofibers grown on graphene sheets by chemical vapor deposition: High-performance anode materials for lithium ion batteries. ACS Nano 5, 2787–2794 (2011). [DOI] [PubMed] [Google Scholar]
- 89.Wen Y., Zhu Y., Langrock A., Manivannan A., Ehrman S. H., Wang C., Graphene-bonded and -encapsulated Si nanoparticles for lithium ion battery anodes. Small 9, 2810–2816 (2013). [DOI] [PubMed] [Google Scholar]
- 90.Liu L., Qiu Y., Mai Y., Wu Q., Zhang H., Influences of neodymium doping on magnetic and electrochemical properties of Li3V2(PO4)3/C synthesized via a sol–gel method. J. Power Sources 295, 246–253 (2015). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/9/e1701010/DC1
fig. S1. Characterizations of graphite, GO, and RGO.
fig. S2. XRD patterns of the Blank cathode treated with hydrazine.
fig. S3. Contact angle measurement results of the hydrazine aqueous solution on the GO-coated and Blank cathodes.
fig. S4. SEM image of the 10RGO cathode obtained by 10 times layer-by-layer deposition of 1RGO.
fig. S5. Rate capability results of the battery with the 10GO cathode (without hydrazine reduction) in comparison with the Blank cathode.
fig. S6. Typical charge-discharge curves of ReHAB with the 10RGO cathode at 0.2 C.
fig. S7. Capacity loss after 1-day float charge of the cathodes with different thicknesses of the G-SEI (1RGO, 5RGO, and 10RGO) and Blank cathodes (dip-coating GO and in situ reduction to RGO).
fig. S8. Capacity loss after 1-day float charge of the 1RGO, 5RGO, 10RGO, and Blank cathodes (directly dip-coating RGO).
fig. S9. Rate capability results of the pristine LMO and RGO-coated LMO particles in the cathode.
fig. S10. Capacity loss after 1-day float charge and rate capabilities of Blank and 10RGO (thermally reduced) cathodes.
fig. S11. The inside and outside of a large-scale battery cell (7 mA·hour) with 9-cm2 cathode surface area for potential applications in the UPS power sources.
fig. S12. Capacity loss after 1-day float charge of the 1RGO, 5RGO, 10RGO, and Blank cathodes in the large-scale cell (fig. S10) (mass loading of active materials, 25 to 28 mg cm−2; L-S method to coat GO followed by water vapor reduction).
fig. S13. Typical charge-discharge curves of the 10RGO-LMO/AC asymmetrical supercapacitor at different current densities.
fig. S14. Cycling stability of the 10RGO-LMO/AC asymmetrical supercapacitor at a current density of 4 A g−1.
fig. S15. Relevant equivalent circuit model for EIS data in Fig. 3.
fig. S16. Rct of the 10RGO and Blank cathodes as a function of time without repeated cycles.
fig. S17. Rs of the 10RGO and Blank cathodes as a function of repeated cycles of charge-discharge.
fig. S18. Rs of the 10RGO and Blank cathodes as a function of time without repeated cycles.
fig. S19. O1s peaks of the 10RGO cathode after the cycling test on the surface (red) and inside the bulk (blue).
fig. S20. Coulombic efficiencies of the 10RGO and Blank samples during 300 cycles at 4 C.
fig. S21. In situ XRD patterns of Blank and 10RGO cathode during discharge process (LixMn2O4, 0.11 < x < 1).
fig. S22. Contact angle results of the aqueous electrolytes on the Blank (left) and 10RGO (right) cathodes.
fig. S23. The cycling properties of the Blank and 10RGO cathodes that are made of inactive silica particles rather than the LMO.
fig. S24. Discharge curves of the 10RGO/silica cathode at a current density of 16 mA g−1.
fig. S25. Typical CVs of the 10RGO cathode at a scan rate of 0.1 to 0.7 mV s−1.
fig. S26. The peak current (ip) in the 10RGO cathode samples (peak 1 in this study) as a function of the square root of the scan rate (v).
Calculations of Li+ diffusivity based on EIS data
Calculations of Li+ diffusivity based on CV plot
Reference (90)