

Abstract
Wnt5a-Ror signaling constitutes a developmental pathway crucial for embryonic tissue morphogenesis, reproduction and adult tissue regeneration, yet the molecular mechanisms by which the Wnt5a-Ror pathway mediates these processes are largely unknown. Using a proteomic screen, we identify the kinesin superfamily protein Kif26b as a downstream target of the Wnt5a-Ror pathway. Wnt5a-Ror, through a process independent of the canonical Wnt/β-catenin-dependent pathway, regulates the cellular stability of Kif26b by inducing its degradation via the ubiquitin-proteasome system. Through this mechanism, Kif26b modulates the migratory behavior of cultured mesenchymal cells in a Wnt5a-dependent manner. Genetic perturbation of Kif26b function in vivo caused embryonic axis malformations and depletion of primordial germ cells in the developing gonad, two phenotypes characteristic of disrupted Wnt5a-Ror signaling. These findings indicate that Kif26b links Wnt5a-Ror signaling to the control of morphogenetic cell and tissue behaviors in vertebrates and reveal a new role for regulated proteolysis in noncanonical Wnt5a-Ror signal transduction.
Research organism: Mouse
Introduction
The Wnt family of extracellular signaling factors orchestrates diverse developmental processes during both embryogenesis and adult tissue homeostasis. Dysfunction of Wnt signaling has been implicated in many human diseases ranging from congenital birth defects to neoplasia (Clevers and Nusse, 2012; Kikuchi et al., 2012). Wnt ligands achieve high functional versatility in part by activating multiple biochemically distinct pathways to regulate diverse cell biological processes (Veeman et al., 2003; Semenov et al., 2007).
Unlike the well-characterized ‘canonical’ Wnt pathway that signals through the transcription co-regulator β-catenin to induce target gene expression, a subset of Wnts signal independently of this Wnt/β-catenin pathway via ‘noncanonical’ mechanisms to control cytoskeleton-driven morphogenetic events such as directed cell movements, changes in cell polarization and cell adhesion (Moon et al., 1993; Veeman et al., 2003). The best-studied noncanonical Wnt, Wnt5a, is critical for the proper morphogenesis of many tissues throughout the developing embryo (Moon et al., 1993; Yamaguchi et al., 1999; Bodmer et al., 2009; Laird et al., 2011; Chawengsaksophak et al., 2012; Miyoshi et al., 2012; Cha et al., 2014). Dysregulation of Wnt5a signaling leads to birth defects and is associated with multiple pathological processes in the adult including cancer metastasis and inflammatory diseases such as atherosclerosis (Kurayoshi et al., 2006; Da Forno et al., 2008; Yamamoto et al., 2010; Jin et al., 2012; Lu et al., 2012; Bhatt and Malgor, 2014; Ford et al., 2014; Lin et al., 2014; Lu et al., 2014; Qin et al., 2015).
A growing consensus suggests that the Ror family of receptor tyrosine kinases mediates Wnt5a-dependent morphogenetic functions in the developing animal (Oishi et al., 2003; Green et al., 2008a; Mikels et al., 2009; Ho et al., 2012). However, how Wnt5a signaling via Ror receptors affects downstream cellular processes remains poorly understood. In a previous study, we found that among the many biochemical activities previously proposed to be downstream of Wnt5a signaling, only the phosphorylation of the cytoplasmic scaffolding protein Dishevelled (Dvl) required the expression of both Wnt5a and Ror proteins (Ho et al., 2012). This finding suggested that Wnt5a-Ror-dependent phosphorylation of Dvl specifically mediates the biological functions of Wnt5a signaling and led us to propose that Ror and Dvl are key components of the noncanonical Wnt5a pathway. The assignment of these proteins to a common pathway is further supported by the observation that human mutations in WNT5A, ROR2, DVL1 and DVL3 can cause Robinow syndrome, a congenital disorder characterized by short-limbed dwarfism and morphological defects in craniofacial and genital structures, demonstrating that the Wnt5a-Ror-Dvl pathway regulates morphogenesis during human development (Afzal et al., 2000; van Bokhoven et al., 2000; Person et al., 2010; Bunn et al., 2015; White et al., 2015, 2016). However, since the function of Dvl phosphorylation is not clear, and Dvl is a common component of several signaling pathways including the canonical Wnt signaling pathway and the planar cell polarity (PCP) pathway, how the Wnt5a-Ror pathway signals to carry out its biological functions remains incompletely understood.
In this study, we conducted a whole phosphoproteome-scale mass spectrometric screen comparing wild-type cells with cells lacking the Ror family of proteins in an effort to identify additional effectors of Wnt5a-Ror signaling. The screen identified a number of candidate proteins whose levels or phosphorylation status was influenced by Wnt5a-Ror signaling, including factors involved in cytoskeletal regulation and cell adhesion, processes crucial for the morphogenesis of tissues. We then focused the remainder of the study on characterizing Kif26b, a member of the kinesin microtubule motor superfamily, which stood out as a particularly promising target of Wnt5a-Ror signaling for the following reasons. Mutations in the C. elegans orthologs of Ror and Kif26b produce similar neuronal migration and axon guidance phenotypes, suggesting that these molecules might function in a common molecular pathway (Wightman et al., 1996; Forrester et al., 1998). Moreover, recent studies demonstrated that Kif26b plays crucial roles in regulating cytoskeleton-driven processes, including cell migration, polarization and adhesion, raising the possibility that Kif26b could function specifically as a cytoskeletal effector of the Wnt5a-Ror pathway (Uchiyama et al., 2010; Guillabert-Gourgues et al., 2016).
Through a series of biochemical studies, we demonstrate that Wnt5a-Ror signaling regulates the steady-state abundance of Kif26b in cells via a mechanism involving the ubiquitin-proteasome system that is independent of the canonical Wnt/β-catenin-dependent pathway. Importantly, gain- and loss-of-function experiments in cultured mesenchymal cells indicate that Wnt5a-Ror-Kif26b signaling modulates mesenchymal cell migration. We also find that perturbation of Kif26b function disrupts a number of Wnt5a/Ror-dependent processes in vivo. For example, in developing zebrafish embryos, mis-expression of Kif26b causes axis and craniofacial malformations, thus mirroring the effects of mis-expression of Wnt5a or Ror in zebrafish. In developing mouse embryos, Kif26b expression is required for primordial germ cells to populate the developing gonad, a process that also requires the expression of Wnt5a or Ror proteins. Taken together, these findings establish Kif26b as a downstream effector of the noncanonical Wnt5a-Ror pathway that regulates cell and tissue behaviors during development.
Results
A phosphoproteomic screen identifies Wnt5a-Ror signaling targets
We sought to discover downstream effectors of Wnt5a-Ror signaling, as these could provide insight into the biochemical regulation and cell biological functions of the pathway. We reasoned that perturbation of upstream pathway components, such as the Ror receptors, would result in alterations in the biochemical regulation of downstream effectors. To test this hypothesis, we took advantage of primary mouse embryonic fibroblasts (MEFs) carrying conditional knockout alleles for the Ror1 and Ror2 genes (Ho et al., 2012) and screened for biochemical changes that occur upon genetic ablation of these genes. We previously showed that embryonic day 12.5 (E12.5) MEFs are a useful physiologically-relevant system for studying Wnt5a-Ror signaling. Not only are these cells derived from mesenchymal tissues that undergo active Wnt5a-Ror signaling in vivo, they continue to express high levels of endogenous Wnt5a, Ror1, Ror2 and Dvl proteins in culture and undergo autocrine/paracrine Wnt5a-Ror signaling without the addition of exogenous Wnt5a (Ho et al., 2012).
Using these conditional knockout MEFs, we performed a phosphoproteome-wide mass spectrometric screen to identify Ror-dependent changes in protein phosphorylation and/or abundance. Our reasoning was that since Wnt5a signaling regulates the phosphorylation state of known downstream components of the Wnt5a-Ror pathway, including Ror1, Ror2 and Dvl proteins (Bryja et al., 2007b; Nishita et al., 2010; Grumolato et al., 2010; Ho et al., 2012) and microarray analysis of primary MEFs lacking both Ror1 and Ror2 proteins failed to identify transcriptional changes relative to wild-type cells, Wnt5a-Ror signaling likely affects cellular functions via a transcription-independent process in MEFs (M.W.S., M.E.G., H.H.H. unpublished data).
To conduct the screen, we employed tandem mass tag (TMT) technology that enables the characterization and quantification of peptides from six experimental conditions in a single, multiplexed mass spectrometric (MS) analysis (Ting et al., 2011). This paradigm enabled the direct comparison of the identity, abundance and post-translational modifications of proteins present in cells in which the Ror proteins have been knocked out relative to wild-type control cells. Specifically, we analyzed phosphopeptides isolated from MEFs derived from E12.5 Ror1f/f; Ror2f/f; CAG-CreER embryos. As we described previously, the Ror1 and Ror2 conditional knockout alleles combined with the 4-hydroxytamoxifen (4-OHT) inducible CAG-CreER allele enable the acute elimination of Ror1 and Ror2 protein expression in vitro (Ho et al., 2012).
For the first four of the six experimental conditions analyzed in the MS screen, we derived MEFs from two separate Ror1f/f; Ror2f/f; CAG-CreER embryos and treated each group with either 4-OHT or a vehicle control (Figure 1A). 4-OHT treatment of Ror1f/f; Ror2f/f; CAG-CreER MEFs effectively eliminated Ror1 and Ror2 protein expression, as measured by western blotting, and reduced the phosphorylation of Dvl2, as measured by a motility shift of this protein on SDS-PAGE gels (Figure 1—figure supplement 1). This result confirmed the acute elimination of Ror1 and Ror2 protein expression and a decrease in Wnt5a-Ror-Dvl signaling in both biological replicates analyzed in the MS screen.
Figure 1. Phosphoproteomic screen identifies putative targets of the Wnt5a-Ror pathway.
(A) Experimental design of the six-plex mass spectrometry analysis to screen for Ror-dependent biochemical cellular changes (see the main text for details of the experimental design). (B) Volcano plot of all phosphopeptides identified and quantified in the experimental group. Significance (y-axis) is plotted against the average fold change (4-OHT/vehicle treated samples) of the two biological replicates (x-axis). Significance values were generated with a two-sample t-test comparing values from the biological replicates. 6498 phosphopeptides were screened in each experiment. ‘Hit’ phosphopeptides, as defined in the text, are marked in blue. All Kif26b phosphopeptides are circled in orange. (C) Volcano plot of all phosphopeptides identified and quantified in the control group. The fold change (4-OHT/vehicle treated samples) is plotted along the x-axis. The position of the phosphopeptides along the y-axis is arbitrary, since there is only one replicate of the control group and no significance value is calculated. Kif26b phosphopeptides are circled in orange.

Figure 1—figure supplement 1. Conditional depletion of Ror1 and Ror2 protein in MEFs.
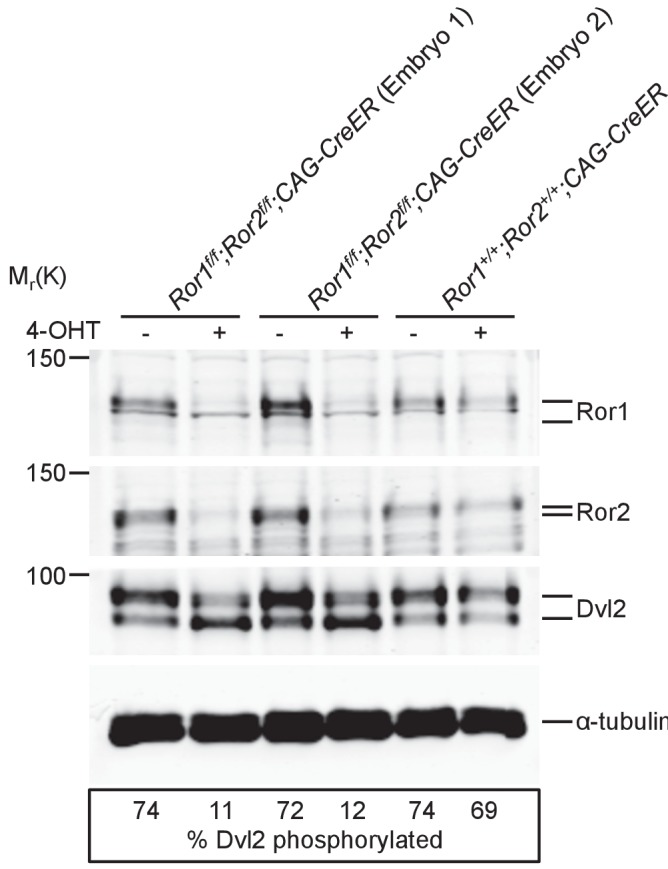
Figure 1—figure supplement 2. Kif26b phosphorylation sites identified in the phosphoproteomic screen.

For the last two of the six experimental conditions in the screen, we cultured control MEFs from a single Ror1+/+; Ror2+/+; CAG-CreER embryo and treated these cells with either 4-OHT or a vehicle control to identify any nonspecific effects due to the addition of 4-OHT and the induction of Cre recombinase expression (Figure 1A). As expected, treatment of these control cells with 4-OHT did not alter the expression of Ror proteins or the phosphorylation of Dvl2, as compared with the vehicle control (Figure 1—figure supplement 1). Together, the two experimental replicates and the control condition allowed us to identify changes in the abundance of phosphorylated proteins and/or specific changes in protein phosphorylation events that are due to the disruption of Ror expression.
6498 unique phosphopeptides, representing 7426 distinct phosphosites, were quantified in the screen (Supplementary file 1). For high confidence identification of biochemical changes that are specific to the cells in which the Ror proteins were inducibly knocked out, phosphopeptides categorized as ‘hits’ had to meet the following criteria: (1) an average of ≥2-fold increase or decrease in the abundance of the phosphopeptide in 4-OHT-treated Ror1f/f; Ror2f/f; CAG-CreER MEFs relative to vehicle-treated Ror1f/f; Ror2f/f; CAG-CreER MEFs; (2) a significant fold change (p<0.05) across two experimental replicates; and (3) a <2-fold increase or decrease in the abundance of the phosphopeptide in 4-OHT-treated Ror1+/+; Ror2+/+; CAG-CreER MEFs relative to vehicle-treated Ror1+/+; Ror2+/+; CAG-CreER MEFs. The 2-fold threshold was chosen to capture candidates whose change in abundance ranked in the top 0.2 percent of all phosphopeptides analyzed in the screen.
Hits in this screen could reflect two possibilities: (1) Ror signaling mediates the phosphorylation or dephosphorylation of a candidate protein, or (2) Ror signaling alters the total level of expression of a candidate protein. The first possibility is more likely when a specific phosphorylated peptide changes in abundance in a Ror-dependent manner while other phosphopeptides from the same protein do not change. The second possibility is more likely when each phosphorylated peptide from a given protein increases or decreases in abundance in a Ror-dependent manner.
A total of fifteen unique phosphopeptides were identified as hits (Figure 1B and Supplementary file 2). Eleven phosphopeptides increased in abundance and four phosphopeptides decreased in abundance upon Ror depletion. Eight of the phosphopeptides that increased in abundance mapped to the same protein—the kinesin superfamily member Kif26b—making it a high-confidence candidate target of Wnt5a-Ror signaling (Figure 1B,C and Figure 1—figure supplement 2). Moreover, all eight Kif26b phosphopeptides exhibited a significant increase in abundance following genetic ablation of Ror expression in both experimental replicates (Figure 1B and Supplementary file 2). Taken together, these observations strongly suggest that Wnt5a-Ror signaling leads to a decrease in the level of Kif26b protein expression.
Kif26b is a downstream target of noncanonical Wnt5a-Ror signaling
Kif26b is a highly conserved atypical kinesin of the Kinesin-11 family, which includes Kif26a and Kif26b, two proteins whose developmental and cellular functions have only begun to be revealed in recent years (Uchiyama et al., 2010; Hirokawa and Tanaka, 2015; Guillabert-Gourgues et al., 2016). To further test the hypothesis that Wnt5a-Ror signaling leads to a decrease in Kif26b protein expression, as suggested by our MS screen, we generated polyclonal antibodies that specifically recognize the Kif26b protein. We validated the specificity of these antibodies by western blotting of protein extracts obtained from wild-type MEFs, multiple Kif26b shRNA-knockdown MEFs or Kif26b-/- MEFs (Figure 2—figure supplement 1A–C). We found that the anti-Kif26b antibodies recognized protein bands at the predicted size of Kif26b (~220 kD) in wild-type MEF lysates but not in the Kif26b knockout or knockdown MEF lysates, confirming that our antibodies specifically recognize endogenous Kif26b.
Using these antibodies, we assessed the expression of Kif26b protein in primary MEFs in which Ror1 and Ror2 proteins had been inducibly knocked out. We found that Kif26b levels were elevated in Ror1f/f; Ror2f/f; CAG-CreER MEFs treated with 4-OHT as compared to MEFs with the same genotype treated with a vehicle control (Figure 2A). This finding suggests that Wnt5a-Ror signaling negatively regulates total Kif26b protein expression, as opposed to selectively catalyzing the dephosphorylation of multiple phosphorylation sites across the Kif26b protein. Moreover, this finding validates that our MS screening approach reliably identifies cellular proteins whose expression is regulated by Wnt5a-Ror signaling.
Figure 2. Kif26b protein levels are regulated by the expression of Wnt5a and Ror proteins in primary mouse embryonic fibroblasts.
(A) Immunoblots of Kif26b protein, Ror1 protein, Ror2 protein and Dvl2 protein in primary MEFs derived from two E12.5 Ror1f/f; Ror2f/f; CAG-CreER mice. 4-OHT or vehicle control was added for 72 hr prior to lysis. Results of two independent biological replicates are shown. (B) Immunoblots of Kif26b protein, Ror1 protein, Ror2 protein and Dvl2 protein in primary MEFs derived from littermate E12.5 Wnt5a+/+ and Wnt5a-/- mice. Results of two independent biological replicates are shown. (C) Immunoblots of Kif26b protein, Ror1 protein and Dvl2 protein in primary MEFs derived from E12.5 Wnt5a-/- mice. Recombinant Wnt5a protein was added for 6 hr prior to lysis at the indicated dose. (D) Immunoblots of Kif26b protein, Ror1 protein and Dvl2 protein in primary MEFs derived from E12.5 Wnt5a-/- mice. 0.1 μg/ml recombinant Wnt5a protein was added for the indicated amount of time prior to lysis. (E) Plot showing relative mRNA expression of Kif26b and β-actin as measured by RT-qPCR in primary MEFs derived from E12.5 Wnt5a-/- mice, with 1 hr or 6 hr of Wnt5a stimulation. The y-axis represents fold change relative to expression levels in unstimulated cells. Error bars represent ± SEM calculated from three technical replicates. t-test (unpaired) was determined for the following comparisons: Kif26b 1 hr vs. 6 hr, p=0.249, not significant; β-actin 1 hr vs. 6 hr, p=0.320, not significant. (F) Immunoblots of Kif26b protein and phospho-Lrp6 (serine 1490) protein in primary MEFs derived from E12.5 Wnt5a-/- mice. Recombinant Dkk-1 (0.1 μg/ml) or vehicle control was added 8 hr prior to lysis. Recombinant Wnt3a protein, Wnt5a protein or vehicle control was added for 6 hr prior to lysis. (G) Immunoblots of Kif26b protein in primary MEFs derived from E12.5 Wnt5a-/- mice. IWR-1 or vehicle control was added 7 hr prior to lysis. Recombinant Wnt3a protein, Wnt5a protein or vehicle control was added for 6 hr prior to lysis. α-tubulin was used for loading controls in all experiments. All immunoblot samples were normalized by BCA assays for total protein.

Figure 2—figure supplement 1. Validation of anti-Kif26b antibodies.

To test directly whether Wnt5a signaling regulates Kif26b protein levels, we assessed the expression of Kif26b in primary MEFs in which Wnt5a had been knocked out. We observed that Wnt5a-/- MEFs also had a higher level of Kif26b protein relative to wild-type control MEFs, indicating that Wnt5a, like Ror1 and Ror2, negatively regulates the steady-state level of Kif26b expression (Figure 2B). This finding suggests that Wnt5a signaling via Ror proteins leads to a decrease in Kif26b protein expression.
We next investigated whether acute activation of Wnt5a-Ror signaling by the addition of exogenous Wnt5a triggers a decrease in Kif26b protein expression in MEFs. We stimulated Wnt5a-/- MEFs with purified, recombinant Wnt5a and found that this treatment led to a decrease in Kif26b protein expression in a dose-dependent manner (Figure 2C). The decrease in Kif26b was accompanied by a commensurate increase in Ror1 and Dvl2 phosphorylation, a readout of Wnt5a-Ror signaling (Figure 2C). These findings suggest that Wnt5a induces the downregulation of endogenous Kif26b expression as the Wnt5a-Ror-Dvl pathway becomes activated.
We found that the Wnt5a-induced downregulation of Kif26b expression is first detected 1 hr after Wnt5a stimulation and that Kif26b expression is maximally decreased after 6 hr (Figure 2D). The kinetics of Kif26b protein downregulation closely paralleled those of Ror1 and Dvl2 phosphorylation in response to acute Wnt5a stimulation (Figure 2D), suggesting that Wnt5a signaling regulates these downstream biochemical events in a coordinated manner. In addition, we found that stimulation with Wnt5a does not change the level of Kif26b mRNA in Wnt5a-/- MEFs, as assayed by reverse transcription-quantitative PCR (RT-qPCR), throughout the course of the experiment (Figure 2E), suggesting that Wnt5a-dependent downregulation of Kif26b protein levels occurs post-transcriptionally. Together, these findings establish that Wnt5a-Ror signaling leads to a decrease in the steady-state levels of Kif26b protein expression in MEFs and validate Kif26b as a bona fide target of the Wnt5a-Ror signaling pathway.
The Wnt5a-Ror pathway is generally thought to operate via a noncanonical, β-catenin-independent mechanism (Green et al., 2008b). To determine whether Wnt5a-Ror signaling induces the downregulation of Kif26b expression via a noncanonical Wnt signaling mechanism, we tested whether blocking the canonical Wnt/β-catenin pathway with Dkk-1, an antagonist of the β-catenin-dependent Wnt pathway that binds and prevents the phosphorylation of the canonical Wnt signaling pathway co-receptors Lrp5 and Lrp6, inhibits the Wnt5a-induced decrease in Kif26b protein in MEFs (Bafico et al., 2001). We found that exposure of Wnt5a-/- MEFs to exogenous Wnt5a protein induced Kif26b downregulation to a similar degree with and without the addition of Dkk-1 (Figure 2F). To ensure that the Dkk-1 protein used in the experiment was active, we assessed whether the same concentration of Dkk-1 was capable of blocking signaling by Wnt3a, a prototypic canonical Wnt that induces the phosphorylation of the Wnt receptor Lrp6. We found that Wnt3a-dependent phosphorylation of Lrp6 was completely blocked in the presence of Dkk-1 (Figure 2F). These findings suggest that Wnt5a-dependent regulation of Kif26b occurs via a noncanonical Wnt signaling mechanism that is independent of the canonical Wnt/β-catenin pathway.
To verify that Wnt5a regulation of Kif26b occurs via a noncanonical Wnt signaling mechanism, we next tested whether inhibiting the Wnt/β-catenin pathway at a more downstream step blocks Wnt5a-dependent downregulation of Kif26b levels. For this purpose, we used IWR-1, a small molecule that inhibits Wnt/β-catenin signaling by stabilizing Axin2, a key component of the β-catenin destruction complex (Lee et al., 2003; Chen et al., 2009). Similar to our findings using Dkk-1 to block the canonical Wnt/β-catenin pathway at the receptor level, we found that pre-treatment of Wnt5a-/- MEFs with IWR-1 did not block the ability of Wnt5a or Wnt3a to induce the downregulation of Kif26b protein expression (Figure 2G). These results further support the conclusion that Wnt5a-Ror signaling leads to the downregulation of Kif26b protein expression via a noncanonical, β-catenin-independent mechanism.
It is interesting to note that the addition of exogenous Wnt3a protein, classically considered a canonical Wnt, also led to a decrease in the expression of Kif26b in Wnt5a-/- MEFs, even in the presence of Dkk-1 or IWR-1 (Figure 2F,G). These findings indicate that exogenous Wnt3a also signals via a noncanonical Wnt signaling mechanism to downregulate the expression of Kif26b and supports the emerging view that the distinction between canonical and noncanonical Wnt signaling is not strictly determined at the level of Wnt ligands (van Amerongen et al., 2008). However, it is also possible that there is some specificity to Wnt3a and Wnt5a signaling in the developing embryo that is lost when these factors are studied using cultured MEFs.
Wnt5a signals the degradation of Kif26b via the ubiquitin-proteasome pathway
We next investigated the biochemical mechanisms by which Wnt5a-Ror signaling leads to decreased Kif26b protein expression. To more accurately quantify Kif26b levels in live cells, we developed a flow cytometry-based reporter assay using NIH/3T3 cell lines stably expressing a GFP-Kif26b fusion protein. We chose NIH/3T3 cells for the reporter assay because these cells express key components of the Wnt5a-Ror pathway, including Ror1, Ror2, Dvl2 and Kif26b, which we found to be similarly regulated by Wnt5a as in MEFs (Figure 3—figure supplement 1A,B). Furthermore, NIH/3T3 cells are an immortalized cell line derived from mouse embryonic mesenchymal cells that undergo morphogenetic movements during development (Todaro and Green, 1963), and they have been used previously to study cell behaviors in the context of Wnt5a-Ror signaling (Endo et al., 2012). Western analysis and immunostaining of these NIH/3T3 cell lines confirmed that the GFP-Kif26b protein is stably expressed in these cells (Figure 3—figure supplement 2A–C).
Given that Wnt5a signaling acutely downregulates the level of endogenous Kif26b expression in MEFs, we tested whether Wnt5a similarly downregulates GFP-Kif26b levels in the reporter NIH/3T3 cells. Treatment of these cells with exogenous recombinant Wnt5a protein induced an approximately 50% decrease in GFP-Kif26b expression as detected by three independent methods: flow cytometry (Figure 3A), western analysis (Figure 3—figure supplement 2A) and time-lapse microscopy (Video 1). These results indicate that Wnt5a treatment leads to a decrease in GFP-Kif26b in NIH/3T3 cells similar to that observed for endogenous Kif26b in MEFs, and that the GFP-Kif26b reporter can be used reliably to monitor the decrease in Kif26b protein expression in response to Wnt5a signaling. To the best of our knowledge, this is the first fluorescence-based reporter for real-time measurement of a non-transcriptional Wnt5a-Ror signaling response in live cells. For the remainder of this article, the Wnt5a-Ror-Kif26b reporter assay will be referred to as the WRK reporter assay.
Figure 3. Wnt5a downregulates Kif26b levels via a ubiquitin/proteasome-dependent mechanism.
(A) Flow cytometry histograms depicting the downregulation of GFP-Kif26b fluorescence in the WRK reporter cell line after Wnt5a stimulation (0.2 μg/ml Wnt5a) for 6 hr. (B) Dose-response curve showing GFP-Kif26b downregulation as a function of Wnt5a concentration, as measured in the WRK reporter assay. (C) The kinetics of GFP-Kif26b turnover in the absence or presence of Wnt5a stimulation, as measured in the WRK reporter assay. Cycloheximide was used to block new protein synthesis in the reporter cells. (D) Flow cytometry histograms depicting the effect of epoxomicin treatment (1 μM) on the ability of Wnt5a (0.2 μg/ml) to downregulate GFP-Kif26b fluorescence in the WRK reporter assay. DMSO was used as the drug vehicle control. (E) Flow cytometry histograms depicting the effect of PYR-41 treatment (50 μM) on the ability of Wnt5a (0.2 μg/ml) to downregulate GFP-Kif26b fluorescence in the WRK reporter assay. DMSO was used as the drug vehicle control. (F) Quantification of the effects of DMSO, epoxomicin and PYR-41 on the ability of Wnt5a to downreagulate GFP-Kif26b fluorescence in the WRK reporter assay, as shown in (D) and (E). Error bars represent ± SEM calculated from three technical replicates. t-tests were determined for the following comparisons: DMSO vs. epoxomicin, p<0.001; DMSO vs. PYR-41, p<0.001.

Figure 3—figure supplement 1. Expression of Wnt5a-Ror signaling components in MEFs and NIH/3T3 cells.

Figure 3—figure supplement 2. Characterization of the GFP-Kif26b (WRK) reporter line.

Video 1. Time-lapse fluorescent confocal microscopy reveals rapid Wnt5a-dependent downregulation of the GFP-Kif26b signal in the NIH/3T3 reporter cell line.
Bright signal reflects GFP-Kif26b fluorescence. Buffer or 0.2 μg/mL recombinant Wnt5a was added at the start of the imaging session. Cells were imaged every 10 min for 16 hr at 40x magnification.
We next used the WRK reporter assay in conjunction with flow cytometry to determine the dose-response relationship between Wnt5a and GFP-Kif26b. We found that nanomolar amounts of Wnt5a induced the downregulation of GFP-Kif26b expression with a calculated EC50 of 79.8 ng/ml (or 2.1 nM; Figure 3B). This response to Wnt5a occurs at a Wnt5a concentration that is similar to that of other previously reported Wnt-induced cellular responses (Bryja et al., 2007a, 2007b; Witze et al., 2008; Ho et al., 2012; van Amerongen et al., 2012; Witze et al., 2013; Park et al., 2015; Connacher et al., 2017), suggesting that Wnt5a-induced Kif26b downregulation is a physiologically relevant response to Wnt5a.
Wnt5a-Ror signaling could downregulate the steady state level of Kif26b protein either by decreasing Kif26b synthesis or by increasing Kif26b turnover. Given that exposure of fibroblasts to Wnt5a leads to a decrease in Kif26b expression over minutes to hours but does not lead to a reduction in the level of Kif26b mRNA (Figure 2D,E), we favored the latter possibility. To directly measure the rate of Kif26b turnover, we treated WRK reporter cells with cycloheximide to block new protein synthesis and then used flow cytometry to measure the effect of Wnt5a treatment on the rate of GFP-Kif26b turnover. Consistent with the hypothesis that Wnt5a treatment leads to the increased degradation of Kif26b, we found that Wnt5a accelerated the turnover of GFP-Kif26b in the reporter cells (Figure 3C). In the absence of Wnt5a stimulation, 80.2% of the GFP-Kif26b signal remained in the cells 6 hr after the initiation of the cycloheximide treatment. By contrast, in the presence of Wnt5a stimulation, only 33.2% of the GFP-Kif26b signal remained in the same period. These results strongly suggest that Wnt5a treatment leads to a decrease in the steady-state levels of Kif26b expression in cells by promoting Kif26b turnover.
We next investigated whether Wnt5a downregulates Kif26b expression by increasing the rate of Kif26b degradation via the ubiquitin-proteasome system (UPS). To test this hypothesis, we asked whether a selective inhibitor of the proteasome, epoxomicin, blocks downregulation of Kif26b expression in response to Wnt5a (Meng et al., 1999). Pre-treatment of the WRK reporter line with epoxomicin strongly blocked Wnt5a-induced Kif26b downregulation (Figure 3D,F). This result suggests that Wnt5a-induced downregulation of Kif26b occurs via proteasome-dependent degradation.
To determine whether protein ubiquitination is specifically required for Wnt5a-induced Kif26b degradation, we tested whether an inhibitor of the E1 enzyme required for ubiquitin activation, PYR-41, also blocks Wnt5a-induced Kif26b downregulation (Yang et al., 2007). Pre-treatment of the WRK reporter cells with PYR-41 blocked Wnt5a-induced Kif26b downregulation to an extent similar to that observed upon epoxomicin treatment (Figure 3E,F). This finding provides further evidence that downregulation of Kif26b expression induced by Wnt5a occurs via the UPS. We conclude from these results that exposure to Wnt5a leads to the downregulation of the steady-state levels of Kif26b expression by promoting Kif26b ubiquitin- and proteasome-dependent degradation.
Frizzled and Dishevelled proteins mediate Wnt5a-induced Kif26b degradation
Our pharmacological inhibitor experiments suggest that the WRK reporter assay might be used to interrogate other molecular components of the Wnt5a-Ror signaling pathway that operate upstream of Kif26b. To test this idea, we first investigated a possible role of Frizzled (Fzd) proteins in Wnt5a-dependent Kif26b degradation. Fzds make up a family of 10 seven-transmembrane domain receptor proteins that function as co-receptors in canonical Wnt/β-catenin signaling and as polarity determinants in the PCP pathway (Vinson and Adler, 1987; Bhanot et al., 1996). Recent work additionally implicates Fzd proteins in aspects of noncanonical Wnt function as well as in the phosphorylation of Ror2 in cultured cells (Habas et al., 2001; Nishita et al., 2010; Grumolato et al., 2010; Sato et al., 2010). Moreover, protein sequence homology analysis and in vivo mouse genetic studies revealed that Fzd1, Fzd2 and Fzd7 form a distinct sub-family that functions redundantly to control tissue morphogenetic events such as convergent extension of embryonic tissues and closure of the palate and ventricular septum (Yu et al., 2010; Yu et al., 2012). As many of these developmental processes also require the Wnt5a-Ror pathway, we hypothesized that certain Fzd proteins, such as those in the Fzd1, Fzd2 and Fzd7 sub-family, might participate in Wnt5a-Ror-Kif26b signaling.
To test whether Fzd proteins mediate Wnt5a-Ror-Kif26b signaling, we first took a loss-of-function approach using Shisa proteins, which are Wnt signaling regulators that inhibit Fzd processing and trafficking by sequestering Fzds in the endoplasmic reticulum (Yamamoto et al., 2005). We found that viral transduction of Shisa2 expression, but not Cas9 expression as a control, partially blocked the ability of Wnt5a to induce Kif26b degradation in the WRK reporter cell line (Figure 4A,B). This observation suggests that the Fzd family plays a role in Wnt5a-dependent regulation of Kif26b.
Figure 4. Involvement of Fzd and Dvl proteins in Kif26b degradation.
(A) Flow cytometry histograms depicting the effect of ectopic Shisa2 expression on the ability of Wnt5a to induce GFP-Kif26b degradation in the WRK reporter assay. Mouse Shisa2 was expressed via lentiviral transduction. A lentivirus expressing Cas9 was used as the control. The effect of Wnt5a or control buffer treatment on the WRK reporter line without virus infection is shown as a reference. (B) Quantification of effects of the control virus (Cas9) and the Shisa2 virus on the ability of Wnt5a to downregulate the GFP-Kif26b fluorescence in the WRK reporter assay, as shown in (A). Error bars represent ± SEM calculated from three technical replicates. t-tests were determined for the following comparisons: control virus vs. Shisa2 virus, p<0.001; control virus vs. no virus, p=0.0957 (not significant). (C) Flow cytometry histograms depicting the effect of ectopic Fzd1 or Fzd7 expression on GFP-Kif26b levels in the WRK reporter assay. Mouse Fzd1 and Fzd7 were expressed via lentiviral transduction. A lentivirus expressing Cas9 was used as the control. (D) Quantification of the median GFP-Kif26b fluorescence in the WRK reporter cell line infected with a Fzd1 virus, a Fzd7 virus, or a Cas9 control virus. Error bars represent ± SEM calculated from six technical replicates. t-tests were determined for the following comparisons: control virus vs. Fzd1 virus, p<0.001; control virus vs. Fzd7 virus, p<0.001. (E) Flow cytometry histograms depicting the effect of ectopic Dvl1 expression on GFP-Kif26b levels in the WRK reporter assay. Dvl1 was expressed via lentiviral transduction. A lentivirus expressing Cas9 was used as the control. (F) Quantification of the median GFP-Kif26b fluorescence in the WRK reporter cell line infected with a Dvl1-expressing virus, or a Cas9-expressing control virus. Error bars represent ± SEM calculated from three technical replicates. t-test was determined for control virus vs. Dvl1 virus, p<0.001.

Figure 4—figure supplement 1. Comparison of the abilities of distinct Fzd subfamily members to induce Kif26b degradation.

To more directly test the involvement of Fzds in Wnt5a-Kif26b signaling, we asked whether overexpression of Fzds could induce Kif26b degradation in the absence of exogenously added Wnt5a. We first focused on members of the Fzd1, Fzd2 and Fzd7 sub-family, as the mouse knockout phenotypes of these Fzds are most consistent with a functional interaction with the Wnt5a-Ror pathway (Yu et al., 2010; Yu et al., 2012). Interestingly, we found that lentiviral transduction of Fzd1 or Fzd7 expression, but not of the negative control Cas9, constitutively induced Kif26b degradation as measured in the WRK assay (Figure 4C,D). This finding, taken together with the decreased Wnt5a-Ror-Kif26b signaling upon expression of the Fzd inhibitor Shisa2, strongly suggests that Fzd family proteins are involved in Wnt5a regulation of Kif26b degradation, possibly as co-receptors together with members of the Ror family of proteins.
We also considered the possibility that functional specificity might exist among the different Fzd sub-families. We therefore used the WRK assay to test multiple members from each Fzd sub-family, as defined previously (Yu et al., 2010; Yu et al., 2012). Interestingly, we found that all the Fzd family members that we tested were able to induce Kif26b degradation (Figure 4—figure supplement 1). However, it is possible that the specificity of Fzd protein function is lost when these proteins are overexpressed.
We next investigated the role of Dvl proteins in Wnt5a-Ror-dependent Kif26b degradation. Our previous study identified Dvl phosphorylation as a specific downstream target of Wnt5a-Ror signaling (Ho et al., 2012), and a recent study demonstrated that the Dvl and Kif26b proteins physically interact (Guillabert-Gourgues et al., 2016). Moreover, we found that Wnt5a-induced Kif26b degradation occurs with similar kinetics as Wnt5a-induced Dvl phosphorylation (Figure 2D). However, it remains unknown whether Dvl proteins are required for Wnt5a-dependent degradation of Kif26b. Since the presence of three Dvl genes in the mammalian genome makes loss-of-function analysis of Dvl proteins challenging, we took a gain-of-function approach to determine if Dvl protein expression affects Wnt5a-Ror-dependent Kif26b degradation. Notably, a similar overexpression approach was previously used to demonstrate a role for Dvl proteins in Wnt/β-catenin signaling, as overexpression of Dvl proteins in Xenopus embryos induces axis duplication recapitulating overexpression of canonical Wnts (Sokol et al., 1995). Using the WRK reporter assay, we found that overexpression of Dvl1, but not overexpression of a control protein Cas9, led to an increase in Kif26b degradation (Figure 4E,F), mimicking the effects of Wnt5a stimulation or Fzd overexpression. Taken together, these findings establish a functional role for both Fzd receptors and Dvl1 (and likely other Dvl proteins) in Wnt5a-Ror-dependent Kif26b degradation.
The Wnt5a-Ror-Kif26b signaling cassette directs the migratory behavior of cells
Genetic studies in C. elegans have shown that mutations in the nematode orthologs of Kif26b (vab-8) and Ror (cam-1) cause similar polarized cell migration and axon guidance phenotypes, and a recent study in human umbilical vein endothelial cells (HUVECs) demonstrates a physical interaction between Kif26b and Dvl3 (Forrester et al., 1998; Wolf et al., 1998; Chien et al., 2015; Guillabert-Gourgues et al., 2016). These findings, together with the observations reported above, raise the possibility that the cell biological effects of noncanonical Wnt signaling are mediated by the Wnt5a-Ror-dependent degradation of Kif26b. To test this hypothesis, we employed both gain- and loss-of-function approaches in cultured NIH/3T3 cells, the same cells used for the WRK assays. Western analysis of the NIH/3T3 cell lines used in the WRK assay showed that GFP-tagged Kif26b protein is overexpressed relative to endogenous Kif26b in NIH/3T3 cells (Figure 5—figure supplement 1A), indicating that these cells could be used to assess the effects of Kif26b overexpression on cell responses. For examining the effects of loss of Kif26b expression on cellular responses in NIH/3T3 cells, we used CRISPR/Cas9-mediated genome editing to generate stable cell lines in which the expression of Kif26b is knocked out (Figure 5—figure supplement 1B and C). Western blot analysis verified complete elimination of Kif26b protein relative to a control cell line expressing the Cas9 endonuclease without a guide RNA (Figure 5—figure supplement 1B). We next confirmed that neither GFP-Kif26b overexpression nor loss of Kif26b affects cell proliferation or survival through mitotic index quantification and TUNEL staining, respectively (Figure 5—figure supplement 1D–G), indicating that these NIH/3T3 cell lines could be used to study other possible Kif26b-dependent cellular responses.
We employed an automated kinetic wound-healing assay that provides a quantitative, integrated readout of cell morphogenesis such as cell polarization, cell motility and cell adhesion by measuring the wound closure efficiency of cells under different experimental conditions (Gujral et al., 2014a). The wound-healing assay has been used previously to assess effects of noncanonical Wnt signaling (Gujral et al., 2014a, 2014b). Using this assay, we asked if the level of Kif26b expression affects wound closure efficiency. We found that Kif26b-knockout cells exhibit a decrease in wound closure efficiency relative to control cells while cell lines overexpressing GFP-Kif26b exhibit an enhanced rate of wound closure efficiency relative to control cells (Figure 5A,B). These findings suggest that Kif26b may promote cell migration and are consistent with previous observations in HUVECs where Kif26b expression was correlated with increased directional cell migration (Guillabert-Gourgues et al., 2016). Taken together, these findings suggest that by controlling the rate of Kif26b protein degradation, Wnt5a-Ror signaling regulates cell migration.
Figure 5. Wnt5a-Kif26b signaling modulates the migratory behavior of NIH/3T3 cells.
(A) Relative wound density of two separate NIH/3T3 cell lines in which Kif26b expression is knocked out using CRISPR/Cas9 genome editing, and one control Cas9-expressing NIH/3T3 cell line in a kinetic wound-healing assay. Data are the mean of five independent samples and error bars indicate SEM. t-tests (unpaired) were determined for the following comparisons: control NIH/3T3 vs. gRNA1, p<0.05; control NIH/3T3 vs. gRNA2, p<0.05. (B) Relative wound density of a GFP-Kif26b expressing NIH/3T3 cell line, treated with or without Wnt5a (0.2 μg/ml, added at the 0 hr time point) and a control NIH/3T3 cell line in a kinetic wound-healing assay. Data are the mean of five independent samples and error bars indicate SEM. t-tests (unpaired) were determined for the following comparisons: control NIH/3T3 vs. Kif26b overexpression, p<0.05 (at every time point except the last time point); Kif26b overexpression vs. Kif26b overexpression +Wnt5a, p<0.05 (from 3 to 38 hr time points).

Figure 5—figure supplement 1. Overexpression of GFP-Kif26b or knockout of Kif26b expression does not affect the proliferation rate or survival of NIH/3T3 cells.

Figure 5—figure supplement 2. Wnt5a downregulates the cellular levels of GFP-Kif26b.

Figure 5—figure supplement 3. Wnt5a treatment has no effect on the wound closure efficiency of Kif26b knockout NIH/3T3 cells.

Given that exposure of NIH/3T3 cells to Wnt5a acutely downregulates Kif26b protein expression, we next tested whether this treatment affects the migration of these cells in the wound-healing assay. We found that Wnt5a treatment decreases the wound closure efficiency of cells overexpressing GFP-Kif26b to a rate approximating that of a control NIH/3T3 cell line in which GFP-Kif26b is not overexpressed (Figure 5B). This Wnt5a-mediated decrease in wound closure efficiency was correlated with a concomitant decrease in the cellular abundance of Kif26b, suggesting that the specific degradation of Kif26b could underlie the decrease in wound closure efficiency (Figure 5—figure supplement 2). Importantly, Wnt5a stimulation of NIH/3T3 cells in which Kif26b expression had been knocked out via CRISPR/Cas9-mediated genome editing resulted in no decrease in wound closure efficiency (i.e. had no effect on cell migration) (Figure 5—figure supplement 3), suggesting that Kif26b expression is required for Wnt5a-dependent changes in cell migration. Taken together, these results indicate that one key function of Kif26b is to promote cell migration, and that Wnt5a signaling may control the extent of cell migration by regulating the degradation of the Kif26b protein.
In vivo perturbation of Kif26b function produces phenotypes characteristic of noncanonical Wnt5a-Ror signaling defects
We next sought to determine whether in vivo perturbation of Kif26b during embryonic development results in phenotypes that are consistent with an important function for Kif26b in Wnt5a-Ror signaling during embryonic development. A previously used approach for determining if a given signaling molecule functions as part of the noncanonical Wnt pathway was to determine if mis-expression of the signaling molecule leads to a phenotype similar to that observed when Wnt5a or Rors are mis-expressed in developing Xenopus or zebrafish embryos (Moon et al., 1993; Hikasa et al., 2002; Bai et al., 2014; Habas et al., 2001). Mis-expression of either Wnt5a or Ror2 in these embryos produces tissue morphogenesis defects, including defective convergent extension movements and a shortened or bent body axis, which are characteristic of disrupted noncanonical Wnt signaling. We therefore asked whether mis-expression of Kif26b protein in developing zebrafish embryos similarly induced phenotypes typical of abnormal noncanonical Wnt signaling.
To mis-express proteins in the developing zebrafish embryo, we microinjected in vitro-transcribed mRNA into one-cell stage embryos. Replicating previous reports (Moon et al., 1993; Bai et al., 2014), microinjection of Wnt5a mRNA caused axis truncation and bending phenotypes (Figure 6A,B). Strikingly, microinjection of Kif26b mRNA into one-cell zebrafish embryos also caused axis truncation and bending phenotypes resembling those caused by mis-expression of Wnt5a (Figure 6A,B). These phenotypes were rarely observed in uninjected embryos or in embryos injected with a negative control mRNA of similar size, Cas9, indicating that the axis truncation and bending phenotypes produced by Kif26b mis-expression are specific (Figure 6A,B). Together, these findings suggest that Kif26b specifically affects morphogenetic movements of cells in developing embryos in a manner similar to that of Wnt5a or Ror2 and support a model whereby Kif26b functions as part of a noncanonical Wnt5a-Ror regulatory cassette that regulates morphogenetic movements during embryogenesis.
Figure 6. In vivo perturbation of Kif26b expression in zebrafish and mouse embryos induces phenotypes characteristic of Wnt5a-Ror signaling defects.

(A) Representative images showing the effects of Wnt5a and Kif26b mis-expression on zebrafish embryonic tissue morphogenesis. Microinjection of Wnt5a mRNA did not produce any embryos that were scored as ‘normal’, so the image of an uninjected embryo is shown to represent normal embryos. (B) Quantification of the effects of Wnt5a and Kif26b mis-expression on zebrafish embryonic tissue morphogenesis. Images were taken from embryos at 50 hr post fertilization (50 hpf). Data for each experimental condition are pooled from at least three independent injection experiments. (C) Representative images showing immunofluorescence staining of SEAA1 to mark PGCs that have successfully entered the gonads of E11.5 Kif26b+/+ or Kif26b-/- mouse embryos. Scale bar represents 100 μm. (D) Quantification of the numbers of PGCs per gonad in E11.5 Kif26b+/+ or Kif26b-/- mouse embryos. Error bars represent ± SD calculated from independent biological samples (Kif26b+/+, n = 2; Kif26b-/-, n = 4). t-test (unpaired) was determined for Kif26b+/+ vs. Kif26b-/- (p<0.01).
To investigate further if Kif26b functions as part of the Wnt5a-Ror pathway during mouse development, we analyzed the development of primordial germ cells (PGCs), a process previously shown to require the Wnt5a-Ror pathway (Laird et al., 2011; Chawengsaksophak et al., 2012). During mouse embryogenesis, PGCs are specified from the epiblast at ~E7.25 and subsequently migrate through the hindgut and dorsal mesentery to populate the gonadal ridges by E11.5. PGCs that fail to enter the gonad are eliminated by programmed cell death (McLaren, 2003; Laird et al., 2008). Loss-of-function mutations in Wnt5a or Ror2 alleles result in a substantial decrease in the number of PGCs that successfully colonize the gonad at E11.5 as compared to wild-type controls (~75% fewer in Wnt5a and ~50% fewer in Ror2 mutants) (Chawengsaksophak et al., 2012; Laird et al., 2011), indicating that Wnt5a-Ror signaling is required for the proper colonization of the gonadal ridges by migrating PGCs. We reasoned that if Kif26b mediates biological activities of the Wnt5a-Ror pathway during PGC development, genetic perturbation of Kif26b might also disrupt PGC colonization of the gonads.
To test this hypothesis, we quantified the number of PGCs in the gonadal ridges of E11.5 Kif26b-/- embryos by whole-mount SSEA1 (a marker of PGCs) staining. The mean number of SSEA1-positive PGCs in the Kif26b-/- gonads (372.8 ± 74.95, n=4) was decreased significantly to 44.9% of Kif26b+/+ littermate controls (829.5 ± 88.553, n=2) (Figure 6C,D). The relative similarity of the PGC depletion phenotypes observed in E11.5 Kif26b-/-, Ror2-/- and Wnt5a-/- mouse embryos suggest that Kif26b functions in a common signaling pathway with Wnt5a and Ror proteins to orchestrate PGC development. This experiment, taken together with the mis-expression analysis in zebrafish, provides in vivo evidence that Kif26b contributes to Wnt5a-Ror signaling in multiple tissues during embryonic development.
Discussion
Since the seminal discovery that certain Wnt proteins can signal independently of β-catenin-mediated transcription to affect organ and tissue morphogenesis during development (Moon et al., 1993), few downstream effectors of this signaling system have been identified and validated in biological systems. In our previous work, we used in vivo mouse genetics to demonstrate that Ror receptors are essential mediators of a core noncanonical Wnt5a pathway crucial for tissue morphogenesis (Ho et al., 2012). In the current study, we integrate conditional mouse genetics with quantitative proteomics to identify new targets of Wnt5a-Ror signaling, and focus on one particularly high confidence target, Kif26b.
The first indication that Kif26b may mediate Wnt5a/Ror-dependent developmental processes came from studies in C. elegans. Orthologs of Kif26b (vab-8) and Ror (cam-1) were among the fourteen genes identified in a forward mutagenesis screen for genes required for the directional migration of the C. elegans canal-associated neuron (Forrester et al., 1998). C. elegans mutants of vab-8 and cam-1 display similar polarized cell migration and axon guidance defects where cell bodies and axons that normally move specifically toward the posterior end of the body become abnormally anteriorly displaced (Wightman et al., 1996; Forrester et al., 1999). In addition, both vab-8 and cam-1 mutants exhibit a lower penetrance withered-tail (Wit) phenotype reminiscent of the posterior A-P body axis truncation phenotype seen in Wnt5a or Ror mutant mice (Forrester et al., 1998). These studies, taken together with the findings in mammalian cells described in this article, suggest that Wnt5a, Ror and Kif26b proteins may function as part of an evolutionarily conserved pathway that orchestrates morphogenetic processes during the development of tissues.
Further evidence suggesting Kif26b may function in noncanonical Wnt signaling came from a recent study that identified Kif26b as a binding partner of Dvl3 in HUVECs (Guillabert-Gourgues et al., 2016), as Dvl proteins are known targets of Wnt5a-Ror signaling (Ho et al., 2012). Interestingly, this study demonstrated that Kif26b promotes the directional cell polarization and growth of HUVECs (Guillabert-Gourgues et al., 2016), consistent with our finding that Kif26b controls the migration of cells.
Our study demonstrates that Wnt5a-Ror signaling regulates Kif26b degradation, thereby influencing dynamic cellular processes such as the migratory behavior of cells. Moreover, by perturbing Kif26b function during embryogenesis, we provide in vivo evidence that Kif26b mediates certain biological effects of the Wnt5a-Ror pathway in body axis elongation and PGC development. Together, these findings suggest that a Wnt5a-Ror-Kif26b pathway comprises a conserved signaling cassette crucial for the execution of noncanonical Wnt functions.
Biochemical mechanisms of Wnt5a-Ror-Kif26b signaling
Our observation that Wnt5a-Ror signaling triggers the ubiquitin/proteasome-dependent degradation of Kif26b demonstrates that UPS-mediated proteolysis is a conserved strategy employed by both the canonical Wnt/β-catenin and the noncanonical Wnt5a-Ror pathways to control the cellular abundance of their respective downstream effectors (Figure 7). In addition, we provide further evidence that Fzd and Dvl proteins are shared functional components of both Wnt signaling branches. Collectively, these observations suggest that the Wnt-Fzd-Dvl signaling module is an ancient and conserved feature common to multiple Wnt signaling systems. During evolution of the pathway, different signaling branches appear to have adopted additional regulatory mechanisms to achieve signaling and functional specificity. For example, while the Wnt/β-catenin pathway uses the Lrp5/6 receptors in conjunction with Fzd receptors to transmit a canonical Wnt signal across the plasma membrane, the Wnt5a-Ror pathway uses Ror1/2 receptors, likely also in conjunction with Fzds, to transmit a noncanonical Wnt signal.
Figure 7. Model of Wnt5a-Ror-Kif26b signaling.

Schematic of canonical Wnt/β-catenin signaling (A) vs. noncanonical Wnt5a-Ror-Kif26b signaling (B). See the main text for details.
At present, we do not understand the biochemical mechanisms by which Ror and/or Fzd receptors function in the pathway. In the conventional paradigm of receptor tyrosine kinase signaling, ligand binding induces receptor dimerization or oligomerization, which in turn enhances the intrinsic kinase activity of the receptor (Lemmon and Schlessinger, 2010). This then triggers receptor autophosphorylation and the subsequent recruitment and/or phosphorylation of downstream cellular effectors. Whether the Ror family of receptor tyrosine kinases possesses catalytically active kinase activity, however, is still under debate. It was reported that Wnt5a can stimulate the ability of full-length Ror2 to phosphorylate a GST-Ror2 kinase domain fusion protein on tyrosine residue(s) in vitro, and that mutagenesis of conserved tyrosine residues within the tyrosine kinase regulatory loop of Ror2 impairs the protein’s ability to repress canonical Wnt/β-catenin signaling (Mikels et al., 2009). Ror1 and Ror2, however, are known to harbor substitutions at amino acid residues generally believed to be critical for normal kinase function, and recent biochemical and structural studies have further suggested that the Ror proteins are pseudokinases (Gentile et al., 2011; Artim et al., 2012; Mendrola et al., 2013). In our phosphoproteomic study, we identified nine distinct Kif26b phosphorylation sites on eight unique phosphopeptides, and all these sites mapped to serine or threonine residues (Figure 1—figure supplement 2 and Supplementary file 1). In general, tyrosine phosphorylation is more dynamic than serine or threonine phosphorylation and tends to be underrepresented in large-scale phosphoproteomic studies (Lombardi et al., 2015). We therefore examined whether Wnt5a-Ror signaling induces tyrosine phosphorylation of Kif26b in NIH/3T3 cells using an anti-phosphotyrosine antibody-based affinity pull-down approach. However, under the various experimental conditions tested, we observed no effect of Wnt5a-Ror signaling on the tyrosine phosphorylation of Kif26b (S.S.C. and H.H.H., unpublished data). Thus, whether Ror receptors are active kinases or pseudokinases, and whether Kif26b is a direct substrate of these receptor tyrosine kinases will require further investigation.
It is also possible that phosphorylation of Kif26b by other cellular kinases is involved in Wnt5a-dependent regulation of Kif26b. Interestingly, a previous study showed that cyclin-dependent kinase (CDK) phosphorylates Kif26b on multiple serine and threonine sites and that these phosphorylation events play a critical role in controlling the stability of Kif26b by recruiting the E3 ubiquitin ligase Nedd4 (Terabayashi et al., 2012). It will be important in future studies to test whether these mechanisms, as well as the additional phosphorylation sites identified in our proteomic screens, are required for Wnt5a regulation of Kif26b degradation.
The functional interplay between Ror and Fzd receptors is also currently unclear. We have demonstrated that overexpression of Fzd family members is sufficient to induce Kif26b degradation in NIH/3T3 cells (Figure 4C,D). Overexpression of Ror2, however, does not have the same effect (E.P.K. and H.H.H., unpublished observation). These observations raise the possibility that Fzd proteins may function as the signaling receptors, while Ror proteins may play a modulatory role. This model is also consistent with our observation that even though endogenously expressed Wnt5a requires Ror1 and Ror2 expression to downregulate levels of Kif26b (Figures 1B and 2A), high concentrations of exogenously added Wnt5a can induce Kif26b degradation in Ror1 and Ror2 double knockout MEFs (M.W.S., M.E.G. and H.H.H., unpublished data), suggesting that Wnt5a can signal through receptors other than Rors. A deeper understanding of how Ror and Fzd proteins mediate Wnt5a-Kif26b signaling will require additional biochemical, structural and functional studies.
Our study also directly implicates Dvl proteins in promoting Kif26b degradation (Figure 4E,F) and further suggests a broader role for Dvl in regulating the stability of Wnt signaling effectors. Interestingly, during canonical Wnt signaling, Dvl functions to inhibit the degradation of β-catenin, whereas during noncanonical Wnt5a signaling, Dvl functions to induce the degradation of Kif26b (Figure 7). How Dvl transmits pathway-specific signals remains to be elucidated, but it likely involves differential post-translational modifications such as phosphorylation, differential protein-protein interactions, or both (Wallingford and Habas, 2005). The identification of Kif26b as a specific target of Dvl in the noncanonical Wnt signaling branch provides a new inroad to address this important question.
There is also emerging evidence that noncanonical Wnt signaling may function more generally to regulate the degradation of multiple proteins. For instance, Wnt5a signaling can induce the degradation of the cell adhesion receptor Syndecan 4 via the proteasome (Carvallo et al., 2010), and noncanonical Wnt5a and Wnt11 signaling has been associated with the proteasomal degradation of the focal adhesion molecule Paxillin (Kurayoshi et al., 2006; Iioka et al., 2007). It is possible that Wnt5a downregulates multiple effector molecules to affect the shape and behavior of cells in a variety of cell types and developmental contexts.
Cellular and developmental functions of Kif26b
Data from our wound-healing assay suggest that one function of the Wnt5a-Ror-Kif26b axis is to modulate the migration of cells via Wnt5a-dependent degradation of Kif26b. How might Kif26b function at a subcellular level to affect cell migration? Mesenchymal cell migration typically involves four reiterative steps: protrusion of the leading edge, formation and maturation of focal adhesions, forward translocation of the cell body and finally retraction and de-adhesion of the trailing edge. Using the GFP-Kif26b NIH/3T3 cell line, we observed that the Kif26b protein is predominantly localized to the trailing edge of the cell, which is most consistent with a role of Kif26b in promoting trailing edge de-adhesion and/or retraction (Figure 3—figure supplement 2D,E; Video 1). This model is supported by a previously reported binding interaction between Kif26b and Myosin IIb, a key regulator of cell trailing edge de-adhesion and contractility (Uchiyama et al., 2010). It may also explain why Wnt5a has been reported in some studies to promote, while in others to suppress, cell migration (Kremenevskaja et al., 2005; Dissanayake et al., 2007; Nomachi et al., 2008; Enomoto et al., 2009; McDonald and Silver, 2009; Jiang et al., 2013; Liu et al., 2014; Zhang et al., 2014; Prasad et al., 2016; Yu et al., 2016; Connacher et al., 2017). It is well established that a delicate balance of cell adhesive and contractile activities dictates the manner by which cells migrate (Parsons et al., 2010; Huttenlocher and Horwitz, 2011). Moreover, this balance is likely to be different in different cell types and subject to further regulation by the extracellular environments to which the cells are exposed. Thus, if the primary function of the Wnt5a-Kif26b signaling axis is to modulate the strength of cell adhesion and/or contractility at the trailing edge, it could potentially manifest in opposing cell migratory behaviors depending on the cell types and the culture conditions used in a given study. Higher resolution imaging studies capable of directly measuring cell adhesion dynamics and contractile forces will be crucial to understanding how Kif26b functions at the trailing edge of the cell, or at other subcellular locations, to affect cell migration.
Although we have shown in a wound-healing assay that higher Kif26b levels promote the migration of NIH/3T3 cells and that Wnt5a signaling negatively regulates the migratory behavior of these cells via the degradation of Kif26b, it is likely that the signaling dynamics of the Wnt5a-Ror-Kif26b pathway are actually considerably more complex in vivo. During embryogenesis, migration and other morphogenetic processes must occur in a highly choreographed fashion where signals that promote and restrict cell movements are finely tuned in time and space. It is therefore possible that the relatively modest Wnt5a-dependent alteration of Kif26b levels we observed in vitro, as measured across entire cell populations, does not accurately reflect the dynamic regulation of the pathway that occurs during the development of tissues. The function of the Wnt5a-Ror pathway may not be simply to degrade Kif26b constitutively within a cell or tissue, but rather to tune the activity of Kif26b with a high degree of temporospatial resolution to achieve proper tissue morphogenesis. Indeed, cells are known to integrate and amplify shallow spatial gradients of extracellular cues into robust changes in behaviors, such as during axon pathfinding and neutrophil chemotaxis (von Philipsborn and Bastmeyer, 2007).
The in vivo significance of Kif26b in noncanonical Wnt5a-Ror function is demonstrated by our gene perturbation experiments. Disruption of Kif26b expression, whether by mis-expression in zebrafish or by loss-of-function gene ablation in mouse embryos, results in developmental defects that are similar to those observed when Wnt5a or Ror expression is perturbed. These findings, taken together with the observation that loss-of-function cam-1 and vab-8 mutants in C. elegans exhibit similar neuronal migration and polarization phenotypes (Forrester et al., 1998), support the model that Wnt5a, Ror and Kif26b comprise a signaling cassette crucial for the morphogenesis of tissues during development. However, it is important to note that while our in vivo experiments focus on comparing the gross morphological phenotypes of Kif26b, Wnt5a and Ror mutants, they do not directly demonstrate that the similarities in the observed phenotypes are caused by common underlying mechanisms. Therefore, it remains crucial to characterize the molecular and cellular basis of the Wnt5a, Ror and Kif26b mutant phenotypes, which will further establish the functional interactions among these molecules.
It is notable that genetic loss-of-function perturbation of Kif26b expression in mice does not closely mimic perturbation of Wnt5a or Ror expression in all developmental contexts. For instance, Kif26b knockout mice do not exhibit the global tissue truncation and craniofacial malformation phenotypes seen in Wnt5a and Ror1/2 double mutants (Yamaguchi et al., 1999; Uchiyama et al., 2010; Ho et al., 2012). In addition, Kif26b knockout mice fail to elicit sprouting of the ureteric bud, resulting in kidney agenesis or severe kidney hypoplasia (Uchiyama et al., 2010), whereas Wnt5a or Ror knockout mice exhibit duplication of the ureteric bud, resulting in pleiotropic kidney defects (Huang et al., 2014; Nishita et al., 2014; Yun et al., 2014).
There are a number of explanations that might reconcile these seemingly discordant findings in different biological contexts. First, given the highly complex temporal and spatial dynamics that Wnt5a-Ror-Kif26b signaling likely undergoes in vivo, it becomes difficult to predict the ultimate phenotypes of embryos that constitutively lack the expression of downstream signaling components across development. Second, Kif26b may not be required in all tissues that undergo Wnt5a-Ror signaling during their morphogenesis. It is likely that other effector molecules may compensate for the genetic loss of Kif26b in certain developing tissues (Rossi et al., 2015). For instance, the closely related Kif26a gene is broadly expressed during mouse embryogenesis, and the Kif26a protein is also a target of Wnt5a-Ror signaling (M.K.S., E.P.K., H.H.H. unpublished data). Thus, one plausible explanation is that the developmental requirement of Kif26b in tissue extension and craniofacial morphogenesis may be masked by the continued expression of Kif26a in Kif26b knockout mice. By RT-qPCR, we verified that Kif26a mRNA is indeed expressed at appreciable levels in both wild-type and Kif26b knockout E12.5 MEFs, although we did not find evidence that Kif26a transcription is specifically upregulated in Kif26b knockout MEFs (E.P.K. and H.H.H., unpublished data). Kif26a single knockout mice also do not exhibit phenotypes typical of noncanonical Wnt5a-Ror signaling defects, instead manifesting with megacolon from enteric nerve hyperplasia (Zhou et al., 2009). Thus, discovering the full extent of developmental processes regulated by the Kinesin-11 family of proteins, as well as their functional relationship to the Wnt5a-Ror pathway, awaits a loss-of-function analysis of both Kif26a and Kif26b together, which may reveal non-overlapping phenotypes not present in either Kif26a or Kif26b single loss-of-function mutants. Lastly, it is also likely that other signaling branches apart from Kif26b exist within the Wnt5a-Ror pathway, and that these branches control distinct cellular processes that do not require the expression of the Kinesin-11 family of proteins. Thus, building a more complete inventory of the pathway components and understanding how these components interact functionally represents an important direction of future investigation.
Materials and methods
Mice
All mouse strains used in the study were described previously: Ror1f/f (Ho et al., 2012), Ror2f/f (Ho et al., 2012), CAG-CreER (Hayashi and McMahon, 2002, stock # 004453, Jackson Laboratory, Bar Harbor, ME), Wnt5a-/- (Yamaguchi et al., 1999, stock # 004758, Jackson Laboratory), and Kif26bf/f (Recuenco et al., 2015, stock # CDB0800K, RIKEN CDB, Japan, http://www2.clst.riken.jp/arg/mutant_list_file/CDB0800K.html). Kif26b-/- mice were produced by crossing Kif26bf/f mice to a ubiquitous Cre deleter line (EIIA-Cre [Lakso et al., 1996], stock # 003724, Jackson Laboratory). Ror1f/f, Ror2f/f and CAG-CreER mice were maintained in a mixed 129 and C57BL/6J background. Wnt5a-/- and Kif26b-/- mice were maintained in a C57BL/6J background. Wild-type mice are C57BL/6J. All animals were used according to institutional and NIH guidelines approved by the Institutional Animal Care and Use Committees at Harvard Medical School and University of California, Davis.
Cell lines
Primary MEFs were isolated directly from mouse embryos as described (Ho et al., 2012) and used within three passages. NIH/3T3 Flp-In (R76107, Thermo Fisher Scientific, Hanover Park, IL) and HEK293T (CRL-3216, ATCC, Manassas, VA) cells were purchased and not re-authenticated. NIH/3T3 Flp-In and HEK293T cells were tested negative for mycoplasma contamination using the Universal Mycoplasma Detection Kit (30–1012K, ATCC). All cell lines were cultured at 37°C and 5% CO2 in Dulbecco’s Modified Eagles Medium (MT15017CV, Corning, Tewksbury, MA) supplemented with 1x glutamine (25–005 CI, Corning), 1x penicillin-streptomycin (30–002 CI, Corning) and 10% fetal bovine serum (16000069, Thermo Fisher Scientific).
TMT/MS3 phosphoproteomic screen
Two E12.5 Ror1f/f; Ror2f/f; CAG-CreER/+ embryos were individually dissected and used to derive primary MEFs. One E12.5 Ror1+/+; Ror2+/+; CAG-CreER/+ embryo was used to derive the control MEFs. The primary passage of cells derived from each embryo was cultured in a 10-cm plate until confluent. The primary passage was then split into two 10 cm plates at six million cells per plate (day 0). 24 hr later (day 1), 4-OHT was added to one plate (0.25 μM final concentration), and the drug vehicle (EtOH) was added to the other plate. On day 2, a media change was performed using fresh media containing 0.25 μM 4-OHT. On day 3, a media change was performed again, but the final 4-OHT concentration was reduced to 0.1 μM. On day 4, a media change was performed without 4-OHT. On day 5, cells were washed once in ice-cold PBS, and cells from each 10-cm plate was scraped into 1 mL of ice-cold lysis buffer (8 M urea, 75 mM NaCl, 50 mM Tris pH 8.2, 1 mM NaF, 1 mM β-glycerophosphate, 1 mM Na3VO4, 10 mM Na4P2O7, 1 mM PMSF, and cOmplete protease inhibitor (-EDTA, 04693159001, Roche, Indianapolis, IN). Cells were homogenized by pipetting up and down using a P-1000 pipettor and then sonicated in a Bioruptor (Diagenode, Denville, NJ; 17 × 30 s ON/OFF cycles). Cell lysates were then centrifuged at 40,000 RPM for 20 min at 4°C. The clarified high-speed supernatants were collected, snap frozen in liquid nitrogen and stored at −80°C until the TMT/MS3 analysis was performed. Protein concentrations were determined using BCA reagents (23225, Thermo Fisher Scientific).
To perform the TMT/MS3 screen, tryptic peptides were prepared from whole cell lysates and the peptide mixtures from the six experimental conditions were individually labeled with the TMT reagents, such that reporter ions at m/z of 126, 127, 128, 129, 130 and 131 would be generated in the tandem spectrometry. Phosphopeptides were enriched by TiO2 chromatography. Liquid chromatography, MS3 tandem mass spectrometry and data analysis were carried out as previously described (Ting et al., 2011; McAlister et al., 2014; Paulo et al., 2015).
Cloning of mouse Kif26b cDNA
A first-strand cDNA library was generated from MEF total RNA using M-MuLV reverse transcriptase (M0253S, New England BioLabs, Ipswich, MA) and the d(T)23VN primer according to the manufacturer’s instructions. This cDNA library was then used as template for PCR amplification of the Kif26b open reading frame with the following primers: Forward: gatcggccggcctaccatgaattcggtagccggaaataaag; Reverse: gatcggcgcgccttatcggcgcctggaggtgatgtc. The PCR product was subcloned into a modified pCS2+ vector using the FseI and AscI restriction sites. The entire Kif26b open reading frame was confirmed by sequencing.
Antibodies
Antibodies against Kif26b were generated using a previously described antigen (Uchiyama et al., 2010). A C-terminal fragment of Kif26b was PCR amplified using the following primers and subcloned into a modified pGEX (28-9546-63, GE Healthcare, Pittsburgh, PA) vector to generate a GST fusion protein in E. coli. Forward: gatcggccggcctaccatgcgaaacgtgcaagagcctgagtcc; Reverse: gatcggcgcgccttatcggcgcctggaggtgatgtc. Protein expression was induced in the E. coli strain BL21(DE3) using IPTG (0.3 mM). To purify the C-terminal Kif26b protein fragment, bacterial pellets were lysed in STE (150 mM NaCl, 1 mM Tris pH 8.0, 1 mM EDTA) supplemented with protease inhibitors (04693159001, Roche) and 0.1 mg/ml lysozyme (L-6876, Sigma-Aldrich, St. Louis, MO) and incubated on ice for 15 min. Just before sonication, DTT was added to a final concentration of 2 mM and pre-diluted sodium lauroyl sarcosinate (in STE) was added to a final concentration of 10%. Lysates were then sonicated in a Bioruptor (5 × 30 s ON/OFF cycles) and then centrifuged at 60,000 RPM for 30 min at 4°C. Triton X-100 was added to the supernatant to a final concentration of 3% and incubated with Glutathione Sepharose 4B beads (17075601, GE Healthcare) for affinity purification. Purified C-terminal Kif26b proteins were dialyzed in PBS and used for immunization of rabbits.
Antibodies against Ror1 and Ror2 were described previously (Ho et al., 2012). The following antibodies were purchased: rabbit anti-Dvl2 (3216, Cell Signaling Technology, Danvers, MA); rabbit anti-EGFR (SC-03, Santa Cruz Biotechnology, Dallas, TX); goat anti-Wnt5A (AF645, R&D Systems, Minneapolis, MN); mouse anti-α-tubulin (clone DM1A, ab7291, Abcam, Cambridge, MA); chicken anti-GFP (GFP-1020, Aves Labs, Tigard, OR); rabbit anti-Myosin IIb (3404, Cell Signaling Technology); mouse anti-GM130 (610822, BD Biosciences, San Jose, CA); rabbit anti-phospho-LRP6 (Ser1490) (2568, Cell Signaling Technology).
Western blotting
Quantitative western blotting was performed using the Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, IL) according to the manufacturer’s instructions. Non-saturated protein bands were quantified using Odyssey software, with the gamma level set at 1. Protein lysates for SDS-PAGE and western blotting were prepared in 1x or 2x LDS sample buffer (NP0008, Thermo Fisher Scientific) supplemented with 2-mercaptoethanol (5% final concentration). If BCA assays were required to quantify the protein lysate concentrations, the lysates were prepared instead in a homemade 1x SDS sample buffer (50 mM Tris pH 6.8, 2% SDS, 10% glycerol) without bromophenol blue or 2-mercaptoethanol. Once the protein concentrations were determined and normalized, the lysates were then mixed with 1/3 volume of 4x SDS sample buffer containing bromophenol blue (0.025%) and 2-mercaptoethanol (20%). Protein lysates used for Kif26b western blotting were not heated, as the Kif26b signal weakens substantially after heating, likely due to heat-induced protein aggregation. All other protein lysates were heated at 90°C for 5 min before SDS-PAGE and western blotting.
Generation of shRNA targeting Kif26b
The lentiviral vector pLLX3.7 was used to generate recombinant lentiviruses expressing shRNA that target mouse Kif26b. The following sequences were targeted: gtgccttgcaaatctttat and gctcgagatacctcagaat.
Generation of stable NIH/3T3 cell lines
To construct the GFP-Kif26b expression plasmid, the eGFP open reading frame was first subcloned into pENTR-2B (11816-014, Thermo Fisher Scientific), and the full-length mouse Kif26b open reading frame was subcloned in frame to the C-terminus of GFP. The resulting construct was verified by sequencing and then recombined with the pEF5-FRT-V5 vector (V602020, Thermo Fisher Scientific) using LR clonase II (11791100, Thermo Fisher Scientific) to create pEF5-GFP-Kif26b-FRT. The pEF5-GFP-Kif26b-FRT plasmid was used to generate stable isogenic cell lines using the Flp-In system and Flp-In NIH/3T3 cell line (Thermo Fisher Scientific). DNA transfection was performed in 10-cm plates with Genjet In Vitro Transfection Reagent (SL100488; SignaGen Laboratories, Rockville, MD). Cells that stably integrated the Flp-In constructs were selected using 200 μg/ml hygromycin B and expanded. A more detailed protocol is described at Bio-Protocol (Karuna et al., 2018).
Lentivirus-mediated protein overexpression
Recombinant lentiviruses were generated using the pLEX_307 (for Dvl1 and all Fzd constructs) or pLVX-EF1α-mCherry-N1 (for Shisa2) vectors. Both vector systems use the EF1 promoter for driving transgene expression. pLEX_307 was a gift from David Root (Plasmid 41392, Addgene, Cambridge, MA) and pLVX-EF1α-mCherry-N1 was purchased (631986, Clontech Laboratories, Mountain View, CA). The human Dvl1 open-reading frame was cloned by PCR from a HeLa cell cDNA pool. The mouse Shisa2 open reading frame was PCR amplified from a Shisa2-containing plasmid (a gift from Xi He). The Fzd open reading frames were PCR amplified from the following Addgene plasmids: 42253, 42259, 42255, 42256, 42267, 42258, 42270 and 42261 (gifts from Chris Garcia and Jeremy Nathans). The open-reading frames in all lentiviral constructs were verified by sequencing. Lentiviruses were packaged and produced in HEK293T cells by co-transfection of the lentiviral vectors with the following packaging plasmids: pRSV-REV, pMD-2-G and pMD-Lg1-pRRE (gifts from Thomas Vierbuchen). 3 ml or 0.3 ml of the viral supernatants was used to infect WRK reporter cells seeded at 50% confluency in six-well plates. Puromycin selection (0.002 mg/ml) was carried out for 3 days. Cells from the viral titer that killed a large proportion of cells (60–90%) were expanded and used for FACS; this ensured that the multiplicity of infection (MOI) is ~1 for all cell lines used in the experiments.
Generation of Kif26b knockout NIH/3T3 cell lines
The mouse Kif26b gene was mutated by CRIPSR/Cas9-mediated genome editing as according to (Ran et al., 2013). Briefly, LentiCRISPR V2 (a gift of Feng Zhang, Addgene plasmid 52961) was used to generate lentiviruses expressing small guide RNAs (sgRNAs). The sgRNA target sequences are: GCTTACGAGGAGTCGCGCGCCGG; GAACTGTAACGCCCGCTTGGTGG.
Following lentivirus infection and puromycin selection, NIH/3T3 cells were passaged for 3 days to allow time for mutagenesis to occur. Individual cell clones were picked from cell populations targeted with each of these sgRNAs, expanded and then screened initially by western blotting. Clones that appeared to lack Kif26b expression were sequenced to confirm genome modification.
Cell proliferation and survival assays
For quantifications of cell proliferation, NIH/3T3 cell lines were plated on glass coverslips 24 hr prior to fixation. Cells were fixed with 4% paraformaldehyde in Cytoskeleton Buffer with sucrose (10 mM MES pH 6.1, 138 mM KCl, 3 mM MgCl, 2 mM EGTA, 0.32 M sucrose) for 20 min at room temperature, permeabilized in TBS-0.5% Triton X-100 for 10 min, then rinsed 3x with TBS-0.1% Triton X-100. Cells were blocked in Antibody Diluting Solution (AbDil) (TBS-0.1% Triton X-100, 2% BSA, 0.1% sodium azide) for 30 min at room temperature, then incubated overnight at 4°C with 1:500 of rabbit anti-phospho-Histone H3 (Ser10, Mitosis Marker) (#3377, Cell Signaling Technology) diluted in AbDil, or a no primary antibody control. After five washes in TBS-0.1% Triton X-100, Alexa dye-conjugated secondary antibodies were added at 1:1000 in AbDil for 45 min at room temperature. After five washes in TBS-0.1% Triton X-100, coverslips were mounted in DAPI Fluoromount-G (0100–20, SouthernBiotech, Birmingham, AL). Images of cells were acquired using a 10x objective at equal exposure, and then analyzed for the presence of nuclear staining of the Mitosis Marker per DAPI-positive nuclei counted.
For quantifications of cell survival, NIH/3T3 cells lines were plated similarly as for the cell proliferation assays. TUNEL staining was performed according to the manufacturer's instructions (In Situ Cell Death Detection Kit, TMR red, 12156792910, Roche), including a DNase-positive control (M0303S, New England BioLabs). Images of cells were acquired using a 10x objective at equal exposure, and then analyzed for the presence of nuclear TUNEL staining per DAPI-positive nuclei counted.
Kinetic wound-healing cell migration assay
Cells were plated on 96-well plates (Essen ImageLock, 4379, Essen Instruments, Ann Arbor, MI). Wnt-C59 (100 nM final concentration) was added to cells 24 hr prior to wound creation with a wound scratcher (Essen Instruments). For Wnt5a treatment, recombinant Wnt5a (100 ng/ml final concentration) was added immediately after creation of wounds. Wound confluence was monitored with Incucyte Live-Cell Imaging System and software (Essen Instruments). Wound closure was observed every 1–2 hr for 48–96 hr by comparing the mean relative wound density of at least four biological replicates in each experiment.
Immunocytochemistry
NIH/3T3 cells were plated at low density and grown on glass coverslips for 24 hr. Cells were rinsed 1x with PBS, then fixed with either ice-cold methanol for 3 min or with 4% paraformaldehyde in Cytoskeleton Buffer with sucrose for 20 min at room temperature. Cells were then rinsed 3x in PBS, permeabilized in TBS-0.5% Triton X-100 for 10 min, then rinsed 3x with TBS-0.1% Triton X-100. Cells were blocked in AbDil for 30 min at room temperature. Primary antibodies were added at the following dilutions in AbDil overnight at 4°C: chicken anti-GFP at 1:1000, mouse anti-α-tubulin at 1:5000, anti-Myosin IIb at 1:200, and anti-GM130 at 1:50. After five washes in TBS-0.1% Triton X-100, Alexa dye-conjugated secondary antibodies were added at 1:1000 in AbDil for 45 min at room temperature. After five washes in TBS-0.1% Triton X-100, coverslips were mounted in DAPI Fluoromount-G.
Recombinant proteins and inhibitors
The following recombinant proteins and drugs were purchased: human/mouse Wnt5a (654-WN-010, R&D Systems); mouse Wnt3a (1324-WN-010, R&D Systems); mouse Dkk-1 (5897-DK-010, R&D Systems); Wnt-C59 (C7641-2s; Cellagen Technology, San Diego, CA); epoxomicin (A2606, ApexBio, Houston, TX); PYR-41 (B1492, ApexBio); cycloheximide (C7698-1G, Sigma-Aldrich).
Reverse transcription and qPCR
Total RNA was isolated from MEFs using the PureLink RNA Mini Kit (121830108A, Thermo Fisher Scientific) according to the manufacturer's instructions. Isolated RNA was treated with DNase I (recombinant, RNase-free; 4716728001, Roche) and a cDNA library was synthesized using the cDNA High Capacity cDNA Reverse Transcription Kit (4368814, Thermo Fisher Scientific). The cDNA was the source of input for qPCR, using 7900 HT FAST and SYBR Green reagents (4329001, Thermo Fisher Scientific). The following qPCR primer pairs were used: mKif26b forward, CAAGTACGAGTGGCTGATGAA; mKif26b reverse, GGACCTGCTCCAAGTCAAAT; β-actin forward, GCTTCTAGGCGGACTGTTACTGA; β-actin reverse, GCGCAAGTTAGGTTTTGTCAAA.
Flow cytometry
NIH/3T3 cells were plated at a density of 0.09 million/well in a poly-D-lysine-coated 48-well plate. 24 hr after plating, the cells were incubated with 10 nM Wnt-C59 and allowed to reach confluency. 48 hr after plating, cells were stimulated with either Wnt proteins or an equivalent volume of the control buffer (PBS with 0.1% BSA and 0.5% (w/v) CHAPS) in the presence of 10 nM Wnt-C59 for 6 hr. Cells were then harvested, resuspended in PBS + 0.5% FBS and analyzed using a flow cytometer (FACScan with a 488 nm laser, Becton Dickinson, San Jose, CA). Raw data were acquired with CellQuest (Becton Dickinson) and processed in FlowJoX (FLOWJO, Ashland, OR). Processing entailed gating out dead cells, calculation of median fluorescence, percent change of medians, and overlay of histograms. Dose-response curves based on percent change were fitted in MATLAB with the doseResponse function (written by Richie Smith and publicly available on Matlab File Exchange, File ID # 33604). A more detailed protocol is described at Bio-Protocol (Karuna et al., 2018).
Live imaging
0.05 million cells were plated in a collagen-coated 35-mm glass bottom plate (P35GCOL-0-10-C, MatTek Corp, Ashland, MA). After adhering to the plate, cells were incubated in culture media supplemented with 25 mM Hepes and 10 nM Wnt-C59 for 24 hr. Cells were stimulated with 200 ng/ml recombinant Wnt5a in the presence of 25 mM Hepes and 10 nM Wnt-C59. Cells were imaged every 10 min for 16 hr, with 500 ms exposure at 40x magnification.
Zebrafish
Wild-type NHGRI-1 fish were bred and maintained using standard procedures (LaFave et al., 2014). Embryos were obtained by natural spawning and staged as described (Kimmel et al., 1995). All zebrafish works were approved by the Institutional Animal Care and Use Committee, Office of Animal Welfare Assurance, University of California, Davis.
In vitro transcribed capped RNAs were prepared using the mMessage mMachine RNA Synthesis Kit (AM1340, Thermo Fisher Scientific) and purified using the RNeasy Mini Kit (74104, Qiagen, Germantown, MD) following manufacturers' instructions.
Microinjection of RNA was performed as described (Jao et al., 2012). In brief, one-cell-stage embryos from wild-type zebrafish intercrosses were injected with the following amounts of in vitro transcribed RNA: Cas9, 400 pg; Kif26b, 400 pg; Wnt5a, 150 pg. Pipettes were pulled on a micropipette puller (Model P-97, Sutter Instruments, Novato, CA). Injections were performed with an air injection apparatus (Pneumatic MPPI-2 Pressure Injector, Eugene, OR). Injected volume (typically ~1 nl) was calibrated with a microruler.
PGC analysis
For PGC analysis, E11.5 embryos were dissected from timed matings. E0.5 is defined as noon of the day when the vaginal plug is detected. To expose the gonadal ridges, the abdominal cavity was opened and the visceral organs removed. The embryos were then cut just anterior to the forelimbs and just posterior to the hindlimbs. The midsection containing the gonadal ridges were washed once in cold (−20°C) methanol:DMSO (4:1), and then stored in the same fixative solution at −20°C until analysis. Genotypes were determined by PCR.
For whole-mount immunofluorescence, fixed embryos were rehydrated and rocked at 4°C overnight in PBSMT (PBS with 2% nonfat dry milk and 0.5% Triton X-100) with antibodies to SSEA1 (mouse IgM, MC-480, Developmental Studies Hybridoma Bank, Iowa City, IA, 1:200). Three PBSMT washes were followed by overnight incubation with secondary antibodies (1:200) and Hoechst (1:1000) in PBSMT. Embryos were then washed three times in PBS, dehydrated in a series of 5 min washes in 50%, 70%, 95%, and two times in 100% ethanol while rocking in the dark, and cleared with methyl salicylate for imaging.
Confocal imaging was carried out at room temperature with a 10x dry objective on a Leica SP5 TCS microscope equipped with 405, 488, 543, 594, and 633 nm lasers. Use of the 10x objective typically required the addition of a 1.5x digital zoom for optimal visualization of PGCs for quantification. Files of 1024 × 1024 pixel images with 2–3 μm distance between z-stacks were captured by a scanner with maximal frame resolution and Leica acquisition software. PGCs were counted on Imaris imaging software (Bitplane, Belfast, UK) using the Spots module. Spots of 7 μm size in the SSEA1 channel were identified by the software and visually inspected to confirm accuracy. All measurements were exported to Excel (Microsoft, Redmond, WA) for calculations and statistical analyses.
Acknowledgements
We thank Andres Lebensohn and Rajat Rohatgi for comments on the manuscript. We thank Laura Borodinsky for help with Xenopus microinjection during the earlier phase of our in vivo study. We thank Gian Garriga and Kermit Carraway for discussions. We thank Xi He for providing the Shisa2 expression construct and discussions. We thank CDB Laboratory for Animal Resources and Genetic Engineering for the source of the Kif26bf/f mice. We thank Chris Garcia and Jeremy Nathans for providing the Fzd expression constructs through Addgene. We thank all members of the Ho, Greenberg and Jao labs for input on the study. This work was supported by: National Institutes of Health grants 1R35GM119574-01 to HH Ho, 5R01NS045500-29 to ME Greenberg, 1R21ES023297-01 and DP2OD007420 to DJ Laird, T32GM007753 to MW Susman; an American Cancer Society grant IRG-95-125-13 to HH Ho; National Science Foundation predoctoral fellowships to AV Cantú and BY Jong. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Funding Statement
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Contributor Information
Hsin-Yi Henry Ho, Email: hyhho@ucdavis.edu.
Jeremy Nathans, Johns Hopkins University School of Medicine, United States.
Funding Information
This paper was supported by the following grants:
National Institutes of Health 1R35GM119574-01 to Hsin-Yi Henry Ho.
National Science Foundation Predoctoral Fellowship to Andrea V Cantú, Brigette Y Jong.
American Cancer Society IRG-95-125-13 to Hsin-Yi Henry Ho.
National Institutes of Health 5R01NS045500-29 to Michael E Greenberg.
National Institutes of Health 1R21ES023297-01 to Diana J Laird.
National Institutes of Health DP2OD007420 to Diana J Laird.
National Institutes of Health T32GM007753 to Michael W Susman.
Additional information
Competing interests
No competing interests declared.
Author contributions
Conceptualization, Resources, Data curation, Formal analysis, Supervision, Validation, Investigation, Visualization, Methodology, Writing—original draft, Project administration, Writing—review and editing.
Conceptualization, Resources, Data curation, Formal analysis, Validation, Investigation, Visualization, Methodology, Writing—original draft, Project administration, Writing—review and editing.
Conceptualization, Resources, Data curation, Formal analysis, Investigation, Methodology, Writing—review and editing.
Resources, Data curation, Formal analysis, Investigation, Methodology.
Resources, Data curation, Formal analysis, Investigation, Methodology, Writing—review and editing.
Data curation, Formal analysis, Investigation, Methodology.
Data curation, Formal analysis, Investigation.
Data curation, Formal analysis, Investigation.
Data curation, Formal analysis, Investigation, Writing—review and editing.
Data curation, Formal analysis, Investigation, Writing—review and editing.
Resources, Investigation, Methodology.
Conceptualization, Resources, Funding acquisition.
Resources, Funding acquisition, Writing—review and editing.
Resources, Formal analysis, Investigation, Visualization, Writing—review and editing.
Resources, Supervision, Funding acquisition, Methodology, Writing—review and editing.
Resources, Data curation, Formal analysis, Investigation, Methodology, Visualization.
Conceptualization, Resources, Data curation, Supervision, Funding acquisition, Methodology.
Conceptualization, Resources, Supervision, Funding acquisition, Project administration, Writing—review and editing.
Conceptualization, Resources, Data curation, Formal analysis, Supervision, Funding acquisition, Validation, Investigation, Visualization, Methodology, Writing—original draft, Project administration, Writing—review and editing.
Ethics
Animal experimentation: All animals were used according to institutional and NIH guidelines approved by the Institutional Animal Care and Use Committees (IACUC) at Harvard Medical School (Protocol # 04572/IS00000074) and University of California, Davis (Protocol # 19465).
Additional files
Columns include: Uniprot protein identification number, gene symbol, protein description/name, phosphosite position, phosphosite motif, localization score, spectral counts, the normalized summed signal to noise for each of the six TMT (126 to 131) channels.
(A) Upregulated phosphopeptides that scored as ‘hits’ as defined in the text. Phosphopeptides above the bold line are ‘hits’ scored using the 2-fold cutoff filter. Phosphopeptides below the bold line are those scored between the 1.5- and 2-fold cutoffs. Columns include: gene name, protein description, fold change (4-OHT/vehicle treated samples), the p value. (B) Downregulated phosphopeptides that scored as ‘hits’ as defined in the text. Phosphopeptides above the bold line are ‘hits’ scored using the 2-fold cutoff filter. Phosphopeptides below the bold line are those scored between the 1.5- and 2-fold cutoffs. Columns include: gene name, protein description, fold change (4-OHT/vehicle treated samples), the p value.
References
- Afzal AR, Rajab A, Fenske CD, Oldridge M, Elanko N, Ternes-Pereira E, Tüysüz B, Murday VA, Patton MA, Wilkie AO, Jeffery S. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nature genetics. 2000;25:419–422. doi: 10.1038/78107. [DOI] [PubMed] [Google Scholar]
- Artim SC, Mendrola JM, Lemmon MA. Assessing the range of kinase autoinhibition mechanisms in the insulin receptor family. Biochemical Journal. 2012;448:213–220. doi: 10.1042/BJ20121365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bafico A, Liu G, Yaniv A, Gazit A, Aaronson SA. Novel mechanism of Wnt signalling inhibition mediated by Dickkopf-1 interaction with LRP6/Arrow. Nature Cell Biology. 2001;3:683–686. doi: 10.1038/35083081. [DOI] [PubMed] [Google Scholar]
- Bai Y, Tan X, Zhang H, Liu C, Zhao B, Li Y, Lu L, Liu Y, Zhou J. Ror2 receptor mediates Wnt11 ligand signaling and affects convergence and extension movements in zebrafish. Journal of Biological Chemistry. 2014;289:20664–20676. doi: 10.1074/jbc.M114.586099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhanot P, Brink M, Samos CH, Hsieh JC, Wang Y, Macke JP, Andrew D, Nathans J, Nusse R. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 1996;382:225–230. doi: 10.1038/382225a0. [DOI] [PubMed] [Google Scholar]
- Bhatt PM, Malgor R. Wnt5a: a player in the pathogenesis of atherosclerosis and other inflammatory disorders. Atherosclerosis. 2014;237:155–162. doi: 10.1016/j.atherosclerosis.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer D, Levine-Wilkinson S, Richmond A, Hirsh S, Kuruvilla R. Wnt5a mediates nerve growth factor-dependent axonal branching and growth in developing sympathetic neurons. Journal of Neuroscience. 2009;29:7569–7581. doi: 10.1523/JNEUROSCI.1445-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryja V, Schulte G, Arenas E. Wnt-3a utilizes a novel low dose and rapid pathway that does not require casein kinase 1-mediated phosphorylation of Dvl to activate beta-catenin. Cellular Signalling. 2007a;19:610–616. doi: 10.1016/j.cellsig.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Bryja V, Schulte G, Rawal N, Grahn A, Arenas E. Wnt-5a induces Dishevelled phosphorylation and dopaminergic differentiation via a CK1-dependent mechanism. Journal of Cell Science. 2007b;120:586–595. doi: 10.1242/jcs.03368. [DOI] [PubMed] [Google Scholar]
- Bunn KJ, Daniel P, Rösken HS, O'Neill AC, Cameron-Christie SR, Morgan T, Brunner HG, Lai A, Kunst HP, Markie DM, Robertson SP. Mutations in DVL1 cause an osteosclerotic form of robinow syndrome. The American Journal of Human Genetics. 2015;96:623–630. doi: 10.1016/j.ajhg.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvallo L, Muñoz R, Bustos F, Escobedo N, Carrasco H, Olivares G, Larraín J. Non-canonical Wnt signaling induces ubiquitination and degradation of Syndecan4. Journal of Biological Chemistry. 2010;285:29546–29555. doi: 10.1074/jbc.M110.155812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha J, Bartos A, Park C, Sun X, Li Y, Cha SW, Ajima R, Ho HY, Yamaguchi TP, Dey SK. Appropriate crypt formation in the uterus for embryo homing and implantation requires Wnt5a-ROR signaling. Cell Reports. 2014;8:382–392. doi: 10.1016/j.celrep.2014.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawengsaksophak K, Svingen T, Ng ET, Epp T, Spiller CM, Clark C, Cooper H, Koopman P. Loss of Wnt5a disrupts primordial germ cell migration and male sexual development in mice. Biology of Reproduction. 2012;86:1–12. doi: 10.1095/biolreprod.111.095232. [DOI] [PubMed] [Google Scholar]
- Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW, Wei S, Hao W, Kilgore J, Williams NS, Roth MG, Amatruda JF, Chen C, Lum L. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nature Chemical Biology. 2009;5:100–107. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien SC, Gurling M, Kim C, Craft T, Forrester W, Garriga G. Autonomous and nonautonomous regulation of Wnt-mediated neuronal polarity by the C. elegans Ror kinase CAM-1. Developmental Biology. 2015;404:55–65. doi: 10.1016/j.ydbio.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Connacher MK, Tay JW, Ahn NG. Rear-polarized Wnt5a-receptor-actin-myosin-polarity (WRAMP) structures promote the speed and persistence of directional cell migration. Molecular Biology of the Cell. 2017;28:1924–1936. doi: 10.1091/mbc.E16-12-0875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Forno PD, Pringle JH, Hutchinson P, Osborn J, Huang Q, Potter L, Hancox RA, Fletcher A, Saldanha GS. WNT5A expression increases during melanoma progression and correlates with outcome. Clinical Cancer Research. 2008;14:5825–5832. doi: 10.1158/1078-0432.CCR-07-5104. [DOI] [PubMed] [Google Scholar]
- Dissanayake SK, Wade M, Johnson CE, O'Connell MP, Leotlela PD, French AD, Shah KV, Hewitt KJ, Rosenthal DT, Indig FE, Jiang Y, Nickoloff BJ, Taub DD, Trent JM, Moon RT, Bittner M, Weeraratna AT. The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. Journal of Biological Chemistry. 2007;282:17259–17271. doi: 10.1074/jbc.M700075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo M, Nishita M, Minami Y. Analysis of Wnt/planar cell polarity pathway in cultured cells. Methods in molecular biology. 2012;839:201–214. doi: 10.1007/978-1-61779-510-7_16. [DOI] [PubMed] [Google Scholar]
- Enomoto M, Hayakawa S, Itsukushima S, Ren DY, Matsuo M, Tamada K, Oneyama C, Okada M, Takumi T, Nishita M, Minami Y. Autonomous regulation of osteosarcoma cell invasiveness by Wnt5a/Ror2 signaling. Oncogene. 2009;28:3197–3208. doi: 10.1038/onc.2009.175. [DOI] [PubMed] [Google Scholar]
- Ford CE, Punnia-Moorthy G, Henry CE, Llamosas E, Nixdorf S, Olivier J, Caduff R, Ward RL, Heinzelmann-Schwarz V. The non-canonical Wnt ligand, Wnt5a, is upregulated and associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Gynecologic Oncology. 2014;134:338–345. doi: 10.1016/j.ygyno.2014.06.004. [DOI] [PubMed] [Google Scholar]
- Forrester WC, Dell M, Perens E, Garriga G. A C. elegans ror receptor tyrosine kinase regulates cell motility and asymmetric cell division. Nature. 1999;400:881–885. doi: 10.1038/23722. [DOI] [PubMed] [Google Scholar]
- Forrester WC, Perens E, Zallen JA, Garriga G. Identification of caenorhabditis elegans genes required for neuronal differentiation and migration. Genetics. 1998;148:151–165. doi: 10.1093/genetics/148.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile A, Lazzari L, Benvenuti S, Trusolino L, Comoglio PM. Ror1 is a pseudokinase that is crucial for Met-driven tumorigenesis. Cancer Research. 2011;71:3132–3141. doi: 10.1158/0008-5472.CAN-10-2662. [DOI] [PubMed] [Google Scholar]
- Green JL, Inoue T, Sternberg PW. Opposing Wnt pathways orient cell polarity during organogenesis. Cell. 2008a;134:646–656. doi: 10.1016/j.cell.2008.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JL, Kuntz SG, Sternberg PW. Ror receptor tyrosine kinases: orphans no more. Trends in Cell Biology. 2008b;18:536–544. doi: 10.1016/j.tcb.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumolato L, Liu G, Mong P, Mudbhary R, Biswas R, Arroyave R, Vijayakumar S, Economides AN, Aaronson SA. Canonical and noncanonical Wnts use a common mechanism to activate completely unrelated coreceptors. Genes & Development. 2010;24:2517–2530. doi: 10.1101/gad.1957710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillabert-Gourgues A, Jaspard-Vinassa B, Bats ML, Sewduth RN, Franzl N, Peghaire C, Jeanningros S, Moreau C, Roux E, Larrieu-Lahargue F, Dufourcq P, Couffinhal T, Duplàa C. Kif26b controls endothelial cell polarity through the dishevelled/daam1-dependent planar cell polarity-signaling pathway. Molecular Biology of the Cell. 2016;27:941–953. doi: 10.1091/mbc.E14-08-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujral TS, Chan M, Peshkin L, Sorger PK, Kirschner MW, MacBeath G. A noncanonical frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell. 2014a;159:844–856. doi: 10.1016/j.cell.2014.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujral TS, Peshkin L, Kirschner MW. Exploiting polypharmacology for drug target deconvolution. PNAS. 2014b;111:5048–5053. doi: 10.1073/pnas.1403080111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habas R, Kato Y, He X. Wnt/Frizzled activation of rho regulates vertebrate gastrulation and requires a novel formin homology protein Daam1. Cell. 2001;107:843–854. doi: 10.1016/S0092-8674(01)00614-6. [DOI] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Developmental Biology. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- Hikasa H, Shibata M, Hiratani I, Taira M. The xenopus receptor tyrosine kinase Xror2 modulates morphogenetic movements of the axial mesoderm and neuroectoderm via Wnt signaling. Development. 2002;129:5227–5239. doi: 10.1242/dev.129.22.5227. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Tanaka Y. Kinesin superfamily proteins (KIFs): various functions and their relevance for important phenomena in life and diseases. Experimental Cell Research. 2015;334:16–25. doi: 10.1016/j.yexcr.2015.02.016. [DOI] [PubMed] [Google Scholar]
- Ho HY, Susman MW, Bikoff JB, Ryu YK, Jonas AM, Hu L, Kuruvilla R, Greenberg ME. Wnt5a-ror-dishevelled signaling constitutes a core developmental pathway that controls tissue morphogenesis. PNAS. 2012;109:4044–4051. doi: 10.1073/pnas.1200421109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Xiao A, Choi SY, Kan Q, Zhou W, Chacon-Heszele MF, Ryu YK, McKenna S, Zuo X, Kuruvilla R, Lipschutz JH. Wnt5a is necessary for normal kidney development in zebrafish and mice. Nephron Experimental Nephrology. 2014;128:80–88. doi: 10.1159/000368411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher A, Horwitz AR. Integrins in cell migration. Cold Spring Harbor Perspectives in Biology. 2011;3:a005074. doi: 10.1101/cshperspect.a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iioka H, Iemura S, Natsume T, Kinoshita N. Wnt signalling regulates paxillin ubiquitination essential for mesodermal cell motility. Nature Cell Biology. 2007;9:813–821. doi: 10.1038/ncb1607. [DOI] [PubMed] [Google Scholar]
- Jao LE, Appel B, Wente SR. A zebrafish model of lethal congenital contracture syndrome 1 reveals Gle1 function in spinal neural precursor survival and motor axon arborization. Development. 2012;139:1316–1326. doi: 10.1242/dev.074344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Crossman DK, Mitchell EH, Sohn P, Crowley MR, Serra R. WNT5A inhibits metastasis and alters splicing of Cd44 in breast cancer cells. PLoS One. 2013;8:e58329. doi: 10.1371/journal.pone.0058329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Zhao C, Han X, Han Y. Wnt5a promotes ewing sarcoma cell migration through upregulating CXCR4 expression. BMC Cancer. 2012;12:480. doi: 10.1186/1471-2407-12-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuna E, Susman M, Ho H-Y. Quantitative Live-cell Reporter Assay for Noncanonical Wnt Activity. Bio-Protocol. 2018;8:e2762. doi: 10.21769/BioProtoc.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi A, Yamamoto H, Sato A, Matsumoto S. Wnt5a: its signalling, functions and implication in diseases. Acta Physiologica. 2012;204:17–33. doi: 10.1111/j.1748-1716.2011.02294.x. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Developmental Dynamics. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Kremenevskaja N, von Wasielewski R, Rao AS, Schöfl C, Andersson T, Brabant G. Wnt-5a has tumor suppressor activity in thyroid carcinoma. Oncogene. 2005;24:2144–2154. doi: 10.1038/sj.onc.1208370. [DOI] [PubMed] [Google Scholar]
- Kurayoshi M, Oue N, Yamamoto H, Kishida M, Inoue A, Asahara T, Yasui W, Kikuchi A. Expression of Wnt-5a is correlated with aggressiveness of gastric cancer by stimulating cell migration and invasion. Cancer Research. 2006;66:10439–10448. doi: 10.1158/0008-5472.CAN-06-2359. [DOI] [PubMed] [Google Scholar]
- LaFave MC, Varshney GK, Vemulapalli M, Mullikin JC, Burgess SM. A defined zebrafish line for high-throughput genetics and genomics: NHGRI-1. Genetics. 2014;198:167–170. doi: 10.1534/genetics.114.166769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird DJ, Altshuler-Keylin S, Kissner MD, Zhou X, Anderson KV. Ror2 enhances polarity and directional migration of primordial germ cells. PLoS Genetics. 2011;7:e1002428. doi: 10.1371/journal.pgen.1002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird DJ, von Andrian UH, Wagers AJ. Stem cell trafficking in tissue development, growth, and disease. Cell. 2008;132:612–630. doi: 10.1016/j.cell.2008.01.041. [DOI] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. PNAS. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Salic A, Krüger R, Heinrich R, Kirschner MW. The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biology. 2003;1:E10. doi: 10.1371/journal.pbio.0000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Liu Y, Zhao W, Sun B, Chen Q. Wnt5A expression is associated with the tumor metastasis and clinical survival in cervical cancer. International journal of clinical and experimental pathology. 2014;7:6072–6078. [PMC free article] [PubMed] [Google Scholar]
- Liu A, Chen S, Cai S, Dong L, Liu L, Yang Y, Guo F, Lu X, He H, Chen Q, Hu S, Qiu H. Wnt5a through noncanonical Wnt/JNK or Wnt/PKC signaling contributes to the differentiation of mesenchymal stem cells into type II alveolar epithelial cells in vitro. PLoS One. 2014;9:e90229. doi: 10.1371/journal.pone.0090229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi B, Rendell N, Edwards M, Katan M, Zimmermann JG. Evaluation of phosphopeptide enrichment strategies for quantitative TMT analysis of complex network dynamics in cancer-associated cell signalling. EuPA Open Proteomics. 2015;6:10–15. doi: 10.1016/j.euprot.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu BJ, Wang YQ, Wei XJ, Rong LQ, Wei D, Yan CM, Wang DJ, Sun JY. Expression of WNT-5a and ROR2 correlates with disease severity in osteosarcoma. Molecular medicine reports. 2012;5:1033–1036. doi: 10.3892/mmr.2012.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Wei W, de Bock GH, Zhou H, Li Q, Shen X. The roles of Wnt5a, JNK and paxillin in the occurrence of metastasis of pancreatic adenocarcinoma. International Journal of Clinical Oncology. 2014;19:1011–1019. doi: 10.1007/s10147-013-0648-0. [DOI] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, Gygi SP. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Analytical Chemistry. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald SL, Silver A. The opposing roles of Wnt-5a in cancer. British Journal of Cancer. 2009;101:209–214. doi: 10.1038/sj.bjc.6605174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren A. Primordial germ cells in the mouse. Developmental Biology. 2003;262:1–15. doi: 10.1016/S0012-1606(03)00214-8. [DOI] [PubMed] [Google Scholar]
- Mendrola JM, Shi F, Park JH, Lemmon MA. Receptor tyrosine kinases with intracellular pseudokinase domains. Biochemical Society Transactions. 2013;41:1029–1036. doi: 10.1042/BST20130104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. PNAS. 1999;96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikels A, Minami Y, Nusse R. Ror2 receptor requires tyrosine kinase activity to mediate Wnt5A signaling. Journal of Biological Chemistry. 2009;284:30167–30176. doi: 10.1074/jbc.M109.041715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, Ajima R, Luo CT, Yamaguchi TP, Stappenbeck TS. Wnt5a potentiates TGF-β signaling to promote colonic crypt regeneration after tissue injury. Science. 2012;338:108–113. doi: 10.1126/science.1223821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon RT, Campbell RM, Christian JL, McGrew LL, Shih J, Fraser S. Xwnt-5A: a maternal Wnt that affects morphogenetic movements after overexpression in embryos of Xenopus laevis. Development. 1993;119:97–111. doi: 10.1242/dev.119.1.97. [DOI] [PubMed] [Google Scholar]
- Nishita M, Itsukushima S, Nomachi A, Endo M, Wang Z, Inaba D, Qiao S, Takada S, Kikuchi A, Minami Y. Ror2/Frizzled complex mediates Wnt5a-induced AP-1 activation by regulating dishevelled polymerization. Molecular and Cellular Biology. 2010;30:3610–3619. doi: 10.1128/MCB.00177-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishita M, Qiao S, Miyamoto M, Okinaka Y, Yamada M, Hashimoto R, Iijima K, Otani H, Hartmann C, Nishinakamura R, Minami Y. Role of Wnt5a-Ror2 signaling in morphogenesis of the metanephric mesenchyme during ureteric budding. Molecular and Cellular Biology. 2014;34:3096–3105. doi: 10.1128/MCB.00491-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomachi A, Nishita M, Inaba D, Enomoto M, Hamasaki M, Minami Y. Receptor tyrosine kinase Ror2 mediates Wnt5a-induced polarized cell migration by activating c-Jun N-terminal kinase via actin-binding protein filamin A. Journal of Biological Chemistry. 2008;283:27973–27981. doi: 10.1074/jbc.M802325200. [DOI] [PubMed] [Google Scholar]
- Oishi I, Suzuki H, Onishi N, Takada R, Kani S, Ohkawara B, Koshida I, Suzuki K, Yamada G, Schwabe GC, Mundlos S, Shibuya H, Takada S, Minami Y. The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes to Cells. 2003;8:645–654. doi: 10.1046/j.1365-2443.2003.00662.x. [DOI] [PubMed] [Google Scholar]
- Park HW, Kim YC, Yu B, Moroishi T, Mo JS, Plouffe SW, Meng Z, Lin KC, Yu FX, Alexander CM, Wang CY, Guan KL. Alternative wnt signaling activates YAP/TAZ. Cell. 2015;162:780–794. doi: 10.1016/j.cell.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nature Reviews Molecular Cell Biology. 2010;11:633–643. doi: 10.1038/nrm2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA, McAllister FE, Everley RA, Beausoleil SA, Banks AS, Gygi SP. Effects of MEK inhibitors GSK1120212 and PD0325901 in vivo using 10-plex quantitative proteomics and phosphoproteomics. PROTEOMICS. 2015;15:462–473. doi: 10.1002/pmic.201400154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Person AD, Beiraghi S, Sieben CM, Hermanson S, Neumann AN, Robu ME, Schleiffarth JR, Billington CJ, van Bokhoven H, Hoogeboom JM, Mazzeu JF, Petryk A, Schimmenti LA, Brunner HG, Ekker SC, Lohr JL. WNT5A mutations in patients with autosomal dominant Robinow syndrome. Developmental Dynamics : An Official Publication of the American Association of Anatomists. 2010;239:327–337. doi: 10.1002/dvdy.22156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad CP, Chaurasiya SK, Guilmain W, Andersson T. WNT5A signaling impairs breast cancer cell migration and invasion via mechanisms independent of the epithelial-mesenchymal transition. Journal of Experimental & Clinical Cancer Research. 2016;35:144. doi: 10.1186/s13046-016-0421-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Yin YT, Zheng FJ, Peng LX, Yang CF, Bao YN, Liang YY, Li XJ, Xiang YQ, Sun R, Li AH, Zou RH, Pei XQ, Huang BJ, Kang TB, Liao DF, Zeng YX, Williams BO, Qian CN. WNT5A promotes stemness characteristics in nasopharyngeal carcinoma cells leading to metastasis and tumorigenesis. Oncotarget. 2015;6:10239–10252. doi: 10.18632/oncotarget.3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recuenco MC, Ohmori T, Tanigawa S, Taguchi A, Fujimura S, Conti MA, Wei Q, Kiyonari H, Abe T, Adelstein RS, Nishinakamura R. Nonmuscle Mmyosin II regulates the morphogenesis of metanephric mesenchyme-derived immature nephrons. Journal of the American Society of Nephrology. 2015;26:1081–1091. doi: 10.1681/ASN.2014030281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Kontarakis Z, Gerri C, Nolte H, Hölper S, Krüger M, Stainier DY. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 2015;524:230–233. doi: 10.1038/nature14580. [DOI] [PubMed] [Google Scholar]
- Sato A, Yamamoto H, Sakane H, Koyama H, Kikuchi A. Wnt5a regulates distinct signalling pathways by binding to Frizzled2. The EMBO Journal. 2010;29:41–54. doi: 10.1038/emboj.2009.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenov MV, Habas R, Macdonald BT, He X. Snapshot: noncanonical wnt signaling pathways. Cell. 2007;131:1378. doi: 10.1016/j.cell.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Sokol SY, Klingensmith J, Perrimon N, Itoh K. Dorsalizing and neuralizing properties of Xdsh, a maternally expressed Xenopus homolog of dishevelled. Development. 1995;121:1637–1647. doi: 10.1242/dev.121.6.1637. [DOI] [PubMed] [Google Scholar]
- Terabayashi T, Sakaguchi M, Shinmyozu K, Ohshima T, Johjima A, Ogura T, Miki H, Nishinakamura R. Phosphorylation of Kif26b promotes its polyubiquitination and subsequent proteasomal degradation during kidney development. PLoS One. 2012;7:e39714. doi: 10.1371/journal.pone.0039714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nature Methods. 2011;8:937–940. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. The Journal of Cell Biology. 1963;17:299–313. doi: 10.1083/jcb.17.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama Y, Sakaguchi M, Terabayashi T, Inenaga T, Inoue S, Kobayashi C, Oshima N, Kiyonari H, Nakagata N, Sato Y, Sekiguchi K, Miki H, Araki E, Fujimura S, Tanaka SS, Nishinakamura R. Kif26b, a kinesin family gene, regulates adhesion of the embryonic kidney mesenchyme. PNAS. 2010;107:9240–9245. doi: 10.1073/pnas.0913748107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amerongen R, Fuerer C, Mizutani M, Nusse R. Wnt5a can both activate and repress Wnt/β-catenin signaling during mouse embryonic development. Developmental Biology. 2012;369:101–114. doi: 10.1016/j.ydbio.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amerongen R, Mikels A, Nusse R. Alternative wnt signaling is initiated by distinct receptors. Science Signaling. 2008;1:re9. doi: 10.1126/scisignal.135re9. [DOI] [PubMed] [Google Scholar]
- van Bokhoven H, Celli J, Kayserili H, van Beusekom E, Balci S, Brussel W, Skovby F, Kerr B, Percin EF, Akarsu N, Brunner HG. Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nature genetics. 2000;25:423–426. doi: 10.1038/78113. [DOI] [PubMed] [Google Scholar]
- Veeman MT, Axelrod JD, Moon RT. A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Developmental cell. 2003;5:367–377. doi: 10.1016/s1534-5807(03)00266-1. [DOI] [PubMed] [Google Scholar]
- Vinson CR, Adler PN. Directional non-cell autonomy and the transmission of polarity information by the frizzled gene of Drosophila. Nature. 1987;329:549–551. doi: 10.1038/329549a0. [DOI] [PubMed] [Google Scholar]
- von Philipsborn A, Bastmeyer M. Mechanisms of gradient detection: a comparison of axon pathfinding with eukaryotic cell migration. International review of cytology. 2007;263:1–62. doi: 10.1016/S0074-7696(07)63001-0. [DOI] [PubMed] [Google Scholar]
- Wallingford JB, Habas R. The developmental biology of Dishevelled: an enigmatic protein governing cell fate and cell polarity. Development. 2005;132:4421–4436. doi: 10.1242/dev.02068. [DOI] [PubMed] [Google Scholar]
- White J, Mazzeu JF, Hoischen A, Jhangiani SN, Gambin T, Alcino MC, Penney S, Saraiva JM, Hove H, Skovby F, Kayserili H, Estrella E, Vulto-van Silfhout AT, Steehouwer M, Muzny DM, Sutton VR, Gibbs RA, Lupski JR, Brunner HG, van Bon BW, Carvalho CM, Baylor-Hopkins Center for Mendelian Genomics DVL1 frameshift mutations clustering in the penultimate exon cause autosomal-dominant Robinow syndrome. The American Journal of Human Genetics. 2015;96:612–622. doi: 10.1016/j.ajhg.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JJ, Mazzeu JF, Hoischen A, Bayram Y, Withers M, Gezdirici A, Kimonis V, Steehouwer M, Jhangiani SN, Muzny DM, Gibbs RA, van Bon BW, Sutton VR, Lupski JR, Brunner HG, Carvalho CM, Baylor-Hopkins Center for Mendelian Genomics DVL3 Alleles resulting in a -1 frameshift of the last exon mediate autosomal-dominant robinow syndrome. The American Journal of Human Genetics. 2016;98:553–561. doi: 10.1016/j.ajhg.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman B, Clark SG, Taskar AM, Forrester WC, Maricq AV, Bargmann CI, Garriga G. The C. elegans gene vab-8 guides posteriorly directed axon outgrowth and cell migration. Development. 1996;122:671–682. doi: 10.1242/dev.122.2.671. [DOI] [PubMed] [Google Scholar]
- Witze ES, Connacher MK, Houel S, Schwartz MP, Morphew MK, Reid L, Sacks DB, Anseth KS, Ahn NG. Wnt5a directs polarized calcium gradients by recruiting cortical endoplasmic reticulum to the cell trailing edge. Developmental Cell. 2013;26:645–657. doi: 10.1016/j.devcel.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witze ES, Litman ES, Argast GM, Moon RT, Ahn NG. Wnt5a control of cell polarity and directional movement by polarized redistribution of adhesion receptors. Science. 2008;320:365–369. doi: 10.1126/science.1151250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FW, Hung MS, Wightman B, Way J, Garriga G. vab-8 is a key regulator of posteriorly directed migrations in C. elegans and encodes a novel protein with kinesin motor similarity. Neuron. 1998;20:655–666. doi: 10.1016/S0896-6273(00)81006-5. [DOI] [PubMed] [Google Scholar]
- Yamaguchi TP, Bradley A, McMahon AP, Jones S. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development. 1999;126:1211–1223. doi: 10.1242/dev.126.6.1211. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Nagano T, Takehara S, Hibi M, Aizawa S. Shisa promotes head formation through the inhibition of receptor protein maturation for the caudalizing factors, Wnt and FGF. Cell. 2005;120:223–235. doi: 10.1016/j.cell.2004.11.051. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Oue N, Sato A, Hasegawa Y, Yamamoto H, Matsubara A, Yasui W, Kikuchi A. Wnt5a signaling is involved in the aggressiveness of prostate cancer and expression of metalloproteinase. Oncogene. 2010;29:2036–2046. doi: 10.1038/onc.2009.496. [DOI] [PubMed] [Google Scholar]
- Yang Y, Kitagaki J, Dai RM, Tsai YC, Lorick KL, Ludwig RL, Pierre SA, Jensen JP, Davydov IV, Oberoi P, Li CC, Kenten JH, Beutler JA, Vousden KH, Weissman AM. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Research. 2007;67:9472–9481. doi: 10.1158/0008-5472.CAN-07-0568. [DOI] [PubMed] [Google Scholar]
- Yu H, Smallwood PM, Wang Y, Vidaltamayo R, Reed R, Nathans J. Frizzled 1 and frizzled 2 genes function in palate, ventricular septum and neural tube closure: general implications for tissue fusion processes. Development. 2010;137:3707–3717. doi: 10.1242/dev.052001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Ye X, Guo N, Nathans J. Frizzled 2 and frizzled 7 function redundantly in convergent extension and closure of the ventricular septum and palate: evidence for a network of interacting genes. Development. 2012;139:4383–4394. doi: 10.1242/dev.083352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Chen L, Cui B, Widhopf GF, Shen Z, Wu R, Zhang L, Zhang S, Briggs SP, Kipps TJ. Wnt5a induces ROR1/ROR2 heterooligomerization to enhance leukemia chemotaxis and proliferation. Journal of Clinical Investigation. 2016;126:585–598. doi: 10.1172/JCI83535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun K, Ajima R, Sharma N, Costantini F, Mackem S, Lewandoski M, Yamaguchi TP, Perantoni AO. Non-canonical Wnt5a/Ror2 signaling regulates kidney morphogenesis by controlling intermediate mesoderm extension. Human Molecular Genetics. 2014;23:6807–6814. doi: 10.1093/hmg/ddu397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A, He S, Sun X, Ding L, Bao X, Wang N. Wnt5a promotes migration of human osteosarcoma cells by triggering a phosphatidylinositol-3 kinase/Akt signals. Cancer Cell International. 2014;14:15. doi: 10.1186/1475-2867-14-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Niwa S, Homma N, Takei Y, Hirokawa N. KIF26A is an unconventional kinesin and regulates GDNF-Ret signaling in enteric neuronal development. Cell. 2009;139:802–813. doi: 10.1016/j.cell.2009.10.023. [DOI] [PubMed] [Google Scholar]