

Abstract
Epigenetic regulation is a key factor in cellular homeostasis. Post-translational modifications (PTMs) are a central focus of this regulation as they function as signaling markers within the cell. Lysine acetylation is a dynamic, reversible PTM that has garnered recent attention due to alterations in various types of cancer. Acetylation levels are regulated by two opposing enzyme families: lysine acetyltransferases (KATs) and histone deacetylases (HDACs). HDACs are key players in epigenetic regulation and have a role in the silencing of tumor suppressor genes. The dynamic equilibrium of acetylation makes HDACs attractive targets for drug therapy. However, substrate selectivity and biological function of HDAC isozymes is poorly understood. This review outlines the current understanding of the roles and specific epigenetic interactions of the metal-dependent HDACs in addition to their roles in cancer.
Keywords: Post-translational modifications: The enzyme-catalyzed changes that occur to proteins following translation; these include the covalent addition of functional groups; Nε-Acetylation: The post-translational modification of lysine residues by enzymatic addition of an acetyl group; Histone deacetylase: Family of enzymes responsible for catalyzing the hydrolysis of Nε-acetyl lysine residues; HDACi: Histone deacetylase inhibitors, primarily small molecules that chelate active site divalent metal ions; SAHA: Suberoylanilide hydroxamic acid, a potent pan-HDAC inhibitor currently used as treatment for T-cell lymphoma; Cancer: A disease involving uncontrolled cell growth
Graphical abstract
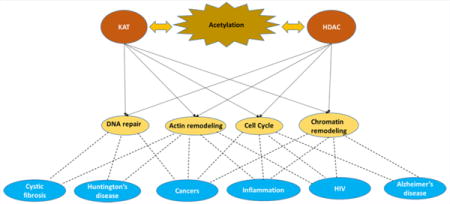
Regulation of gene expression by post-translational modifications (PTMs) is important. Many of these modifications are dynamic and reversible. PTMs include methylation, phosphorylation, ubiquitination, acetylation, and biotinylation, among others. Some of these PTMs, such as acetylation and methylation, were originally identified as modifications on the tails of core histones to regulate access to DNA by alterations of the chromatin structure.1 Lysine acetylation has garnered increasing interest in recent years, with a trend in publication rate that rivals that of phosphorylation.2
A major advance in the acetylation field has been the transition from analysis of acetylation (hyper- and hypoacetylation) of specific sites in histone tails to defining and understanding the numerous proteins that are modified through acetylation events. Acetylation has been shown to compete and cooperate with other PTMs, such as ubiquitination,3 and affect specific protein–protein interactions, protein stability, and protein–DNA interactions.4 Identifying these interactions will lead to a better understanding of the acetylome, the collection of nonhistone proteins that undergo acetylation/deacetylation, and the role of acetylation in cell regulation, growth, and homeostasis (Figure 1).
Figure 1.
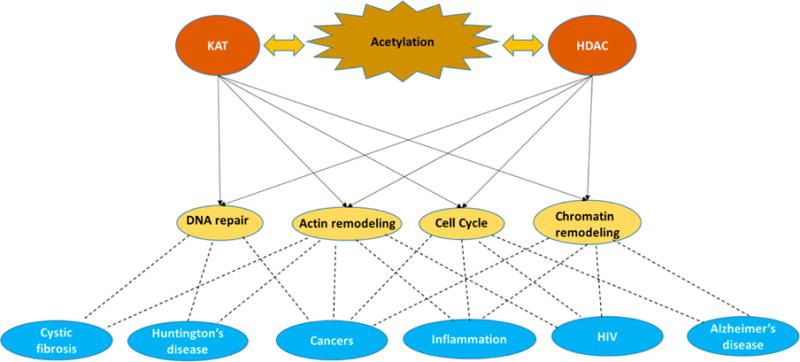
Acetylation, critical for regulation and proper cellular function. Lysine acetyltransferases (KATs) and histone deacetylases (HDACs) maintain acetylation at an optimal level. HATs and HDACs regulate essential processes such as DNA repair, chromatin and actin remodeling, and proteins that serve as checkpoints during the cell cycle.
Acetylation is an enzymatically catalyzed and reversible PTM in which an acetyl group is attached to the Nε-position of a lysine side chain. Lysine acetyltransferases catalyze acetylation of lysine side chains using acetyl-CoA as a cofactor, and metal-dependent histone deacetylases (HDACs) catalyze hydrolysis of the acetyl moiety to regenerate lysine and free acetate. Traditionally, HDACs have been described as transcriptional repressors since they change the recruitment and intramolecular interactions of many proteins, from bromodomain-containing proteins, MEF2-binding proteins, and domains to histone tails, in addition to aiding in local chromatin compaction.1 However, acetylation and deacetylation of many nonhistone proteins has now been observed. In fact, currently, over 3600 acetylation sites have been identified in mammalian proteins, with many more being discovered through proteomics and computational and modeling analyses.5 In light of these many nonhistone HDAC targets, a more suitable name for these enzymes is acetyl-lysine deacetylases, or acKDACs. Aberrant regulation of protein acetylation has been observed in various types of cancers including prostate,8 breast,9 and colon10 among others, in addition to a variety of diseases, such as Cornelia de Lange Syndrome (CdLS),6 Huntington’s disease,7 and inflammation.17
The increasingly evident role and overexpression of HDACs in cancer has made them an interesting anticancer target. Current research shows that there are multiple mechanisms by which HDACs affect cancer development, such as inducing growth arrest, differentiation, senescence, and death of cancerous cells.14,19 Several HDAC inhibitors (HDACi) have been developed to combat these mechanisms; however, the current clinically used compounds do not possess isozyme selectivity. Three pan-HDAC inhibitors—suberoylanilide hydroxamic acid (SAHA),11 Romidepsin (cyclic peptide),11 and belinostat (hydroxamic acid)12—have been approved by the FDA for the treatment of T-cell lymphomas, and a fourth inhibitor, Panobinostat (hydroxamic acid), has recently been approved for multiple myeloma treatment.13
Studies have recently demonstrated that pan-HDACi can also be used to increase the effectiveness of anticancer immunotherapy treatments. In one mechanism, T-cell survival is enhanced due to prevention of activation-induced cell death by lymphocytes.14,15 However, these effects vary significantly and can produce a variety of nondesirable side effects as a consequence. Additionally, HDACi’s have been used to enhance vaccine strategies; namely, mice vaccinated with melanoma cells that have been pretreated with trichostatin A (TSA) show an increase in immune response toward additional tumors, effectively enhancing their tumor specific immunity mechanisms.16,17
HDACs are divided into four different classes based on their sequence homology to yeast orthologs.18 Class I, which shares homology with Rpd3, consists of HDACs 1, 2, 3, and 8. Class II, with homology to Hda1, can be divided into two subclasses—IIa (HDACs 4, 7 and 9) and IIb (HDACs 6 and 10). Class III, with homology to the Sir2 family, is known as the sirtuins and utilizes NAD+ as a cofactor. Class IV, which shares homology with both class I and class II, consists of HDAC11. Classes I, II, and IV are metal-dependent HDACs that use a metal–water as the nucleophile during catalysis, which is activated via a general acid–base mechanism (Figure 2).19 In addition, the activity of HDACs can be regulated by monovalent metal cations.20 Metal-dependent HDACs are the focus of this review.
Figure 2.
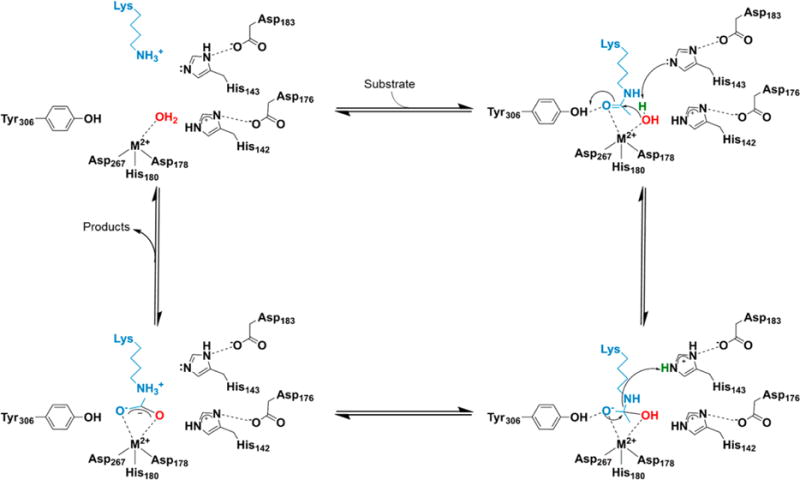
General acid/base mechanism of catalysis utilized by metal-dependent deacetylases.19 The conserved Asp–Asp–His triad coordinates a divalent metal ion that coordinates the metal–water nucleophile. His143 acts as both a general acid and a general base, while Tyr306 and His142 stabilize the oxyanion intermediate.
Metal-dependent HDACs share common sequence motifs (Figure 3), including a deacetylase domain that is comprised of an arginase-deacetylase fold consisting of a multistrand β-sheet surrounded by α-helices and a divalent metal ion cofactor coordinated by an Asp–Asp–His triad.23 Class I HDACs possess a deacetylase domain with little sequence variation and are localized mainly in the nucleus,18 with the exception of HDAC8, which has been observed in the cytoplasm of smooth muscle cells.21 Class II HDACs are shuttled between the nucleus and the cytoplasm and possess additional domains, such as MEF2 binding domains. HDAC6 has the largest array of domains with two deacetylase domains and a zinc finger protein binding domain.18 Class II HDACs are expressed ubiquitously through the cell and generally have lower catalytic activity in vitro when compared to class I enzymes. Finally, HDAC11, the only class IV metal-dependent deacetylase, possesses characteristics from both class I and II enzymes and is expressed in higher abundance in specific tissues such as brain, heart, and kidney.22
Figure 3.
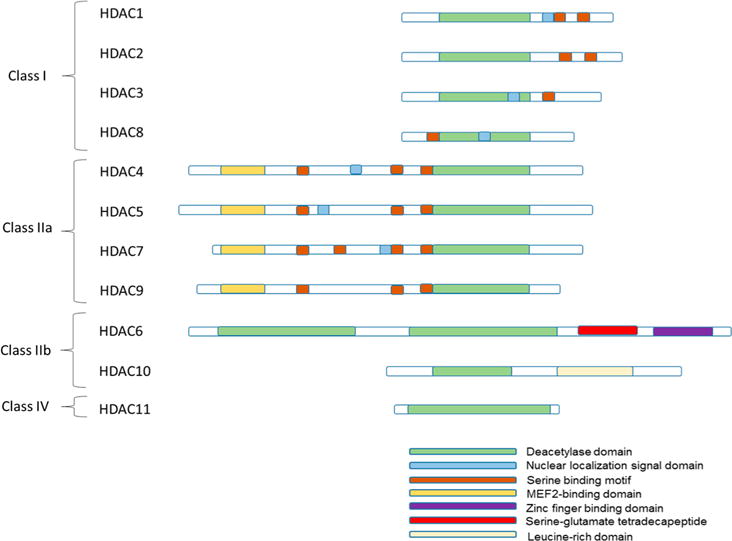
Schematic comparison of the metal-dependent deacetylases. All isozymes possess a common deacetylase domain. Class I isozymes are highly conserved and small. Class IIa isozymes have specific MEF2-binding domains in addition to their conserved deacetylase domains. Class IIb contain unique domains unlike the other classes. Only the deacetylase domain has been identified in class IV.
Crystal structures have been solved for HDAC1 (PDB: 4BKX), HDAC2 (PDB: 3MAX), HDAC3 (PDB: 4A69), HDAC4 (PDB: 2VQM), HDAC6 (PDB: 3PHD), HDAC7 (PDB: 3C0Y; catalytic domain only), HDAC8 (PDB: 2V5W), and HDAC9 (PDB: 1TQE). The isozymes lacking crystal structures are HDAC5, HDAC10, and HDAC11.
One of the most prevalent questions in the HDAC field is the substrate selectivity of each isozyme. There are over 3600 validated mammalian acetylation sites5 and 18 deacetylase isozymes. Thus, defining the substrate pool for each isozyme is essential for understanding the biological functions of acetylation. Additionally, the substrate specificity of HDACs might be regulated by oxidative stress, protein–protein interactions, and other PTMs. Elucidating HDAC substrate specificity and regulation will provide insight into the function and control of acetylation sites in proteins. Much research in the field has focused on class I HDACs; thus, only the most recent work on that class is cited in this review. Classes IIa, IIb, and IV have fewer published studies, and the literature is therefore covered more extensively.
In this review, we highlight functions of metal-dependent deacetylases with regard to epigenetic regulation and homeostasis and how these modifications play a role in cell proliferation and growth in various cancers, in addition to other, as of now, unknown roles.
CLASS I HDACS
The class I HDAC subfamily is disregulated in cancers and is the best studied subfamily of the metal-dependent deacetylases. Overexpression of this subclass has been observed in a variety of cancers such as gastric,24 breast,9 prostate,8 and colon,10 as well as T-cell25 and Hodgkin’s lymphoma.26 Class I protein–protein interactions are currently the most well understood (Table 1).
Table 1.
Summary of Cancer and Disease Related HDAC–Protein Interactions and Associated Phenotypes for Class I (1, 2, 3, and 8) and Class IV (11) HDACs
| HDAC isozyme | potential interactions | disease/condition | observable phenotype |
|---|---|---|---|
| HDAC1 | plasmogenic activators28 | breast cancer27 | cell cycle arrest, growth inhibition and apoptosis |
| IKF229 | |||
| transcription factor BBX29 | |||
| ADNP29 | |||
| ARID5B29 | |||
| C16orf8729 | |||
| estrogen and progesterone receptors30 | acute promyelocytic lymphoma30 | lower genetic stability and increase of progenitor cells | |
| PML-RAR30 | |||
| carcinomas31 | |||
| HDAC231 | |||
| leukemogenesis acceleration rate | |||
| SNAIL131 | leukemia31 | ||
| E-cadherin31 | thymocyte accumulation | ||
| P5330 | |||
| c-myc30 | |||
| HDAC2 | P53/DNA30 | apoptosis induction in cells | |
| blocking of cell proliferation | |||
| Bax36 | |||
| Bcl236 | |||
| Cyclin E237 | |||
| Cyclin D137 | |||
| CDK237 | |||
| HDAC139 | chronic lymphocytic leukemia39 | TRAIL-induced apoptosis | |
| Peryone’s disease40 | regression of fibril formation | ||
| growth inhibition | |||
| insulin-like growth factors34 | decreased body size in mice | ||
| HDAC3 | P5344 | pancreatic cancer44 | |
| P2744 | |||
| Bax44 | |||
| NCoR45 | promyelocytic leukemia45 | restoration of retinoic acid gene expression | |
| PML-RARalpha45 | |||
| HDAC8 | T-cell lymphoma25 | affects skull morphology and growth | |
| childhood neuroblastoma50 | cell apoptosis | ||
| SMC36 | cell cycle inhibition | ||
| Cornelia de Lange syndrome6 | improper chromatid separation | ||
| ARID1A52 | |||
| stops cell proliferation | |||
| CSRP2BP52 | lung cancer50 | ||
| cervical cancer50 | |||
| colon cancer50 | |||
| HDAC11 | Cytokine-interleukin 10105 | inflammation105 | |
| ductal breast carcinoma100 | |||
| Cdt1102 | cell cycle affector |
In the majority of cases, upregulation of HDAC1 is associated with poor cancer prognosis.91 Silencing of HDAC1 using siRNA knockouts results in cell cycle arrest, growth inhibition, and induction of apoptosis in breast cancer cells27 and induction of a plasminogen activator in neuroblastoma cells, increasing their invasive capacity.28 Mass proteomic analyses have revealed additional HDAC1 protein–protein interactions, ranging from short-lived interaction proteins, such as IKF2, HMG box transcription factor BBX, and activity-dependent neuroprotector homeobox protein ADNP to proteins with methylation-related functions such as ARID5B to previously uncharacterized zinc-binding proteins and domains, such as C16orf87.29 Additionally, HDAC1 has been demonstrated to interact with the oncogene fusion protein PML-RAR, a protein involved in the pathogenesis of acute promyelocytic lymphoma (APL); in particular, HDAC1 diminishes the tumorigenic activity of PML-RAR by blocking differentiation, impairing genetic stability, and increasing the renewal of progenitor cells. However, HDAC1 expression enhances cell survival once they are differentiated, thus leading to a dual role in cancerous tissue.30
Immunodeficient mice have been used to evaluate the role of HDAC1 in tumor formation using teratomas. In these models, HDAC1 deficiency leads to partially undifferentiated carcinomas, upregulation of HDAC2, elevated levels of SNAIL1 expression, and delocalization of E-cadherin.31 Knockouts of HDAC1 and -2 show dramatic acceleration of leukemogenesis in preleukemic mice. HDAC1 knockouts also led to deletion of p53 and c-myc overexpression.30 Additionally, Dovey and collaborators demonstrated that knockouts of key components of the HDAC1/2 deacetylase complex (Sin3A and Mi2) that decrease HDAC activity in T-cells perturb the differentiation of thymocytes into mature T lymphocytes.32 Similarly, mice knockouts of HDAC1/2 demonstrate that the loss of HDAC activity leads to the accumulation of thymocytes in addition to blocking early thymic development.33
The previously described protein–protein interactions have led to the proposal that some HDACs function in large deacetylase complexes. HDAC1 and HDAC2, together with histone binding proteins RBBP4 and RBBP7, DNA/chromatin recognition motifs, and transcription factors form the core deacetylase complexes that help localize HDACs 1 and 2 to chromatin35 Expression of HDAC1 and HDAC3 correlate with both estrogen and progesterone receptor expression and have been proposed as prognostic markers in breast cancer tumors.35
HDAC2 is overexpressed in lung cancer tissue and mesenchymal tumors, suggesting that it is an effector for the disease. Silencing of HDAC2 via siRNA leads to an increase in p53 DNA binding activity, Bax activation, and Bcl2 suppression36 These changes in Bax activation and Bcl2 suppression are consistent with suppressed expression of cyclin E2, cyclin D1, and CDK2, blocking cell proliferation and inducing apoptosis.37 Truncations of HDAC2 have been detected in a large number of cancers38 and knockouts of both HDAC1 and HDAC2 prompt TRAIL-induced apoptosis in chronic lymphocytic leukemia (CLL), indicating a possible level of cooperativity between these two isozymes.39 Recent studies have also shown that both HDAC2 silencing and inhibition induce regression of fibrotic formation in Peyronie’s disease models.40,41 Using mutant fibroblasts that are HDAC2-deficient, Zimmerman and collaborators demonstrated a lack of response to insulin-like growth factors (IGFs) when compared to wild type cells, showing a potential link between HDACs and IGFs.34 Mice models lacking HDAC1 and with a single HDAC2 allele develop a lethal pathology within 3 months, likely due to neoplastic transformation of immature T cells.32 Additionally, mutant mice with an inactive HDAC2 mutant exhibit a 25% decrease in body size and reduced cell number and thickness of intestinal mucosa.34
HDAC3, along with HDAC1 and HDAC2, is often expressed in high levels in renal, colorectal, and gastric cancer.42,43 High expression of HDAC3 has also been observed in eight different pancreatic cancer cell lines and potentially generates a postinduction repression of p53, p27, and Bax genes through deacetylation of K9 of histone H3.44 Knockouts of HDAC3 in promyelocytic leukemia cells restore retinoic acid dependent gene expression, primarily due to the loss of the interactions between HDAC3, the nuclear corepressor NCoR, and PML-RARalpha fusion protein. HDAC3 interactions with the nuclear corepressor NCoR to block transcription are enhanced by PML-RARalpha binding to DNA.45 The best understood example of HDAC3 function is repression of retinoic acid and thyroid hormone receptors, which can modulate p53 expression.44 Additionally, HDAC3 depletion in mouse liver upregulates lipogenic genes and causes histone hyperacetylation, leading to hepatostaetosis.46 However, expression of inactive HDAC3 mutant proteins in these knockout mice almost completely rescues the metabolic and gene transcription alterations, suggesting that HDAC3 plays important non-catalytic roles, such as protein–protein interactions. Consistent with this, mice knockouts of the nuclear corepressor NcoR, an essential part of the HDAC3 deacetylase complex, exhibit metabolic and transcriptional effects resembling those of mice without hepatic HDAC3, demonstrating that interaction with NcoR is essential for the deacetylase-independent function of HDAC3.46
HDAC8 is the best biochemically characterized HDAC isozyme to date.19,20,47,48 HDAC8 is the only class I isozyme that is localized to both the cytoplasm and the nucleus and that is not observed in large, multiprotein complexes in vivo.49 HDAC8 is overexpressed in childhood neuroblastoma50 and T-cell lymphoma.25 Knockouts of HDAC8 produce skull morphology and growth complications in mice models18 and stop cell proliferation in lung, colon, and cervical cancer cells.50 Point mutations in HDAC8 have been observed in patients with symptoms similar to the Cornelia de Lange Syndrome (CdLS). Lack of deacetylation of SMC3 in the cohesin complex has been implicated as a contributor to this disease, inhibiting the cell cycle, disrupting proper chromatid separation, and causing debilitating mental and physical abnormalities.6 Currently, work on HDAC8 has focused on mass spectrometry and coimmunoprecipitation studies using the HDAC8 specific inhibitor – PCI-34051–and have provided insight into potential protein substrates and interaction partners.29,52 Knockouts and inhibition of HDAC8 have been shown to induce apoptosis in T-cell lymphoma and leukemia cell lines.25 Using mice xenograft models of oncogene-amplified neuroblastoma, Rettig and collaborators demonstrated that selective inhibition of HDAC8 shows antineuroblastoma activity without significant toxicity and induces cell cycle arrest and differentiation both in vivo and in vitro.53 Additionally, the combined treatment with HDACi and retinoic acid enhanced cell differentiation, demonstrating that inhibition of HDAC isozymes can be combined with differentiation-inducing agents to target tumors.53
Investigations of HDAC8 specificity are poised to provide insight into the role of protein–protein interactions in determining substrate specificity of metal-dependent deacetylases.
CLASS II HDACS
Class II HDACs were discovered in the early 2000s.54–57 These proteins are significantly larger than both class I and class IV HDACs due to N-terminal and C-terminal tails and/or domains attached to the canonical deacetylase domain. This class is subdivided into two subfamilies: Classes IIa and IIb. Class IIa consists of HDAC 4, 5, 7, and 9, and class IIb is comprised of HDAC 6 and 10. These isozymes differ from class I HDACs in that the additional N-terminal domains interact with transcription factors and target genes, such as the MEF2 proteins, a family of transcription factors that are key regulators of cellular differentiation.56 Recruitment of class II HDACs by MEF2 proteins to protein complexes can heavily alter the protein acetylation landscape due to blocking interactions with acetylation complexes such as CREBBP/EP300 in non-Hodgkin lymphoma.58,59 Class II HDACs are localized to both the cytoplasm and the nucleus. Additionally, both upregulation and downregulation of these enzymes have severe repercussions in various types of cancers (Table 2).
Table 2.
Summary of Cancer and Disease Related HDAC-Protein Interactions and Associated Phenotypes for Class IIa (4, 5, 7, and 9) and Class IIb (6 and 10) HDACs
| HDAC isozyme | potential interactions | disease/condition | observable phenotype |
|---|---|---|---|
| HDAC4 | p21WAF/Cip162 | breast cancer68 | affects cell proliferation |
| miR-1 and miR-2270,71 | |||
| Waldenstrom’s | |||
| macroglobulinemia68 | |||
| melanoma69 | |||
| STAT164 | |||
| ovarian cancer64 | increased cellular survival | ||
| HIF-173 | |||
| chondrosarcoma74 | growth and metastasis of cells | ||
| VEGF74 | |||
| cell growth regulation | |||
| androgen receptor75 | B-cell differentiation is reduced | ||
| miR-15576 | solid and hematological malignancies76 | ||
| Bcl-676 | |||
| MEF276 | |||
| HDAC5 | caspase-3-like protein77 | high-risk medulloblastoma77 | cell death |
| hepatocellular carcinoma78 | induces cell proliferation | ||
| P14/TBX378 | |||
| breast cancer78 | transport of HDAC5 | ||
| protein kinase C79 | |||
| dephosphorylation of HDAC5 | |||
| protein phosphatase PP2A83 | |||
| MEF276 | chromatin remodeling | ||
| 14–3–3 cytoplasmic anchoring proteins83 | |||
| HDAC7 | estrogen receptor alpha84 | breast cancer84 | cell growth |
| Reprimo84 | |||
| ACTN487 | increased estrogen receptor alpha activity | ||
| Nur7787 | T-cell and B-cell programing87 | inhibition of apoptosis | |
| HDAC9 | T-cell programing90 | limits T-cell response activity | |
| decreased T-cell proliferation | |||
| ATCD/TRI2991 | acute myeloma leukemia91 | increased cell proliferation | |
| HDAC6 | HSP9091 | prostate cancer91 | growth and transcriptional activation |
| multiple myeloma91 | |||
| acute myeloma leukemia93 | |||
| nitric oxide synthases95 | breast cancer94 | ||
| α-tubulin95 | Alzheimer’s disease97 | protein aggregation and misfolding | |
| HDAC10 | TXNIP98 | gastric cancer98 | regulation of reactive oxygen species and apoptosis trigger |
CLASS IIA
Class IIa HDACs are expressed in specific tissues and organs, demonstrating their importance in growth and development.18 The N-terminal domains of these proteins have conserved serine residues that can undergo phosphorylation by kinases and regulate their cellular location.60 A unique feature of class IIa HDACs is their lack of measurable catalytic activity toward acetylated histones, despite their highly conserved deacetylase domain. The enzymatic activity of these HDACs can be enhanced through recruitment into multiprotein complexes, some containing HDAC3.61 Class IIa HDACs have been proposed to be regulators and adaptors of repressor proteins, and several studies have established a link between class IIa HDAC expression and various types of cancers.62–64 Additionally, HDACs 4, 5, and 7 promote protein sumoylation, and these isozymes have been suggested to have small ubiquitin-like modifier (SUMO) E3 ligase activity.65–67
HDAC4 expression is highly upregulated in breast cancer samples and, along with HDAC9, overexpressed in patients with Waldenstrom’s macroglobulinemia.68 However, downregulation and misfunction of HDAC4 has also been related to cancer development; deletions of HDAC4 have been observed in melanoma cell lines.69 HDAC4 has been proposed as a target for microRNAs (miRNAs) due to the overexpression in the absence of miR-1 and miR-22.70,71 HDAC4, along with other class I and class II HDACs, has also been shown to help to directly regulate the expression of cyclin-dependent kinase inhibitors p21WAF/Cip162 and affect cell proliferation. In ovarian cancer cells, HDAC4 is overexpressed and catalyzes deacetylation of the transcription factor STAT1, leading to increased cell survival.64 HDAC4 overexpression also increases survival of prostate cancer cells by stabilizing HIF-1, a transcription factor that responds to decreases in oxygen and mediates the cellular response to hypoxia.73 Downregulation of HDAC4 has been linked to increased levels of VEGF expression in chondrosarcoma cells, enabling growth and metastasis of these cells.74 Additionally, HDAC4 repression has been linked to repression of transcription of the androgen receptor, which inhibits the induction of prostate specific antigens that regulate cell growth.75 Also, studies of transgenic mice models with deletion of miRNA155, one of the most overexpressed miRNAs in solid and hematological malignancies, have shown that miR-155 targets HDAC4. Particularly, miR-155 blocks the interaction between HDAC4 and Bcl6, a proto-oncogene and transcriptional suppressor that downregulates miR-155 in mice. In support of this, B-cell differentiation is highly reduced when HDAC4 is expressed.76
HDAC5 and HDAC9 are both overexpressed in high-risk medulloblastoma patients, showing a likely relationship between their expression levels and patient survival.77 Knockouts of both HDAC5 and HDAC9 promote cell death by increasing the activity of caspase-3-like.77 HDAC3 and HDAC5 have been linked to hepatocellular carcinoma by inhibition of these enzymes with the HDAC pan-inhibitor panobinostat.78 In breast cancer cells, HDAC5 regulates the expression of p14 in association with TBX3, which in turn induces cell proliferation.78 HDAC5 shuttling between the cytoplasm and the nucleus via phosphorylation catalyzed by protein kinase C has been implicated in axon regeneration. Export out of the nucleus allows for increased nuclear acetylation and activation of pro-regenerative genes.79,80 HDAC5 has also been linked to the behavioral response to cocaine in mice models; activation of the protein phosphatase PP2A leads to dephosphorylation of HDAC5 at S279, creating a transient accumulation of HDAC5 in the nucleus. The development of the cocaine reward behavior in animal models is suppressed by chromatin remodeling caused by HDAC5 enhanced interaction with 14–3–3 cytoplasmic anchoring proteins and disrupted binding interaction with MEF2 transcription factors.83 In addition, HDAC5 has been demonstrated to be necessary for replication fork formation as well as maintaining and assembling heterochromatin structure in cancer cells.82 Finally, loss of HDAC5 activity impairs memory function but has little to no effect on Alzheimer’s disease pathogenesis in mouse models for amyloid pathology.83
HDAC7 contributes to cell growth by associating with the estrogen receptor alpha (ERα) and inhibiting the expression of Reprimo, a cell cycle inhibitor in breast cancer cells.84 Studies have shown that siRNA silencing of HDAC7 enhanced the expression of Reprimo and induced apoptosis leading to the proposal that interaction of ERα with HDAC7 promotes cancer cell proliferation and growth. Other studies have reported association of HDAC7 with actinin alpha 4 (ACTN4), a nuclear receptor gene coactivator, increasing ERα transcriptional activity. Phosphorylation and localization of HDAC7 are critical for activation and regulation of the transcriptional machinery activation and regulation.87 HDAC7 has also been linked to B-cell and T-cell programming, as well as autoimmune disease in humans.85 HDAC7 is highly expressed in the nucleus of thymocytes at the CD4+ and CD8+ dual stage, altering both positive and negative growth regulators.86 For example, HDAC7 represses transcription of Nur77, an orphan nuclear receptor responsible for apoptosis activation due to recruitment of HDAC7 by MEF2 proteins.87 HDAC7 is highly abundant in mature B-cells but significantly downregulated upon conversion into macrophages.88 The mechanism of HDAC7 regulation by protein–protein interactions remains to be established.
HDAC9 has also been linked to the immune system. In particular, it is highly overexpressed in regulatory T-cells, which limit the T-cell immune response. Regulatory T-cells from HDAC9-deficient mice have more repressive activity than wild type.90 Additionally, HDAC9-deficient mice exhibit decreased T-cell proliferation and higher inflammation. HDAC9 has also been implicated in obesity and adipogenesis, as expression is downregulated in fat storing cells and tissues. Finally, HDAC9 catalyzes deacetylation of ATDC/TRI29 protein, an ataxia telangiectasia group D-complementing protein in acute myeloma patients which inhibits p53 activity, leading to the upregulation of genes implicated in cell proliferation.91 In this case, HDAC9 acts as a tumor suppressor, though the full extent of its function is not fully understood.
CLASS IIB
HDAC6 contains two deacetylase catalytic domains and a C-terminal zinc finger and is located exclusively in the cytoplasm. HDAC6 has been implicated in catalysis of the deacetylation of Hsp90, leading to inhibition of transcriptional activation and growth in prostate cancer cells.91 Inhibition of HDAC6 has antiproliferation effects in a variety of cancerous tissues, including multiple myeloma,91 and pediatric acute myeloid leukemia.93 Increased expression of HDAC6 has been observed in advanced stage cancers as compared to early stage,94 particularly in breast cancer. This suggests that HDAC6 levels could be used as an indicator for breast cancer progression and prognosis. Recently, HDAC6 has been observed to be a target for nitrosylation catalyzed by nitric oxide synthases. Nitrosylation repressed the catalytic activity of HDAC6, leading to increased acetylation of α-tubulin, the best validated HDAC6 substrate.95 Deacetylation of α-tubulin affects protein–protein interactions and has been implicated in the cellular response to protein aggregation and misfolding.96 Interestingly, mice models lacking HDAC6 are cognitively normal. In Alzheimer’s disease models, reduction of endogenous HDAC6 levels leads to a restoration of learning and memory along with α-tubulin acetylation. Loss of HDAC6 renders neurons resistant to amyloid-β-mediated impairment.97
Additionally, our current level of knowledge about HDACs and inhibitors has allowed researchers to begin using targeted approaches in developing new HDACi based therapies. A recent example of this is seen in the coupling of HDACi’s to genetically engineered viruses in an effort to target and attack tumor cells.107 The use of engineered viruses, known as oncolytic viral (OV) therapy, has shown potential in FDA trials;107 however, such an approach inherently relies on the effectiveness with which the virus is able to enter host cells and replicate. HDACi coupled to OVs has demonstrated an increase in both the initial phase of OV infection and subsequent viral replication.107 This has been taken a step further with researchers linking HDAC6, known to interact with tubulin, to the success of this therapy.108 Inhibition of HDAC6 leads to an increase in the acetylation profile of tubulin and down-regulation of beta interferon (IFN-β), potentially hindering the cell’s antiviral machinery.108,109 Indeed, upon inhibition of HDAC6, OV trials on glioma cells showed enhanced replication and entry of the virus into the nucleus.108
HDAC10, one of the least studied deacetylases, can be shuttled between the nucleus and the cytoplasm;18 however, its substrate set remains unknown. Recent studies show that HDAC10 is important for the regulation of reactive oxygen species in gastric cancer cells; inhibition of HDAC10 using siRNA causes an increase in reactive oxygen species, triggering the apoptotic pathway via the induced released of cytochrome c.98 Additionally, knockouts of HDAC10 in human gastric cancer cells significantly increase mRNA expression levels of thioredoxin-interaction protein (TXNIP) which acts as a cellular antioxidant in gastric cancer cells. However, the mechanism of TXNIP regulation by HDAC10 requires further investigation.
CLASS IV HDACS
HDAC11, the most recently discovered isozyme, is the sole member of the class IV HDAC subfamily.22 At 39 kDa, HDAC11 is the smallest isozyme. Sequence alignments suggest that HDAC11 is more closely aligned with class I HDACs (28% sequence identity to HDAC8) than class II with retention of highly conserved residues in the active site and in the mono-and divalent metal binding sites seen in other metal-dependent deacetylases. HDAC11 sequence alignments identify one significant sequence change, an aspartate (D101 in HDAC8) to asparagine, in a residue located on the flexible L2 loop, near the entrance to the active site tunnel.104
HDAC11 expression is tissue specific, with the greatest expression occurring in the brain, heart, kidneys, skeletal muscle, and testis.22 Studies of murine brain development suggest a role for HDAC11 in the formation of mature oligodendrocytes.99 Overexpression of HDAC11 in RAW264.7 cells is associated with a decrease in mRNA levels of the anti-inflammatory cytokine interleukin-10, indicating a possible role for HDAC11 in inflammatory and autoimmune diseases.105 Furthermore, mRNA analysis uncovered a link between HDAC11 and cancer; mRNA levels for HDAC11 are in the top 1% of differentially overexpressed genes in ductal breast carcinoma when compared to healthy breast tissue.100 Finally, the DNA replication factor Cdt1 is a potential HDAC11 substrate. Cdt1 is integral in recruiting mini-chromosome maintenance (MCM) helicase to DNA, which is required for DNA replications during the cell cycle. To maintain a single copy of DNA per cell, Cdt1 must be inhibited after loading MCM in the G1 phase.102 Cdt1 is an acetylated protein that coimmunoprecipitates with HDAC11.102 Finally, HDAC11 knockout mice are viable, but they exhibit increased cell proliferation and secrete higher levels of IL-2, TNF, and IFN-y than WT mice.106
CONCLUSIONS
Lysine acetylation/deacetylation is a dynamic, reversible post-translational modification with a defined role in histone modification and a growing pool of nonhistone substrates that are critical for epigenetic regulation, DNA repair, cell cycle regulation, and cancer growth and proliferation. The HDAC family of enzymes catalyzes deacetylation of both histones and nonhistone proteins to maintain acetylation homeostasis that is critical for cell regulation and survival. Changes in acetylation patterns have become a key feature of various types of cancer, and many of these changes in acetylation levels are due to increased expression and/or misregulation of HDACs. Due to the link between HDACs and cancer prognosis and survival, these enzymes have become an attractive target for drug development, as shown by the development and FDA approval of pan-HDAC inhibitors and the current development of novel immuno- and OV therapies. To further our understanding of the cellular function of HDACs, huge strides are currently being made in discovering HDAC-specific binding partners and substrates using a variety of methods, from knockouts and mass spectrometric techniques to in vivo mammalian cell work. The current body of literature on HDACs has shed light on the multiple roles of these enzymes in both transcriptional regulation and in protein–protein interactions, particularly with respect to their roles in disease, including multiple cancers, Alzheimer’s disease, and many others.
However, a major limitation with the current understanding of HDACs is still the lack of a full profile of specific inhibitors for each isozyme. New isozyme-specific inhibitors will continue to provide insight into the regulation and cellular role of each isozyme, potentially without compromising cellular viability. We predict that future work will incorporate varied approaches such as mass spectrometry, coimmunoprecipitation, in vivo perturbation, in vitro functional studies, etc., to advance our understanding of the cellular role of each isozyme. Understanding the specificity of each HDAC will provide further insight into both their role in various diseases as well as their function as epigenetic regulators and markers.
Acknowledgments
We would like to acknowledge Dr. Carol Ann Pitcairn and Katherine R. Leng for their input in the text and context of this review. This review was supported by the National Institutes of Health (Grants NIGMS GM40602 (C.A.F.), F31-GM-116619 (J.E.L.)) and the Rackham Graduate School (Rackham Merit Fellowship (J.E.L.)).
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Parbin S, Kar S, Shilpi A, Sengupta D, Deb M, Rath SK, Patra SK. Histone deacetylases: a saga of perturbed acetylation homeostasis in cancer. J Histochem Cytochem. 2014;62:11–33. doi: 10.1369/0022155413506582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. 2014;16:258–64. doi: 10.1038/nrm3931. [DOI] [PubMed] [Google Scholar]
- 3.Caron C, Boyault C, Khochbin S. Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. BioEssays. 2005;27:408–15. doi: 10.1002/bies.20210. [DOI] [PubMed] [Google Scholar]
- 4.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 5.Khoury GA, Baliban RC, Floudas CA. Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci Rep. 2011;1:90. doi: 10.1038/srep00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, Cole KE, De Baere E, Decroos C, Di Donato N, Ernst S, Francey LJ, Gyftodimou Y, Hirashima K, Hullings M, Ishikawa Y, Jaulin C, Kaur M, Kiyono T, Lombardi PM, Magnaghi-Jaulin L, Mortier GR, Nozaki N, Petersen MB, Seimiya H, Siu VM, Suzuki Y, Takagaki K, Wilde JJ, Willems PJ, Prigent C, Gillessen-Kaesbach G, Christianson DW, Kaiser FJ, Jackson LG, Hirota T, Krantz ID, Shirahige K. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012;489:313–7. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jia H, Morris CD, Williams RM, Loring JF, Thomas EA. HDAC inhibition imparts beneficial transgenerational effects in Huntington’s disease mice via altered DNA and histone methylation. Proc Natl Acad Sci U S A. 2015;112:E56–E64. doi: 10.1073/pnas.1415195112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE, Robson CN. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate. 2004;59:177–89. doi: 10.1002/pros.20022. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Z, Yamashita H, Toyama T, Sugiura H, Omoto Y, Ando Y, Mita K, Hamaguchi M, Hayashi SI, Iwase H. HDAC6 expression is correlated with better survival in breast cancer. Clin Cancer Res. 2004;10:6962. doi: 10.1158/1078-0432.CCR-04-0455. [DOI] [PubMed] [Google Scholar]
- 10.Wilson AJ, Byun DS, Popova N, Murray LB, L’Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH, Mariadason JM. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281:13548–58. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- 11.Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898–941. doi: 10.3390/molecules20033898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poole RM. Belinostat: first global approval. Drugs. 2014;74:1543–54. doi: 10.1007/s40265-014-0275-8. [DOI] [PubMed] [Google Scholar]
- 13.Fenichel MP. FDA approves new agent for multiple myeloma. J Natl Cancer Inst. 2015;107:djv165. doi: 10.1093/jnci/djv165. [DOI] [PubMed] [Google Scholar]
- 14.Gallagher S, Tiffen J, Hersey P. Histone Modifications, Modifiers and Readers in Melanoma Resistance to Targeted and Immune Therapy. Cancers. 2015;7:1959–1982. doi: 10.3390/cancers7040870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao K, Wang G, Li W, Zhang L, Wang R, Huang Y, Du L, Jiang J, Wu C, He X, Roberts AI, Li F, Rabson AB, Wang Y, Shi Y. Histone deacetylase inhibitors prevent activation-induced cell death and promote anti-tumor immunity. Oncogene. 2015;34:5960–70. doi: 10.1038/onc.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khan AN, Magner WJ, Tomasi TB. An epigenetically altered tumor cell vaccine. Cancer Immunol Immunother. 2004;53:748–754. doi: 10.1007/s00262-004-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan AN, Magner WJ, Tomasi TB. An epigenetic vaccine model active in the prevention and treatment of melanoma. J Transl Med. 2007;5:64. doi: 10.1186/1479-5876-5-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gantt SML, Decroos C, Lee MS, Gullett LE, Bowman CM, Christianson DW, Fierke CA. General Base-General Acid Catalysis in Human Histone Deacetylase 8. Biochemistry. 2016;55:820–832. doi: 10.1021/acs.biochem.5b01327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gantt SL, Joseph CG, Fierke CA. Activation and Inhibition of Histone Deacetylase 8 by Monovalent Cations. J Biol Chem. 2010;285:6036–6043. doi: 10.1074/jbc.M109.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waltregny D, Glénisson W, Tran SL, North BJ, Verdin E, Colige A, Castronovo V. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J. 2005;19:966–8. doi: 10.1096/fj.04-2303fje. [DOI] [PubMed] [Google Scholar]
- 22.Gao L, Cueto MA, Asselbergs F, Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem. 2002;277:25748–55. doi: 10.1074/jbc.M111871200. [DOI] [PubMed] [Google Scholar]
- 23.Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications. Biochim Biophys Acta, Gene Regul Mech. 2009;1789:45–57. doi: 10.1016/j.bbagrm.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spiegel S, Milstien S, Grant S. Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene. 2012;31:537–51. doi: 10.1038/onc.2011.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia. 2008;22:1026–34. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- 26.Adams H, Fritzsche FR, Dirnhofer S, Kristiansen G, Tzankov A. Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin’s lymphoma. Expert Opin Ther Targets. 2010;14:577–84. doi: 10.1517/14728221003796609. [DOI] [PubMed] [Google Scholar]
- 27.Senese S, Zaragoza K, Minardi S, Muradore I, Ronzoni S, Passafaro A, Bernard L, Draetta GF, Alcalay M, Seiser C, Chiocca S. Role for histone deacetylase 1 in human tumor cell proliferation. Mol Cell Biol. 2007;27:4784–95. doi: 10.1128/MCB.00494-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pulukuri SMK, Gorantla B, Rao JS. Inhibition of histone deacetylase activity promotes invasion of human cancer cells through activation of urokinase plasminogen activator. J Biol Chem. 2007;282:35594–603. doi: 10.1074/jbc.M705867200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Joshi P, Greco TM, Guise AJ, Luo Y, Yu F, Nesvizhskii AI, Cristea IM. The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol. 2013;9:672. doi: 10.1038/msb.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santoro F, Botrugno OA, Dal Zuffo R, Pallavicini I, Matthews GM, Cluse L, Barozzi I, Senese S, Fornasari L, Moretti S, Altucci L, Pelicci PG, Chiocca S, Johnstone RW, Minucci S. A dual role for Hdac1: oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood. 2013;121:3459–68. doi: 10.1182/blood-2012-10-461988. [DOI] [PubMed] [Google Scholar]
- 31.Lagger S, Meunier D, Mikula M, Brunmeir R, Schlederer M, Artaker M, Pusch O, Egger G, Hagelkruys A, Mikulits W, Weitzer G, Muellner EW, Susani M, Kenner L, Seiser C. Crucial function of histone deacetylase 1 for differentiation of teratomas in mice and humans. EMBO J. 2010;29:3992–4007. doi: 10.1038/emboj.2010.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dovey OM, Foster CT, Conte N, Edwards SA, Edwards JM, Singh R, Vassiliou G, Bradley A, Cowley SM. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood. 2013;121:1335–44. doi: 10.1182/blood-2012-07-441949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heideman MR, Wilting RH, Yanover E, Velds A, de Jong J, Kerkhoven RM, Jacobs H, Wessels LF, Dannenberg JH. Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood. 2013;121:2038–50. doi: 10.1182/blood-2012-08-450916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zimmermann S, Kiefer F, Prudenziati M, Spiller C, Hansen J, Floss T, Wurst W, Minucci S, Göttlicher M. Reduced body size and decreased intestinal tumor rates in HDAC2-mutant mice. Cancer Res. 2007;67:9047–54. doi: 10.1158/0008-5472.CAN-07-0312. [DOI] [PubMed] [Google Scholar]
- 35.Yao YL, Yang WM. The metastasis-associated proteins 1 and 2 form distinct protein complexes with histone deacetylase activity. J Biol Chem. 2003;278:42560–8. doi: 10.1074/jbc.M302955200. [DOI] [PubMed] [Google Scholar]
- 36.Krusche CA, Wülfing P, Kersting C, Vloet A, Böcker W, Kiesel L, Beier HM, Alfer J. Histone deacetylase-1 and –3 protein expression in human breast cancer: a tissue microarray analysis. Breast Cancer Res Treat. 2005;90:15–23. doi: 10.1007/s10549-004-1668-2. [DOI] [PubMed] [Google Scholar]
- 37.Jung KH, Noh JH, Kim JK, Eun JW, Bae HJ, Xie HJ, Chang YG, Kim MG, Park H, Lee JY, Nam SW. HDAC2 overexpression confers oncogenic potential to human lung cancer cells by deregulating expression of apoptosis and cell cycle proteins. J Cell Biochem. 2012;113:2167–77. doi: 10.1002/jcb.24090. [DOI] [PubMed] [Google Scholar]
- 38.Ropero S, Fraga MF, Ballestar E, Hamelin R, Yamamoto H, Boix-Chornet M, Caballero R, Alaminos M, Setien F, Paz MF, Herranz M, Palacios J, Arango D, Orntoft TF, Aaltonen LA, Schwartz S, Esteller M. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat Genet. 2006;38:566–9. doi: 10.1038/ng1773. [DOI] [PubMed] [Google Scholar]
- 39.Inoue S, Mai A, Dyer MJS, Cohen GM. Inhibition of histone deacetylase class I but not class II is critical for the sensitization of leukemic cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2006;66:6785–92. doi: 10.1158/0008-5472.CAN-05-4563. [DOI] [PubMed] [Google Scholar]
- 40.Kwon KD, Choi MJ, Park JM, Song KM, Kwon MH, Batbold D, Yin GN, Kim WJ, Ryu JK, Suh JK. Silencing histone deacetylase 2 using small hairpin RNA induces regression of fibrotic plaque in a rat model of Peyronie’s disease. BJU Int. 2014;114:926–36. doi: 10.1111/bju.12812. [DOI] [PubMed] [Google Scholar]
- 41.Ryu JK, Kim WJ, Choi MJ, Park JM, Song KM, Kwon MH, Das ND, Kwon KD, Batbold D, Yin GN, Suh JK. Inhibition of histone deacetylase 2 mitigates profibrotic TGF-β1 responses in fibroblasts derived from Peyronie’s plaque. Asian J Androl. 2013;15:640–5. doi: 10.1038/aja.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams H, Fritzsche FR, Dirnhofer S, Kristiansen G, Tzankov A. Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin’s lymphoma. Expert Opin Ther Targets. 2010;14:577–84. doi: 10.1517/14728221003796609. [DOI] [PubMed] [Google Scholar]
- 43.Fritzsche FR, Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, Scholman K, Denkert C, Dietel M, Kristiansen G. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer. 2008;8:381. doi: 10.1186/1471-2407-8-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiao F, Hu H, Yuan C, Jin Z, Guo Z, Wang L, Wang L. Histone deacetylase 3 promotes pancreatic cancer cell proliferation, invasion and increases drug-resistance through histone modification of P27, P53 and Bax. Int J Oncol. 2014;45:1523–30. doi: 10.3892/ijo.2014.2568. [DOI] [PubMed] [Google Scholar]
- 45.Atsumi A, Tomita A, Kiyoi H, Naoe T. Histone deacetylase 3 (HDAC3) is recruited to target promoters by PML-RARalpha as a component of the N-CoR co-repressor complex to repress transcription in vivo. Biochem Biophys Res Commun. 2006;345:1471–80. doi: 10.1016/j.bbrc.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 46.Sun Z, Feng D, Fang B, Mullican SE, You SH, Lim HW, Everett LJ, Nabel CS, Li Y, Selvakumaran V, Won KJ, Lazar MA. Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol Cell. 2013;52:769–82. doi: 10.1016/j.molcel.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gantt SL, Gattis SG, Fierke CA. Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry. 2006;45:6170–8. doi: 10.1021/bi060212u. [DOI] [PubMed] [Google Scholar]
- 48.Dowling DP, Gattis SG, Fierke CA, Christianson DW. Structures of metal-substituted human histone deacetylase 8 provide mechanistic inferences on biological function. Biochemistry. 2010;49:5048–56. doi: 10.1021/bi1005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–18. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oehme I, Deubzer HE, Wegener D, Pickert D, Linke JP, Hero B, Kopp-Schneider A, Westermann F, Ulrich SM, von Deimling A, Fischer M, Witt O. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin Cancer Res. 2009;15:91–9. doi: 10.1158/1078-0432.CCR-08-0684. [DOI] [PubMed] [Google Scholar]
- 51.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkühler C, Di Marco S. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci U S A. 2004;101:15064–9. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olson DE, Udeshi ND, Wolfson NA, Pitcairn CA, Sullivan ED, Jaffe JD, Svinkina T, Natoli T, Lu X, Paulk J, McCarren P, Wagner FF, Barker D, Howe E, Lazzaro F, Gale JP, Zhang YL, Subramanian A, Fierke CA, Carr SA, Holson EB. An unbiased approach to identify endogenous substrates of “histone” deacetylase 8. ACS Chem Biol. 2014;9:2210–6. doi: 10.1021/cb500492r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rettig I, Koeneke E, Trippel F, Mueller WC, Burhenne J, Kopp-Schneider A, Fabian J, Schober A, Fernekorn U, von Deimling A, Deubzer HE, Milde T, Witt O, Oehme I. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 2015;6:1657. doi: 10.1038/cddis.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci U S A. 1999;96:4868–73. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang AH, Bertos NR, Vezmar M, Pelletier N, Crosato M, Heng HH, Th’ng J, Han J, Yang XJ. HDAC4, a human histone deacetylase related to yeast HDA1, is a transcriptional corepressor. Mol Cell Biol. 1999;19:7816–27. doi: 10.1128/mcb.19.11.7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dequiedt F, Kasler H, Fischle W, Kiermer V, Weinstein M, Herndier BG, Verdin E. HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity. 2003;18:687–98. doi: 10.1016/s1074-7613(03)00109-2. [DOI] [PubMed] [Google Scholar]
- 57.Mahlknecht U, Schnittger S, Ottmann OG, Schoch C, Mosebach M, Hiddemann W, Hoelzer D. Chromosomal organization and localization of the human histone deacetylase 5 gene (HDAC5) Biochim Biophys Acta, Gene Struct Expression. 2000;1493:342–8. doi: 10.1016/s0167-4781(00)00191-3. [DOI] [PubMed] [Google Scholar]
- 58.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, Jackman S, Krzywinski M, Scott DW, Trinh DL, Tamura-Wells J, Li S, Firme MR, Rogic S, Griffith M, Chan S, Yakovenko O, Meyer IM, Zhao EY, Smailus D, Moksa M, Chittaranjan S, Rimsza L, Brooks-Wilson A, Spinelli JJ, Ben-Neriah S, Meissner B, Woolcock B, Boyle M, McDonald H, Tam A, Zhao Y, Delaney A, Zeng T, Tse K, Butterfield Y, Birol I, Holt R, Schein J, Horsman DE, Moore R, Jones SJM, Connors JM, Hirst M, Gascoyne RD, Marra MA. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Han A, He J, Wu Y, Liu JO, Chen L. Mechanism of recruitment of class II histone deacetylases by myocyte enhancer factor-2. J Mol Biol. 2005;345:91–102. doi: 10.1016/j.jmb.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 60.Parra M, Verdin E. Regulatory signal transduction pathways for class IIa histone deacetylases. Curr Opin Pharmacol. 2010;10:454–60. doi: 10.1016/j.coph.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 61.Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell. 2002;9:45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 62.Mottet D, Pirotte S, Lamour V, Hagedorn M, Javerzat S, Bikfalvi A, Bellahcène A, Verdin E, Castronovo V. HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene. 2009;28:243–56. doi: 10.1038/onc.2008.371. [DOI] [PubMed] [Google Scholar]
- 63.Wilson AJ, Byun DS, Nasser S, Murray LB, Ayyanar K, Arango D, Figueroa M, Melnick A, Kao GD, Augenlicht LH, Mariadason JM. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol Biol Cell. 2008;19:4062–75. doi: 10.1091/mbc.E08-02-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stronach EA, Alfraidi A, Rama N, Datler C, Studd JB, Agarwal R, Guney TG, Gourley C, Hennessy BT, Mills GB, Mai A, Brown R, Dina R, Gabra H. HDAC4-regulated STAT1 activation mediates platinum resistance in ovarian cancer. Cancer Res. 2011;71:4412–22. doi: 10.1158/0008-5472.CAN-10-4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grégoire S, Tremblay AM, Xiao L, Yang Q, Ma K, Nie J, Mao Z, Wu Z, Giguère V, Yang XJ. Control of MEF2 transcriptional activity by coordinated phosphorylation and sumoylation. J Biol Chem. 2006;281:4423–33. doi: 10.1074/jbc.M509471200. [DOI] [PubMed] [Google Scholar]
- 66.Zhao X, Sternsdorf T, Bolger TA, Evans RM, Yao TP. Regulation of MEF2 by histone deacetylase 4- and SIRT1 deacetylase-mediated lysine modifications. Mol Cell Biol. 2005;25:8456–64. doi: 10.1128/MCB.25.19.8456-8464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gao C, Ho CC, Reineke E, Lam M, Cheng X, Stanya KJ, Liu Y, Chakraborty S, Shih HM, Kao HY. Histone Deacetylase 7 Promotes PML Sumoylation and Is Essential for PML Nuclear Body Formation. Mol Cell Biol. 2008;28:5658–5667. doi: 10.1128/MCB.00874-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun JY, Xu L, Tseng H, Ciccarelli B, Fulciniti M, Hunter ZR, Maghsoudi K, Hatjiharissi E, Zhou Y, Yang G, Zhu B, Liu X, Gong P, Ioakimidis L, Sheehy P, Patterson CJ, Munshi NC, O’Connor OA, Treon SP. Histone deacetylase inhibitors demonstrate significant preclinical activity as single agents, and in combination with bortezomib in Waldenström’s macroglobulinemia. Clin Lymphoma Myeloma Leuk. 2011;11:152–6. doi: 10.3816/CLML.2011.n.036. [DOI] [PubMed] [Google Scholar]
- 69.Stark M, Hayward N. Genome-wide loss of heterozygosity and copy number analysis in melanoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 2007;67:2632–42. doi: 10.1158/0008-5472.CAN-06-4152. [DOI] [PubMed] [Google Scholar]
- 70.Datta J, Kutay H, Nasser MW, Nuovo GJ, Wang B, Majumder S, Liu CG, Volinia S, Croce CM, Schmittgen TD, Ghoshal K, Jacob ST. Methylation mediated silencing of MicroRNA-1 gene and its role in hepatocellular carcinogenesis. Cancer Res. 2008;68:5049–58. doi: 10.1158/0008-5472.CAN-07-6655. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 71.Zhang J, Yang Y, Yang T, Liu Y, Li A, Fu S, Wu M, Pan Z, Zhou W. microRNA-22, downregulated in hepatocellular carcinoma and correlated with prognosis, suppresses cell proliferation and tumourigenicity. Br J Cancer. 2010;103:1215–20. doi: 10.1038/sj.bjc.6605895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Song B, Wang Y, Xi Y, Kudo K, Bruheim S, Botchkina GI, Gavin E, Wan Y, Formentini A, Kornmann M, Fodstad O, Ju J. Mechanism of chemoresistance mediated by miR-140 in human osteosarcoma and colon cancer cells. Oncogene. 2009;28:4065–74. doi: 10.1038/onc.2009.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Geng H, Harvey CT, Pittsenbarger J, Liu Q, Beer TM, Xue C, Qian DZ. HDAC4 protein regulates HIF1α protein lysine acetylation and cancer cell response to hypoxia. J Biol Chem. 2011;286:38095–102. doi: 10.1074/jbc.M111.257055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sun X, Wei L, Chen Q, Terek RM. HDAC4 represses vascular endothelial growth factor expression in chondrosarcoma by modulating RUNX2 activity. J Biol Chem. 2009;284:21881–90. doi: 10.1074/jbc.M109.019091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang Y, Tse AKW, Li P, Ma Q, Xiang S, Nicosia SV, Seto E, Zhang X, Bai W. Inhibition of androgen receptor activity by histone deacetylase 4 through receptor SUMOylation. Oncogene. 2011;30:2207–2218. doi: 10.1038/onc.2010.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sandhu SK, Volinia S, Costinean S, Galasso M, Neinast R, Santhanam R, Parthun MR, Perrotti D, Marcucci G, Garzon R, Croce CM. miR-155 targets histone deacetylase 4 (HDAC4) and impairs transcriptional activity of B-cell lymphoma 6 (BCL6) in the Eμ-miR-155 transgenic mouse model. Proc Natl Acad Sci U S A. 2012;109:20047–52. doi: 10.1073/pnas.1213764109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Milde T, Oehme I, Korshunov A, Kopp-Schneider A, Remke M, Northcott P, Deubzer HE, Lodrini M, Taylor MD, von Deimling A, Pfister S, Witt O. HDAC5 and HDAC9 in medulloblastoma: novel markers for risk stratification and role in tumor cell growth. Clin Cancer Res. 2010;16:3240–52. doi: 10.1158/1078-0432.CCR-10-0395. [DOI] [PubMed] [Google Scholar]
- 78.Lachenmayer A, Toffanin S, Cabellos L, Alsinet C, Hoshida Y, Villanueva A, Minguez B, Tsai HW, Ward SC, Thung S, Friedman SL, Llovet JM. Combination therapy for hepatocellular carcinoma: additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J Hepatol. 2012;56:1343–50. doi: 10.1016/j.jhep.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yarosh W, Barrientos T, Esmailpour T, Lin L, Carpenter PM, Osann K, Anton-Culver H, Huang T. TBX3 is overexpressed in breast cancer and represses p14 ARF by interacting with histone deacetylases. Cancer Res. 2008;68:693–9. doi: 10.1158/0008-5472.CAN-07-5012. [DOI] [PubMed] [Google Scholar]
- 80.Cho Y, Cavalli V. HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J. 2012;31:3063–78. doi: 10.1038/emboj.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cho Y, Sloutsky R, Naegle KM, Cavalli V. Injury-Induced HDAC5 Nuclear Export Is Essential for Axon Regeneration. Cell. 2013;155:894–908. doi: 10.1016/j.cell.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Taniguchi M, Carreira MB, Smith LN, Zirlin BC, Neve RL, Cowan CW. Histone deacetylase 5 limits cocaine reward through cAMP-induced nuclear import. Neuron. 2012;73:108–20. doi: 10.1016/j.neuron.2011.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Agis-Balboa RC, Pavelka Z, Kerimoglu C, Fischer A. Loss of HDAC5 impairs memory function: implications for Alzheimer’s disease. J Alzheimers Dis. 2013;33:35–44. doi: 10.3233/JAD-2012-121009. [DOI] [PubMed] [Google Scholar]
- 84.Peixoto P, Castronovo V, Matheus N, Polese C, Peulen O, Gonzalez A, Boxus M, Verdin E, Thiry M, Dequiedt F, Mottet D. HDAC5 is required for maintenance of pericentric heterochromatin, and controls cell-cycle progression and survival of human cancer cells. Cell Death Differ. 2012;19:1239–52. doi: 10.1038/cdd.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Malik S, Jiang S, Garee JP, Verdin E, Lee AV, O’Malley BW, Zhang M, Belaguli NS, Oesterreich S. Histone deacetylase 7 and FoxA1 in estrogen-mediated repression of RPRM. Mol Cell Biol. 2010;30:399–412. doi: 10.1128/MCB.00907-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu JZ, Hov JR, Folseraas T, Ellinghaus E, Rushbrook SM, Doncheva NT, Andreassen OA, Weersma RK, Weismüller TJ, Eksteen B, Invernizzi P, Hirschfield GM, Gotthardt DN, Pares A, Ellinghaus D, Shah T, Juran BD, Milkiewicz P, Rust C, Schramm C, Müller T, Srivastava B, Dalekos G, Nöthen MM, Herms S, Winkelmann J, Mitrovic M, Braun F, Ponsioen CY, Croucher PJP, Sterneck M, Teufel A, Mason AL, Saarela J, Leppa V, Dorfman R, Alvaro D, Floreani A, Onengut-Gumuscu S, Rich SS, Thompson WK, Schork AJ, Næss S, Thomsen I, Mayr G, König IR, Hveem K, Cleynen I, Gutierrez-Achury J, Ricaño-Ponce I, van Heel D, Björnsson E, Sandford RN, Durie PR, Melum E, Vatn MH, Silverberg MS, Duerr RH, Padyukov L, Brand S, Sans M, Annese V, Achkar JP, Boberg KM, Marschall HU, Chazouillères O, Bowlus CL, Wijmenga C, Schrumpf E, Vermeire S, Albrecht M, Rioux JD, Alexander G, Bergquist A, Cho J, Schreiber S, Manns MP, Färkkilä M, Dale AM, Chapman RW, Lazaridis KN, Franke A, Anderson CA, Karlsen TH. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet. 2013;45:670–5. doi: 10.1038/ng.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Youn HD, Liu JO. Cabin1 represses MEF2-dependent Nur77 expression and T cell apoptosis by controlling association of histone deacetylases and acetylases with MEF2. Immunity. 2000;13:85–94. doi: 10.1016/s1074-7613(00)00010-8. [DOI] [PubMed] [Google Scholar]
- 88.Kasler HG, Verdin E. Histone deacetylase 7 functions as a key regulator of genes involved in both positive and negative selection of thymocytes. Mol Cell Biol. 2007;27:5184–200. doi: 10.1128/MCB.02091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barneda-Zahonero B, Román-González L, Collazo O, Rafati H, Islam ABMMK, Bussmann LH, di Tullio A, De Andres L, Graf T, López-Bigas N, Mahmoudi T, Parra M. HDAC7 is a repressor of myeloid genes whose downregulation is required for transdifferentiation of pre-B cells into macrophages. PLoS Genet. 2013;9:e1003503. doi: 10.1371/journal.pgen.1003503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yuan Z, Peng L, Radhakrishnan R, Seto E. Histone deacetylase 9 (HDAC9) regulates the functions of the ATDC (TRIM29) protein. J Biol Chem. 2010;285:39329–38. doi: 10.1074/jbc.M110.179333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gao L, Alumkal J. Epigenetic regulation of androgen receptor signaling in prostate cancer. Epigenetics. 2010;5:100–4. doi: 10.4161/epi.5.2.10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, Jarpe M, van Duzer JH, Mazitschek R, Ogier WC, Cirstea D, Rodig S, Eda H, Scullen T, Canavese M, Bradner J, Anderson KC, Jones SS, Raje N. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119:2579–89. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xu X, Xie C, Edwards H, Zhou H, Buck SA, Ge Y. Inhibition of Histone Deacetylases 1 and 6 Enhances Cytarabine-Induced Apoptosis in Pediatric Acute Myeloid Leukemia Cells. PLoS One. 2011;6:e17138. doi: 10.1371/journal.pone.0017138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sakuma T, Uzawa K, Onda T, Shiiba M, Yokoe H, Shibahara T, Tanzawa H. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int J Oncol. 2006;29:117–24. [PubMed] [Google Scholar]
- 95.Okuda K, Ito A, Uehara T. Regulation of Histone Deacetylase 6 Activity via S-Nitrosylation. Biol Pharm Bull. 2015;38:1434–7. doi: 10.1248/bpb.b15-00364. [DOI] [PubMed] [Google Scholar]
- 96.Li G, Jiang H, Chang M, Xie H, Hu L. HDAC6 α-tubulin deacetylase: a potential therapeutic target in neuro-degenerative diseases. J Neurol Sci. 2011;304:1–8. doi: 10.1016/j.jns.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 97.Govindarajan N, Rao P, Burkhardt S, Sananbenesi F, Schlüter OM, Bradke F, Lu J, Fischer A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol Med. 2013;5:52–63. doi: 10.1002/emmm.201201923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee JH, Jeong EG, Choi MC, Kim SH, Park JH, Song SH, Park J, Bang YJ, Kim TY. Inhibition of histone deacetylase 10 induces thioredoxin-interacting protein and causes accumulation of reactive oxygen species in SNU-620 human gastric cancer cells. Mol Cells. 2010;30:107–12. doi: 10.1007/s10059-010-0094-z. [DOI] [PubMed] [Google Scholar]
- 99.Liu H, Hu Q, Kaufman A, D’Ercole AJ, Ye P. Developmental expression of histone deacetylase 11 in the murine brain. J Neurosci Res. 2008;86:537–43. doi: 10.1002/jnr.21521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Deubzer HE, Schier MC, Oehme I, Lodrini M, Haendler B, Sommer A, Witt O. HDAC11 is a novel drug target in carcinomas. Int J Cancer. 2013;132:2200–2208. doi: 10.1002/ijc.27876. [DOI] [PubMed] [Google Scholar]
- 101.Wong PG, Glozak MA, Cao TV, Vaziri C, Seto E, Alexandrow M. Chromatin unfolding by Cdt1 regulates MCM loading via opposing functions of HBO1 and HDAC11-geminin. Cell Cycle. 2010;9:4351–63. doi: 10.4161/cc.9.21.13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Glozak MA, Seto E. Acetylation/deacetylation modulates the stability of DNA replication licensing factor Cdt1. J Biol Chem. 2009;284:11446–53. doi: 10.1074/jbc.M809394200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Minamiya Y, Ono T, Saito H, Takahashi N, Ito M, Mitsui M, Motoyama S, Ogawa J. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer. 2011;74:300–4. doi: 10.1016/j.lungcan.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 104.Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfí A, De Francesco R, Steinkühler C, Di Marco S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8–substrate complex. EMBO Rep. 2007;8:879–884. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, Nguyen D, Wright KL, Atadja PW, Bhalla K, Pinilla-Ibarz J, Seto E, Sotomayor EM. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol. 2009;10:92–100. doi: 10.1038/ni.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Woods D, Woan K, Wang D, Yu Y, Powers J, Sahakian E, Cheng F, Wang H, Rock-Klotz J, Villagra A, Pinilla-Ibarz J, Yu XZ, Sotomayor E. Histone deacetylase 11 is an epigenetic regulator of cytotoxic T-lymphocyte effector function and memory formation (P1404) J Immunol. 2013;190(117):2. [Google Scholar]
- 107.Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res. 2014;2:295–300. doi: 10.1158/2326-6066.CIR-14-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nakashima H, Kaufmann JK, Wang PYY, Nguyen T, Speranza MCC, Kasai K, Okemoto K, Otsuki A, Nakano I, Fernandez S, Goins WF, Grandi P, Glorioso JC, Lawler S, Cripe TP, Chiocca EA. Histone deacetylase 6 inhibition enhances oncolytic viral replication in glioma. J Clin Invest. 2015;125:4269–80. doi: 10.1172/JCI80713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nusinzon I, Horvath CM. Positive and negative regulation of the innate antiviral response and beta interferon gene expression by deacetylation. Mol Cell Biol. 2006;26:3106–13. doi: 10.1128/MCB.26.8.3106-3113.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]