

Abstract
Acyl carrier proteins (ACPs) are central hubs in polyketide and fatty acid biosynthetic pathways, but the fast motions of the ACP’s phosphopantetheine (Ppant) arm make its conformational dynamics difficult to capture using traditional spectroscopic approaches. Here we report that converting the terminal thiol of E. coli ACP’s Ppant arm into a thiocyanate activates this site to form a selective crosslink with the active site cysteine of its partner ketoacyl synthase (KS; FabF). The reaction releases a cyanide anion, which can be detected by infrared spectroscopy. This represents a practical and generalizable method to obtain and visualize ACP-protein complexes relevant to biocatalysis and will be valuable in future structural and engineering studies.
Keywords: Acyl carrier protein, ketoacyl synthase, polyketide biosynthesis, fatty acid synthase, mechanism-based crosslink, vibrational spectroscopic probes, thiocyanate
TOC image
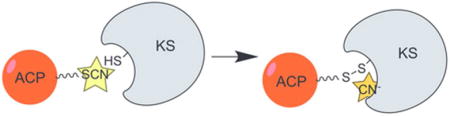
Polyketide synthases (PKSs) and fatty acid synthases (FASs) produce structurally diverse molecules with pharmacologically relevant properties. Acyl carrier proteins (ACPs) play a central role in these synthases by using their 18 Å-long 4′-phosphopantetheine (Ppant) arm to shuttle building blocks and intermediates to appropriate catalytic partners to carry out the chemistries programmed by the synthase (Figure 1A). The fast motions of ACPs enable the protein to protect and present its molecular cargo as needed, but also make it difficult to study its structure and its interactions with other proteins.1 Here we report a practical method to obtain and visualize ACP-protein interactions relevant to catalysis. We anticipate that this method will be valuable in future structural and engineering studies.
Figure 1.
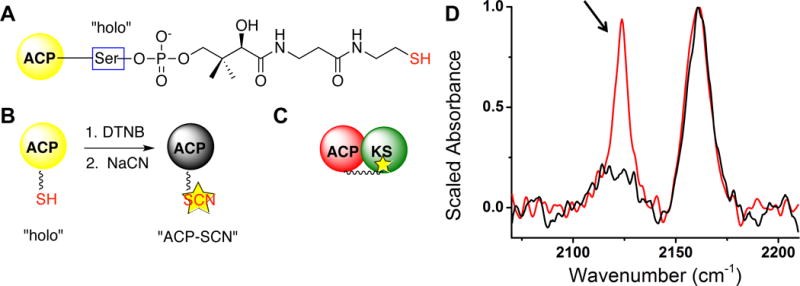
The thiol of the ACP Ppant arm (A) is converted into thiocyanate via a one-pot cyanylation reaction (B). We hypothesized that the labeled Ppant arm would gain access to the active site of cognate ketoacyl synthase (KS) enzymes (C), and that this change in solvation could be monitored by vibrational spectroscopy. IR analysis of cyanylated E. coli ACP, AcpP-SCN, titrated with its cognate KS (FabF) supports this hypothesis (D). The infrared CN stretching band of AcpP-SCN (black) absorbs at 2161 cm−1, a frequency consistent with the label being water exposed.2 When AcpP SCN was titrated with FabF a new peak at 2120 cm−1 (arrow) was observed.
We recently reported that converting the ACP Ppant arm’s thiol to a thiocyanate (ACP-SCN) turns the reactive end of the arm into a probe that reports on hydrogen bonding and local dynamics (Figure 1B).2 The frequency of the thiocyanate band is sensitive to the local solvation environment: when the Ppant arm of ACP-SCN is solvent exposed it absorbs at ~2163 cm−1, but when the arm is buried the probe band exhibits a red-shift of 5–10 cm−1.2 Since the Ppant arm can enter the hydrophobic active site of catalytic partners,1,3 we hoped that this catalytically relevant change of solvation environment could be traced with the solvato-sensitive thiocyanate probe (Figure 1C).
To test this hypothesis, we identified a model ACP/partner system: the E. coli fatty acid ACP, AcpP, and one of its cognate KSs, FabF. During the fatty acid elongation cycle in E. coli, AcpP collaborates with FabF to catalyze a Claisen condensation reaction that extends the growing alkyl chain by two carbon units. While characterizing the AcpP-FabF interaction is paramount to understanding and manipulating FASs, the AcpP-FabF interaction is difficult to study due to its transient nature. To overcome this obstacle, Burkart and coworkers used solvatochromatic fluorescence and crosslinking studies to visualize the movement of the AcpP Ppant arm into the hydrophobic active site of FabF.4,5 That work confirmed that the AcpP Ppant arm experiences a change in solvation environment (from solvent exposed to solvent excluded) upon its interaction with FabF, and validate the use of the AcpP/FabF pair to test our hypothesis.
We labeled AcpP with the thiocyanate probe to make the product AcpP-SCN and found that cyanylation did not perturb the protein’s helical structure (Figure S1). AcpP-SCN exhibited a mode frequency of 2161 cm−1, which based on prior results from model molecules6 and proteins2 suggests that the Ppant arm is mainly solvent exposed in the absence of a catalytic partner (Figure 1D).
We predicted that the interaction between AcpP-SCN and FabF would induce a red shift of 6–7 cm−1 in the CN frequency of AcpP-SCN when the probe entered the solvent-excluded FabF active site. To our surprise, the introduction of FabF resulted in a decrease in intensity of the 2161 cm−1 band and an appearance of a new peak at 2120 cm−1 (Figure 1D). The 40 cm−1 shift in the CN stretching band indicated that the interaction of AcpP-SCN with FabF chemically transformed the CN-containing functional group.
We hypothesized that the band at 2120 cm−1 was due to free CN− released from the SCN group. To test this theory, we repeated the titration experiment using AcpP-S13CN, where the isotopic mass of carbon-13 would lead to a lower vibrational frequency of any bonds containing the carbon atom of the thiocyanate. Both the AcpP-S13CN alone and AcpP-S13CN treated with FabF displayed 13C isotope frequency shifts of −50 cm−1 vs natural abundance (Figure S2), indicating that the new band at 2120 cm−1 in Figure 1D is due to a mode containing the thiocyanate carbon atom.7 Further support for assignment of the 2120 cm−1 band to CN− released from the SCN group includes the observation of this peak upon titration of FabF with NaCN in the absence of AcpP (data not shown).
We postulated that CN− was released upon the formation of a crosslinked FabF-AcpP complex, and therefore analyzed the reaction mixture using size exclusion chromatography (SEC; Figure 2A). For AcpP-SCN, we observed two peaks in the chromatogram (18 and 22 min), corresponding to the known equilibrium of the monomer and dimer AcpP states under non-reducing conditions.8 Titration of AcpP-SCN with FabF resulted in a decrease in these peaks and the appearance of a complex larger than FabF alone eluting at 16 minutes, consistent with the formation of a FabF-AcpP adduct.
Figure 2.

Size-exclusion chromatography (280 nm) shows that when AcpP-SCN (black) is titrated with FabF (green) a higher molecular weight complex is formed (orange) (A). AcpP partly forms a dimeric complex under non-reducing conditions,8 giving rise to two peaks in the chromatogram. The complex eluting at 16 mL was collected and analyzed via non-reducing (B) and reducing (C) SDS PAGE gels (lane 1 = AcpP; lane 2 = FabF; lane 3 = complex at 16 mL). The disappearance of the high molecular weight complex (red box) and the increase in intensity of AcpP/FabF bands observed under reducing conditions, support that AcpP and FabF are linked via a disulfide bond.
The most likely covalent link between AcpP and FabF is via a disulfide bond resulting from the FabF active site (Cys163) attacking AcpP-SCN’s sulfur atom, releasing free CN−, as shown in Figure 2A. To test this model, we titrated AcpP-SCN with FabF and analyzed the product via gel electrophoresis in the presence and absence of a disulfide reducing agent. We observed the covalent crosslinked product via gel electrophoresis under non-reducing conditions, but not reducing conditions (Figures 2B,C and S3).
To determine if the disulfide crosslink occurred at Cys163, we titrated AcpP-SCN with FabF pre-incubated with cerulenin, an inhibitor of the FabF KS active site cysteine.9 SDS PAGE analysis showed that the inhibited FabF does not form a crosslink with AcpP-SCN (Figure S4). The 2120 cm−1 IR band was not observed, indicating that the CN− release requires that AcpP’s Ppant arm enter the FabF active site (Figure S4). The attachment point between AcpP and FabF was further confirmed by tandem proteolysis mass spectrometry: digestion of the crosslinked product with trypsin and chymotrypsin yields a product consistent with a FabF peptide fragment tethered to an AcpP peptide fragment via a disulfide bond between the FabF Cys163 active site and AcpP Ppant arm (Figures S5–S10).
Reactions of cysteine sulfenyl thiocyanates with thiols are known to afford unsymmetrical disulfides.10 While the resultant disulfides are typically only stable in protic solvents under very acidic conditions, it appears that this reaction can take place within the active site of FabF, and that the local environment facilitates the formation of the FabF-AcpP disulfide bond. Since unlabeled holo-AcpP does not readily form a crosslink with FabF in vitro (Figure S11), we speculate that the cyanylation of AcpP either mimics a nascent substrate and induces a conformational change that alters the orientation of AcpP with respect to KS,11 or facilitates crosslinking via an SN2-type reaction by providing a robust leaving group. Once the arm is “activated”, a stable Ppant-Cys crosslink can be formed, as observed previously in vivo,12 and in engineered nonribosomal peptide synthetase systems.13
The simple installation of a thiocyanate probe on the AcpP Ppant arm provides a reactive and spectroscopically unique handle that facilitates the creation of a mechanistically relevant FabF-AcpP crosslinked product. IR spectroscopy using relatively conventional commercial instrumentation can be used to monitor formation of the crosslinked product via release of CN−. To extend the potential impact of these findings, we investigated whether other ACPs could be crosslinked to FabF via this cyanylation approach, and if the appearance of an IR peak at 2120 cm−1 could serve as a general readout for productive FabF-ACP interactions. Previous studies showed that FabF productively interacts with the polyketide ACP from frenolicin biosynthesis (ACPfren), but not the polyketide ACP from actinorhodin biosynthesis (ACPact).14 Thus, if FabF-ACP crosslinks were selectively formed when FabF is titrated with a functional cyanylated ACP, we would expect to see evidence of a covalent crosslink and release of CN− for ACPfren but not ACPact. Indeed, titration of ACPfren-SCN with FabF led to a crosslinked product (Figure S12) also marked by CN− in the IR spectrum (Figure S13). Titration of ACPact-SCN with FabF neither afforded a crosslink (Figure S12) nor the release of CN− (Figure S13). Similarly, no crosslink or 2120 cm−1 peak was observed upon titration of ACP-SCN with a bovine serum albumin (BSA) protein control (data not shown). These data show that the crosslinking reaction between ACP-SCN and FabF is selective to functional interactions and could potentially be used to monitor ACP interactions with other cognate catalytic partners.
The thiocyanate-facilitated crosslinking of ACPs to FabF is a practical method to obtain protein complexes relevant to biocatalysis. Previous efforts to visualize AcpP-protein interactions required isotopic labeling, multi-step synthesis of pantetheine-based probes, and/or chemoenzymatic loading of synthetic probes onto AcpP.15,16 While these approaches can target ACP interactions with diverse catalytic partners, the thiocyanylation with IR method offers an alternative in which the simple bioorthogonal modification of AcpP activates the protein for covalent linkage to FabF, and possibly other enzymes containing an active site cysteine.
Future work will focus on using this method to obtain crystal structures of FabF-ACP complexes and identify residues that guide functional interactions. The ability to visualize functional FabF-ACP interactions in situ via the presence/absence of a peak in the IR at 2120 cm−1 will also be explored as a low-barrier tool to screen functional unnatural biosynthetic partners. Given that cyanylation of ACPs and IR spectroscopy are amenable to high throughput approaches, these methods could enable rapid progress towards building hybrid synthases capable of creating new chemical diversity.
Supplementary Material
Acknowledgments
We thank Dr. Hsin-Yao Tang at the Wistar Institute Proteomics and Metabolomics Facility for analysis of proteomics data, and Prof. Chaitan Khosla for generously providing FabF and AcpP expression plasmids.
Funding Sources
We gratefully acknowledge the Research Corporation for Scientific Advancement Cottrell College Science Award (L.K.C.), NIH R15GM120704 (L.K.C), NSF CAREER grant CHE-1652424 (L.K.C.), NSF CAREER grant CHE-1150727 (CHL), a Henry Dreyfus Teacher-Scholar Award (CHL), summer research fellowships from Haverford College (K.J.S.T., G.A.T., C.P.F) and B.A. Rudolph Foundation (K.J.S.T.). The content is solely the responsibility of the authors and does not necessarily represent the views of the NIH or NSF.
ABBREVIATIONS
- ACP
acyl carrier protein
- FAS
fatty acid synthase
- IR
infrared
- KS
ketoacyl synthase
- PKS
polyketide synthase
- Ppant
phoshopantetheine
- SEC
size exclusion chromatography
Footnotes
Supporting information (methods and figures) is available free of charge at http://pubs.acs.org.
Notes
No competing financial interests have been declared.
References
- 1.Cronan JE. Biochem J. 2014;460:157–163. doi: 10.1042/BJ20140239. [DOI] [PubMed] [Google Scholar]
- 2.Johnson MNR, Londergan CH, Charkoudian LK. J Am Chem Soc. 2014;136:11240–11243. doi: 10.1021/ja505442h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nguyen C, Haushalter RW, Lee DJ, Markwick PRL, Bruegger J, Caldara-Festin G, Finzel K, Jackson DR, Ishikawa F, O’Dowd B, McCammon JA, Opella SJ, Tsai SC, Burkart MD. Nature. 2014;505:427–431. doi: 10.1038/nature12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beld J, Cang H, Burkart MD. Angew Chem Int Ed. 2014;53:14456–14461. doi: 10.1002/anie.201408576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Worthington AS, Porter DF, Burkart MD. Org Biomol Chem. 2010;8:1769. doi: 10.1039/b925966j. [DOI] [PubMed] [Google Scholar]
- 6.Maienschein-Cline MG, Londergan CH. J Phys Chem A. 2007;111:10020–10025. doi: 10.1021/jp0761158. [DOI] [PubMed] [Google Scholar]
- 7.Yoshikawa S, O’Keefe DH, Caughey WS. J Biol Chem. 1985;260:3518–3528. [PubMed] [Google Scholar]
- 8.Keating DH, Cronan JE. J Biol Chem. 1996;271:15905–15910. doi: 10.1074/jbc.271.27.15905. [DOI] [PubMed] [Google Scholar]
- 9.Moche M, Schneider G, Edwards P, Dehesh K, Lindquist Y. J Biol Chem. 1999;274:6031–6034. doi: 10.1074/jbc.274.10.6031. [DOI] [PubMed] [Google Scholar]
- 10.Alguindigue Nimmo SL, Lemma K, Ashby MT. Heteroatom Chem. 2007;18:467–471. [Google Scholar]
- 11.Whicher JR, Dutta S, Hansen DA, Hale WA, Chemler JA, Dosey AM, Narayan ARH, Håkansson K, Sherman DH, Smith JL, Skiniotis G. Nature. 2014;510:560–564. doi: 10.1038/nature13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rock CO. J Bacteriol. 1982;152:1298–1300. doi: 10.1128/jb.152.3.1298-1300.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tarry MJ, Schmeing TM. Protein Eng Des Sel. 2015;28:163–170. doi: 10.1093/protein/gzv009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bruegger J, Haushalter B, Vagstad A, Shakya G, Mih N, Townsend CA, Burkart MD, Tsai SC. Chem Biol. 2013;20:1135–1146. doi: 10.1016/j.chembiol.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finzel K, Lee DJ, Burkart MD. ChemBioChem. 2015;16:528–547. doi: 10.1002/cbic.201402578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beld J, Lee DJ, Burkart MD. Mol BioSyst. 2014;11:38–59. doi: 10.1039/c4mb00443d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.