


Abstract
The mode of action of regulated promoters is largely determined by kinetic parameters which govern the interaction between promoters and proteins involved in induction and repression of transcription. To gain insight into the interplay between positively and negatively acting transcriptional regulators, in this case AraC and LacR, we have generated a panel of promoter sequences derived from Plac, the promoter of the Escherichia coli lac operon. The function of these promoters is limited at different steps and to various extents within the pathway of RNA polymerase (RNAP)/promoter interaction. Moreover, in all promoters the cAMP receptor protein binding site was replaced by the binding motif of AraC to prevent pleiotropic effects in vivo upon activation. Analyzing the activation of these promoters by AraC in vivo under conditions of repression by LacR and derepression yielded a three step model of transcription initiation which reveals mechanisms of AraC and LacR action. Our data show three distinct rate limiting steps at which AraC can exert its function. In general, the activator accelerates the formation of the first stable complex between RNAP and promoter. At most promoter sequences, however, its main impact is on the conversion of the closed to the open complex. However, AraC is also capable of eliminating limitations at steps following open complex formation.
INTRODUCTION
To start transcription in Escherichia coli an RNA polymerase (RNAP) molecule has to follow a multistep pathway. The most prominent steps of this pathway are formation of a closed complex (RPc), isomerization of this complex into an open complex (RPo), initiation of RNA synthesis and finally, upon formation of a ternary RNA producing complex, clearance of the promoter region (RPcl) (reviewed in 1–3). The rate at which productively transcribing complexes clear the promoter region defines promoter strength (4). In the E.coli system promoters differ not only in strength by orders of magnitude (5), their activity is generally regulated by various positively and/or negatively acting elements (reviewed in 6). Promoters, which are controlled by regulatory proteins, have undergone an adaptation process which has made their interaction with RNAP susceptible to regulation. For example, promoters that are controlled by activators must encode one or several rate limiting steps within their functional program in order to be amenable to activation. A priori, any step within the transcription initiation process may be rate limiting and thus constitute a target for activator proteins (2). On the other hand, effective control of promoter activity by repressors requires an optimization as well. Thus, repressors that compete directly with RNAP for their mutual binding sites are most effective when the respective promoter sequence is recognized by RNAP relatively slowly (7). As the transcriptional activity of many operons is governed by more than one physiological parameter, the functional program of the respective promoters is optimized accordingly.
The promoter of the E.coli lac operon, Plac, is controlled negatively by the Lac repressor (LacR) and positively by the cAMP receptor protein (CRP), a principle which conveys a high degree of regulation to the lac operon in vivo. How is the susceptibility of Plac towards these two regulatory proteins brought about? Inspection of the Plac sequence reveals interesting deviations from consensus promoter sequences as derived by Mulligan et al. (8) and Harley and Reynolds (9). They concern base pair changes in both highly conserved hexamers at –10 and –35 (+1 being the first nucleotide transcribed) as well as in the distance between the two hexamers. Can these sequence deviations be correlated with specific properties that make Plac controllable and, if so, which steps in the RNAP promoter interaction are affected?
To approach these questions, we have converted Plac stepwise into a sequence with a high homology score [as defined by Mulligan et al. (8)], thereby generating a panel of promoters with distinctly altered functional programs. Moreover, we have replaced the CRP binding site of these promoters with the target sequence of the activator protein AraC to avoid the pleiotropic effects characteristic of CRP–cAMP action in vivo. The resulting set of promoters has allowed us to measure in vivo the effect of AraC binding on the activity of promoters encoding different functional programs. We were able to discriminate between the influence of the activator on the formation and release of open complexes by analyzing in vivo KMnO4 footprints. Moreover, the impact of the activator on early or late steps of RNAP/promoter interaction was revealed by monitoring AraC stimulation under conditions of repression and derepression of Plac via LacR. Our results show that activators like AraC are capable of releasing at least three different rate limiting steps within the pathway of transcription initiation. Moreover, they provide new insights into the role of LacR on the kinetics of RNAP/promoter interaction. In particular, we show that LacR decreases the forward rate constant of all promoters of a lac-type architecture by a defined increment. Thus, these studies contribute equally to our understanding of how transcriptional regulator proteins function and how functional programs of regulated promoters are optimized.
MATERIALS AND METHODS
DNA, plasmids and E.coli strains
The regulatory regions containing promoter/operator sequences of the lac operon as well as the binding site for AraC (I1 and a portion of I2) were assembled from synthetic oligonucleotides via the cleavage sites XhoI, HindIII, HinfI and EcoRI, respectively (Fig. 1). After insertion into pDS12Luci (10,11), all sequences were verified according to Sanger et al. (12). In pDS12Luci, these regulatory regions control transcription of the luciferase gene of Photinus pyralis (13). For in vivo studies, plasmids were transferred into E.coli strains DH5α (araC+, laci–) or DH5αZ1 (araC+, laciq) (14). For footprint analysis with KMnO4, E.coli strains TR322 (araC+, laciq) and TR321 (araC–, laciq) (15) were used in addition.
Figure 1.

The sequences of Plac including the CRP binding site and the Plar variants fused to araI1I2. The sequences are aligned at the conserved hexamers which are boxed and the transcriptional start site (+1). Restriction sites XhoI (upstream of the promoters) and EcoRI (downstream of the promoters) were used to clone the sequence variants into the pDS12Luci plasmid system (10,11). For exchanging the core promoter sequences (position +1 to –36) and for creating the Plar derivatives, a HindIII cleavage site upstream of the –35 region and a HinfI cleavage site downstream of the –10 region were generated. The base pair exchanges resulting in the respective cleavage sites are underlined. In the core regions of the Plar variants only the bases of the conserved hexamers are shown. Bases which deviate from Plar are printed in bold. Δ indicates the deletion of a CG base pair in the spacer at position –22. The LacR binding site is designated lacO. araI1I2 indicates the binding site of AraC. To place the primary AraC binding site araI2 in an upstream position corresponding to the araBAD promoter and to maintain at the same time the sequence of the –35 hexamer of Plac the araI2 binding site was shortened by 5 bp.
Determination of in vivo promoter activities
Promoter activities were determined by monitoring luciferase activity in vivo. Overnight cultures of E.coli cells DH5α or DH5αZ1 respectively harboring plasmids of the pDS12Luci family which carried the respective promoters were grown at 37°C in LB and diluted 1:100 in the same medium (100 µg/ml ampicillin) either in the presence or absence of 0.2% l(+)-arabinose. After 3 h, the OD600 was measured and the cultures were kept at room temperature for 15–20 min. Aliquots of 100 µl were removed from the cultures, 100 µl of luciferin solution (125 µM) was added and luciferase activity was measured (10 s, delay 0 s) in a Lumat type LB9501 (Berthold, Germany). Activities are given as relative light units (RLU) after subtraction of the instrumental background and normalization to the number of viable cells (10).
Kinetics of RNAP/promoter complex formation in vitro
Kinetics of complex formation between RNAP and promoters were measured in competition assays as described (5). Equal amounts of 32P-end-labeled DNA fragments (∼1000 c.p.m./promoter/binding reaction) of different lengths carrying different promoters were incubated with decreasing amounts of RNAP under standard conditions (25 mM Tris–HCl, pH 7.9, 100 mM KCl, 10 mM MgCl2, 2 mM DTT, 0.2 mM EDTA, 2.5% glycerol) for 5 min at 37°C. After addition of 100-fold excess of M13 ssDNA as competitor for 90 s, specific complexes were monitored by nitrocellulose adsorption followed by electrophoretic analysis of the retained DNA in 6% PAA gels (46% urea, 1× TBE). At each RNAP concentration the fraction of stable RNAP/promoter complexes was quantified. The rate constants were determined from the fraction of bound RNAP/promoter complexes as described previously (5). RNAP holoenzyme was prepared by J. Gamer in our group using the method described by Burgess and Jendrisak (16). The concentration of transcriptionally active RNAP was 2 pmol/µl.
Kinetics of RNAP/promoter complex dissociation in vitro
The stability of RNAP/promoter complexes in vitro was measured according to Brunner and Bujard (5). End-labeled DNA fragments carrying the synthetic promoters were incubated with a 10-fold molar excess of RNAP in the presence of 50 mM KCl for 10 min at 37°C. At time point 0 a 100-fold excess of competitor M13 ssDNA was added and the incubation was continued for another 90 s before aliquots of the reaction mixture were withdrawn at defined time intervals and subjected to filtration through nitrocellulose filters. The radioactivity retained on the filters was quantified in a scintillation counter and plotted versus time.
In vivo KMnO4 footprint analysis
KMnO4 footprints were carried out essentially as described previously (17,18). Escherichia coli strains TR322, TR321 (19) or DH5α carrying the various plasmids were grown to mid-log phase in M9 minimal medium supplemented with 0.5% glycerol (w/v), 0.2% casaminoacids (w/v), thiamine (0.05 mg/ml) and ampicillin (100 µg/ml). Whenever needed, l(+)-arabinose was added to a final concentration of 0.2%. Oxidation reactions were performed by adding 150 µl of KMnO4 (370 mM) to 10 ml of the bacterial culture for 2 min. Whenever required, 40 µl of rifampicin (50 mg/ml in methanol) was added 5 min prior to the addition of KMnO4. The reaction was terminated by placing the bacterial cultures in Corex-tubes chilled to 0°C. Cells were harvested by spinning the tubes at 5000 g for 5 min. Plasmid DNA was isolated according to Birnboim and Doly (20). Around 0.7 µg of DNA was subjected to a primer extension reaction by Klenow enzyme using a 32P-end-labeled primer (5′-GGATAGAATGGCGCCGGGCC-3′), which annealed ∼100 bp downstream of the transcriptional start site. Primer extension products were subjected to gel electrophoresis in a 6% PAA sequencing gel and made visible by autoradiography. The hypersensitive bands were quantitated densitometrically and normalized versus a region located outside the open complex as described (17).
RESULTS
Construction of variant sequences of Plac
To carry out functional studies with a highly defined set of promoter sequences, where the individual promoters would differ only by defined alterations, we synthesized first the regulatory region of the E.coli lac operon spanning from –81 to +20, however, with 5 bp exchanges (Fig. 1). Four alterations at positions –1, –2, –4 and –5 of Plac introduced a HinfI cleavage site between the –10 region and the transcriptional start site. A single G→A transition at position –39 created a HindIII site partly overlapping the –35 region. By utilizing the HinfI and HindIII sites the core region of the promoter could be readily replaced by altered sequences. In particular, the HindIII site was used to fuse the araI1I2 site to Plac centered around position –53 as found in the PBAD promoter of the ara operon (21). The resulting promoter Plar exhibited identical in vitro and in vivo properties (kON, t and promoter strength) compared to Plac (data not shown). By substituting 1 bp within the –10 and the –35 region and by deleting 1 bp within the spacer, the sequence of Plar was stepwise converted towards consensus (Fig. 1). This was reflected by the homology score (8) calculated for the different constructs which increase from 58% for Plar to 81% for PlarconS17 (Table 1). Primer extension analysis of mRNAs produced from the various promoters revealed that the sequence changes did not alter the transcriptional start sites (data not shown), i.e. all Plar constructs gave rise to identical transcripts. Therefore, measurement of luciferase activity in vivo directly reflected promoter activity.
Table 1. Kinetic in vitro parameters and in vivo promoter activities of Plar variants.
| Promoter |
Homology score (%) |
kON (× 107 M–1 s–1) |
t (min) |
| Plar | 58 | 0.08 | 18 |
| PlarS17 | 63 | 0.20 | 48 |
| Plar-8A | 61 | 0.24 | 56 |
| Plar-33G | 64 | 0.18 | 54 |
| Plar-8A-9A | 67 | 1.5 | 80 |
| PlarconS18 | 74 | 4.5 | 27 |
| PlarconS17 | 81 | 20 | 105 |
Effect of sequence alterations on the in vitro properties of Plar
We have shown previously that rates of complex formation between RNAP and promoters measured in vitro reflect well the situation prevailing in vivo (7). Therefore, forward rate constants for all promoters under study were determined. As all promoters formed stable complexes (t ≥ 18 min; Table 1) the forward rate constants (kON) could be determined under pseudo-first order conditions (Fig. 2 and Table 1). Single base pair changes introduced into the core region of Plar between +1 and –35 affected the homology scores and the kON values in the same sense, which is in good agreement with earlier observations (2,5,18). Thus, generating PlarS17, Plar-8A or Plar-33G increased the homology score of all three sequences by 3–6 U resulting in a 2–3-fold increase in the rate of complex formation. When such single sequence alterations were combined more drastic effects were observed. Plar-8A-9A exhibits a kON value ∼14 times higher than Plar and binds RNAP four times more stably. As expected, the consensus-like promoter PlarconS17 shows the highest homology score and exhibits a kON value that is close to the highest value of 30 × 107 M–1 s–1 reported for PN25 (5). The increase in kON values at PlarS17 compared to Plar and at PlarconS17 compared to PlarconS18, respectively, is due to the differences in spacer length. The same phenomenon was observed at the consensus-like promoters TAC17 and TAC18, respectively (22).
Figure 2.

Relative rates of complex formation between RNAP and Plar variants. A mixture of 32P-end-labeled DNA fragments carrying the various promoters was incubated under standard conditions with RNAP at the molar ratios indicated. Specific complexes were recovered by nitrocellulose filter adsorption and subjected to PAGE. The intensity of the bands was analyzed via a phosphoimager. The relative rate constants for complex formation were derived by plotting the fraction of unoccupied promoters 1 – FP versus the corresponding fractions of PL from phage Lambda which served as a standard promoter as described (5). Lane C1, an aliquot of the reaction mixture that was not subjected to filtration; lane C2, competitor DNA was added prior to RNAP. PlarconS17 is not included in this figure but was measured analogously.
Intrinsic promoter strength and activation potential of AraC
We next examined how the individual alterations in our panel of promoters affected (i) the intrisic strength, i.e. the non-activated and derepressed state of the Plar variants, and (ii) the activation potential of AraC. To exclude any influence of AraC on promoter activity in the absence of arabinose we first determined the various promoter activities in the araC+ strain TR322 in the absence of arabinose. The data obtained were indistinguishable from those obtained in araC– strain TR321 (data not shown; 19) indicating that AraC activation could be completely abolished by omitting arabinose from the growth medium. Therefore, promoter activities were determined in araC+ strains DH5α or DH5αZ1. The data provided in Table 2 show that the intrinsic strengths of the Plar variants differ over an almost 600-fold range and increase to different degrees when Plar is mutated towards consensus. Interestingly, the mutations leading to promoters PlarS17, Plar-8A and Plac-33G affect their in vitro properties almost identically. By contrast, the in vivo promoter strength of PlarS17 gives rise to only one-fifth to one-sixth of the other two constructs indicating that PlarS17 is limited at a step following RNAP binding. The two consensus-like promoters PlarconS18 and PlarconS17 exhibit a nearly 600-fold higher promoter activity than Plar and thus are the strongest promoters in this family. They nevertheless differ 4-fold in their rate of RNAP binding, indicating that PlarconS18 is not limited at this level. By contrast, Plar-8A-9A initiates productive transcription with a 6-fold lower rate than the consensus-like promoters but appears nevertheless unlimited in RNAP binding for reasons addressed in the Discussion. Its limitation must therefore occur at steps following this event. The data provided in Table 2 show that the susceptibility of the various promoters for activation by AraC is highly dependent on the sequence context within the core region. Whereas Plar is activated ∼70-fold by AraC, the activator has no measurable effect on the activity of the two consensus-like promoters (PlarconS17 and PlarconS18). Particularly interesting differences are observed in comparing promoters PlarS17, Plar-8A and Plar-33G, which all bind RNAP with about the same efficiency and with comparable stability (Table 1). PlarS17, which exhibits a 5–6-fold lower promoter strength than Plar-8A and Plar-33G, is activated by ∼60-fold whereas the activity of Plar-8A and Plar-33G is increased only 19-fold and 8-fold, respectively (Table 2). Therefore, AraC must abolish a rate limitation of PlarS17, which follows the formation of the first stable complex. Comparing the stimulation of Plar-8A and Plar-33G, two promoters with practically the same intrinsic promoter strength and the same in vitro RNAP binding properties, reveals that the latter promoter cannot be activated to the same extent as the former. Like Plar it must therefore contain a limitation which cannot be overcome by AraC. Interestingly, even if activated by AraC, Plar reaches only about one-eighth of the promoter strength of the consensus-like promoters demonstrating that AraC cannot fully compensate for all rate limitations encoded in this promoter sequence. The same holds true for PlarS17 and Plar-33G although to a lesser extent (Tables 1 and 2). By contrast, under conditions of activation, Plar-8A and Plar-8A-9A nearly reach the activity of the consensus-like promoters.
Table 2. Promoter activities of Plar variants in E.coli DH5α in the presence or absence of 0.2% arabinose.
| Promoter | Promoter activity | Activation factor | |
| |
–arabinose |
+arabinose |
|
| Plar |
1.1 ± 0.2 |
77 ± 8 |
70 |
| PlarS17 |
5.2 ± 0.8 |
303 ± 31 |
58 |
| Plar-8A |
25 ± 2 |
478 ± 81 |
19 |
| Plar-33G |
29 ± 3 |
225 ± 23 |
7.8 |
| Plar-8A-9A |
88 ± 10 |
578 ± 84 |
6.6 |
| PlarconS18 |
595 ± 80 |
605 ± 122 |
1.0 |
| PlarconS17 | 591 ± 92 | 608 ± 105 | 1.0 |
Luciferase activities, which were determined as described in Materials and Methods, are given as RLU/cell × 104 and are the mean values of five independent experiments.
Activation of Plac variants under conditions of repression
To determine the impact of AraC on the overall activation potential and in particular on RNAP binding in the presence of LacR promoter activities of the Plar variants were also measured in strain DH5αZ1 (Table 3) either in the presence or absence of arabinose. Plotting promoter activities of the repressed but non-activated Plar variants versus the respective kON values obtained in vitro revealed an almost perfect inverse correlation (Fig. 3). By contrast, in the non-repressed state, the kON values and the activities of the promoters correlate only qualitatively (Table 2), in accordance with previous studies (5,18,23,24). From this correlation we conclude that under conditions of repression kON becomes the major rate limiting step at all promoters. Accordingly, the AraC-mediated promoter activation in the presence of LacR has to be primarily attributed to an increase in kON. Thus, unlike in the non-repressed state, AraC is able to increase the rates of complex formation with RNAP at all Plar variants under conditions of repression by LacR. This becomes most obvious with the consensus promoters PlarconS18 and PlarconS17 where a 8-fold and 3-fold activation by AraC is observed in the repressed but none in the derepressed state (Tables 2 and 3). Likewise, the activation factors obtained at Plar and PlarS17 differ significantly under conditions of repression and derepression. In the presence of LacR these promoters are activated by only 6-fold and 20-fold, respectively, by AraC compared to a 70-fold and 58-fold activation in the derepressed state. This result implies that besides increasing kON, AraC also accelerates at least one subsequent kinetic step at these promoters. Finally, Plar-8A, Plar-33G and Plar-8A-9A are activated to roughly the same extent either in the presence or absence of LacR (Tables 2 and 3).
Table 3. Activatibility of Plar variants in E.coli DH5αZ1.
| Promoter | Promoter activity | Activation factor | |
| |
–arabinose |
+arabinose |
|
| Plar |
0.4 ± 0.1 |
2.6 ± 0.3 |
6.5 |
| PlarS17 |
0.6 ± 0.1 |
12 ± 3 |
20 |
| Plar-8A |
1.1 ± 0.2 |
23 ± 2 |
21 |
| Plar-33G |
1.2 ± 0.4 |
10 ± 1 |
8.5 |
| Plar-8A-9A |
12 ± 2 |
85 ± 6 |
7.1 |
| PlarconS18 |
57 ± 9 |
445 ± 73 |
7.8 |
| PlarconS17 | 145 ± 35 | 450 ± 92 | 3.1 |
All details are essentially as described in Table 2. DH5αZ1 harbors a chromosomal copy of the laciq gene (14).
Figure 3.

Correlation between repressibility, i.e. promoter strength under conditions of repression and forward rate constants (kON) between RNAP and Plar variants. The promoter activities of the non-activated lac promoter variants measured in the repressed state were plotted logarithmically versus the inverse of the respective kON values.
KMnO4 in vivo footprinting of Plar variants in the presence and absence of AraC
To distinguish between rate limitations that affect either the formation of open complexes or their release we performed permanganate footprints of the Plar variants in vivo under steady state conditions either in the presence or absence of arabinose (Fig. 4). In general, KMnO4 footprints visualize the fraction of promoters where RNAP has formed an open complex during the time of permanganate treatment. The intensity of the footprint signal correlates with the amount of time a promoter is open and available to react with KMnO4 (18). Strong footprint signals reflect limitations in RNA chain initiation and/or release of open complexes. At the Plar variants some hyper-reactive bands typically occurring at lac-type promoters show up at positions +4 to –2 (Fig. 4). To calculate the fraction of open complexes, rifampicin was added to the footprint reaction of PlarconS17 (Fig. 4, lane 13). Due to the high kON value and the high activity of this promoter we can assume that all available DNA is trapped in the open complex after 5 min of rifampicin treatment. Compared to PlarconS17 footprint intensities at Plar, PlarS17, Plar-8A and Plar-33G are very weak in the absence of arabinose. Only 1–3% (Fig. 4, lanes 1, 3, 5 and 7) of these promoters are engaged in open complexes under steady state conditions. Thus, transcription from these promoters is limited in the formation of open complexes. By contrast, promoters Plar-8A-9A and PlarconS17 are engaged in open complexes for ∼45 and 85% of the initiation time, respectively, indicating that both promoters are limited on the release of open complexes. Interestingly, the footprint pattern of these two promoters did not change, irrespective of whether AraC was active or not (Fig. 4). In the case of Plar-8A-9A, activation by AraC resulted in a ∼7-fold activation of the overall transcription process (Table 2) as well as a 7-fold increase in kON (Table 3). The unchanged footprint intensity therefore suggests that the overall activation is equally composed of an acceleration of both formation and release of open complexes. Likewise, we have demonstrated that AraC increases kON at PlarconS17 (Table 3); however, since initial binding of RNAP to this promoter is not the rate limiting step the footprint intensity remains the same in the presence of AraC. In contrast to Plar-8A-9A and PlarconS17, AraC increased the footprint signals at Plar (5-fold), PlarS17 (10-fold), Plar-33G (8-fold) and Plar-8A (20-fold). This demonstrates that AraC accelerates the formation of open complexes (Fig. 4); however, under these conditions a new rate limiting step becomes apparent at the release of the open complex.
Figure 4.

In vivo KMnO4 footprint of the non-template strands of Plar variants. Promoters Plar-8A, Plar-33G, Plar-8A-9A and PlarconS17 were footprinted either in strain TR321 (araC–) in the absence of arabinose or in strain TR322 (araC+) in the presence of 0.2% arabinose and 1 mM IPTG. Footprints of promoters Plar and PlarS17 were obtained in strain DH5α in the absence or presence of 0.2% arabinose. Control experiments revealed no difference in the footprint patterns between these strains (data not shown). The hypersensitive signals reminiscent of single-stranded DNA regions indicate open complexes around the transcriptional start site (–4 to +2). To calculate the fraction of open complexes, rifampicin was added to the footprint reaction of PlarconS17 in order to trap all available DNA in the open complex (lane 13). This intensity was set as 100% and served as reference for all other footprint signals for the calculation of the fraction of promoters that are open and available to react with KMnO4 (17,18). The footprint intensities were normalized to an internal standard (S) to allow their direct comparison as described (17). In lanes A and T sequencing reactions of Plar-8A-9A using the same primer as used for the primer extension reactions are shown. Lane C shows a primer extension reaction of promoter Plar-8A which was modified in vitro in the absence of RNAP.
Calculating the kinetics of transcription initiation
The results obtained in this study allow us to dissect the onset of transcription in vivo of Plar and its variants into three distinct steps and to estimate the time required to reach each of these steps: tRPc for the formation of the closed complex, tRPo for the isomerization of the closed into the open complex and tRPcl for the release of the open complex followed by mRNA initiation and promoter clearance. We calculate the real times required for RNAP to proceed to the various steps based on the following considerations. (i) PlacL8UV5, the core sequence of which is identical to Plar-8A-9A, initiates a productive transcript every ∼30 s (2). This initiation time was taken as reference point to calculate the time required for starting a productive transcription (tpt) at Plar and its variants using their in vivo activities as compiled in Tables 2 and 3. (ii) Quantitation of the footprint intensities around the transcription start point enabled us to estimate the time periods that the various promoters were open or closed (17) in the absence or presence of arabinose, reconciling that in the ‘unmelted’ state promoters might be either unoccupied or occupied by RNAP while engaged in a closed complex (RPc). (iii) kON describes the kinetics of stable and transcription-competent RNAP/promoter complex formation which irreversibly removes RNAP from the equilibrium. This complex is capable of prohibiting LacR from binding to its operator site. To calculate the time for RNAP binding to PlarconS17, we used Pspc, the promoter of the spc ribosomal protein operon, as a reference. The latter promoter binds RNAP every 0.4 s under growth conditions comparable to those used in this study (25). Since Pspc exhibits a lower homology score and thus presumably lower kON value compared to PlarconS17 we assume that RNAP binding to PlarconS17 occurs within 0.2 s. The times required to form RNAP/promoter complexes at the other Plar variants were calculated based on the respective kON values. (iv) Under conditions of repression the activities of the Plar variants depend primarily on the rate of complex formation with RNAP indicating that kON becomes the major rate limiting step and the main determinant of the overall promoter activity (Fig. 3). Interestingly, the same correlation can be found at a series of synthetic promoters of the same architecture but different core sequences (7). The observation is also in good accordance with data provided by Schlax et al. (26) who demonstrated that LacR acts by decreasing the apparent rate of complex formation between RNAP and PlacUV5 but leaving later kinetic steps unaffected. Based on this reasoning, tRPc, tRPo, tRPcl and tpt were calculated for, for example, Plar-8A as follows (Table 4). Plar-8A gives rise to one functional transcript every 100 s. According to the footprint data the release of the open complex (tRPcl) takes ∼2 s. tRPc, which we derive from kON, equals 16 s. Given these values, tRPo can be calculated according to tRpo = tpt – tRPc – tRPcl, which is 83 s. LacR action increases tpt to ∼2400 s. Activation of Plar-8A by AraC results in a 20-fold acceleration of ti (from 100 to 5 s). At the same time the footprint indicates that the promoter is engaged in an open complex for ∼35% of the respective time. Taken together these data suggest that tRPcl remains practically unchanged compared to the non-activated situation. Under conditions of repression AraC’s impact on kON is 20-fold (accelerating tRPc from 2315 to 121 s). We can therefore calculate that AraC accelerates tRPc in the non-repressed state from 15 to ∼0.8 s. Using the same term as above it follows that AraC accelerates tRPo from 83 to 2 s.
Table 4. Kinetics of RNAP/promoter interaction at various Plar variants under different conditions of repression and/or activation.
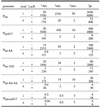 |
‘a’ indicates that AraC is active, ‘i’ indicates that AraC is inactive. ‘+’ indicates the repressed state, ‘–’ indicates the non-repressed state. tRPc, time required for closed complex formation; tRPo, time required for open complex formation; tRPcl, time required for RNA initiation and promoter clearance; tpt, time required for starting a productive transcription. ‘R’ denotes RNAP and ‘P’ denotes promoter. All initiation times and tRPc of the unregulated promoters were calculated from the promoter activities and kON values. tRPcl equals the time the promoters are open and available to react with KMnO4. It was calculated on the basis of the footprint intensities either in the presence or absence of arabinose. AraC’s influence on tRPc was derived from the activation factors determined under conditions of repression (+LacR). At PlarconS17 the AraC-mediated increase in kON is ∼2-fold higher than its overall impact on promoter activity since the time required to release the open complex contributes significantly to the overall transcription process. All values are given in seconds and are rounded up to their closest power. The error is up to 30%. See Discussion for further details.
DISCUSSION
Based on the sequence of Plac of the lac operon and the topography of PBAD of the ara operon we have generated a panel of hybrid promoters which were analyzed in vitro and in vivo. Replacing the CRP binding site with the recognition sequence for AraC enabled us to study promoter activation in vivo highly specifically as, unlike CRP, AraC does not produce relevant pleiotropic effects. We are nevertheless confident that the data obtained with the panel of Plar hybrid promoters describe a paradigmatic situation particularly since a limited set of analogous experiments with Plac and CRP confirm our findings with Plar (27). The seven Plar variants investigated behaved identically with regard to selection of the transcription start point. Moreover, the correlation found between their homology scores and the forward rate constants of polymerase binding was predictable and in good accordance with previously published reports (5,18). By contrast, no correlation was found between the levels of transcription and the match of the conserved hexamers to consensus. This lack of predictability, which has originally been described for PL of phage Lambda (23), indicates that the functional program of the Plar variants has been altered at different levels and to various extents. The set of promoters appeared therefore suited to investigate the impact of regulatory proteins such as LacR and AraC on the RNAP/promoter interaction.
Rate limitations of the Plar variants
The single base pair changes introduced into the spacer, the –10 region or the –35 region of Plar, generating promoters PlarS17, Plar-8A and Plar-33G, had only minor and rather predictable effects on RNAP binding. Although these promoters bind RNAP relatively slowly our calculations indicate that none of the promoters is limited by kON. However, since the rates of complex formation did not correlate with the respective promoter activities, these promoters are obviously limited to different extents at steps following stable complex formation. The permanganate footprints indicate that their major rate limitation occurs at open complex formation. The times required to release the open complexes also differ significantly among these promoters: from ∼50 s at Plar to ∼2 s at Plar-8A and Plar-33G. These values, however, are somewhat imprecise as the rather weak footprint signals obtained in the absence of AraC were not optimal for quantitation. At Plar-8A-9A formation and release of open complexes contribute equally to the overall promoter activity. It has been shown previously (28,29) that the closely related PlacUV5 is not limited at the level of RNAP binding but at a step of isomerization from an initial (RPc1) to an intermediate (RPc2) closed complex and at the onset of mRNA initiation. Although our approach does not permit to distinguish between two closed complexes our results are in good agreement with these studies. Finally, PlarconS17, and probably also PlarconS18, are primarily limited at the release of open complexes as the permanganate footprints prove unambiguously.
AraC action at the Plar variants
The impact of AraC on the enzyme’s kON can be quantified in vivo by monitoring the activation level of a given construct under conditions of repression. This analysis reveals that the activator accelerates stable complex formation of all promoters of our collection. However, as demonstrated in Table 4, its main role is to increase the rate of open complex formation, i.e. transition from RPc to RPo. According to the footprint data the relative rate of open complex release at Plar, PlarS17 and Plar-8A-9A is significantly slower compared to the other promoters. They furthermore indicate that AraC can relieve this limitation to a degree corresponding to the rates calculated for the other Plar variants. Interestingly, the activator seems incapable of increasing kcl substantially beyond this value (tRPcl ≈ 2–3 s; Table 4). At promoters PlarconS17, and presumably PlarconS18, where clearance becomes the kinetic bottleneck, the initiation rate is ∼0.25 transcripts/s. This is noticeably below the rate of the very strong E.coli promoter rrnP1 which initiates 1.5–2 transcripts/s during logarithmic growth (2,30). Since we know that the lacO sequence, when located in the downstream region, i.e. between +1 and +20, slows down promoter clearance at a variety of promoters without affecting kON (7,23,31) we suppose that this sequence is also responsible for the relatively slow release of open complexes of Plar variants. In this context it is interesting to note that the very strong promoter PL of phage Lambda, which is highly optimized towards promoter clearance (23), cannot be activated by AraC (10). Interestingly, when Plar-8A-9A was activated by AraC, the footprint signals remained unchanged (Fig. 4) regardless of the 7-fold activation of the overall transcription process (Table 2). This result can be explained by assuming that the overall activation observed is composed of an enhanced formation of open complexes as well as of their accelerated release. At the same time, our data indicate that AraC increases kON at Plar-8A-9A 7-fold. This activation, however, is qualitatively different from the activation observed under conditions of derepression as here kON has become the limiting term. Thus, as with Plar and PlarS17, AraC is able to accelerate three distinct steps at this promoter. Likewise, the AraC-mediated increase in kON at PlarconS17 became apparent only under conditions of repression, whereas it had no effect on promoter activity in the non-repressed state. This observation is also in accordance with the unchanged footprint signal (Fig. 4).
LacR decreases the enzyme’s kON by a defined increment
The data provided in Table 4 show that the impact of LacR on kON differs drastically from its effect on the overall promoter activity [compare rows LacR (+) and (–)]. The difference is due to the fact that kON is not the major rate limiting step at any of the Plar variants in the absence of LacR. In order to slow down the overall transcription process significantly LacR has to decrease kON to such an extent that it becomes the major rate limiting step of the overall process. Regardless of whether AraC is active or not, the actual quantitative impact of LacR decreases the enzyme’s kON by a defined increment which is in the range of 75–150-fold for all promoters examined in this study. Interestingly, it appears to be rather independent of the rate of RNAP/promoter complex formation. Since these observations can also be made at a number other synthetic promoters of the same architecture (7) we conclude that this mechanism of repression is generally applicable for repressors which directly interfere with RNAP binding.
Principles of Plar regulation
Plar and, accordingly, Plac are examples of negatively controlled promoters where the repressor interferes directly with the binding of RNAP. Our data show that the repressor affects the rate of RNAP binding and decreases the apparent kON by a defined increment. However, whether this deceleration of RNAP binding results in an efficient reduction of promoter activity depends on the rate of polymerase binding to the individual promoter in the non-repressed state, i.e. on the competition of the two proteins for their mutual binding site. Moreover, the regulatory range, i.e. promoter activity of the repressed compared to the non-repressed state, is also dependent on the rate of transcript initiation and promoter clearance by RNAP. Interestingly, while Plar interacts with RNAP with a low forward rate constant and thus fulfills one criterion for efficient repression, it initiates transcription only slowly. This limits the regulatory range of the Lac repressor/operator system. Also not too obvious is the fact that upon derepression the activity of Plar increases only 3-fold even though the rate of closed complex formation increases 150-fold (Table 4) revealing that the derepressed Plar is strongly limited at later steps of the promoter/RNAP interaction, namely at the formation and release of open complexes. These limitations are candidates for the action of the activator and indeed AraC, although increasing the rate of complex formation by another factor of 5 has its main impact on accelerating open complex formation and, to a lesser extent, steps following this event. The regulatory range of Plar under our in vivo conditions is ∼200-fold and our data indicate that this range may not be further expanded by activation via AraC. However, the rather modest repression factor may still be increased as it is directly dependent on the occupancy of the operator. The latter may be enhanced by higher intracellular concentrations or, more elegantly, by just increasing the local repressor concentration. In the lac as well as in several other operons, nearby secondary operators indeed contribute to higher occupancy of the main operator by this mechanism (32), which at the same time makes the system highly sensitive to low concentrations of inducer. In conclusion, the functional program of Plar reflects an interesting optimization for both negative and positive control. Obviously activators like AraC can have an impact on several limiting steps of the enzyme/promoter interaction. On the other hand, optimizing a promoter for efficient repression is primarily directed towards deceleration of the rate of complex formation between promoter and RNAP. Possibly this deceleration can only partially be brought about by modification of the promoter sequence since below a certain threshold the promoter may not become active any more in the derepressed state. Our analysis of the mode of action of AraC as a transcriptional activator and LacR as a directly interfering repressor has revealed an intricate balance between the elements interacting at a start site of transcription. The functional program which governs this finely tuned balance is embedded in a rather simple promoter sequence. It may give a taste for the great complexity we have to expect when studying quantitative parameters of transcription regulation in more sophisticated systems where a multitude of factors participate in a combinatorial mode.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Schleif for E.coli strains TR322 and TR321 and Dr Frank and co-workers for the synthesis of oligonucleotides. This work was supported by the Deutsche Forschungsgemeinschaft SFB229 and by the Fonds der Chemischen Industrie Deutschlands.
References
- 1.McClure W.R. (1985) Mechanism and control of transcription initiation in prokaryotes. Annu. Rev. Biochem., 54, 171–204. [DOI] [PubMed] [Google Scholar]
- 2.Knaus R. and Bujard,H. (1990) Priniciples governing the activity of E. coli promoters. In Eckstein,F. and Lilley,D.M. (eds), Nucleic Acids and Molecular Biology. Springer Verlag, Heidelberg, Germany, Vol. 4, pp. 110–122.
- 3.DeHaseth P., Zupancic,M.L. and Record,M.T.,Jr (1998) RNA polymerase-promoter interactions: the comings and goings of RNA polymerase. J. Bacteriol ., 180, 3019–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deuschle U., Kammerer,W., Gentz,R. and Bujard,H. (1986) Promoters of Escherichia coli: A hierarchy of in vivo strength indicates alternate structures. EMBO J., 5, 2987–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunner M. and Bujard,H. (1987) Promoter recognition and promoter strength in the Escherichia coli system. EMBO J., 6, 3139–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collado-Vides J., Magasanik,B. and Gralla,J.D. (1991) Control site location and transcriptional regulation in Escherichia coli. Microbiol. Rev., 55, 371–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lanzer M. and Bujard,H. (1988) Promoters largely determine the efficiency of repressor action. Proc. Natl Acad. Sci. USA, 85, 8973–8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mulligan M.E., Hawley,D.K. and McClure,W.R. (1984) Escherichia coli promoter sequences predict in vitro RNAP selectivity. Nucleic Acids Res., 12, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harley C.B. and Reynolds,R.P. (1987) Analysis of E. coli promoter sequences. Nucleic Acids Res., 15, 2343–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berlin M. (1993) Untersuchung zur Aktivierung von Escherichia coli Sigma70-Promotoren durch cAMP/CRP und AraC. PhD thesis. University of Heidelberg, Germany.
- 11.Stueber D. and Bujard,H. (1982) Transcription from efficient promoters can interfere with plasmid replication and diminish expression of plasmid specified genes. EMBO J., 1, 1399–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanger F., Nicklen,S. and Coulsen,A.R. (1977) DNA sequencing with chain-terminating inhibitors, Proc. Natl Acad. Sci. USA, 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Wet J.R., Wood,K.V., de Luca,M., Helinski,D.R. and Subramani,S. (1987) Firefly luciferase gene: structure and expression in mammalian cells. Mol. Cell. Biol., 7, 725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lutz R. and Bujard,H. (1997) Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res., 25, 1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hahn S., Dunn,T. and Schleif,R. (1984) Upstream repression and CRP stimulation of the Escherichia coli l-arabinose operon. J. Mol. Biol., 180, 61–72. [DOI] [PubMed] [Google Scholar]
- 16.Burgess R.R. and Jendrisak,J.J. (1975) A procedure for the rapid, large-scale purification of Escherichia coli DNA-dependent RNAP involving Polymin P precipitation and DNA-cellulose chromatography. Biochemistry, 14, 4634–4638. [DOI] [PubMed] [Google Scholar]
- 17.Sasse-Dwight S. and Gralla,J.D. (1989) KMnO4 as a probe for lac promoter DNA melting and mechanism in vivo. J. Biol. Chem., 264, 8074–8081. [PubMed] [Google Scholar]
- 18.Ellinger T., Behnke,D., Bujard,H. and Gralla,J.D. (1994) Stalling of Escherichia coli RNAP in the +6 to +12 region in vivo is associated with tight binding to consensus promoter elements. J. Mol. Biol., 239, 455–465. [DOI] [PubMed] [Google Scholar]
- 19.Reeder T. and Schleif,R. (1993) AraC protein can activate transcription from only one position and when pointed in only one direction. J. Mol. Biol., 231, 205–218. [DOI] [PubMed] [Google Scholar]
- 20.Birnboim H.C. and Doly,J. (1979) A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res., 7, 1513–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brunelle A. and Schleif,R. (1989) Determining residue-base interactions between AraC protein and araI DNA. J. Mol. Biol., 209, 607–622. [DOI] [PubMed] [Google Scholar]
- 22.Mulligan M.E., Brosius,J. and McClure,W.R. (1985) Characterization in vitro of the effect of spacer length on the activity of Escherichia coli RNAP at the tac promoter. J. Biol. Chem., 260, 3529–3538. [PubMed] [Google Scholar]
- 23.Knaus R. and Bujard,H. (1988) PL of coliphage lambda: an alternative solution for an efficient promoter. EMBO J., 7, 2910–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grana D., Gardella,T. and Susskind,M.M. (1988) The effects of mutations in the ant promoter of phage P22 depend on context. Genetics, 120, 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang S.T., Bipatnath,M., Xu,Y.-C., Chen,S.-L., Dennis,P., Ehrenberg,M. and Bremer,H. (1999) Activities of constitutive promoters in Escherichia coli. J. Mol. Biol., 292, 19–37. [DOI] [PubMed] [Google Scholar]
- 26.Schlax P.J., Capp,M.W. and Record,M.T.,Jr (1995) Inhibition of transcription initiation by lac repressor. J. Mol. Biol., 245, 331–350. [DOI] [PubMed] [Google Scholar]
- 27.Lutz R. (1996) Untersuchungen zur Aktivierung und Repression von Sigma70 Promotoren in Escherichia coli. PhD thesis. University of Heidelberg, Germany.
- 28.Buckle M., Pemberton,I.K., Jacquet,M.-A. and Buc,H. (1999) The kinetics of sigma subunit directed promoter recognition by E. coli RNA polymerase. J. Mol. Biol., 285, 955–964. [DOI] [PubMed] [Google Scholar]
- 29.Carpousis A.J. and Gralla,J.D. (1985) Interaction of RNAP with lacUV5 promoter DNA during mRNA initiation and elongation. J. Mol. Biol., 183, 165–177. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X. and Bremer,H. (1996) Effects of Fis on ribosome synthesis and activity and on rRNA promoter activities in Escherichia coli. J. Mol. Biol., 259, 27–40. [DOI] [PubMed] [Google Scholar]
- 31.Kammerer W., Deuschle,U., Gentz,R. and Bujard,H. (1986) Functional dissection of Escherichia coli promoters: information in the transcribed region is involved in late steps of the overall process. EMBO J., 5, 2995–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oehler S., Eismann,E.R., Kraemer,H. and Mueller-Hill,B. (1990) The three operators of the lac operon cooperate in repression. EMBO J., 9, 973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]