

Abstract
Management of comorbidities and medications is complex in HIV-1 infected patients. The overall objective of this project was to develop separate physiologically-based pharmacokinetic (PBPK) substrate models for the protease inhibitors darunavir and lopinavir. These protease inhibitors are used in the treatment of HIV infection. Both darunavir and lopinavir are coadministered with another medication that inhibits cytochrome (CYP) 3A. The current project focused on PBPK modeling for darunavir and lopinavir coadministered with ritonavir. Darunavir and lopinavir PBPK models that accounted for ritonavir CYP3A inhibition effects (linked PBPK models) were developed. The linked PBPK models were then used to predict the effect on darunavir or lopinavir exposure from CYP modulators. In the next step, the predicted effect of hepatic impairment was evaluated. Additional exploratory analyses predicted CYP3A inhibition effects on darunavir or lopinavir exposure in simulated hepatically impaired subjects. The linked PBPK models reasonably predict darunavir or lopinavir exposure based on simulations with CYP inhibitors or inducers. Exploratory simulations using the linked darunavir or lopinavir PBPK models indicate CYP3A inhibition may further increase darunavir or lopinavir exposure in patients with hepatic impairment.
Keywords: darunavir, drug interactions, hepatic impairment, lopinavir, protease inhibitors, physiologically based pharmacokinetic modeling
1. Introduction
In HIV-1 infected patients, antiretroviral therapy has transformed HIV-1 infection into a chronically managed medical condition. HIV-1 infected patients frequently receive both antiretrovirals and medications for managing comorbidities. Therefore, the assessment of potential drug-drug interactions (DDIs) is a critical part of the treatment paradigm for HIV-1 infection. Additionally, HIV-1 infected patients may also have liver disease resulting in hepatic impairment subsequent to different etiologies, including Hepatitis B or Hepatitis C co-infection. Hepatic impairment can alter the systemic exposure of the patient’s medications, including antiretrovirals. Potential clinical implications of changes in systemic exposure for antirertrovirals include therapeutic failure or adverse events. Therefore, the appropriate management of antiretroviral DDIs in HIV-1 infected patients with hepatic impairment is complex and clinically relevant.
Currently, for hepatically impaired HIV-1 infected patients taking concomitant medications, information is not available for healthcare providers regarding whether the current recommendations for managing drug-drug interactions are appropriate. During the drug development process, clinical trials are typically conducted in order to determine whether safety or efficacy issues exist resulting from extrinsic or intrinsic factors that can change the exposure of a medication. The trials are usually designed to provide information on changes in exposure with one specific extrinsic factor (e.g. concomitant administration of two medications) or one intrinsic factor (e.g. hepatic impairment, renal impairment) at a time to determine whether dosage adjustments are necessary. However, a limitation of these trials is that the impact of anticipated multiple factors affecting drug exposure in real world scenarios, such as drug-drug interactions and hepatic impairment, are not evaluated and the effects are not adequately characterized. Subsequently, appropriate dosing recommendations are not available for patients with multiple factors that can affect the safety or efficacy of a medication.
One method to derive drug exposure data regarding multiple factors that can affect the safety or efficacy of a medication in the absence of obtaining exposure data is to utilize physiologically-based pharmacokinetic (PBPK) modeling. PBPK modeling integrates information related to disposition by various human organ systems as well as drug specific clinical pharmacology information (e.g. absorption, metabolism, transport) to simulate exposure data in a virtual population1,2. When appropriate, the simulated data can be used to fill in knowledge gaps to provide important clinically relevant information that is not otherwise available, such as supporting dosing recommendations for managing DDIs3. As an example, PBPK modeling was used during the New Drug Application review for eliglustat to evaluate drug-drug interaction scenarios with different CYP2D6 genotypes4,5.
HIV treatment involves using antiretrovirals from multiple classes. HIV protease inhibitors, including darunavir and lopinavir, are an important option as part of antiretroviral therapy in maintaining HIV virologic suppression. It is necessary to coadminister both of these antiretroviral medications with another medication to increase the antiretroviral medication’s systemic exposure through cytochrome P450 (CYP)3A inhibition. The project focused on darunavir and lopinavir because historically these medications have been extensively used in the treatment of HIV-1 infection (see the 2016 U.S. Department of Health and Human Services’ “Guidelines for the use of antiretroviral agents in HIV-1-Infected adults and adolescents” that is available at http://aidsinfo.nih.gov/guidelines) and understanding how to manage DDIs involving HIV protease inhibitors is important for maintaining therapeutic effects. PBPK substrate models for darunavir and lopinavir were developed accounting for ritonavir CYP3A inhibition (subsequently referred to as the linked PBPK model). These linked PBPK models were then used to explore the feasibility of evaluating the effects of different factors (e.g. drug-drug interactions, hepatic impairment) on darunavir or lopinavir exposure in subjects receiving darunavir/ritonavir or lopinavir/ritonavir.
2. Methods
A population-based PBPK software (Simcyp®, v13.2, Simcyp Ltd, Sheffield, UK) was used for all PBPK modeling and simulations described in this work. Unless otherwise specified, Simcyp’s built-in healthy volunteer population and population for subjects with different degrees of hepatic impairment (e.g. Child-Pugh classification) were used. All simulations were performed using the “Healthy Volunteers” population provided by the software, with 10 trials and 10 subjects per trial. Virtual subjects were aged 20 years to 50 years, with a proportion of 50% females (the software’s default Healthy Volunteers population values).
Figure 1 provides a workflow of this project. Darunavir and lopinavir PBPK substrate models were developed using physicochemical, in vitro, and human data. Clinically, another medication is coadministered with darunavir or lopinavir to increase the antiretroviral medication’s systemic exposure through CYP3A inhibition. Although darunavir and lopinavir are administered in combination with ritonavir, initially the darunavir and lopinavir PBPK substrate models were developed without ritonavir (stand-alone models). Subsequently, in the development step, the effects of ritonavir on darunavir or lopinavir exposure were evaluated using a ritonavir CYP3A inhibitory PBPK model to construct the linked PBPK model. In the next steps, the linked darunavir or lopinavir PBPK substrate models were verified by comparing simulated and observed effects of different CYP modulators. After constructing acceptable linked darunavir and lopinavir PBPK substrate models in the verification step, the next part of the project involved using the linked PBPK models to simulate various clinical scenarios (application step), such as the effect of hepatic impairment on darunavir or lopinavir exposure. For the steps outlined in Figure 1, the simulated darunavir or lopinavir exposure data were compared to the observed darunavir or lopinavir data obtained from darunavir/ritonavir or lopinavir/ritonavir administration (see Figure 2 and Tables 1 through 3 for further details).
Figure 1.
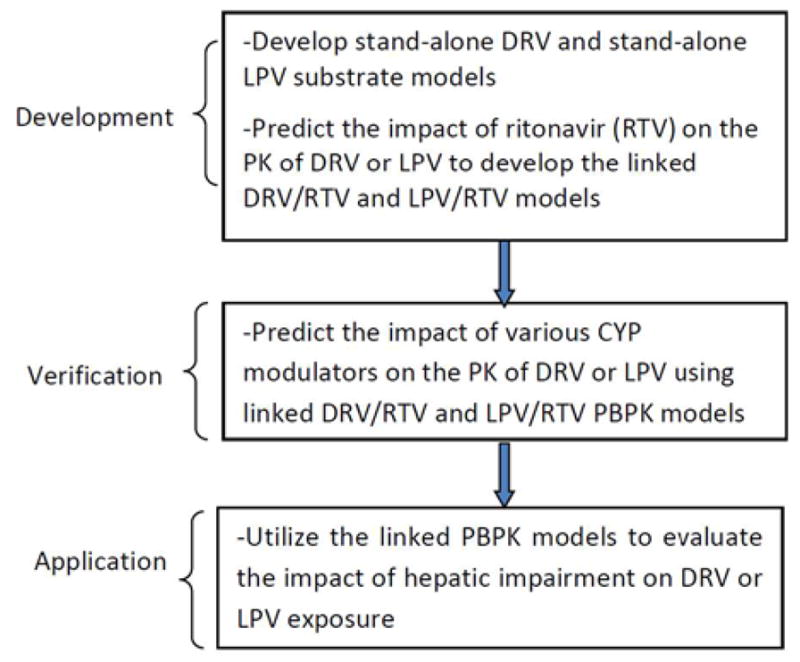
Overview of the strategy used to predict the impact of CYP modulators on the exposure of boosted LPV and DRV, respectively
Abbreviations: CYP=cytochrome P450, DRV=darunavir, LPV=lopinavir, PBPK= physiologically based pharmacokinetic modeling, PK=pharmacokinetic, RTV=ritonavir
Figure 2.

Predicted and observed mean arithmetic DRV and LPV concentration profiles.
A.1: LPV/RTV, 800 mg/200 mg QD. A.2. LPV/RTV, 400 mg/100 mg BID. B.1: DRV/RTV 800 mg/100 mg QD. B.2: DRV/RTV, 600 mg/100 mg BID. The observed concentration profiles were obtained using the Plot Digitizer program to estimate the concentration values (digitized). Green, solid lines: Predicted LPV or DRV (without RTV coadministration) plasma concentration-time profiles. Black, solid lines: Predicted LPV and DRV (with RTV coadministration) plasma concentration-time profiles. Red symbols: observed LPV (A.113, A.213,14,15) and observed DRV B.19,16 and B29) with concomitant use of RTV. Red and brown symbols: observed DRV (B.28)
Abbreviations: DRV=darunavir, hr=hour, LPV=lopinavir, μg=microgram
Table 1.
Comparison between predicted and observed mean arithmetic AUC for DRV or LPV (stand-alone substrate models)
| Regimen | Predicted AUC ± standard deviation [μg*h/mL] | Observed AUC ± standard deviation [μg*h/mL] | AUC Ratio (predicted/observed) |
|---|---|---|---|
| DRV | |||
| 150 mg, IV, SD | 4.90 ± 1.33 | 4.78 ± 1.109 | 1.03 |
| 400 mg, PO, SD † | 6.40 ± 2.91 | 2.6210 | 2.44 |
| 600 mg, PO, SD | 9.60 ± 4.36 | 10.99 ± 4.609 | 0.87 |
| 800 mg, PO, SD † | 12.80 ± 5.81 | 12.1210 | 1.06 |
| 1200 mg, PO, SD † | 19.20 ± 8.72 | 18.0910 | 1.06 |
| 400 mg, PO, day 7 ‡ | 6.50 ± 3.00 | 6.48 ± 3.3411 | 1.00 |
| LPV | |||
| 200 mg, PO, SD | 0.40 ± 0.21 | 0.07 ± 0.0512 | 5.71 |
| 400 mg, PO, SD | 0.82 ± 0.43 | 0.67 ± 0.6812 | 1.22 |
| 800 mg, PO, SD | 1.71 ± 0.90 | 2.50 ± 1.9312 | 0.68 |
Abbreviations: AUC=area under the plasma concentration-time curve, DRV=darunavir, hr=hour, IV=intravenous, LPV=lopinavir, mL=milliliter, PO=oral, SD=single dose, μg=microgram
For Boffito et al (reference 10), the observed concentration profiles were obtained using the Plot Digitizer program to estimate the concentration values (digitized) and standard deviations were not derived.
AUC over 12 hours
Superscripted numbers: see the corresponding reference citation
Table 3.
Comparison of changes in LPV exposure with CYP inhibitors or inducers
| CYP modulator | CYP modulator regimen [mg] | LPV/RTV regimen [mg] | Ratio pred/obs, AUCSS† | Ratio pred/obs, Cmax† | Ratio pred/obs, Ctrough,SS† |
|---|---|---|---|---|---|
| Omeprazole | 40 QD | 400/100 BID | 0.98 | 0.96 | 1.03 |
|
| |||||
| Ketoconazole | 200 SD | 400/100 BID | 1.74 | 1.70 | 1.94 |
|
| |||||
| 400/100 BID | 2.65 | 1.42 | 59.0 | ||
| Rifampin | 600 QD | 400/400 BID | 1.04 | 1.09 | 0.99 |
| 800/200 BID | 1.08 | 0.88 | 2.14 | ||
|
| |||||
| RTV | 100 BID | 400/100 BID | 1.01 | 1.09 | 0.74 |
Abbreviations: AUCss=area under the plasma concentration-time curve (steady state), BID=twice daily, Cmaxss=maximum plasma concentration, CYP=cytochrome P450, Ctroughss= trough plasma concentration (steady state), LPV= lopinavir, mg=milligram, obs=observed, pred=predicted, QD=once daily, RTV=ritonavir
Ratio/pred/obs= simulated ratio of exposure with concomitant use of the CYP modulator compared to exposure alone divided by the observed ratio of exposure with concomitant use of the CYP modulator compared to exposure alone.
Predicted ratios were derived from arithmetic means; the type of means for the observed ratios is not specified in the lopinavir/ritonavir U.S. prescribing information.
2.1 Development of the Stand-Alone and Linked Darunavir and Lopinavir PBPK Substrate Models
The model input parameters are displayed in supplemental digital content 1. For darunavir, model development involved the optimization of metabolism through CYP3A (fm [CYP3A4]). The development of the lopinavir PBPK model involved the optimization of steady state volume of distribution (Vss) and parameters for time-dependent CYP3A inhibition.
In order to develop the linked darunavir/ritonavir and lopinavir/ritonavir models, the steady-state pharmacokinetics of darunavir or lopinavir with ritonavir for once and twice daily dosing regimens were predicted. For this purpose, the ritonavir PBPK model was taken from the Simcyp® software’s drug model library (V13.2).
The predicted exposure of darunavir or lopinavir was compared to the observed data from publically available information for the stand-alone (see Table 1) and linked (see Figure 2) darunavir and lopinavir models.
2.2 Verification of the Linked PBPK Model-Predicting the Impact of Various CYP Modulatorson Darunaviror Lopinavir Exposure
The effects of various CYP modulators were simulated using the linked darunavir/ritonavir and lopinavir/ritonavir models. For this purpose, PBPK models for the selected CYP modulators provided by the Simcyp® compound library were used without further modification. The predicted exposure ratio was calculated as the ratio of darunavir or lopinavir exposure (AUC, Cmax, and Ctrough) with concomitant use of the CYP modulator compared to darunavir or lopinavir exposure alone by itself (data not presented). These predicted ratios were compared to the observed ratios reported in the darunavir and lopinavir/ritonavir U.S. prescribing information 6,7 to derive the displayed ratio (see Table 2 and Table 3). Please also see tables 7 and 8 for the predicted and observed ratios for darunavir and lopinavir AUC, Cmax, and Ctrough.
Table 2.
Comparison of changes in DRV exposure with CYP inhibitors or inducers
| CYP modulator | CYP modulator regimen [mg] | DRV/RTV regimen [mg] | Ratio pred/obs, AUCSS† | Ratio pred/obs, Cmax† | Ratio pred/obs, Ctrough,SS† |
|---|---|---|---|---|---|
| Carbamazepine | 200 BID | 600/100 BID | 0.76 | 0.74 | 0.84 |
| Clarithromycin | 500 BID | 400/100 BID | 1.21 | 1.26 | 1.05 |
| Ketoconazole | 200 BID | 400/100 BID | 0.78 | 0.90 | 0.66 |
| Omeprazole | 20 QD | 400/100 BID | 0.97 | 0.98 | 0.93 |
| Paroxetine | 20 QD | 400/100 BID | 0.99 | 1.04 | 0.94 |
| Saquinavir | 1000 BID | 400/100 BID | 1.38 | 1.23 | 1.77 |
Abbreviations: AUCss=area under the plasma concentration-time curve (steady state), BID=twice daily, Cmax=maximum plasma concentration, CYP=cytochrome P450, Ctrough,ss= trough plasma concentration (steady state), DRV=darunavir, mg=milligram, obs=observed, pred=predicted, QD=once daily, RTV=ritonavir
Ratio/pred/obs= simulated ratio of exposure with concomitant use of the CYP modulator compared to exposure alone divided by the observed ratio of exposure with concomitant use of the CYP modulator compared to exposure alone.
Predicted ratios were derived from arithmetic means; observed ratios were derived from least square means6.
Table 7.
Predicted, observed and predicted/observed ratios for AUC, Cmax and Ctrough: comparison of changes in DRV exposure with CYP inhibitors or inducers
| CYP modulator | CYP modulator regimen [mg] |
DRV/RTV regimen [mg] |
Pred AUC ratio |
Obs AUC ratio |
Ratio pred/obs, AUC† |
Pred Cmax ratio |
Obs Cmax ratio |
Ratio pred/obs, Cmax† |
Pred Ctrough ratio |
Obs Ctrough ratio |
Ratio pred/obs, Ctrough† |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Carbamazepine | 200 BID | 600/100 BID | 0.75 | 0.99 | 0.76 | 0.77 | 1.04 | 0.74 | 0.72 | 0.85 | 0.84 |
| Clarithromycin | 500 BID | 400/100 BID | 1.05 | 0.87 | 1.21 | 1.04 | 0.83 | 1.26 | 1.06 | 1.01 | 1.05 |
| Ketoconazole | 200 BID | 400/100 BID | 1.11 | 1.42 | 0.78 | 1.09 | 1.21 | 0.90 | 1.15 | 1.73 | 0.66 |
| Omeprazole | 20 QD | 400/100 BID | 1.00 | 1.04 | 0.97 | 1.00 | 1.02 | 0.98 | 1.01 | 1.08 | 0.93 |
| Paroxetine | 20 QD | 400/100 BID | 1.00 | 1.02 | 0.99 | 1.00 | 0.97 | 1.04 | 1.01 | 1.07 | 0.94 |
| Saquinavir | 1000 BID | 400/100 BID | 1.02 | 0.74 | 1.38 | 1.02 | 0.83 | 1.23 | 1.03 | 0.58 | 1.77 |
Abbreviations: AUC= area under the plasma concentration-time curve, BID=twice daily, Cmax=maximum plasma concentration, Ctrough=trough plasma concentration, CYP= cytochrome P450, DRV=darunavir, mg=milligram, obs=observed, pred=predicted, QD=once daily, RTV=ritonavir
Predicted ratios were derived from arithmetic means; observed ratios were derived from least square means6.
Table 8.
Predicted, observed and predicted/observed ratios for AUC, Cmax and Ctrough: Comparison of changes in LPV exposure with CYP inhibitors or inducers
| CYP modulator | CYP modulator regimen [mg] |
LPV/RTV regimen [mg] |
Pred AUC ratio |
Obs AUC ratio |
Ratio pred/obs, AUC† |
Pred Cmax ratio |
Obs Cmax ratio |
Ratio pred/obs, Cmax |
Pred Ctrough ratio |
Obs Ctrough ratio |
Ratio pred/obs, Ctrough† |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Omeprazole | 40 QD | 400/100 BID | 1.04 | 1.07 | 0.98 | 1.03 | 1.08 | 0.96 | 1.06 | 1.03 | 1.03 |
|
| |||||||||||
| Ketoconazole | 200 SD | 400/100 BID | 1.52 | 0.87 | 1.74 | 1.52 | 0.89 | 1.70 | 1.45 | 0.75 | 1.94 |
|
| |||||||||||
| 400/100 BID | 0.66 | 0.25 | 2.65 | 0.64 | 0.45 | 1.42 | 0.59 | 0.01 | 59.0 | ||
| Rifampin | 600 QD | 400/400 BID | 1.02 | 0.98 | 1.04 | 1.01 | 0.93 | 1.09 | 1.02 | 1.03 | 0.99 |
| 800/200 BID | 0.91 | 0.84 | 1.08 | 0.90 | 1.02 | 0.88 | 0.92 | 0.43 | 2.14 | ||
|
| |||||||||||
| RTV | 100 BID | 400/100 BID | 1.47 | 1.46 | 1.01 | 1.39 | 1.28 | 1.09 | 1.59 | 2.16 | 0.74 |
Abbreviations: AUC= area under the plasma concentration-time curve, BID= twice daily, Cmax= maximum plasma concentration, Ctrough= trough plasma concentration, CYP= cytochrome P450, LPV= lopinavir, obs=observed, mg=milligram, pred=predicted, QD=once daily, SD=single dose, RTV=ritonavir
Predicted ratios were derived from arithmetic means; the type of means for the observed ratios is not specified in the lopinavir/ritonavir U.S. prescribing information.
2.3 Application of the Linked PBPK Model-Predicting the Effect of Hepatic Impairment (HI)
Using the linked darunavir/ritonavir PBPK model, the effects of mild (Child-Pugh A [CP A]) and moderate ([CP B]) HI on the pharmacokinetics of darunavir were simulated. The simulations were compared to observed data.7,8
2.4 Predicting theEffect of Hepatic Impairment Plus CYP3A Inhibitionon Darunaviror Lopinavir Exposure
An additional exploratory analysis was conducted simulating the effect of ketoconazole, a strong CYP3A inhibitor, on darunavir exposure in hepatically impaired subjects using the linked darunavir/ritonavir PBPK model. In this scenario, the effects of multiple CYP3A inhibitors plus hepatic impairment on darunavir exposure were simulated. The same analysis was conducted for lopinavir using the linked lopinavir/ritonavir PBPK model.
To evaluate model performance, we calculated the ratio (R) for the model predicted exposure parameter and the observed exposure parameter (predicted/observed). The prediction is considered reasonable if the ratio (predicted/observed) is within an arbitrarily defined 2-fold range (e.g., 0.5≤R≤2.0).
3. Results
3.1 Performance of Optimized Stand-Alone Darunavir and Lopinavir PBPK Substrate Models
Table 1 compares the predicted and observed AUC of darunavir or lopinavir using the darunavir and lopinavir stand-alone PBPK substrate models. The model parameters used for the simulations are presented in Supplemental Digital Content 1, Supplemental Table 1.
The stand-alone darunavir PBPK substrate model predicts observed single and multiple dose pharmacokinetics reasonably well, with the majority of the AUC ratios within 2-fold of the observed data.
With the lopinavir 200 mg dose, the stand-alone lopinavir PBPK substrate model overpredicts by more than 5-fold the observed lopinavir AUC without ritonavir (see Table 1). However, for the clinically relevant doses of 400 mg and 800 mg, the predictions are within 2-fold of the observed data. For both the darunavir and lopinavir stand-alone substrate models, the variability between predicted and observed values was comparable.
3.2 Predicting the Effect of Ritonaviron the Exposure of Darunaviror Lopinavir
Figure 2 compares the predicted and observed plasma concentration-time profiles for once and twice daily dosing regimens of darunavir/ritonavir or lopinavir/ritonavir, using the linked darunavir/ritonavir and lopinavir/ritonavir PBPK models that were developed.
Based on Figure 2, in general, using the linked darunavir and lopinavir PBPK models, the simulated darunavir or lopinavir concentration-time profiles seem to reasonably predict the observed darunavir and lopinavir concentration values. The results support the use of the linked darunavir and lopinavir PBPK models for the analyses described in sections 3.3 and 3.4. Additional information regarding the predicted and observed AUC for the linked darunavir and lopinavir PBPK models is displayed in Table 6.
Table 6.
Comparison between predicted and observed mean arithmetic AUC (except where noted) for DRV or LPV (linked substrate models)
| Regimen, study | Predicted AUC ± SD [μg*h/mL] | Observed AUC ± SD [μg*h/mL] | AUC Ratio (pred/obs) |
|---|---|---|---|
| LPV/RTV, 800/200 mg QD | 155.52 ± 189.15 | 164.9 ± 67.513 | 0.94 |
| LPV/RTV, 400/100 mg BID | 81.51 ± 97.04 | 96.79 ± 21.7915 | 0.84 |
| 75.4 (61.8–82.8)A,14 | 1.08 | ||
| 185.2 ± 73.4B,13 | 0.88 | ||
| DRV/RTV, 800/100 mg QD | 63.86 ± 24.63 | 64.23 ± 18.21 (week 4)16 | 0.99 |
| 66.95 ± 18.61 (week 24) 16 | 0.95 | ||
| 75.62 ±26.44 (week 48)16 | 0.84 | ||
| NA | 44.63 (day 1)C,9 | NA | |
| DRV/RTV, 600/100 mg BID | 51.43 ± 17.71 | 52.31 ± 15.90 (group A)8 | 0.98 |
| 37.88 ± 13.82 (group B)8 | 1.36 | ||
| 62.35±16.14D,9 | 0.82 |
Abbreviations: AUC= area under the plasma concentration-time curve, BID= twice daily, DRV= darunavir, hr=hour, LPV =lopinavir, mL=milliliter, NA= not applicable, obs=observed, pred=predicted, QD=once daily, RTV=ritonavir, sd=standard deviation, μg=microgram
Median and interquartile range data were included in the reference
AUC from two 12h dosing intervals
Based on concentration profiles obtained using the Plot Digitizer program
Geometric mean is presented
3.3. Verification of Linked Darunavir/Ritonavir and Lopinavir/Ritonavir PBPK Models: Predicting the Effect of CYP Modulatorson Darunavir/Ritonaviror Lopinavir/Ritonavir Exposure
The predicted and observed changes with various CYP modulators on darunavir or lopinavir exposure are shown in Table 2 and Table 3. In general, when the effect of CYP modulators on the exposure of darunavir or lopinavir was evaluated using the linked darunavir/ritonavir and lopinavir/ritonavir models, the simulated changes in darunavir or lopinavir exposure are consistent with the observed changes in darunavir or lopinavir exposure. When combined with ritonavir, for both darunavir and lopinavir, for the CYP2C19 inhibitor omeprazole, there were minimal differences in the predicted exposure ratio compared to the observed exposure ratio, consistent with the available information regarding darunavir or lopinavir metabolism through CYP2C19. For darunavir combined with ritonavir, a similar finding was observed for paroxetine, a CYP2D6 inhibitor. For CYP3A inhibitors or inducers, with the exception of the CYP3A inducer rifampin (which may be attributed to a need for further rifampin model optimization17), there was a less than two-fold difference in predicted versus observed exposure with darunavir or lopinavir AUC, Cmax, and Ctrough values.
3.4. Application of Linked Darunavir/Ritonavir and Lopinavir/Ritonavir PBPK Models: Predicting the Effect of Hepatic Impairmenton Darunavir/Ritonaviror Lopinavir/Ritonavir Exposure
The predicted and observed changes with mild to moderate hepatic impairment on darunavir or lopinavir exposure are shown in Table 4 and Table 5.
Table 4.
Comparison of changes in DRV exposure with hepatic impairment
| Child-Pugh A | Child-Pugh B | |||
|---|---|---|---|---|
|
| ||||
| Parameter | Observed8,† | Predicted† | Observed8,† | Predicted† |
| AUCSS | −6% | +9% | +20% | +18% |
| Cmax | −12% | +6% | +22% | +13% |
| Cmin,SS | −17% | +16% | +27% | +29% |
Abbreviations: AUCss= area under the plasma concentration-time curve (steady state), Cmax=maximum plasma concentration, Cmin,ss = minimum plasma concentration (steady state), DRV=darunavir, HI=hepatic impairment
Predicted changes in darunavir exposure were derived from arithmetic means; observed changes in darunavir exposure were derived from least square means8.
Table 5.
Comparison of changes in LPV exposure with hepatic impairment
| Observed† | Predicted† | ||
|---|---|---|---|
|
| |||
| Parameter | Mild to moderate HI | Child-Pugh A | Child Pugh B |
| AUCSS | +30% | +60% | +143% |
| Cmax | +20% | +49% | +118% |
Abbreviations: AUCss= area under the plasma concentration-time curve (steady state), Cmax=maximum plasma concentration, HI=hepatic impairment, LPV=lopinavir
Predicted changes in lopinavir exposure were derived from arithmetic means, the type of means for the observed changes in lopinavir exposure is not specified in the lopinavir/ritonavir U.S. prescribing information.
Using the linked darunavir/ritonavir model, the simulated darunavir exposure in hepatically impaired subjects was reasonably predicted when compared to the observed darunavir exposure data with regard to the magnitude of change (the absolute percentage differences). In contrast, for lopinavir, using the linked lopinavir/ritonavir model, the simulated lopinavir exposure in hepatically impaired subjects was overpredicted when compared to the observed lopinavir exposure data.
3.5 Predicting the Effect of Hepatic Impairment (HI) and CYP3 A Inhibitionon Darunavir/Ritonaviror Lopinavir/Ritonavir Exposure
Additional exploratory analyses were conducted using the linked darunavir/ritonavir and lopinavir/ritonavir models to simulate the effect of both hepatic impairment and ketoconazole (a strong CYP3A inhibitor) on darunavir or lopinavir exposure using the PBPK models. Based on the information from the linked PBPK models, the exploratory analyses suggest that ketoconazole CYP3A inhibition may further increase darunavir or lopinavir exposure in hepatically impaired patients. The Discussion section provides further details regarding the current limitations of these analyses.
4. Discussion
The linked PBPK substrate models (darunavir/ritonavir or lopinavir/ritonavir) that incorporate the CYP3A effects of ritonavir reasonably predicted darunavir and lopinavir exposure (Figure 2). Based on the simulations in the presence of various CYP inhibitors or inducers, the linked PBPK models appear to capture the majority of exposure changes for CYP inhibitors or inducers (Tables 2 and 3). One potential utility of the linked models is for evaluating the changes in darunavir or lopinavir exposure in the presence of intrinsic factors such as hepatic impairment. Such simulations are useful in determining whether a potential safety or efficacy issue exists under these complex clinical scenarios as discussed in the Introduction.
Although the linked models can be used to simulate drug exposure in subjects with hepatic impairment, reliable predictions of the effect of hepatic impairment on darunavir or lopinavir exposure cannot be obtained. Current limitations in simulating drug exposure in a hepatically impaired population using PBPK modeling impact the ability to obtain reliable predictions. Subsequently, it was determined that presenting quantitative results of the simulations evaluating the effect of hepatic impairment and CYP3A inhibition on darunavir or lopinavir exposure was premature. The hepatic impairment populations (CP A, CP B, and CP C) of the PBPK software that was used for the analyses (Simcyp ® v13.2) include the following adjustments: reduced hepatic CYP expression, reduced liver size, reduced plasma protein binding (albumin and α-1 acid glycoprotein), and decreased renal function (e.g. adjusted serum creatinine values), depending on the severity of liver disease 18,19. While these changes are currently reflected in the virtual hepatic impairment populations, further work is necessary to verify that these changes are accurately reflected and to implement additional physiological changes that may impact drug absorption and disposition. Subsequently, the ability of PBPK models to predict the effects of both DDIs and hepatic impairment is currently limited by knowledge gaps in the hepatic impairment population. Therefore, these models cannot be used to develop dosing recommendations in the U.S. prescribing information to address drug-drug interactions in a hepatic impairment population.
There are other limitations associated with our PBPK modeling analyses, which further limit the ability to reliably predict protease inhibitor exposure when co-administered with ritonavir in subjects with hepatic impairment. The first limitation is the lack of inclusion of potential transporter mediated interactions. Both darunavir6 and lopinavir20 are substrates for p-glycoprotein. Although incorporating transporter interactions into the model is expected to improve model performance, there is limited PBPK modeling experience with this approach. As such, there is low confidence in predicting transporter-mediated drug interactions using PBPK modeling.21 Among several doses which were evaluated (Table 1), the darunavir stand-alone model over predicted the darunavir AUC for a single oral dose of 400 mg by more than 2-fold, and the predicted AUC appears to be consistent with the model assumption that darunavir pharmacokinetics is dose-independent (Table 1). However, if an intestinal efflux transporter plays a significant role in the oral absorption of darunavir, because the model does not account for transporter mechanisms, potential dose-dependent nonlinear pharmacokinetics caused by saturation of an intestinal efflux transporter at higher oral doses are not accounted for. Similarly, for the three lopinavir doses (Table 1), the stand-alone model failed to describe drug exposure at the lowest dose (200 mg single oral dose). The observed dose-dependent pharmacokinetics of lopinavir may also be attributed to time-dependent inhibition of CYP3A (auto-inhibition).
The second limitation is the use of the rifampin PBPK model to predict rifampin’s effect on lopinavir in the presence of ritonavir. As shown in Table 3, PBPK simulations tend to underpredict the magnitude of induction under certain scenarios (predicted/observed ratio >2 for AUC or Ctrough). The under-prediction of CYP3A induction effect has been noticed in our recent analyses17. Because rifampin is also an inducer of other CYPs and P-glycoprotein17, the absence of these mechanisms in our PBPK analyses can also contribute to under-prediction of induction effects in evaluating drug-drug interaction scenarios between rifampin and lopinavir/ritonavir.
5. Conclusions
The linked PBPK substrate models appear to reasonably simulate the impact of concomitant use of other medications on the exposure of darunavir or lopinavir when combined with ritonavir. Currently, reliable, quantitative predictions regarding changes in drug exposure with hepatic impairment cannot be made using PBPK modeling. Simulations in hepatically impaired patients suggested that darunavir or lopinavir exposure may be further increased in these patients concomitantly taking ritonavir. However, further work is needed to address the knowledge gaps with PBPK modeling in the virtual hepatic impairment populations prior to being able to use PBPK modeling to evaluate the clinical relevance of the effects of both drug-drug interactions and hepatic impairment on the exposure of antiretroviral medications, including darunavir/ritonavir and lopinavir/ritonavir.
Subsequently, the limitation of the PBPK modeing approach is that the currently available linked darunavir or lopinavir PBPK models cannot derive appropriate dosing recommendations in the U.S. prescribing information to address drug-drug interactions in a hepatic impairment population and are not suitable for applying to decision making under such scenarios. The information presented in this manuscript provides a foundation to further improve and optimize the linked darunavir or lopinavir PBPK models and to evaluate the feasibility of developing clinically relevant hepatic impairment PBPK models.
Supplementary Material
Acknowledgments
Funding
U.S Food and Drug Administration, Center for Drug Evaluation and Research (CDER) Critical Path program
C.W. contributed to the data analysis, review of the results, and the manuscript submission. P.Z, V.A, and S.A contributed to the Critical Path proposal, review of the results and the manuscript submission. K.S. and C.M. contributed to the Critical Path proposal and the manuscript submission. All authors have read and approved the manuscript text.
C.W. was supported by an appointment administered by ORISE through an interagency agreement between the U.S. Department of Energy and the U.S. FDA funded through CDER’s
Footnotes
Disclosures
None of the authors have any relevant disclosures to declare.
This work was presented in part in poster form at the 16th International Workshop on Clinical Pharmacology of HIV & Hepatitis Therapy, May 26 – 28, 2015, Washington, DC and the 2015 Annual Meeting of the American College of Clinical Pharmacology, September 27–29, 2015, San Francisco, CA.
Disclaimer
The contents of this manuscript do not reflect the view or policies of the U.S. Food and Drug Administration (FDA) or its staff. No official support or endorsement by the FDA is intended or should be inferred.
References
- 1.Zhao P, Zhang L, Grillo JA, et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin Pharmacol Ther. 2011 Feb;89(2):259–67. doi: 10.1038/clpt.2010.298. [DOI] [PubMed] [Google Scholar]
- 2.Rostami-Hodjegan A. Physiologically based pharmacokinetics joined with in vitro–in vivo extrapolation of ADME: a marriage under the arch of systems pharmacology. Clin Pharmacol Ther. 2012 Jul;92(1):50–61. doi: 10.1038/clpt.2012.65. [DOI] [PubMed] [Google Scholar]
- 3.Wagner C, Zhao P, Pan Y, et al. Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: report of an FDA public workshop on PBPK. CPT Pharmacometrics Syst Pharmacol. 2015 Apr;4(4):226–230. doi: 10.1002/psp4.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cerdelga (eliglustat) [U.S. prescribing information] Waterford, Ireland: Genzyme Corporation; Aug, 2014. Available at: http://www.accessdata.fda.gov. [Google Scholar]
- 5.U.S. Food and Drug Administration. New Drug Application 205494 (eliglustat): clinical pharmacology review. 2014 Aug 19; Available at: http://www.accessdata.fda.gov.
- 6.Prezista (darunavir) [U.S. prescribing information] Titusville, NJ: Janssen Therapeutics; May, 2015. Available at: http://www.accessdata.fda.gov. [Google Scholar]
- 7.Kaletra (lopinavir/ritonavir) [U.S. prescribing information] North Chicago, IL: AbbVie Inc; Nov, 2015. Available at: http://www.accessdata.fda.gov. [Google Scholar]
- 8.Sekar V, Spinosa-Guzman S, De Paepe E, et al. Pharmacokinetics of multiple-dose darunavir in combination with low-dose ritonavir in individuals with mild-to-moderate hepatic impairment. Clin Pharmacokinet. 2010 May;49(5):343–50. doi: 10.2165/11530690-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 9.U.S. Food and Drug Administration. New Drug Application 21976 (darunavir): clinical pharmacology review. 2006 Jun 23; Available at: http://www.accessdata.fda.gov.
- 10.Boffito M, Miralles D, Hill A. Pharmacokinetics, efficacy, and safety of darunavir/ritonavir 800/100 mg once-daily in treatment-naïve and -experienced patients. HIV Clin Trials. 2008 Nov-Dec;9(6):418–27. doi: 10.1310/hct0906-418. [DOI] [PubMed] [Google Scholar]
- 11.Sekar VJ, Lefebvre E, De Pauw M, et al. Pharmacokinetics of darunavir/ritonavir and ketoconazole following co-administration in HIV-healthy volunteers. Br J Clin Pharmacol. 2008 Aug;66(2):215–21. doi: 10.1111/j.1365-2125.2008.03191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.U.S. Food and Drug Administration. New Drug Application 21226 (lopinavir/ritonavir): clinical pharmacology review. 2000 Sep 15; Available at: http://www.accessdata.fda.gov.
- 13.Eron JJ, Feinberg J, Kessler HA, et al. Once-daily versus twice-daily lopinavir/ritonavir in antiretroviral-naive HIV-positive patients: a 48-week randomized clinical trial. J Infect Dis. 2004 Jan 15;189(2):265–72. doi: 10.1086/380799. [DOI] [PubMed] [Google Scholar]
- 14.Crommentuyn KM, Mulder JW, Mairuhu AT, et al. The plasma and intracellular steady-state pharmacokinetics of lopinavir/ritonavir in HIV-1 infected patients. Antivir Ther. 2004 Oct;9(5):779–85. [PubMed] [Google Scholar]
- 15.Schöller-Gyüre M, Kakuda TN, Witek J, et al. Steady-state pharmacokinetics of etravirine and lopinavir/ritonavir melt extrusion formulation, alone and in combination, in healthy HIV-negative volunteers. J Clin Pharmacol. 2013 Feb;53(2):202–10. doi: 10.1177/0091270012445205. [DOI] [PubMed] [Google Scholar]
- 16.Kakuda TN, Brochot A, Tomaka FL, et al. Pharmacokinetics and pharmacodynamics of boosted once-daily darunavir. J Antimicrob Chemother. 2014 Oct;69(10):2591–605. doi: 10.1093/jac/dku193. [DOI] [PubMed] [Google Scholar]
- 17.Wagner C, Pan Y, Hsu V, Sinha V, et al. Predicting the effect of CYP3A inducers on the pharmacokinetics of substrate drugs using physiologically based pharmacokinetic (PBPK) modeling: an analysis of pbpk submissions to the us fda. Clin Pharmacokinet. 2016 Apr;55(4):475–83. doi: 10.1007/s40262-015-0330-y. [DOI] [PubMed] [Google Scholar]
- 18.SIMCYP Limited. SIMCYP Version 15 help pages (cirrhosis populations) SIMCYP, Limited; Shefield UK: 2015. [Google Scholar]
- 19.Johnson TN, Boussery K, Rowland-Yeo K, et al. A semi-mechanistic model to predict the effects of liver cirrhosis on drug clearance. Clin Pharmacokinet. 2010 Mar;49(3):189–206. doi: 10.2165/11318160-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 20.Lopinavir monograph. University of Washington, School of Pharmacy Drug Interaction Database Program; [Accessed January 5, 2017]. Available at: https://www.druginteractioninfo.org/ [Google Scholar]
- 21.Pan Y, Hsu V, Grimstein M, et al. The application of physiologically based pharmacokinetic modeling to predict the role of drug transporters: scientific and regulatory perspectives. J Clin Pharmacol. 2016 Jul;56(Suppl 7):S122–31. doi: 10.1002/jcph.740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.