

Summary
Immune checkpoint blockade is able to achieve durable responses in a subset of patients, however we lack a satisfying comprehension of the underlying mechanisms of anti-CTLA-4 and anti-PD-1 induced tumor rejection. To address these issues we utilized mass cytometry to comprehensively profile the effects of checkpoint blockade on tumor immune infiltrates in human melanoma and murine tumor models. These analyses reveal a spectrum of tumor infiltrating T cell populations that are highly similar between tumor models and indicate that checkpoint blockade targets only specific subsets of tumor infiltrating T cell populations. Anti-PD-1 predominantly induces the expansion of specific tumor infiltrating exhausted-like CD8 T cell subsets. In contrast, anti-CTLA-4 induces the expansion of an ICOS+ Th1-like CD4 effector population in addition to engaging specific subsets of exhausted-like CD8 T cells. Thus, our findings indicate that anti-CTLA-4 and anti-PD-1 checkpoint blockade induced immune responses are driven by distinct cellular mechanisms.
Graphical abstract
Introduction
Immunotherapy is assuming a role as a pillar of cancer treatment, but the remarkable immune mediated responses are limited to a minority of patients. Immune checkpoint blockade (ICB) is able to elicit durable responses in a fraction of cancer patients. For example, 22% of advanced melanoma patients treated with anti-CTLA-4 have durable responses extending beyond 10 years(Hodi et al., 2010; Schadendorf et al., 2015). Similarly, blockade of the PD-1/PD-L1 signaling axis is also sufficient to induce significant responses in multiple tumor types(Brahmer et al., 2012; Topalian et al., 2012). Despite such tremendous clinical progress we still lack a detailed understanding of the fundamental mechanisms that underlie anti-CTLA-4 and anti-PD-1 induced tumor immune rejection, which is necessary for the improvement of current therapies and for the rational design of combination therapy approaches. The aspects of the host immune response and the tumor intrinsic properties that define therapeutic sensitivity to ICB therapy remain to be elucidated(Sharma and Allison, 2015; Topalian et al., 2015). Despite evidence that tumor properties such as mutational load(Hugo et al., 2016; McGranahan et al., 2016) and genetic lesions(Gao et al., 2016; Spranger et al., 2015; Zaretsky et al., 2016) can influence therapeutic response to ICB, we do not fully understand why different tumor types display such a range of therapeutic sensitivity. Conceptually such differences could arise because different tumor types elicit fundamentally distinct immune responses or alternatively, because the magnitude of host immune responses varies between different tumor types.
A critical unresolved question is whether anti-tumor immune responses induced by anti-CTLA-4 and anti-PD-1 antibodies are mediated through distinct, non-redundant mechanisms. A wealth of studies have demonstrated that CTLA-4 and PD-1 attenuate T cell activation through distinct mechanisms(Pardoll, 2012). CTLA-4 is upregulated immediately following TCR ligation and outcompetes CD28 for B7 ligand binding, thus attenuating positive costimulation by CD28(Krummel and Allison, 1995; Walunas et al., 1994). PD-1 is induced later during T cell activation, and upon engagement with PD-L1 or PD-L2, attenuates TCR signaling via recruitment of tyrosine phosphatases(Chemnitz et al., 2004; Freeman et al., 2000; Latchman et al., 2001). In addition to utilizing distinct molecular mechanisms of action, CTLA-4 and PD-1 attenuate T cell activity through mechanisms that are separated spatially and temporally. Whereas CTLA-4 primarily attenuates T cell activation in the priming phase through cell intrinsic and extrinsic mechanisms, PD-1 primarily attenuates T cell activity in peripheral tissues through cell intrinsic mechanisms(Pardoll, 2012; Walker and Sansom, 2011). This distinction is highlighted by the fact that the cellular sources of the ligands of PD-1 and CTLA-4 are different and serve different physiological functions. Thus, we hypothesized that anti-CTLA-4 and anti-PD-1 induced anti-tumor immune responses are mediated by distinct cellular mechanisms.
To address this hypothesis we utilized mass cytometry to comprehensively profile the immune infiltrates of solid tumors following ICB. Mass cytometry allows for the interrogation of greater than 40 analytes at single cell resolution and enables systematic identification of complex cellular populations using high-dimensional analyses(Newell and Cheng, 2016; Tanner et al., 2013). Mass cytometry driven approaches have been utilized to characterize cellular processes including hematopoiesis, immune cell differentiation, and leukemic disease progression(Bendall et al., 2011; Spitzer and Nolan, 2016); and more recently, to analyze the immune infiltrates of solid tumors(Chevrier et al., 2017; Lavin et al., 2017; Leelatian et al., 2017; Spitzer et al., 2017). Here, we leverage mass cytometry to comprehensively characterize the cellular mechanisms of ICB in human melanoma and murine syngeneic transplantable tumor models. Comparisons of murine tumor models indicate that the phenotypes of infiltrating T cell populations and mechanisms of ICB are tumor type independent. Both anti-PD-1 and anti-CTLA-4 only target a subset of tumor infiltrating T cell populations, inducing the expansion of exhausted-like CD8 T cells. Notably, anti-CTLA-4 but not anti-PD-1 modulates the CD4 effector compartment, specifically inducing the expansion of an ICOS+ Th1-like CD4 effector subset. Together these pre-clinical and clinical analyses indicate that anti-tumor immune responses induced by CTLA-4 and PD-1 blockade are driven by distinct cellular mechanisms.
Results
Identification of checkpoint blockade responsive MC38 tumor infiltrating T cell subsets
To identify ICB responsive tumor infiltrating T cell populations we profiled tumors by mass cytometry and utilized a well-validated data-driven unsupervised clustering approach to classify cellular populations (Levine et al., 2015; Melchiotti et al., 2017; Shekhar et al., 2016). We further validated this approach for quantitative de novo classification of tumor infiltrating lymphocyte (TIL) populations using spike-in experiments (Figure S1, see STAR Methods). To comprehensively characterize tumor infiltrating T cell populations we designed a staining panel with 33 surface and 10 intracellular markers. This panel included non-T cell lineage markers (e.g. CD11 b, CD11 c, CD19), T cell differentiation and activation markers (e.g. PD-1, ICOS, TIM3, KLRG1, CD127), and importantly, T cell lineage transcription factors (e.g. TBET, EOMES, GATA3, BCL6, RORγT, FOXP3). Using this approach we analyzed immunogenic MC38 colorectal tumors from mice treated with anti-CTLA-4 or anti-PD-1. To enable analysis of TILs we empirically defined a tumor inoculation dose and treatment schedule (using standard antibody dosages) such that tumors were not completely rejected at time of analysis despite induction of an effective immune response. Treatment was initiated only after tumors became palpable and thus also more closely reflected the clinical context. We focused our analyses on the T cell compartment given our current understanding of CTLA-4 and PD-1 biology, the design of our staining panel, and analyses of the total CD45+ compartment (Figure S1). Analysis of the T cell compartment revealed dramatic population shifts in response to anti-CTLA-4 and anti-PD-1 (Figure 1 A, 1B). These observations are consistent with an increase in CD8/Treg ratio following both treatments, as determined by manual gating analyses and reflect the induction of an effective immune response by ICB (Figure 1C).
Figure 1. Identification of checkpoint blockade responsive MC38 tumor infiltrating T cell populations.
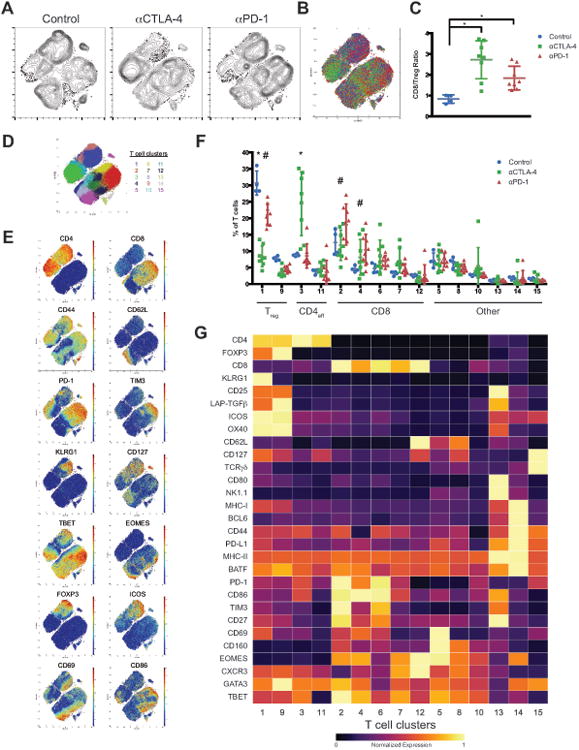
(A) Density t-SNE plots of an equal number of CD3ε+ MC38 tumor infiltrating T cells from each treatment group.
(B) Overlaid t-SNE plot displaying equal number of events from each treatment group (control, blue; anti-CTLA-4, green; anti-PD-1, red).
(C) Plot of CD8/Treg ratios displayed on a per mouse basis with mean ± SD (*, P<0.05, unpaired T-test).
(D) t-SNE plot of MC38 infiltrating T cells overlaid with color-coded clusters.
(E) t-SNE plot of infiltrating T cells overlaid with the expression of selected markers.
(F) Frequency of T cell clusters displayed on a per mouse basis with mean ± SD (*, control v.s. anti- CTLA-4; #, control v.s. anti-PD-1; p<0.05, Dunnett's multiple comparison). T cell compartments are denoted including CD8, Treg, and CD4 effector (CD4eff).
(G) Heat map displaying normalized marker expression of each T cell cluster.
Representative data from three independent experiments is shown.
See also Figure S1 and STAR Methods.
To gain a more in depth understanding of the mechanisms that underlie ICB we generated a high-resolution map of phenotypically defined tumor infiltrating T cell populations using unsupervised clustering. 15 distinct MC38 tumor infiltrating T cell clusters of greater than 0.5% relative frequency were identified, including 5 CD8, 2 Treg, and 2 CD4 effector clusters (Figure 1D-F). This approach focused specifically on tumor infiltrating populations, and thus many canonical T cell subsets present in other tissues would not be expected present (e.g. naïve); as such this represents an extensive catalog of infiltrating T cell subsets. Notably, ICB did not modulate the frequency of any NKT, γδ T cell, or low frequency (<0.5%) clusters. Thus, we focused our analyses on CD4 and CD8 T cell subsets, which displayed a range of activation and exhaustion phenotypes (Figure 1G). Both anti-CTLA-4 and anti-PD-1 treatment led to an expansion of CD8 T cells, however not all CD8 T cell subsets expanded following ICB. Surprisingly, a phenotypically exhausted PD-1hiTIM3+ population expanded the most among CD8 populations (Figure 1F). To address if T cell expansion results from increased proliferation or infiltration, we assessed short-term incorporation of 5-iodo-deoxyuridine (IdU). ICB responsive CD8 clusters incorporated IdU suggesting that these cells are proliferating within the tumor microenvironment (TME) (Figure S1I). Given the timing of IdU treatment and retention of IdU in daughter cells, this approach may detect extratumoral blasting T cells that subsequently infiltrate the tumor, in addition to cells proliferating within the TME. Nonetheless, these observations indicate that ICB sensitive T cells retain proliferative capacity even after multiple rounds of therapy and that ICB leads to the expansion of only specific intratumoral T cell subsets.
We next assessed the effect of ICB on CD4 T cell populations. Within the Treg compartment, 2 clusters were identified that are largely distinguished by KLRG1 expression (Figure 1G). Relative Treg frequency decreased following both anti-PD-1 and anti-CTLA-4, consistent with a shift between effector and regulatory T cell populations (Quezada et al., 2006). The magnitude of this decrease was greater following CTLA-4 blockade; consistent with findings that treatment with anti-CTLA-4 leads to intratumoral Treg depletion in murine tumor models(Selby et al., 2013; Simpson et al., 2013). Two CD4 effector T cell subsets were identified in the TME, which both display an activated phenotype, but differ in their expression of key markers including PD-1 and TBET (Figure 1G). Most notably, treatment with anti-CTLA-4 but not anti-PD-1 was associated with a significant expansion of a TBET+ Th1-like CD4 effector subset (Figure 1F). This population was proliferative, but to a lesser degree than other clusters (Figure S1I). We denote this subset as distinct from canonical Th1 cells because of expression of PD-1 and ICOS, which are defining characteristics of T follicular helper (TFH) cells, despite expression of TBET but not BCL6 (Th1 and TFH lineage transcription factors, respectively). Neither therapy led to an expansion of un-skewed activated CD4 effectors. These observations suggest that specific T cell subsets are targeted by ICB and that anti-CTLA-4 leads to expansion of CD4 effector T cells.
Identification of checkpoint blockade responsive B16BL6 tumor infiltrating T cell subsets
We then sought to determine whether these findings reflect a generalizable mechanism of ICB responses. For this purpose, we performed similar experiments in the poorly immunogenic B16BL6 melanoma model to contrast the relatively high immunogenicity of MC38, allowing us to distinguish ICB response phenomena from tumor type specific observations. Due to low baseline T cell tumor infiltration and the lack of response to anti-CTLA-4 monotherapy(van Elsas et al., 1999), we treated mice with a single dose of the GVAX tumor vaccine in order to boost overall T cell infiltration. As in the MC38 system, we empirically defined the B16BL6 tumor inoculate and treatment schedule (using standard antibody dosages) such that tumors were not completely rejected at time of analysis despite induction of an effective immune response. Analysis of CD45+ TILs revealed significant therapy induced changes in immune composition (Figure S2). We focused our analyses on the T cell compartment to identify ICB responsive T cell populations.
Reflective of induction of an effective immune response, significant shifts in T cell populations in the TME were observed following treatment with anti-CTLA-4 and anti-PD-1 (Figure 2A, 2B), which mirrored an increase in CD8/Treg ratio as determined by manual gating (Figure 2C). Clustering identified 13 clusters of frequency greater than 0.5% including 5 CD8, 3 Treg, 2 CD4 effector, NKT, and γδ T cell clusters (Figure 2D, 2E). ICB did not affect the frequencies of any NKT, γδ T cell, or low frequency subsets. Remarkably, despite analysis of a different tumor type, time point of tumor progression, and the addition of a GM-CSF expressing tumor vaccine, the T cell clusters identified in B16BL6 tumors were nearly identical to those identified in MC38 tumors. Of the 5 identified CD8 clusters, only a subset were responsive to ICB with PD-1+TIM3+ exhausted CD8 T cells expanding the most (Figure 2E, 2F). Of the 3 Treg subsets identified, 2 contracted significantly following ICB. These populations differ primarily in their expression of KLRG1, with KLRG1+ Treg decreasing in relative frequency most dramatically. Of the 2 CD4 effector populations identified, both displayed an activated CD44+CD62Llo phenotype but were distinguished by expression of PD-1, CD127, and TBET (Figure 2F). Notably, the frequency of TBET+ Th1-like CD4 effector T cells increased following anti-CTLA-4 but not anti-PD-1 (Figure 2E). Thus, as observed in MC38 tumors, both anti-CTLA-4 and anti-PD-1 induce the expansion of specific T cell subsets and differentially affect CD4 effector T cells.
Figure 2. Identification of checkpoint blockade responsive B16BL6 tumor infiltrating T cellpopulations.
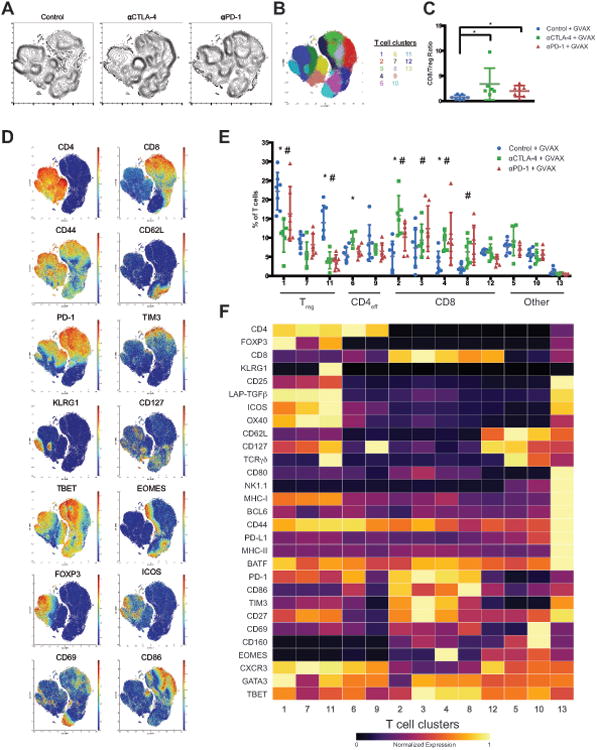
(A) Density t-SNE plots of an equal number of CD3ε+ B16BL6 tumor infiltrating T cells from eachtreatment group.
(B) t-SNE plot of infiltrating T cells overlaid with color-coded clusters.
(C) Plot of CD8/Treg ratios displayed on a per mouse basis with mean ± SD (*, P<0.05, unpaired T-test).
(D) t-SNE plot of tumor infiltrating T cells overlaid with the expression of selected markers.
(E) Frequency of T cell clusters displayed on a per mouse basis with mean ± SD (*, control v.s. anti- CTLA-4; #, control v.s. anti-PD-1; p<0.05, Dunnett's multiple comparison).
(F) Heat map displaying normalized marker expression of each T cell cluster.
Representative data from three independent experiments is shown.
See also Figure S2.
MC38 and B16BL6 tumor infiltrating T cell populations are fundamentally similar
The remarkable similarity in T cell populations identified by unsupervised clustering in MC38 and B16BL6 tumors suggests that the mechanisms governing responses to ICB are tumor type independent. Conceptually, this implies that the same types of T cells are involved in anti-tumor T cell responses to different tumor types, at least in the context of transplantable murine tumor models. To explicitly address this possibility, we analyzed the multivariate profiles of infiltrating T cell populations from MC38 and B16BL6 tumors simultaneously in order to identify any significant associations between T cell phenotype and tumor type. Projecting these phenotypes into the coordinate axes defined by their principal components, we asked whether the distributions along each component differed significantly between MC38- and B16BL6-derived T cell populations (see STAR methods). In other words, we asked whether any of the phenotypic variance among all T cell populations observed in all treatments (independent of frequency) was attributable to the tumor model source.
Comparison along each principal component axis revealed that MC38 and B16BL6 tumor infiltrating T cell subpopulations are phenotypically indistinguishable (Figure 3, Table S1A). The distribution of T cell subpopulations derived from MC38 and B16BL6 tumors did not differ along 38 of 39 principal components, which together explain 95% of the variance of the data (Table S1A). In the one case where a significant difference was detected (PC6), the discrepancy was attributable to contaminating CD19+ subpopulations in several MC38 samples and likely represents a technical artifact rather than a biological effect. This analysis indicates that there is no association between tumor model and the vast majority of phenotypic variance among the T cells identified in these models. This observation is confirmed visually by the overlap of MC38- and B16BL6-derived T cell populations plotted on biaxial pairs of the largest principal component projections (Figure 3).
Figure 3. B16BL6 and MC38 tumor infiltrating T cell populations are quantitatively similar.
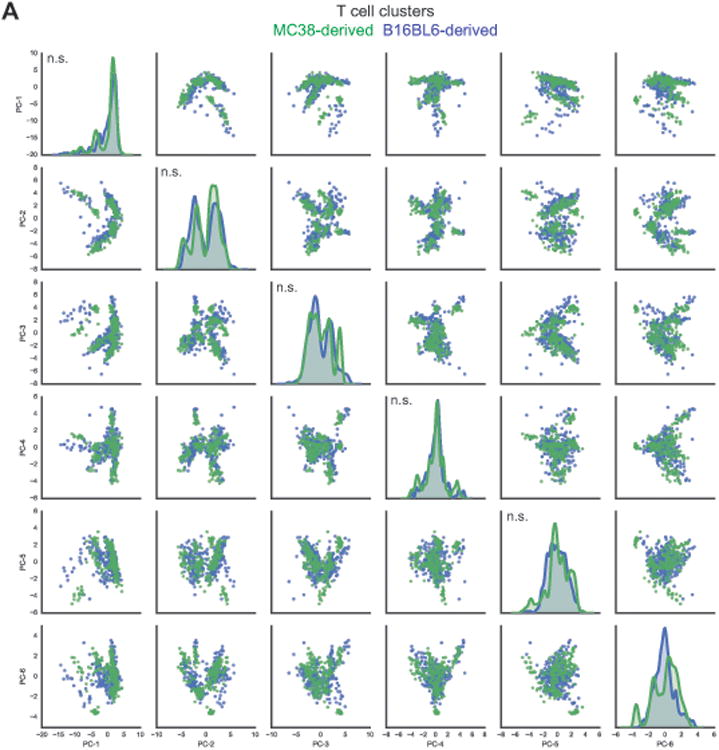
(A) PCA was applied to T cell clusters identified on a per mouse basis from MC38 and B16BL6 mass cytometry datasets. Projections of MC38 and B16BL6 infiltrating T cell clusters on to the first 6 principal components (PC), which together account for 78% of the phenotypic variance, are displayed in a pair wise fashion (MC38, green; B16BL6, blue). Univariate distributions of T cell clusters along each of the first 6 principal components are displayed along the diagonal. The Kolmogorov-Smirnov test was applied to test whether distributions of MC38 and B16BL6 derived T cell clusters along each PC are different (n.s., not significant).
See also Table S1.
Thus, the multivariate phenotypes of T cell subsets from MC38 and B16BL6 tumors are quantitatively similar. This finding is striking given the use of the GVAX tumor vaccine only with the B16BL6 model and the difference in immunogenicity of these models. Consistent with MC38 being highly immunogenic and B16BL6 being poorly immunogenic, MC38 has more than 2-fold more nonsynonymous single nucleotide variants (SNV) than B16BL6 (2327 and 1107, respectively; Table S1B). These data indicate that the types of T cells that infiltrate transplantable murine tumors are tumor type independent and suggest that differences in immunogenicity between tumor types arise due to tumor intrinsic properties that modulate the magnitude (e.g. subset frequency), but not type, of anti-tumor T cell responses. Combined with the observation that similar T cell subsets are regulated in response to ICB in both tumor models, this suggests that the cellular mechanisms of CTLA-4 and PD-1 blockade are tumor-type independent.
Identification of B16BL6 infiltrating T cell populations that correlate with tumor growth
We then sought to identify T cell populations whose frequencies correlate with tumor growth to gain insight into their functional relevance. For this purpose, we leveraged our B16BL6 datasets and combined three independent biological replicate cohorts, which together displayed a robust response to ICB (Figure 4A, 4B). Using a metaclustering approach, in which populations first identified at the individual mouse level using PhenoGraph are then allowed to merge across cohorts (see STAR Methods), 14 T cell populations were identified. The phenotypes and responses of these subsets to ICB were consistent with findings from single cohort analyses. Because ICB only modulated the frequencies of CD4 and CD8 T cell subsets, we focused our analyses on the 10 metaclusters within these compartments (Figure 4C, 4D, S3). Expectedly, the frequency of major Treg subsets correlated positively with tumor growth (Figure 4E, S3). The 2 major Treg populations are primarily distinguished by KLRG1 expression, with the frequency of KLRG1+ Treg (MC4) correlating more strongly with tumor growth than KLRG1- Treg (MC0) or manually gated Treg (Figure S3, Table S2A, S2B). Whether this difference reflects differences in functionality or response to ICB is unclear, however both subsets significantly correlated with tumor growth, suggesting that both retain suppressive activity.
Figure 4. Identification of B16BL6 tumor infiltrating T cell populations that correlate with tumor growth.
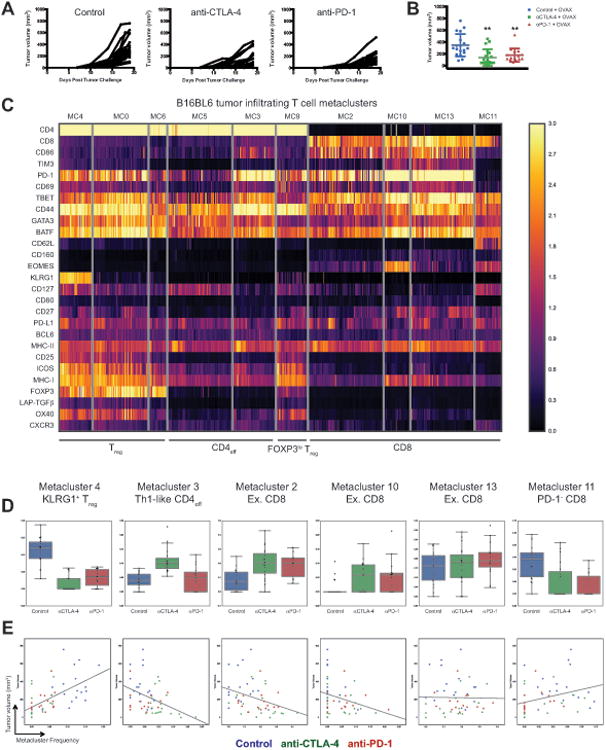
(A) B16BL6 tumor growth curves in each treatment group.
(B) Final tumor volume in each treatment group displayed on a per mouse basis with mean ± SD (**, control v.s. treatment, p<0.01, unpaired T-test).
(C) Metaclustering analysis of B16BL6 tumor infiltrating T cell clusters. Two-way hierarchical clustering of T cell metaclusters and individual parameters displayed as a heat map. Only CD4 and CD8 T cell metaclusters are displayed.
(D) The frequencies of T cell metaclusters in individual mice plotted as a fraction of total tumor infiltrating T cells and displayed as a box plot.
(E) The frequencies of T cell metaclusters in individual mice plotted as a function of B16BL6 tumor volume with linear regression best-fit lines displayed.
See also Figure S3 and Table S2.
Surprisingly, the frequency of only 2 of the 4 tumor infiltrating CD8 T cell subsets negatively correlated with tumor growth (Figure 4E). These populations displayed an activated phenotype and increased frequencies following anti-CTLA-4 and anti-PD1 treatment (Figure 4C, 4D). Metacluster 2 (MC2) displayed a PD-1+TIM3loTBET+EOMES- phenotype while metacluster 10 (MC10) displayed a PD-1hiTIM3+TBET+EOMES+ phenotype. The frequency of a third CD8 population (MC13), which displayed a PD-1hiTIM3+TBET+EOMES- exhausted phenotype, did not correlate with tumor growth (Table S2A). Thus, subtle multivariate phenotypic differences between metaclusters 2, 10, and 13 distinguish T cell populations that significantly differ in their correlation with tumor growth, which likely reflects functional differences between these populations. Moreover, these data suggest that fully exhausted non-terminally differentiated T cells (MC13) may not contribute significantly to tumor rejection in the context of ICB, at least during later stages of response. In contrast, less exhausted non-terminally differentiated (MC2) and fully exhausted terminally differentiated (MC10) appear to provide the bulk of the functional anti-tumor T cell response.
Unexpectedly, the frequency of a non-proliferative CD44+CD62L+PD-1- CD8 T cell subset, metacluster 11 (MC11), positively correlated with tumor growth (Figure 4E, Table S2A). This population may be tumor irrelevant central memory CD8 T cells, raising the possibility that infiltration of antigen irrelevant CD8 T cells is not only ineffective, but may in fact dampen the anti-tumor immune response. In terms of the proliferative capacity of effective CD8 T cell subsets, MC10 incorporated IdU at almost four times the rate of MC2 (Figure S3C). In contrast, despite being highly proliferative, the frequency of MC13 does not correlate with tumor growth. This suggests that high proliferative capacity of CD8 T cells in the TME during later stages of responses to ICB is neither necessary nor sufficient for effective anti-tumor responses. Whether effective CD8 T cell subsets of low (MC2) and high (MC10) proliferative capacity contribute through distinct functions remains unclear.
The 2 CD4 effector T cell metaclusters identified include a PD-1hiTBET+ Th1-like subset (MC3) and a PD-1loCD44intCD127int subset (MC5). Only the frequency of MC3 negatively correlated with tumor growth (Figure 4E, Table S2A). Notably, this correlation is driven by the specific expansion of this population following anti-CTLA-4 treatment. Interestingly, the Th1-like population displayed a low proliferation rate in both B16BL6 and MC38 tumor models (Figure S1, S3), raising the possibility that modulation of this population by anti-CTLA-4 may primarily occur at earlier time points or in secondary lymphoid organs. Together these data indicate that only specific populations of tumor infiltrating CD4 and CD8 T cells mediate responses to ICB, and suggest that the quantification of these phenotypically defined T cell subsets will provide improved predictive value compared to assessment of bulk compartments (e.g. CD8 T cells).
These findings reinforce the notion that data-driven multivariate analyses enable unbiased comprehensive cellular classification and robust de novo discovery of biologically relevant populations. It is important to note that while we ascribe key phenotypic features to identified clusters (Figure 4, Table S2A), quantitative multivariate analyses provide vastly improved subset assignment compared to manual gating. We sought to determine if the insights provided by high-dimensional analyses would enable approximation of these subsets by manual gating. Using a limited number of key parameters derived from multivariate analyses, manual gating is able to discriminate relevant T cell subsets, albeit with significantly reduced fidelity (Figure S3E, Table S2B). Consistent with the importance of lineage transcription factor factors for robust subset identification, expression of TBET but not individual surface markers was sufficient to identify CD4 effector subpopulations that significantly negatively correlate with tumor growth.
Differential transcriptional regulation in tumor infiltrating CD4 T cells following anti-CTLA-4 and anti-PD-1 checkpoint blockade
Next we investigated if anti-CTLA-4 and anti-PD-1 induce different transcriptional changes in tumor infiltrating CD4 T cells, as has been observed in CD8 T cells in preclinical and clinical contexts(Das et al., 2015; Gubin et al., 2014). Gene expression analyses of MC38 tumor infiltrating ICOS+ CD4 T cells revealed significant, but largely non-overlapping, transcriptional responses induced by anti-CTLA-4 and anti-PD-1 (Figure S4). Of the top 15 cellular pathways regulated by each treatment, only 3 were shared. Mitochondrial and oxidative phosphorylation pathways were among the most significantly modulated by anti-PD-1, consistent with findings that these pathways can restrict T cell activity in the TME(Bengsch et al., 2016; Gubin et al., 2014). CTLA-4 blockade led to an engagement of largely distinct pathways, which included pathways involved in cell cycle regulation. These observations indicate that anti-CTLA-4 and anti-PD-1 induce differential transcription effects in tumor infiltrating CD4 T cells and support the paradigm that these therapies act through distinct mechanisms.
Anti-CTLA-4 and anti-PD-1 therapies modulate specific T cell populations in human melanoma
Finally, we sought to determine whether distinct cellular mechanisms also underlie anti-CTLA-4 and anti-PD-1 tumor rejection in humans. Using a similarly designed human T cell mass cytometry panel, we analyzed surgically resected melanoma tumors from patients being treated with ipilimumab (ipi), anti-PD-1 (nivolumab, nivo; or pembrolizumab, pembro), or ipi plus nivo (Table S3). This approach enables direct interrogation of tumor infiltrating T cell populations that may not be fully represented in peripheral blood. t-SNE analysis revealed striking differences between normal donor blood and tumor infiltrating T cells, as well as treatment-specific effects (Figure 5A-C, S5). To more deeply interrogate the effects of anti-CTLA-4 therapy, we compared samples from patients being treated with ipi (alone or in combination with nivo) or anti-PD-1 monotherapy. This approach enabled more robust statistical analyses given the rarity of ipi monotherapy tumor samples in the current landscape of standard of care therapy for patients with metastatic melanoma. Unsupervised clustering of tumor and normal donor blood samples identified 19 distinct T cell subsets, including 5 CD8 and 11 CD4 clusters (Figure 5D-E; see STAR Methods). The increased number of T cell subsets compared to our murine TIL data likely reflects the identification of canonical subsets in blood that are not present in tumors, and as such would be absent from our preclinical analyses. Consistent with this notion, naïve T cell subsets were specific to blood while many of the T cell subsets were observed at similar frequencies in normal donor blood and tumors.
Figure 5. Identification of checkpoint blockade responsive tumor infiltrating T cell populationsin human melanoma.
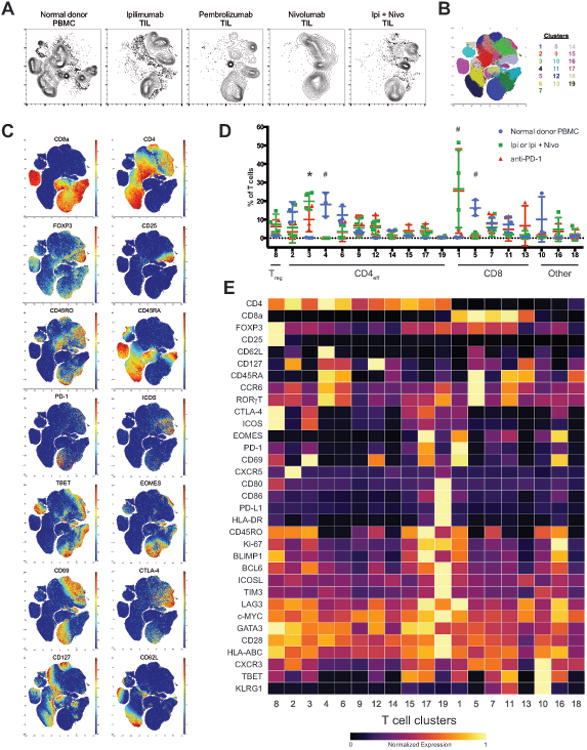
(A) Density t-SNE plots of CD3+ tumor infiltrating T cells from melanoma patients being treated withindicated ICB therapies and T cells from normal donor peripheral blood.
(B) t-SNE plot of total T cells from all samples overlaid with color-coded clusters.
(C) t-SNE plots of total T cells from all samples overlaid with the expression of selected markers.
(D) Frequency of T cell clusters displayed on a per sample basis with mean ± SD (*, ipi/ipi plus nivov.s. anti-PD-1; #, anti-PD-1 and ipi/ipi plus nivo v.s. normal PBMC; p<0.05, Tukey's multiplecomparison).
(E) Heat map displaying normalized marker expression of T cell clusters.
See also Figure S5 and Table S3.
Surprisingly, of the 19 T cell subsets identified, only 2 were significantly expanded in ICB treated tumors compared to normal donor blood. Although most melanoma infiltrating T cell subsets were actively proliferating, only clusters 1 and 3 significantly expanded, suggesting that they are functionally distinguished by as yet unidentified mechanisms (Figure S5D). The CD8 T cell population expanded in ICB treated tumors displayed a CD45RO+PD-1+TBET+EOMES+ phenotype (Figure 5E, cluster 1); analogous to the exhausted-like terminally differentiated CD8 T cell subset identified in murine tumor models as important for tumor rejection. The CD4 T cell population expanded in ICB treated tumors displayed a CD45RO+ICOS+PD-1loTBET+ effector phenotype (Figure 5E, cluster 3); analogous to the activated Th1-like CD4 effector subset identified in murine tumor models that expands in response to CTLA-4 blockade but not PD-1 blockade.
Notably, the only ICB treatment specific effect observed was an increased frequency of Th1-like T cells in melanomas treated with anti-CTLA-4 compared to those treated with anti-PD-1 (Figure 5D, cluster 3). Thus remarkably, despite the presence of confounding variables (e.g. diverse treatment histories) and small sample size, these analyses suggest that anti-CTLA-4 and anti-PD-1 therapies modulate only specific tumor infiltrating T cell subsets and that anti-CTLA-4 induces a more robust CD4 effector response; observations consistent with our preclinical findings. Future studies are needed to validate these findings in a larger patient cohort and determine whether the discrepancy in the number of ICB responsive CD8 T cell subsets in mouse and human tumors reflects a difference in underlying biology or rather a technical aspect of our analyses. In both mouse and human, the CD4 effector response is defined by expansion of an ICOS+ TBET+ Th1-like subset. Notably, despite qualitative (e.g. CD44 versus CD45RO) and quantitative (e.g. levels of ICOS and PD-1) differences in phenotypic profiles of T cells infiltrating human and murine tumors, unsupervised clustering enabled robust detection of biologically analogous populations (Figure S5E-F). Together these data indicate that the cellular mechanisms of CTLA-4 and PD-1 blockade are distinct and that the hallmarks of these mechanisms are largely conserved between mouse and human.
Discussion
Here we systematically classify tumor infiltrating T cells from murine tumor models and human melanomas in the context of ICB using mass cytometry and unsupervised analyses. These studies provide insight into several key concepts: i) ICB only induces the expansion of specific tumor infiltrating T cell subsets, ii) PD-1 blockade primarily induces expansion of exhausted-like tumor infiltrating CD8 T cells, iii) CTLA-4 blockade induces expansion of ICOS+ Th1-like CD4 effector as well as exhausted-like CD8 T cells, iv) the frequency of only specific tumor infiltrating CD4 and CD8 T cell populations correlate with tumor growth, and v) the phenotypes of tumor infiltrating T cell subsets in different transplantable murine tumor models are fundamentally similar.
Together these observations indicate that anti-CTLA-4 and anti-PD-1 induced anti-tumor responses are driven by distinct cellular mechanisms, primarily differing on the expansion of the CD4 effector compartment induced by anti-CTLA-4. Given that we profiled anti-tumor immune responses in the context of partial regression by design, it remains to be determined if the same mechanisms mediate complete tumor rejection in the context of resolution of antigen burden. The similarity of findings in the MC38 and B16BL6 systems despite analyses of different time points (2 and 10 days after treatment, respectively) suggests that these mechanisms persist and may be independent of the phase of tumor rejection. Our findings are consistent with clinical observations that increased CD8, but not CD4, T cell activity is associated with anti-PD-1 therapy in melanoma(Daud et al., 2016), and with the fundamental understanding that PD-1 and CTLA-4 attenuate T cell activation through distinct molecular and cellular mechanisms. It is likely that dual engagement of these distinct cellular mechanisms underlies, at least in part, the enhanced efficacy of combination anti-CTLA-4 and anti-PD-1 therapy that has been observed in preclinical and clinical contexts (Curran et al., 2010; Wolchok et al., 2013).
Additional mechanistic investigation of anti-CTLA-4 and anti-PD-1 is also warranted. For example, the necessity and sufficiency of specific ICB responsive tumor infiltrating T cell subsets identified in our study remains to be definitively tested. Furthermore, recent studies have shown that anti-PD-1 therapy leads to a dynamic expansion of proliferating PD-1+ CD8 T cells in peripheral blood of melanoma and lung cancer patients(Huang et al., 2017; Kamphorst et al., 2017). Whether expansion of ICB responsive exhausted-like CD8 T cells is driven by therapeutic engagement of peripheral or tumor infiltrating populations is unknown. Furthermore, the degree to which anti-tumor T cell subsets are equally represented in tumor and peripheral blood remains unclear. Analyses of paired tumor and blood samples from patients being treated with ICB therapy may provide critical insight into these issues. Examination of additional parameters, such as costimulatory molecules, may offer additional clarity by providing an even finer resolution catalog of T cell subsets, as in recent analyses of renal cell carcinoma(Chevrier et al., 2017).
It remains unclear what functionally distinguishes ICB responsive and nonresponsive CD8 T cell populations. ICB responsive subsets may represent the bulk of tumor-antigen specific T cells or alternatively, represent a functionally distinct subset thereof. Distinguishing between these possibilities may inform the development of therapeutic strategies. Likewise, future studies are required to determine if ICB responsive CD8 T cell subsets are functionally as well as phenotypically exhausted and moreover, if they are functionally distinct from each other. The maintenance of PD-1 on responsive CD8 T cells despite prolonged anti-PD-1 therapy suggests that PD-1 blockade is sufficient to reinvigorate these populations but not to reprogram them into a non-exhausted state, consistent with epigenetic regulation(Pauken et al., 2016).
Although our findings indicate that CTLA-4 blockade induces an expansion of tumor infiltrating Th1-like CD4 T cells, the definitive source (anatomical and temporal) and precise function of this expansion remain open questions. It is possible that expansion of specific tumor infiltrating T cell subsets in response to ICB results from engagement of distinct progenitor populations in secondary lymphoid organs, analogous to findings in viral models(Im et al., 2016). With respect to function, it is tempting to speculate that expansion of Th1-like CD4 effectors by anti-CTLA-4 improves anti-tumor responses by enhancing CD8 infiltration, cytolytic CD8 activity, and T cell memory formation. Addressing these possibilities is of great interest given that expansion of ICOS+ CD4 T cells following ipi treatment has been observed in multiple tumor types(Chaput et al., 2017; Chen et al., 2009; Liakou et al., 2008), expansion of ICOS+ CD4 T cells is associated with overall survival following ipi therapy(Carthon et al., 2010), and that genetic loss of Icos attenuates the efficacy of anti-CTLA-4 in preclinical tumor models(Fan et al., 2014). Our findings suggest that expansion of the CD4 effector compartment by anti-CTLA-4 differentiates its mechanism of action from that of PD-1 blockade. Such insights will inform the rational design of combinatorial approaches, particularly given the fundamental understanding that CD4 help is critical for the development of robust T cell responses, as well as recent findings that CD4 T cells are critical for effective immunotherapy(Spitzer et al., 2017).
In conclusion, we comprehensively profiled T cells in preclinical and clinical tumor samples using a mass cytometry based systems approach. We identify specific tumor infiltrating T cell populations that expand in response to ICB and demonstrate that anti-PD-1 and anti-CTLA-4 operate through distinct cellular mechanisms. These findings highlight the utility of unsupervised systems-based analyses for in-depth mechanistic investigation.
Supplementary Material
Acknowledgments
We thank Duncan Mak for expert advice and technical assistance with mass cytometry analysis. Mass cytometry and cell sorting were performed at the MDACC Flow Cytometry and Cellular Imaging Core Facility, which is in part funded by NCI Cancer Center Support Grant (CCSG) P30CA16672. Sequencing was performed at the MDACC Sequencing and Microarray Facility, which is funded by P30CA016672(SMF). This work was supported by a grant from Cancer Prevention and Research in Texas to J.P.A. (R1203), the MDACC CCSG Bioinformatics Shared Resource, NIH grants to D.P. (DP1-HD084071 and R01CA164729), and a MSK Cancer Center Support Grant/Core Grant to D.P. (P30CA008748). S.C.W. is an MDACC Odyssey postdoctoral fellow. J.P.A. and P.S. are Co-Directors and J.A.W. is a member of the Parker Institute for Cancer Immunotherapy. P.S. has consulted for AstraZeneca, Amgen, GlaxoSmithKline (GSK), and Bristol-Myers Squibb (BMS). P.S. serves on the scientific advisory boards for Jounce, Neon, Constellation, and Kite. P.S. is a co-founder of Jounce Therapeutics. P.S. received funding on NIH R01CA163793. M.C.A. has attended speakers' bureau with travel support from BMS and MSD. J.A.W. has received honoraria from speakers' bureau of Dava Oncology, BMS, and Illumina, and is an advisory board member for GSK, Novartis and Roche/Genentech. J.P.A. is a co-founder of Jounce and Neon Therapeutics, is an advisory board member for Jounce, Neon, Amgen, and Kite Pharmaceuticals, and has received royalties from intellectual property licensed to BMS and Merck.
Footnotes
Author Contributions: S.C.W. and J.P.A. conceived the project and wrote the manuscript. S.C.W. performed experiments. J.H.L. developed computational approaches for mass cytometry data analysis. N.A. provided technical assistance. S.C.W., J.H.L., and Y.Z. analyzed data. A.P.C, M.C.A., and J.A.W. acquired and annotated clinical samples. S.C.W., J.H.L., J.W., P.S., J.A.W., D.P., and J.P.A. interpreted data. All authors contributed to the revision of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe'er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31:545–552. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behbehani GK, Bendall SC, Clutter MR, Fantl WJ, Nolan GP. Single-cell mass cytometry adapted to measurements of the cell cycle. Cytometry A. 2012;81:552–566. doi: 10.1002/cyto.a.22075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, Stelekati E, McLane LM, Paley MA, Delgoffe GM, et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity. 2016;45:358–373. doi: 10.1016/j.immuni.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carthon BC, Wolchok JD, Yuan J, Kamat A, Ng Tang DS, Sun J, Ku G, Troncoso P, Logothetis CJ, Allison JP, et al. Preoperative CTLA-4 blockade: tolerability and immune monitoring in the setting of a presurgical clinical trial. Clin Cancer Res. 2010;16:2861–2871. doi: 10.1158/1078-0432.CCR-10-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, Boselli L, Routier E, Cassard L, Collins M, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2017;28:1368–1379. doi: 10.1093/annonc/mdx108. [DOI] [PubMed] [Google Scholar]
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173:945–954. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- Chen H, Liakou CI, Kamat A, Pettaway C, Ward JF, Tang DN, Sun J, Jungbluth AA, Troncoso P, Logothetis C, et al. Anti-CTLA-4 therapy results in higher CD4+ICOShi T cell frequency and IFN-gamma levels in both nonmalignant and malignant prostate tissues. Proc Natl Acad Sci USA. 2009;106:2729–2734. doi: 10.1073/pnas.0813175106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, Ries CH, Ailles L, Jewett MAS, Moch H, et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell. 2017;169:736–749 e718. doi: 10.1016/j.cell.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, Callahan M, Wolchok JD, Halaban R, Dhodapkar MV, et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol. 2015;194:950–959. doi: 10.4049/jimmunol.1401686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daud AI, Loo K, Pauli ML, Sanchez-Rodriguez R, Sandoval PM, Taravati K, Tsai K, Nosrati A, Nardo L, Alvarado MD, et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest. 2016;126:3447–3452. doi: 10.1172/JCI87324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Quezada SA, Sepulveda MA, Sharma P, Allison JP. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J Exp Med. 2014;211:715–725. doi: 10.1084/jem.20130590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016;167:397–404. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60–65. doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537:417–421. doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, Sica GL, Yu K, Koenig L, Patel NT, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci USA. 2017;114:4993–4998. doi: 10.1073/pnas.1705327114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell. 2017;169:750–765 e717. doi: 10.1016/j.cell.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leelatian N, Doxie DB, Greenplate AR, Mobley BC, Lehman JM, Sinnaeve J, Kauffmann RM, Werkhaven JA, Mistry AM, Weaver KD, et al. Single cell analysis of human tissues and solid tumors with mass cytometry. Cytometry B Clin Cytom. 2017;92:68–78. doi: 10.1002/cyto.b.21481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amir el AD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liakou CI, Kamat A, Tang DN, Chen H, Sun J, Troncoso P, Logothetis C, Sharma P. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc Natl Acad Sci USA. 2008;105:14987–14992. doi: 10.1073/pnas.0806075105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei HE, Leipold MD, Maecker HT. Platinum-conjugated antibodies for application in mass cytometry. Cytometry A. 2016;89:292–300. doi: 10.1002/cyto.a.22778. [DOI] [PubMed] [Google Scholar]
- Melchiotti R, Gracio F, Kordasti S, Todd AK, de Rinaldis E. Cluster stability in the analysis of mass cytometry data. Cytometry A. 2017;91:73–84. doi: 10.1002/cyto.a.23001. [DOI] [PubMed] [Google Scholar]
- Newell EW, Cheng Y. Mass cytometry: blessed with the curse of dimensionality. Nat Immunol. 2016;17:890–895. doi: 10.1038/ni.3485. [DOI] [PubMed] [Google Scholar]
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354:1160–1165. doi: 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pounds S, Morris SW. Estimating the occurrence of false positives and false negatives in microarray studies by approximating and partitioning the empirical distribution of p-values. Bioinformatics. 2003;19:1236–1242. doi: 10.1093/bioinformatics/btg148. [DOI] [PubMed] [Google Scholar]
- Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest. 2006;116:1935–1945. doi: 10.1172/JCI27745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J Clin Oncol. 2015;33:1889–1894. doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1:32–42. doi: 10.1158/2326-6066.CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, et al. Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell. 2016;166:1308–1323 e1330. doi: 10.1016/j.cell.2016.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210:1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell. 2017;168:487–502 e415. doi: 10.1016/j.cell.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer MH, Nolan GP. Mass Cytometry: Single Cells, Many Features. Cell. 2016;165:780–791. doi: 10.1016/j.cell.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- Tanner SD, Baranov VI, Ornatsky OI, Bandura DR, George TC. An introduction to mass cytometry: fundamentals and applications. Cancer Immunol Immunother. 2013;62:955–965. doi: 10.1007/s00262-013-1416-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11 10 11–33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L, Hinton GE. Visualizing High-Dimensional Data Using t-SNE. J Mach Learn Res. 2008;9:2579–2605. [Google Scholar]
- van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190:355–366. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol. 2011;11:852–863. doi: 10.1038/nri3108. [DOI] [PubMed] [Google Scholar]
- Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.