

Summary
Three-prime repair exonuclease I (TREX1) is an anti-viral enzyme that cleaves nucleic acids in the cytosol, preventing accumulation and a subsequent type-I interferon-associated inflammatory response. Autoimmune diseases, including Aicardi-Goutières syndrome (AGS) and systemic lupus erythematosus, can arise when TREX1 function is compromised. AGS is a neuroinflammatory disorder with severe and persistent intellectual and physical problems. Here, we generated a human AGS model that recapitulates disease-relevant phenotypes using pluripotent stem cells lacking TREX1. We observed abundant extrachromosomal DNA in TREX1-deficient neural cells, of which endogenous Long Interspersed Element-1 retrotransposons were a major source. TREX1-deficient neurons also exhibited increased apoptosis and formed three-dimensional cortical organoids of reduced size. TREX1-deficient astrocytes further contributed to the observed neurotoxicity through increased type-I interferon secretion. In this model, reverse transcriptase inhibitors rescued the neurotoxicity of AGS neurons and organoids, highlighting their potential utility in therapeutic regimens for AGS and related disorders.
Keywords: Disease modeling, TREX1, neuroinflammation, Aicardi-Goutières syndrome, LINE-1, Type-I IFN
eTOC Blurb
Thomas, Tejwani et al. used human pluripotent stem cells to dissect the contribution of neurons and glia to the neuroinflammatory disorder Aicardi-Goutières syndrome (AGS). They found that mutant cells accumulate retroviral-like extrachromosomal nucleic acids that trigger a neurotoxic response, and suggest that anti-retrovirals could potentially provide therapy for this disease.
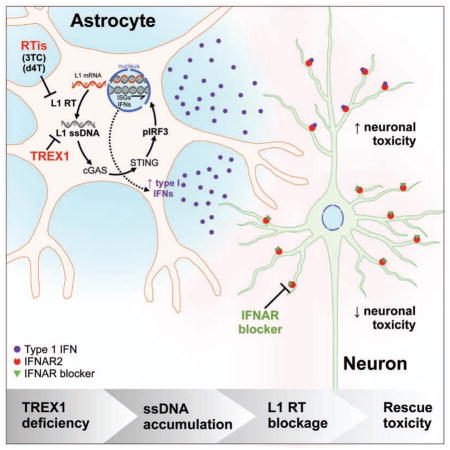
Introduction
Aicardi-Goutières syndrome (AGS) is an autosomal recessive, severe, progressive inflammatory disorder with onset in early infancy (Crow et al., 2015). Mutations in anti-viral genes related to nucleic acid processing, such as the Three-prime Repair Exonuclease 1 (TREX1), can cause the disorder (Crow and Manel, 2015). TREX1 is a DNase that breaks down both single-stranded (ss) and double-stranded (ds) DNA in a 3′ to 5′ fashion (Mazur and Perrino, 1999). TREX1 has a transmembrane domain that anchors the protein to the endoplasmic reticulum, permitting the protein to degrade DNA species in the cytosol (Richards et al., 2007). Early studies on AGS suggested that inflicted patients exhibit clinical outcomes reminiscent of congenital infection, involving an innate immune response and type-I interferon (IFN) induction (Lebon et al., 1988). The absence of pathogenic infection in AGS patients led to a hypothesis that IFN induction could be trigged by accumulation of endogenous nucleic acids, and aberrantly sensed as non-self by the innate immune system (Crow and Rehwinkel, 2009; Yang et al., 2007).
One of the more common repeated sequences in the human genome is the retrotransposon Long INterspersed Element-1 (LINE-1 or L1), which comprises ~17% of the human genome (Cordaux and Batzer, 2009). Although almost all L1 elements are evolutionarily inactive, surviving active elements exist and encode two proteins, one of which has reverse-transcription function (Cost et al., 2002). Interestingly, L1 elements are expressed and highly active in the nervous system (Coufal et al., 2009; Muotri et al., 2005; Muotri et al., 2010; Perrat et al., 2013). To combat the high activity of L1, human cells have developed several regulatory mechanisms that inhibit a specific process during L1 retrotransposition (Heras et al., 2014; Muotri et al., 2005; Muotri et al., 2010). One key protein that may be involved in the inhibition of L1 is TREX1. Loss of function of this enzyme has been suggested to lead to DNA accumulation (Stetson et al., 2008), by mechanisms that are not yet entirely understood. Recently, TREX1 has been suggested to deplete L1 ORF1p activity by altering its cellular localization. This finding, however relies entirely on the use of exogenous overexpression of TREX1 and ORF1p fusion proteins and does not consider endogenous TREX1 (Li et al., 2017).
The endogenous relationship between type-I IFN-mediated inflammation, the L1 DNA element, and TREX1 has not been adequately explored, particularly in the context of the human nervous system. While the Trex1 knockout AGS mouse model recapitulates certain key aspects of the human disease, these mice do not exhibit the neuroinflammation prominent in AGS (Gall et al., 2012). Thus, we sought to explore the role of TREX1 and L1 in the progression of neural autoinflammation using a human stem cell model. To create the stem cell model, we mutated the TREX1 gene in two locations in embryonic stem cells (ESCs) using CRISPR/Cas9 genome editing. In addition, we obtained fibroblasts from a patient with a naturally occurring homozygous mutation in TREX1 and induced pluripotency (de Silva et al., 2007). We differentiated the TREX1-deficient pluripotent cells into neural precursor cells (NPCs), neurons and astrocytes to examine DNA accumulation, toxicity, and IFN induction. We also explored the structural consequences of TREX1 deficiency using a stem cell-derived organoid model of the developing human cerebral cortex.
TREX1-deficient NPCs, neurons and astrocytes demonstrated a significant increase of intracellular DNA species, which correlated with neuronal toxicity. We show that L1 retroelements are a major source of the accumulated DNA in TREX1-deficient neural cells, and that inhibition of L1 reverse transcription leads to a reduction of extrachromosomal DNA and rescue of the associated neurotoxicity. We also determined that TREX1-deficient astrocytes express increased levels of type-I IFNs to further exacerbate the neurotoxicity in a non-cell autonomous fashion. Finally, we were able to block the astrocyte-induced toxicity with a type-I IFN receptor antagonist. Our data reveal a novel molecular and cellular mechanism to explain the pathology of AGS and reveal potential treatments for AGS by repurposing FDA-approved drugs.
Results
Generation of TREX1-deficient neural cells
To model AGS with human neural cells we developed a pluripotent cell model system with three different cell lines, each carrying a distinct TREX1 mutation (Figure 1A and 1B). For two of the cell lines we mutagenized H9 human embryonic stem cells with the CRISPR/Cas9 genome-editing system, using guide RNAs directed to the TREX1 DNA loci corresponding to the amino acids valine 63 (V63) and glutamate 83 (E83) (Mali et al., 2013). Isolated Cas9-expressing H9 ESCs showed robust nuclease activity with each guide RNA (Figures S1A and S1B). After clonal expansion of several mutated cell lines, we chose two lines with uniform frameshift (fs) mutations for further experimentation, which we refer to as V63fs and E83fs, respectively (Figures 1B and S1C). The V63fs and E83fs lines carry a homozygous single-nucleotide insertion in both alleles of the TREX1 gene, resulting in frame-shift mutations (Figure 1A and 1B) and an early stop codon at amino acid 100, rendering the TREX1 protein nonfunctional. Because there is only one coding exon in the TREX1 gene, the TREX1 RNA of the TREX1-deficient cell lines does not undergo nonsense-mediated decay, and regular TREX1 expression is maintained (Figure S1D) (Zhang et al., 1998). In addition to the V63fs and E83fs mutant lines, we selected two other H9 ESC-derived and clonally expanded lines that underwent CRISPR/Cas9 endonuclease cleavage, but repaired the DNA loci correctly and thus did not carry TREX1 mutations (Figure 1B). These wild-type TREX1 lines were named WT63 and WT83, respective of the cleavage site, and used as isogenic controls.
Figure 1. TREX-1-deficient neural cells exhibit higher levels of ssDNA in the cytosol (see also Figures S1–S3).

A, Schematic representation of the TREX1 gene showing the mutations in the derived pluripotent lines. B, DNA sequence chromatogram displaying the nucleotide changes in the TREX1 sequence in the mutant lines. A golden box denotes a nucleotide mutation. The amino acid sequence is provided in the white ribbon beneath the nucleic acid sequence. C, Schematic representation of the generation of cell lines and differentiation into neural cells. D, Phase-contrast images showing differentiation in the neural lineage. Scale bar, 200μm. E, Representative fluorescence images showing differentiation in the neural lineage. Scale bar, 20μm. F, Representative images of ssDNA immunofluorescence in NPCs, neurons and astrocytes. Scale bar, 20μm. G, Quantification of ssDNA puncta in the cytosol of NPCs, neurons and astrocytes. All ssDNA images were blindly acquired and ssDNA puncta were blindly quantified. The numbers of puncta per cell for each line were averaged and graphed according to genotype (n = 3 cell lines). Each mutant line was chronically treated with nucleoside analogue reverse-transcriptase inhibitors 3TC and d4T (RTi), then subjected to imaging, and the results were graphed (n = 3 cell lines). The V63fs line was also chronically treated with the non-nucleoside analogue reverse-transcriptase inhibitor NVP (n = 1 cell line). The presented values are the means ± SD. Student’s t-tests with Welch’s correction were performed to compare genotypes. *P<0.05; **P<0.01. H, Normalized TREX1 expression across differentiation using RNA sequencing data from control cell lines (n = 3 cell lines). The presented values are the means ± SEM. I, Fluorescence images showing subcellular localization of TREX1 in control NPCs. Scale bar, 20μm. J, Western blot on astrocyte protein lysates measuring levels of TREX1.
Additionally, we generated a third TREX1-deficient cell line by inducing pluripotency in fibroblasts derived from an AGS patient homozygous for the stereotypical V201D mutation in TREX1 (Figure 1B) (Crow et al., 2006a). In summary, we obtained three TREX1-deficient lines, V63fs, E83fs, and V201D, and three control lines WT63, WT83 and naïve H9 cells. Exome sequencing of genomic DNA of these lines did not identify any off-target mutations created by the Cas9 endonuclease (Tables S1 and S2).
Once we confirmed the differentiation potential of the pluripotent stem cell lines (PSCs) (Figures 1C, 1D, 1E, S1E–H and S2A–E), we differentiated these lines into NPCs, neurons and astrocytes using established protocols (Figures S2A–S2E) (Chailangkarn et al., 2016; Griesi-Oliveira et al., 2015; Marchetto et al., 2010). The resultant NPCs, neurons and astrocytes derived from each cell line expressed the expected specific markers (Figures S2F–S2K). Thus, we concluded that the mutagenized and control neural lines were suitable for assessing neuroinflammation in the context of TREX1 deficiency.
TREX1 degrades reverse-transcribed extranuclear DNA species
A fundamental role of TREX1 is to degrade DNA species in the cytosol (Mazur and Perrino, 1999; Yang et al., 2007). We therefore hypothesized that TREX1-deficient neural cells might accumulate cytosolic ssDNA. To test this hypothesis, we stained neural cells with an antibody specific to ssDNA. To ensure that the antibody sensitively, robustly and specifically detects ssDNA, we performed multiple auxiliary control experiments. In all ssDNA immunostaining experiments, the fixed and permeabilized cells were treated with RNase A to remove RNA and thus prevent any contaminant detection of this nucleotide species. We were able to conclude the antibody was sensitive to varying amounts of ssDNA when we stained NPCs transfected with increasing amounts of single-stranded oligomers (0.1 to 2μg) and found the amount of puncta correlated with the amount of ssDNA transfected (Figures S3A and S3B). Furthermore, we were assured the antibody robustly detected ssDNA puncta because transfected cells treated with the ssDNA nuclease S1 had few to no puncta (Figure S3B). We also confirmed that the ssDNA antibody was specific for ssDNA, and not dsDNA (Figure S3C). In accordance with our results and previous studies, we concluded that the antibody was well suited to detect an accumulation of ssDNA in the extranuclear space (Yang et al., 2007).
We examined ssDNA levels in TREX1-deficient and control cells. In agreement with previous studies, TREX1-deficient fibroblasts displayed higher levels of ssDNA compared to control fibroblasts (Figures S3D and S3E) (Yang et al., 2007). A similar increase was observed in TREX1-deficient PSCs (Figures S3F and S3G). We also found TREX1-deficient NPCs exhibited significantly greater numbers of ssDNA puncta per cell (Figures 1F, 1G, S3H and S3I). This increase of puncta can be restored to near-control levels by exogenously re-expressing TREX1 in TREX1-deficient NPCs (Figures S3J and S3K).
We next tested the hypothesis that increased levels of ssDNA arise due to reverse transcriptase. Accordingly, we maintained some of the TREX1-deficient cell lines with the nucleoside analog reverse-transcriptase inhibitors (RTi) Lamivudine (3TC) and Stavudine (d4T). These inhibitors are well known anti-HIV-1 drugs that have been shown to inhibit both HIV and L1 reverse transcription (Jones et al., 2008). In addition, as a control, we maintained one TREX1-deficient cell line in the presence of a different anti-HIV-1 drug called Nevirapine (NVP), which has no notable effect on L1 reverse transcription (Dai et al., 2011; Jones et al., 2008). Treatment of TREX1-deficient NPCs with 3TC and d4T, but not NVP, reduced the levels of ssDNA puncta to near-control levels (Figures 1F, 1G, S3H and S3I). We also investigated TREX1-deficient neurons and astrocytes for ssDNA and found that both exhibited significantly more ssDNA puncta per cell than their respective controls (Figures 1F, 1G, and S3L–S3P). These results suggest that in healthy neural cells, the bulk of extranuclear ssDNA species are created through reverse transcription and their accumulation is prevented by TREX1.
TREX1 mRNA expression was assessed using RNA sequencing data from multiple control cell lines, and was normalized for comparison across the different stages of neural differentiation (Figure 1H). All examined control cell types showed detectable expression of TREX1, with levels being highest in astrocytes. Subcellular localization of the TREX1 protein in NPCs was shown to be extranuclear (Figure 1I). Additionally, western blotting confirmed the presence of the TREX1 protein in control astrocyte lines, while TREX1 was absent in all mutant astrocyte lines (Figure 1J). These data confirm that TREX1 is normally present in the cytosol of neural cells to degrade extranuclear DNA and prevent aberrant accumulation.
TREX1-deficient neural cells have increased L1 species in the extrachromosomal fraction of cell lysates
We further analyzed the accumulated DNA species by deep sequencing and qPCR. We extracted and sequenced the NPC extrachromosomal DNA of a TREX1-deficient line, its control isogenic pair, and the TREX1-deficient line treated with 3TC and d4T (Figure 2A). The DNA was extracted using a modified Hirt protocol (Arad, 1998). As expected, the sequencing results of the extrachromosomal extraction revealed mitochondrial DNA to be the most abundant nucleic acid species (Figure S4A), with repetitive elements also corresponding to a substantial proportion. When comparing the retroelements of the mutant line (V63fs) line and its isogenic control (WT63), we observed the TREX1-deficient line to contain an increase of L1 elements (Figures 2B and S4C). More specifically, the most active subfamily of L1, human-specific L1 (L1Hs), was approximately 70% more abundant in the TREX1-deficient line (Figures 2C, S4D and S4E). The sequencing results also revealed low overall levels of HERVs, little to no change in relative amounts of Alu DNA, and that 3TC and d4T treatment reduced L1 and L1Hs copies to near-control levels. Accordingly, these results suggest that the primary source of DNA accumulation in TREX1-deficient cells can be attributed to cis-L1 reverse transcription (Figures 2B, 2C, S4C and S4F). The relative presence of L1s compared to other transposable elements, such as HERVs and Alu, in the Hirt DNA far exceeded its genomic representation (Figure S4B) (Cordaux and Batzer, 2009; Xing et al., 2013), confirming the observed results were a true accumulation of L1 ssDNA and not an artifact of genomic DNA contamination. These sequencing results were corroborated by qPCR, using a distinctly different extraction of NPC extrachromosomal DNA (Figures 2D, S4G, S4H, and Table S3).
Figure 2. TREX-1-deficient NPCs exhibit higher levels of L1 ssDNA in the extrachromosomal fraction (see also Figure S4).

A, Schematic protocol to extract extrachromosomal ssDNA for deep sequencing and qPCR. B, Relative quantity of LINE species in the extrachromosomal fraction, as determined by deep sequencing. C, Relative quantity of the youngest subfamily L1Hs in the extrachromosomal fraction, as determined by deep sequencing. D, L1 copies in the extrachromosomal fraction were quantified with three different primers corresponding to different regions of L1 and then graphed. L1 copies were acquired in duplicate and normalized to the cell number at the time of extrachromosomal DNA extraction. The L1 copy numbers in each line were averaged and graphed according to genotype (n = 6; 3 cell lines, 2 individual Hirt extractions each). Each mutant line was chronically treated with RTi (3TC and d4T), and then subjected to quantification in duplicate, and the results were graphed (n = 6; 3 cell lines, 2 individual Hirt extractions each). The V63fs line was chronically treated with NVP, then subjected to quantification, and the results graphed (n = 2; 1 cell line, 2 individual Hirt extractions). The presented values are the means ± SD. Student’s t-tests with Welch’s correction were performed to compare genotypes. E, Fluorescence images showing L1 ORF1p and L1 ORF2p in the cytosol of TREX1-deficient NPCs. Scale bar, 20μm. F, Representative images of ssDNA immunofluorescence in TREX1-deficient NPCs with or without shLINE1 transfection. Scale bar, 20μm. G, Quantification of ssDNA puncta in TREX1-deficient NPCs with or without shLINE1. The presented values are the means ± SEM (V63fs, n = 75 cells) (shLINE1, n = 25 cells). H, Schematic of retrotransposition assay. NPCs are transfected with p99-GFP-LRE3-mCherry construct and analyzed by FACS 9 days post-transfection. I, Schematic map of p99-GFP-LRE-mCherry construct. J, Quantification of NPCs with de novo retrotransposition events (eGFP-positive cells) normalized by live cell number and transfection efficiency (n = 3; n = separate transfections). *P<0.05; **P<0.01.
Previous reports have demonstrated that L1 elements are expressed and highly active in the nervous system (Coufal et al., 2009; Muotri et al., 2005; Muotri et al., 2010; Perrat et al., 2013), including hESC-derived post-mitotic neurons (Macia et al., 2017). We were able to validate that L1 expression is relatively high in TREX1-deficient NPCs as well. We found L1 mRNA levels were similar in wild-type and TREX1-deficient NPCs and neurons but elevated in TREX1-deficient astrocytes compared to their wild-type counterparts (Figures S4J–S4L). In addition, both L1 ORF1p and ORF2p were highly abundant in NPCs (Figure 2E). The foci containing both L1 ORF1p and ORF2p suggest functional L1 ribonucleoproteins, crucial intermediates in the retrotransposition cycle (Doucet et al., 2010).
We sought to explore whether modulation of L1 expression would result in alteration of the observed level of ssDNA in TREX1-deficient NPCs. Accordingly, we lowered expression of L1 using a previously validated short-hairpin RNA (shRNA) (Philippe et al., 2016). We found that reduction of L1 RNA with shRNA significantly reduced ssDNA in TREX1-deficient NPCs (Figures 2F, 2G and S4I). Additionally, we examined retrotransposition levels in TREX1-deficient NPCs using an engineered L1 retrotransposition reporter construct (Figures 2H and 2I). We discovered that de novo retrotransposition was higher in TREX1-deficient NPCs compared to their isogenic wild-type control, corroborating a systematic loss of retroelement regulation (Figures 2J and S4M).
Reverse-transcriptase activity increases toxicity in TREX1-deficient neurons, but not NPCs or astrocytes
We observed an increased level of cell sickness when we differentiated the TREX1-deficient NPCs and purified the resultant neurons, not apparent in the control lines (Figures 1E, 3A and S2H). Furthermore, we noticed a significant downregulation of canonical neuronal markers in purified TREX1-deficient neuron lines compared to control lines (Figures 3B and S2I). Based on these observations, we hypothesized that TREX1-deficient neurons were experiencing an increase of toxicity.
Figure 3. TREX1 deficiency promotes neurotoxicity (see also Figure S5).

A, Schematic protocol of neuronal purification by FACS. B, Expression of neuronal markers determined by qPCR. The presented values are the means ± SD (n = 3 cell lines). Expression was normalized to zero, and B2M and HPRT1 were used as dual internal references. C–F, Representative images of CC3 and TUNEL staining, along with the corresponding graphs. CC3 and TUNEL images of purified neurons were acquired, and the percentages of apoptotic cells were calculated, averaged, and graphed according to genotype (n = 3 cell lines). Each mutant line was chronically treated with RTi (3TC and d4T), and then subjected to imaging, and the results were graphed (n = 3 cell lines). The V63fs line was chronically treated with NVP, and then subjected to imaging, and the results were graphed. White arrowheads point out CC3 or TUNEL-positive cells. Scale bar, 20μm. The presented values are the means ± SD. G, Representative images of CC3 immunofluorescence in 2 week-old TREX1-deficient neurons with or without shLINE1 transduction. Scale bar, 20μm. H, Quantification of percentages of CC3-positive TREX1-deficient neurons with or without shLINE1. The presented values are the means ± SEM. Student’s t-tests were performed to compare genotypes. *P<0.05; **P<0.01.
We assessed toxicity in purified neuron lines and discovered significantly greater percentages of cleaved caspase-3 (CC3) (Figures 3C, 3D and S5A) and TUNEL-positive cells in the TREX1-deficient neuronal lines (Figures 3E, 3F and S5B), indicating significantly greater neurotoxicity compared to control lines, but this toxicity was not seen in other cell types (Figures S5C–F). This result corresponds with the unique observation of sickness in purified TREX1-deficient neuronal lines and suggests that neurons, but not other cell types, are particularly susceptible to the loss of TREX1, possibly due to high ssDNA density in neurons (Figure S3P).
To investigate if the toxicity of TREX1-deficient neurons was related to accumulated reverse-transcribed DNA species, we differentiated NPCs and purified the resultant neurons in the presence of 3TC and d4T. TREX1-deficient neuronal lines treated with 3TC and d4T appeared healthier, with more neurites and less cell body clumping compared to untreated TREX1-deficient neuron lines (Figure S2H). Furthermore, CC3 and TUNEL assays revealed 3TC and d4T lowered the toxicity to near-control levels. Reverse-transcriptase inhibitor NVP, which has no effect on L1, did not rescue the neurotoxicity (Figures 3C–3F, S5A and S5B), suggesting that the rescue observed with 3TC and d4T was specific to L1 reverse-transcriptase activity. 3TC and d4T treatment also restored the level of neuronal marker expression in the TREX1-deficient neurons (Figures 3B and S2I), but had no significant effect on the toxicity of other TREX1-deficient cell types, likely because the intrinsic cytotoxicity levels were very low (Figures S5E and S5F). Furthermore, shRNA knockdown of L1 expression in TREX1-deficient neurons also significantly reduced toxicity as detected by CC3 (Figures 3G and 3H). Thus, we suggest that reduction of L1 levels or its reverse-transcriptase activity ameliorates the toxicity associated with TREX1 deficiency.
TREX1-deficient derived cortical organoids demonstrate a microcephaly-like phenotype
AGS is highly associated with profound microcephaly (Lanzi et al., 2003; Rice et al., 2012). Based on documented clinical data of microcephaly and the observed increases in neuronal toxicity in TREX1-deficient neurons in our study, we sought to explore cytoarchitectural alterations resulting from TREX1 deficiency using cerebral organoids with a cortical fate. Stem cell-derived cortical organoids are self-assembled spheres with the internal cytoarchitecture reminiscent of a laminated human neocortex and transcriptionally equivalent to a mid-fetal prenatal human brain, and have been previously used as a tool to study microcephaly (Cugola et al., 2016).
We generated organoids through sequential media changes containing factors to promote neural induction, NPC proliferation, and neuronal maturation and assessed their size through various stages of maturation (Figures 4A–4F and S5G–H). TREX1-deficient organoids exhibited no significant difference in diameter 24 hours after induction, however, displayed a significant reduction in diameter during the later stages of differentiation (Figures 4F and S5H). To explore the cellular changes that cause the reduction in size, we assessed proliferation and apoptosis by staining for Ki67 and CC3, respectively. TREX1-deficient organoids demonstrated a higher percentage of CC3-postive cells (Figures 4H and 4I), with organoid apoptosis occurring primarily in regions containing MAP2-positive neurons (Figure S5I), suggesting that reduction of organoid size is primarily due to neuron cell death. We also found that treatment of the TREX1-deficient organoids with 3TC and d4T rescued both size and the percentage of CC3-positive cells (Figures 4C–4I and S5H), corroborating the toxicity experimental results observed in the monolayer culture.
Figure 4. TREX1-deficient cortical organoids demonstrate a microcephaly-like reductionin size (see also Figure S5).

A, Schematic protocol of cortical organoid generation. B, Images of immunostaining showing presence of neural cells within control 3D organoids. Scale bar, 200μm. C, Representative images of organoids used to measure diameter over stages of maturation. Scale bar, 200μm. D, Histogram showing distribution of organoids for each genotype/treatment after neuronal maturation. E, Scatter plot showing quantification of organoid diameter (WT, n = 84 organoids) (ΔTREX1, n = 282 organoids) (RTi, n = 112 organoids) (NVP, n = 161 organoids). The error bars shown represent SEM. F, Line graph quantifying organoid diameter 24 hours after formation (24 h), after neural induction/proliferation (NI/P, day 15), and after neuronal maturation (NM, day 35). The error bars shown represent SEM. G, Representative immunofluorescence images of organoids used to examine proliferation using Ki67. H–I, Representative images of CC3 staining of organoid, along with the corresponding graph. CC3 images of organoids were acquired, and the percentages of apoptotic cells were calculated, averaged, and graphed according to genotype (n = 5 organoids). The V63fs mutant organoids were chronically treated with RTi, and then subjected to imaging, and the results were graphed (n = 5 organoids). The V63fs organoids were chronically treated with NVP, and then subjected to imaging, and the results were graphed (n = 5 organoids). The presented values are the means ± SD. Student’s t-tests with Welch’s correction were performed to compare genotypes. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
Reverse-transcriptase activity in TREX1-deficient astrocytes correlates with type-I IFN and the IFN stimulated gene response
Astrocytes are known to modulate immune function in the nervous system (Farina et al., 2007). Furthermore, it is well established that DNA species can induce a type-I IFN response (Stetson and Medzhitov, 2006). Considering the high level of extranuclear ssDNA, we hypothesized that TREX1-deficent astrocytes might produce type-I IFNs in response to reverse-transcribed DNA species. Accordingly, we examined Interferon-β (IFNb) and Interferon-α (IFNa) mRNA levels in astrocytes by targeting IFNb and several IFNa genes, including IFNa 1, 2, 4, 6, 13, and 21. Compared to control astrocyte lines, TREX1-deficient lines expressed higher levels of IFNb and significantly higher amounts of IFNa13 (Figure 5A). Treatment with 3TC and d4T reduced expression to near control levels, suggesting that the accumulation of reverse-transcribed DNA species can stimulate type-I IFN expression in astrocytes. Transfection of ssDNA into wild-type astrocytes similarly increased IFNa13 and IFNb expression (Figure 5B). Thus, we concluded that accumulation of ssDNA in the cytosol was sufficient to produce a type-I IFN response.
Figure 5. TREX1-deficient astrocytes upregulate interferon and interferon-stimulated genes (see also Figure S6).

A, Expression of IFNα13 and IFNβ1 determined by qPCR (n = 3 cell lines, RTi, n = 1 cell line, NVP, n = 1 cell line). B, Expression of IFNα13 and IFNβ1 determined by qPCR in WT astrocytes transfected with ssDNA (n = 3 replicates). C, Western blot on astrocyte protein lysates measuring levels of phosphorylated IRF3, IRF3, and β-Actin. D, Quantification of phosphorylated IRF3/total IRF3 from western blot using protein lysates from isogenic cell lines and their treatments, normalized to V63fs (n = 4; 2 cell lines, with 2 technical replicates; RTi, n = 3, 2 cell lines with technical replicate; NVP, n = 3; 2 cell lines with technical replicate). E, Expression of ISGs determined by qPCR. The presented values are the means ± standard deviation (n = 3 cell lines; RTi, n = 3 cell lines; NVP, n = 1 cell lines). Student’s t-tests were performed to compare genotypes. Expression was normalized to zero, and B2M and HPRT1 were used as dual internal references. *P<0.05; **P<0.01; ***P<0.001.
The cGAS/STING pathway can sense cytosolic DNA and transduce a signal to stimulate inflammation (Ishikawa et al., 2009). It has been reported that stimulation of cGAS and immunogenicity is enhanced when unpaired guanosine nucleotides in Y-form secondary structures of ssDNA are detected (Herzner et al., 2015). Because reverse-transcribed L1Hs ssDNA is predicted to produce these immunogenic secondary structures (Figure S6A), we investigated if a similar response mechanism may have a role in inducing the astrocytic type-I IFN. We discovered that TREX1-deficient astrocytes exhibited a higher ratio of phosphorylated IRF3 to total IRF3 compared to their respective isogenic controls (Figures 5C, 5D, S6B and S6C). This activation was reduced to control levels in astrocytes treated with 3TC and d4T, but not with NVP treatment, suggesting that reverse-transcribed species are involved. In conjunction with the IFN gene expression data, we suggest the cGAS/STING DNA-sensing pathway is likely to be activated in TREX1-deficient astrocytes, further activating an immunogenic response.
It has been shown that TREX1-deficient cells undergo a STING-dependent IFN stimulated gene (ISG) upregulation (Hasan et al., 2013), corroborating expression profiles in patient peripheral blood mononuclear cells (Rice et al., 2013). We sought to examine whether TREX1-deficient astrocytes also exhibit ISG upregulation. Accordingly, we examined six well-characterized interferon stimulated genes (ISGs) and found that the expression of each gene was upregulated in TREX1-deficient astrocytes (Figure 5E). Treatment with 3TC and d4T, but not NVP, significantly reduced the expression of three of these six ISGs. From these data, we suggest that IRF3 activation by extranuclear DNA induces both type-I IFN and ISG upregulation in TREX1-deficient astrocytes.
Reverse-transcriptase activity in TREX1-deficient astrocytes induces secretion of neurotoxic type-I IFNs
Astrocytic secretion of IFN-α has been shown to have a toxic effect in the nervous system (Akwa et al., 1998). We thus investigated if TREX1-deficient astrocyte lines might also confer a toxic effect on neurons by overlaying astrocyte-conditioned media (ACM) onto healthy neurons and organoids (Figure 6A). Toxicity assays on the neurons demonstrated that ACM derived from TREX1-deficient astrocytes confer significantly greater toxicity (Figures 6B–6E, S6D and S6E). Furthermore, treatment with 3TC and d4T on the TREX1-deficient astrocyte lines reduced the toxicity of the conditioned media to near-control levels. Cortical organoids treated with ACM derived from TREX1-deficient astrocytes resulted in a significant reduction of organoid size and increased percentage of CC3-positive cells compared to organoids treated with control ACM (Figures 6F–I).
Figure 6. TREX1 deficiency in astrocytes exacerbates neurotoxicity through type-I interferons (see also Figure S6).

A, Schematic protocol of astrocyte-conditioned media experiments (ACM) on H9 purified neurons. B–E, Representative images of CC3 and TUNEL images of H9 purified neurons overlaid with ACM of different genotypes, along with corresponding graphs (n = 3 cell lines). Each mutant line was chronically treated with RTi (3TC and d4T), and RTi was removed during media conditioning (n = 3 cell lines). The V63fs line was chronically treated with NVP, and NVP was removed during media conditioning (n = 1 cell lines). Scale bar, 20μm. The presented values are the means ± standard deviation. F, Representative images of control organoids treated with conditioned media from control or V63fs mutant astrocytes. G, Scatter plot showing quantification of organoid diameter after 14 days of treatment with ACM. Each point represents one organoid (WT, n = 121 organoids, ΔTREX1, n = 145 organoids). The error bars shown represent SEM. Scale bar, 200μm. H–I, Representative images of CC3 staining of organoid after treatment with V63fs mutant ACM for 14 days, along with the corresponding graph. CC3 images of organoids were acquired, and the percentages of apoptotic cells were calculated, averaged, and graphed (WT, n = 5 organoids, ΔTREX1, n = 6 organoids). Total of 35 days of organoid differentiation. The presented values are the means ± SD. Student’s t-tests were performed to compare treatments. Scale bar, 200μm. J–K, Representative images of CC3 and images of H9 purified neurons overlaid with ACM of different genotypes with or without the addition of IFNAR2 neutralizing antibody, along with corresponding graphs (n = 3 cell lines, RTi, n = 1 cell line, NVP, n = 1 cell line). The V63fs mutant line was chronically treated with RTi (3TC and d4T), and RTi was removed during media conditioning (n = 1 cell line). The V63fs line was chronically treated with NVP, and NVP was removed during media conditioning (n = 1 cell line). Scale bar, 20μm. The presented values are the means ± SD. Student’s t-tests with Welch’s correction were performed to compare genotypes. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
These results suggest that L1 reverse-transcriptase activity induces TREX1-deficient astrocytes to produce a more toxic environment for neurons via secretion of factors (e.g. type-I IFNs). To test if type-I IFNs are modulating neurotoxicity, we repeated the ACM toxicity assay, but in the presence of a neutralizing antibody (nAb) specific to the type-I IFN alpha/beta receptor chain 2 (IFNAR2) required for type-I IFN signaling. Adding IFNAR2 nAb to the conditioned media derived from TREX1-deficient astrocyte lines significantly rescued its neurotoxicity (Figures 6J, 6K, and S6F). Therefore, we propose that the secretion of type-I IFNs by astrocytes is sufficient to generate neurotoxicity via neuroinflammation in TREX1-deficient neural systems.
Discussion
We have generated a neurodevelopmental model to study the mechanisms of AGS pathology by eliminating the function of TREX1 in human PSCs. Differentiation and isolation into neurons and astrocytes enabled us to dissect out the role of different neural cell types and their interplay in an inflammatory setting, generating new knowledge about type-I IFN-related neuroinflammation.
TREX1-deficient neural cells exhibited increased amounts of extranuclear DNA species,, which could be reduced by lowering L1 mRNA expression or inhibiting reverse-transcriptase activity. Because the L1 element encodes a reverse transcriptase, these results are consistent with the hypothesis that much of the extranuclear DNA arises primarily by reverse transcription of L1 nucleic acid species (Crow et al., 2006b; Crow and Manel, 2015; Stetson et al., 2008). It is possible that other reverse-transcribed species may contribute to the accumulated ssDNA, however, our results suggest that contribution from other elements, such as HERVs, is minimal.
The canonical model of L1 retrotransposition is by target-primed reverse transcription in the nucleus. Our data in neural cells, however, suggests that reverse-transcribed DNA species can also exist in the cytosol. Accordingly, we propose two possible mechanisms for the cytosolic accumulation of L1 DNA. In one, L1 RNA and L1 proteins exist at high levels in the cytosol, and thus reverse transcription occurs without a standard DNA template and results in elevated ssDNA elements in the cytosol. In the alternative mechanism, the L1 RNA and L1 proteins move into the nucleus, as is typical for retrotransposition. Then, as L1 begins to reverse transcribe using chromosomal DNA as its template, the resultant ssDNA does not integrate, but instead cleaves from the chromosomal DNA and exits the nucleus into the cytosol by a currently unrecognized process. These proposed mechanisms may explain such behavior of the L1 element, thus accounting for an increase of extranuclear DNA in TREX1-deficient cells. These models are not mutually exclusive, and further investigation is necessary to fully understand the molecular basis of L1 retrotransposition in human neural cells.
The results of the toxicity experiments suggest that extranuclear ssDNA may cause morbidity in neurons, but not in astrocytes or NPCs, despite all cell types exhibiting high levels of extranuclear ssDNA. The specificity to neurons is intriguing, and may be explained by the higher density of ssDNA due to the extremely small cell body size in mature neurons. Alternatively, it is well documented that neurons are often more sensitive than their glial counterparts to toxicity associated with spurious accumulations of various endogenous biomolecules, such as Aβ in Alzheimer’s and α-synuclein in Parkinson’s (Bloom, 2014; Wong and Krainc, 2017). Thus, it is not entirely surprising that anomalous accumulation of ssDNA species also results in neuronal specific toxicity.
Although the TREX1-deficient astrocytes did not display toxicity, they exhibited an increase in type-I IFNs and ISGs. Moreover, astrocyte-derived secretions conferred a toxicity phenotype on neurons, which could be abrogated by IFN-receptor blockade. Considering the toxic effects of TREX1 deficiency in neurons, in combination with the exacerbation of cell death induced by mutant astrocyte secretions, a TREX1-deficient nervous system would likely exhibit extreme neurotoxicity. Further experiments are necessary to determine if this combination is synergistic or merely additive.
The observed astrocytic response is consistent with the human type I interferonopathy AGS, in which patients with mutations in TREX1 invariably manifest increased levels of IFN activity in the cerebrospinal fluid in the context of a severe microcephaly and profound development delay (Crow et al., 2006a; Crow et al., 2000; Lebon et al., 1988). Additionally, using cortical organoids, we were able to not only validate our findings from monolayer cultures, but also translate these molecular and cellular abnormalities into structural consequences in a self-organizing structure reminiscent of a developing cortex, revealing the microcephaly-like size deficiencies are likely due to increased apoptosis during neuronal maturation.
Our conclusions rely upon the fact that accumulation of reverse-transcribed DNA species results in an inflammatory phenotype that can be reversed by inhibiting reverse transcription with 3TC and d4T, however, these drugs have been reported to provide an intrinsic anti-inflammatory effect (Fowler et al., 2014). We demonstrated that the cGAS/STING pathway, which is known to sense accumulation of nucleic acids and induce an inflammation response, was activated. As of yet, any intrinsic anti-inflammatory activity of 3TC or d4T has not been associated with the cGAS/STING pathway. Thus, while we cannot conclude that 3TC and d4T had no intrinsic anti-inflammatory effect, we do suggest that the accumulation of ssDNA species was the trigger of inflammation and prevention of this accumulation provides the primary anti-inflammatory response. Any intrinsic anti-inflammatory effect provided by 3TC and d4T, even if minor, would be a benefit in a therapeutic regime.
While our results pertain to a particular AGS genotype involving the TREX1 gene, it bears mentioning that our results may be extrapolated to other AGS genotypes. In addition to loss of function of TREX1, AGS can arise from mutations in RNASEH, SAMHD1, ADAR, or IFIH1 (Crow and Manel, 2015; Livingston and Crow, 2016). Each of these proteins has a function related to either nucleic acid processing or sensing, and are thus at least responsible in part for clearance of nucleic acids. Furthermore, a few of these proteins have been discovered to be able to suppress retroelement activity (Goodier, 2016; Zhao et al., 2013). It is possible that these other AGS genotypes might be triggered from overabundance of retroelement species as well, but further research is necessary to establish a causal link.
In summary, we have created a human stem cell model of neuroinflammation that recapitulates many clinical features observed in neuroinflammatory disease related to TREX1 deficiency, such as in AGS. Our results demonstrate that human neurons are susceptible to the accumulation of endogenous reverse-transcribed L1 DNA species and to exogenous type-I IFNs secreted from astrocytes, explaining the molecular underpinnings of AGS. By targeting various steps along this pathway, we were able to rescue many deleterious molecular, cellular, and structural phenotypes induced when the ability to degrade extranuclear species is compromised. This system provides a platform to further investigate the pathology of neuroinflammation, and for drug discovery. Furthermore, these findings suggest that reverse-transcriptase inhibition and neutralization of type-I IFNs represent potential therapeutic strategies to treat patients suffering from AGS and other forms of IFN-driven neuroinflammation.
STAR★Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-Nanog | R&D Systems | AF1997 |
| Rabbit anti-Lin28 | Abcam | ab46020 |
| Rabbit anti-Nestin | Millipore | AB5922 |
| Rabbit anti-Nestin | Millipore | ABD69 |
| Mouse anti-Sox2 | Abcam | ab75485 |
| Rabbit anti-Sox2 | Cell Signaling | 2748S |
| Chicken anti-GFAP | Abcam | ab4674 |
| Rabbit anti-GFAP | Dako | Z033429 |
| Mouse IgM anti-ssDNA | Millipore | MAB3299 |
| Mouse anti-S100 | Abcam | ab4066 |
| Chicken anti-Map2 | Abcam | ab5392 |
| Rabbit anti-Syn1 | Millipore | AB1543 |
| Rabbit anti-TREX1 | Abcam | ab185228 |
| Mouse anti-dsDNA | Abcam | ab27156 |
| Rabbit anti-Cleaved Caspase 3 | Cell Signaling | 9664S |
| Rat anti-CTIP2 | Abcam | ab18465 |
| Rabbit anti-Ki67 | Abcam | ab15580 |
| Mouse anti-NeuN | Millipore | MAB377 |
| Mouse anti-Tuj1 | Sigma | T8578 |
| Mouse anti-LINE-1 ORF1p | Millipore | MABC1152 |
| Chicken anti-LINE-1 ORF2p | Abcam | ab106004 |
| Rabbit anti-IRF3 | Cell Signaling | 4302S |
| Rabbit anti-phospho-IRF3 | Cell Signaling | 4947S |
| Mouse anti-β-Actin | Abcam | ab8226 |
| Donkey anti-Goat IgG-Alexa Fluor 488 | ThermoFisher | A-11055 |
| Donkey anti-Rabbit IgG- Alexa Fluor 488 | ThermoFisher | R37118 |
| Donkey anti-Mouse IgG- Alexa Fluor 488 | ThermoFisher | R37114 |
| Donkey anti-Rat IgG- Alexa Fluor 488 | ThermoFisher | A-21208 |
| Goat anti-Chicken IgY- Alexa Fluor 488 | ThermoFisher | A-11039 |
| Donkey anti-Mouse IgG- Alexa Fluor 555 | ThermoFisher | A-31570 |
| Donkey anti-Rabbit IgG- Alexa Fluor 555 | ThermoFisher | A-31572 |
| Donkey anti-Mouse IgG- Alexa Fluor 647 | ThermoFisher | A-31571 |
| Donkey anti-Rabbit IgG- Alexa Fluor 647 | ThermoFisher | A-31573 |
| Goat anti-Chicken IgY- Alexa Fluor 647 | ThermoFisher | A-21449 |
| Goat anti-Mouse IgM- Alexa Fluor 488 | ThermoFisher | A-21042 |
| Goat anti-Mouse IgM- Alexa Fluor 647 | ThermoFisher | A-21238 |
| Mouse anti-CD184 APC | Fisher Scientific | BDB555976 |
| Mouse anti-CD44 PE | Fisher Scientific | BDB555479 |
| Mouse anti-CD24 FITC | Fisher Scientific | BDB555427 |
| IRDye® 680RD Donkey anti-Rabbit IgG | LI-COR | P/N 926-68073 |
| IRDye® 680RD Donkey anti-Mouse IgG | LI-COR | P/N 926-68072 |
| IRDye® 800CW Donkey anti-Rabbit IgG | LI-COR | P/N 926-32213 |
| IRDye® 800CW Donkey anti-Mouse IgG | LI-COR | P/N 926-32212 |
| Mouse anti-Human IFNAR2 nAB (MMHAR-2) | PBL Assay Science | 21385-1 |
| Bacterial and Virus Strains | ||
| DH5-α | Life Technologies | 18265017 |
| TOP10 | Life Technologies | K450001 |
| Biological Samples | ||
| AGS1 c.602T>A/V201D fibroblasts | Dr. Yanick Crow | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lamivudine (3TC) | Sigma-Aldrich | L1295-50mg |
| Stavudine (d4T) | Sigma-Aldrich | D1413-50mg |
| Nevirapine (NVP) | Toronto Research | N391275 |
| Dorsomorphin | R&D Systems | 3093 |
| Stemolecule SB431542 | StemGent | 04-0010-10 |
| Basic fibroblast growth factor (bFGF) | Life Technologies | PHG0263 |
| ROCK inhibitor (Ri) Y-27632 dihydrochloride | Tocris | 125410 |
| dsDNase | Thermo Fisher | FEREN0771 |
| PhosSTOP phosphatase inhibitor | Roche | 4906837001 |
| cOmplete ULTRA mini protease inhibitor | Roche | 5892791001 |
| Critical Commercial Assays | ||
| Click-iT TUNEL assay kit- Alexa Fluor 488 | Life Technologies | C10245 |
| TaqMan Universal Master Mix II | Life Technologies | 4440040 |
| Quantitect Reverse Transcription Kit | Qiagen | 205313 |
| Surveyor Nuclease Kit | Transgenomic | 706025 |
| DNeasy Blood and Tissue Kit | Qiagen | 69504 |
| RNeasy Plus Mini Kit | Qiagen | 74136 |
| Amaxa Rat Neuronal Stem Cell Nucleofection Kit | Lonza | VPG-1005 |
| Amaxa hESC Nucleofetion Kit 2 | Lonza | VPH-5022 |
| Deposited Data | ||
| Exome sequencing Data | This paper | Bioproject: PRJNA393747 |
| Experimental Models: Cell Lines | ||
| H9 embryonic stem cells | N/A | N/A |
| HEK293 | N/A | N/A |
| Experimental Models: Organisms/Strains | ||
| Nude mice | N/A | N/A |
| Oligonucleotides (see Table S3 for complete list) | ||
| TREX1 gRNA1 - GAGAGCTTGTCTACCACACGCGG | IDT | N/A |
| TREX1 gRNA2 - GCTCAGACCTGTGATCTCGCTGG | IDT | N/A |
| L1ORF1 probe- ATGGGGAAAAAACAGAACAGAAAAACTGGAAACTCTAAAACGCAGAGCGCCTCTCCTCCTCCAAAGGAACGCAGTTCCTC | Life Technologies | N/A |
| L1ORF2 probe- GCTCATGGGTAGGAAGAATCAATATCGTGAAAATGGCCATACTGCCCAAGGTAATTTACAGATTCAATGCCATCCCCATC | Life Technologies | N/A |
| L1ORF2-3′UTR probe- TGGAAACCATCATTCTCAGTAAACTATCGCAAGAACAAAAAACCAAACACCGCATATTCTCACTCATAGGTGGGAATTGA | Life Technologies | N/A |
| TREX1 V201D For- CAGCGAGATCACAGGTCTGAG | IDT | N/A |
| TREX1 V201D Rev- GCCAGGGATAGTCCATACAGT | IDT | N/A |
| TREX1 V63, E83 For- CTTCGGATCTTAACACTGGGC | IDT | N/A |
| TREX1 V63, E83 Rev- CCACACAGAAGGCACCATCC | IDT | N/A |
| Recombinant DNA | ||
| CMV::Cas9-2A-eGFP | Addgene | #44719 |
| pUC57 | N/A | N/A |
| p99-GFP-LRE3-Cherry | N/A | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software, Inc. | N/A |
| ImageJ | N/A | https://imagej.nij.com/ij/ |
| BWA-MEM | N/A | http://bio-bwa.sourceforge.net/ |
| SAMTOOLS | N/A | http://samtools.sourceforge.net/ |
| HTSeq | N/A | http://www-huber.embl.de/users/anders/HTSeq/doc/overview.html |
| ANNOVAR | N/A | http://annovar.openbioinformatics.org/en/latest/ |
| Bowtie | N/A | http://bowtie-bio.sourceforge.net/index.shtml |
| CRISPR Design Tool | N/A | http://www.genome-engineering.org/crispr/ |
CONTACT FOR REAGENTS AND RESOURCE SHARING DETAILS
Requests should be addressed to and will be fulfilled by Corresponding Contact Alysson R. Muotri (muotri@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
PSC generation
Patient characterization: This AGS male patient was born from consanguineous parents with no relevant family history. He was considered to demonstrate abnormal neurological behavior from the time of birth and was identified as showing intracranial calcification and white matter disease through brain imaging at the age of 1 month. At the age of 9 years, he was profoundly developmentally delayed. He was identified as carrying a homozygous c.602T>A/p.V201D mutation in TREX1. Skin fibroblasts from the patient were reprogrammed using episomal Yamanaka factors as described previously (Yu et al., 2009). Cells transfected with reprogramming transgene-containing plasmids were plated onto mouse embryonic fibroblasts and iPS clones were isolated upon emergence onto feeder-free matrigel-coated plates. Five isolated clones were selected and expanded. Three clones were used for further differentiation and experimentation. This study involving human fibroblasts was approved by the Leeds (East) Research Ethics Committee (reference number 10/H1307/132). Additionally, female H9 embryonic stem cells were obtained for generation of isogenic PSC lines.
METHOD DETAILS
Composition of tissue culture media
Pluripotent stem cell medium (MT): mTeSR1 (Stem Cell Technologies). Differentiation medium for embryoid Bodies (N2): Dulbecco’s Modified Eagle’s Medium/Ham’s F12 (DMEM/F12 50/50; Corning Cellgro) with 1× HEPES, 1× penicillin-streptomycin, Glutamax (Life Technologies), and N2 NeuroPlex (Gemini Bio-products), supplemented with 1μM dorsomorphin (Tocris) and 10μM Stemolecule SB431542 (StemGent). Neural progenitor cell medium (NGF): DMEM/F12 50/50 with 1× HEPES, 1× penicillin-streptomycin, Glutamax (Life Technologies), N2 NeuroPlex (Gemini Bio-products), Gem21 NeuroPlex (Gemini Bio-products), supplemented with 20ng/mL basic fibroblast growth factor (bFGF; Life Technologies). Neuronal medium (NG): DMEM/F12 50/50 with 1x HEPES, 1x penicillin-streptomycin, Glutamax (Life Technologies), N2 NeuroPlex (Gemini Bio-products), Gem21 NeuroPlex (Gemini Bio-products); cAMP, GDNF, and BDNF were added to the neuronal medium for purified neurons. Astrocytes were cultured in Astrocyte Growth Medium (Lonza). For treatment with reverse-transcriptase inhibitors 1μM Stavudine (D4T), 10μM Lamivudine (3TC) (Sigma-Aldrich), and 400nM Nevirapine (NVP) were used. Astrocyte-conditioned media were generated by overlaying Neurobasal with B27 and 5% FBS over astrocytes for 48 hours.
Maintenance of iPSC and hESC cultures
Reprogrammed iPSCs, H9 ESCs, and mutagenized H9 ESCs were propagated in mTeSR and passaged manually as small colonies on Matrigel (BD Biosciences)-coated plates.
Mutagenesis of H9 ESCs with CRISPR/Cas9
The CMV::Cas9-2A-eGFP plasmid was purchased from Addgene (#44719). The guide RNA (gRNA) sequences correspond to two loci of the TREX1 gene. The gRNA vector was generated according to the gRNA synthesis protocol (Mali et al., 2013) using DNA Strings (Life Technologies) cloned into the pUC57 plasmid. The guide RNA sequences are below:
gRNA1 – GAGAGCTTGTCTACCACACGCGG
gRNA2 – GCTCAGACCTGTGATCTCGCTGG
To transfect the H9 ESCs, the cells were lifted off the plate with accutase (Stem Cell Technologies) and passed through a 40μM nylon mesh (BD Biosciences) to ensure that they existed as single cells. Using the hESC Kit 2 and the B16 program of Amaxa Nucleofector (Lonza), 1.5 million cells were transfected with 7μg of the CMV::Cas9-2A-eGFP vector and 3μg of the U6::gRNA vector. Transfected cells were plated with mTeSR and 5μM Rock Inhibitor (Ri) (Tocris) and incubated for 48 hours before FAC sorting. eGFP-positive cells were collected using a BD Influx cell sorter then plated communally and allowed to recover for 48 hours with 5μM Ri. After 48 hours, 40,000 cells were seeded individually onto 10 cm plates with 5μM Ri and allowed to grow into colonies (3–4 weeks). Genomic DNA from the unused transfected cells was assayed to determine endonuclease efficiency using the Surveyor Nuclease Kit (Transgenomic). Isolated colonies were selected and expanded, and genomic DNA was extracted for sequencing with the DNeasy Blood and Tissue Kit (Qiagen). To ensure that the mutations were stable, RNA was collected from early- and late-passage ESCs and NPCs and reverse transcribed into cDNA. The cDNA was TOPO cloned, and 6 clones from each passage were sequenced.
Primers for V63 and E83 locus sequencing and Surveyor Nuclease:
For – CTTCGGATCTTAACACTGGGC
Rev – CCACACAGAAGGCACCATCC
Primers for V201 locus sequencing:
For – CAGCGAGATCACAGGTCTGAG
Rev - GCCAGGGATAGTCCATACAGT
Exome sequencing data analysis
Genomic DNA was extracted from passage 42 ESCs from the H9 line and passage 3 ESCs from the WT63, WT83, V63fs, and E83fs lines with the DNeasy Blood & Tissue Kit (Qiagen). The exome DNA sequencing was outsourced and was performed by the UCSD IGM Genomics Center. Raw exome data were filtered to recover high-quality (Phred-scaled quality score higher than 22) sequencing reads using IlluQC software (Patel and Jain, 2012). High-quality reads for each individual sequenced cell line were aligned to the human reference genome (build Hg19) using BWA-MEM software (Li and Durbin, 2009) with default parameters. Unrelated alignments to the human exome were then filtered out of the analysis. Applying SAMTOOLS software (Li et al., 2009) over the valid alignments, duplicated reads as well as redundant alignments were removed to produce sorted alignment files for each sequenced exome library. We then applied FreeBayes over the sorted alignments to perform genomic InDel detection for each H9 control and H9-induced CRISPR/Cas9 mutations. For InDel detection, the alignments for the sequenced cell lines were restricted to a minimum of 50 non-repetitive reads of coverage. All InDel identified mutations were considered homozygous when a minimum of 95% of covered reads showed the same mutation or heterozygous when a minimum of 40% of covered reads showed the same mutation. Only those mutations that were not shared by the H9-induced CRISPR/Cas9-mutated cell lines and the H9 control were considered valid variations between the H9 control and mutated cell lines. We used ANNOVAR (Wang et al., 2010) and a reference transcriptome annotation (UCSC Hg19) of the human genome (build Hg19). To detect off-target genomic mutations caused by CRISPR/Cas9 in the transformed H9 cell lines, gDNA reads were aligned against human reference genome (build Hg19) with Bowtie software (Langmead et al., 2009), allowing up to 13 mismatches but requiring the GG dinucleotide of the PAM motif. A list of in silico genomic off-target sites detected by the CRISPR Design Tool (http://www.genome-engineering.org/) (Hsu and Zhang, 2013) was compiled. The sequences of the predicted off-target sites were examined in the exome data.
The exome sequencing was used to confirm the mutagenesis of the V63fs and E83fs lines. All the reads, over one hundred total, of the exome data across the mutated loci of both V63fs and E83fs indicated single-nucleotide insertions were present. Thus, V63fs and E83fs lines each contained a pure population of cells with a homozygous frame-shift mutation.
The exome data was used to check off-target lesions predicted by the CRISPR design website (Hsu et al., 2013; Hsu and Zhang, 2013). None of the predicted off-target sites were mutagenized in any of the clonally derived lines (Table S1). An unbiased approach was used to identify all insertions and deletions (InDels), including additional potential off-target lesions, in the gDNA of each line. Comparing the V63fs, E83fs, WT63, and WT83 exomes against the parental H9 exome, there are between 13 and 18 homozygous InDels in each line that would lead to frame-shift mutations (Table S2). We examined the sequence of each InDel loci and found that none of the sequences of these homozygous InDels loci matched the sequence of the guide RNA, except for the expected TREX1 locus in the V63fs and E83fs lines. Therefore, each nonspecific InDel already existed in subpopulations of the parental H9 hESCs and arose during clonal expansion. The off-target analysis data are consistent with previous findings (Smith et al., 2014; Veres et al., 2014). Accession codes for exome sequencing data are available under BioProject ID number PRJNA393747.
Immunocytochemistry
Unless otherwise noted, the cells were fixed in 4% paraformaldehyde (PFA, Electron Microscopy Sciences) for 20 minutes, permeabilized with 0.25% Triton X-100 for 15 minutes, blocked with 3% bovine serum albumin (BSA) (Gemini Bio) and incubated overnight at 4°C with primary antibodies diluted in 3% BSA. The following day, the cells were incubated with the secondary antibodies and then DAPI before mounting. Primary antibody dilutions were used as follows: anti-Nanog (R&D, AF1997, 1:500), anti-Lin28 (Abcam, ab46020, 1:500), anti-Nestin (Millipore, AB5922, 1:1,000; Millipore, ABD69, 1:2000), anti-Sox2 (Abcam, ab75485, 1:250; Cell Signaling, 2748S, 1:200), anti-GFAP (Abcam, ab4674, 1:2,000; Dako, Z033429, 1:2000), anti-S100 (Abcam, ab4066, 1:200), anti-Map2 (Abcam, ab5392, 1:2,000), anti-Syn1 (Millipore, AB1543, 1:500), anti-TREX1 (Abcam, ab185228, 1:250), anti-ssDNA (Millipore, MAB3299, 5μg/mL), anti-dsDNA (Abcam, ab27156, 1:1000), anti-Cleaved Caspase-3 (Cell Signaling, 9664S, 1:400), anti-CTIP2 (Abcam, ab18465, 1:500), anti-Ki67 (Abcam, ab15580, 1:1000), anti-NeuN (Millipore, MAB377, 1:500), anti-Tuj1 (Sigma, T8578, 1:1000), anti-LINE-1 ORF1p, (Millipore, MABC1152, 1:500), and anti-LINE-1 ORF2p (Abcam, ab106004, 1:500). Secondary antibodies conjugated to Alexa Fluor 488, 555, and 647 were used at a dilution of 1:1000 (Life Technologies). TUNEL assays were performed using the Click-iT TUNEL assay kit (Life Technologies). To conduct ssDNA staining, cells were fixed on ice with 4% PFA for 20 minutes, then with methanol at −20°C overnight. The next day, the cells were treated with 200μg/mL RNase (Sigma R4642) at 37°C for 4 hours. Then, the cells were blocked with 3% BSA and stained as indicated above. Images of ssDNA were blindly acquired, and ssDNA puncta were blindly quantified.
RT-qPCR analysis
RNA was obtained from cells using the RNeasy Plus Mini Kit (Qiagen). One microgram of RNA was used to generate cDNA using Qiagen’s Quantitect Reverse Transcription Kit, and 10ng of cDNA was employed in each qPCR assay using TaqMan probes and TaqMan Universal Master Mix II (Life Technologies). The reactions were performed in triplicate.
Expression of pluripotency markers
The expression of pluripotency markers in the mutagenized and control pluripotent lines was assessed by immunofluorescence and qPCR. The immunofluorescence images show the colonies robustly express the cytosolic Lin28 and nuclear Nanog. Likewise, the qPCR results show the pluripotent genes LIN28, NANOG, MYC, POU5F1, and SOX2 are expressed in all lines.
Teratoma formation and karyotyping
Teratomas were formed, extracted and stained as described previously (Marchetto et al., 2010). Briefly, when 1×106 cells were injected subcutaneously into anesthetized nude mice, each pluripotent line was able to generate teratomas containing cells from the three germ layers, as determined by hematoxylin and eosin staining on teratomas extracted two months following PSC injection. Karyotyping was outsourced and performed by Children’s Hospital Los Angeles. The karyotype of each cell line was normal.
Differentiation of pluripotent cells into NPCs, neurons and astrocytes
Pluripotent cells were differentiated into NPCs as described previously (Griesi-Oliveira et al., 2015). PSCs were grown for 7 days following passaging in MT and then replaced with N2 supplemented with 1μM dorsomorphin (Tocris) and 10μM Stemolecule SB431542 (StemGent) for 2 days. Colonies were then lifted manually using a cell lifter and allowed to grow as embryoid bodies in shaking suspension culture with EB media for 7 days. Embryoid bodies were plated onto matrigel-coated plates and cultured in NGF media for 7 days. Neuroectodermal rosettes were then manually isolated and re-plated on poly-ornithine and laminin coated plates in NGF media for 7 days prior to dissociation with accutase to yield NPC cultures. As determined by immunofluorescence, all of the lines robustly expressed the NPC markers Nestin and SOX2. Furthermore, qPCR results showed that all of the NPC lines expressed Nestin, SOX2, PAX6, SOX1, and Musashi1.
The NPCs were expanded, further differentiated into neurons, and were purified via FACS as described elsewhere (Yuan et al., 2011). In brief, the three-week-old differentiated neuronal cultures were lifted gently by accutase and accumax, fluorescently labeled for CD184, CD44, and CD24, and run through the BD Influx, collecting the CD184-negative, CD44-negative and CD24-positive cells. The purified neurons were plated onto a 96-well plate coated with poly-ornithine and laminin at a density of one hundred thousand cells per well. The sorted neurons exhibited robust expression of the neuronal markers of MAP2 and Synapsin as determined by immunofluorescence. The qPCR results show expression of MAP2, Synapsin 1, DLG4, RBFOX3, and TUBB3.
For differentiation into astrocytes, NPCs were lifted into suspension and maintained on a shaker (95 rpm) to form neurospheres for three weeks. For the first week, the spheres were grown with neuronal media. The neurospheres were overlaid with the astrocyte media for the remaining two weeks. The neurospheres were plated onto poly-ornithine- and laminin-coated plates and expanded for two to three passages before experimentation. The resulting astrocytes expressed astrocytic markers such as GFAP and S100 as determined by immunofluorescence. qPCR detected expression of GFAP, S100β, chondroitin sulfate, and vimentin.
ssDNA immunocytochemistry, image acquisition and puncta quantification
To conduct ssDNA staining, cells were fixed on ice with 4% PFA for 20 minutes, then with methanol at -20°C overnight. The next day, the cells were treated with 200 μg/mL RNase (Sigma R4642) at 37°C for 4 hours. Then, the cells were blocked with 3% BSA and incubated overnight at 4°C with the anti-ssDNA primary antibody (Millipore, MAB3299, 5μg/mL) and stage-specific cell markers for NPCs, neurons, or astrocytes, (anti-Nestin, Millipore, AB5922, 1:1000; anti-Map2, Abcam, ab3392, 1:2000; anti-GFAP, Abcam, ab4674, 1:2000) diluted in 3% BSA. The following day, the cells were incubated with the secondary antibodies and then DAPI before mounting. Images of ssDNA were blindly acquired with consistent acquisition parameters using a Zeiss AxioObserver.Z1 Apotome microscope and a Plan-Apochromat 100x/1.40 Oil M27 objective. ssDNA puncta were blindly quantified by manually counting puncta per nucleus, excluding puncta farther than 5μm away from the cell outline, indicated by Nestin, MAP2, and GFAP for NPCs, neurons, and astrocytes, respectively. Cell areas for normalization were measured by tracing cell outlines using ImageJ.
Cortical organoid generation
Human PSC-derived cortical organoids were generated as previously described, with some modifications (Pasca et al., 2015). Briefly, PSC colonies were gently dissociated using Accutase in PBS (1:1) (Life Technologies). Cells were transferred to 6-well plates and kept under suspension. For neural induction, media containing DMEM/F12, 15mM Hepes, 1× Glutamax, 1× N2 NeuroPlex (Gemini), 1× MEM-NEAA, 1μM dorsomorphin (R&D System), 10μM SB431542 (Stemgent) and 100 U/ml penicillin-streptomycin was used during 6 days. NPC proliferation was obtained in the presence of Neurobasal media supplemented with 2× Gem21 NeuroPlex, 1×NEAA, 1× Glutamax, 20ng/ml EGF and 20ng/ml bFGF. Next, cells were kept in the same media in the absence of growth factors for neuronal maturation. Organoid results are combined from three separate batches of differentiation.
Mycoplasma Testing
All tissue culture samples were tested for mycoplasma by PCR. 1mL of media supernatants were collected of all cell lines, spun down, and resuspended in TE buffer. 10μL of each sample was used in PCR reaction with the following primers:
For— GGCGAATGGGTGAGTAAC
Rev—CGGATAACGCTTGCGACCT
Any positive sample was immediately discarded.
Oligonucleotide transfection and S1 nuclease treatment
First, 5μg of a random 60-mer oligonucleotide was transfected into one million H9 NPCs using 20μg of polyethylenimine. The cells were fixed at 30 minutes post-transfection. For S1 nuclease treatment, 500 U/mL of nuclease was added during RNase treatment with the supplied S1 buffer (Promega M5761). The NPCs transfected with oligonucleotide displayed high levels of ssDNA puncta, whereas the NPCs transfected with oligonucleotide and subsequently treated with the S1 nuclease showed little to no puncta.
Extrachromosomal DNA extraction, sequencing, and qPCR
Extrachromosomal DNA was extracted using the modified Hirt protocol (Arad, 1998). NPCs were lifted using accutase, pelleted, and resuspended in Buffer 1 (50mM Tris-HCl, 10mM EDTA, supplemented with 100μg/mL RNase A). Cells were then lysed with Buffer 2 (1.2% sodium-dodecyl sulfate) and mixed by inverting. The lysate was then incubated at room temperature for 5 minutes to ensure complete lysis. Then, cellular debris and high-molecular weight chromosomal DNA was precipitated with addition of Buffer 3 (3M CsCl, 1M potassium acetate, and 0.67M acetic acid). The solution was chilled on ice for 15 minutes and the extrachromosomal DNA-containing supernatant was then collected and column purified (Macherey-Nagel). Extrachromosomal DNA for sequencing was treated with dsDNase (ThermoScientific) and cleaned by running through a column (Macherey-Nagel). A single-cycle PCR was performed to convert ssDNA into dsDNA to be sequenced. Samples were sequenced at the UCSD IGM Genomics Center. The Hirt protocol extrachromosomal DNA was also used for the quantitative PCR. Each qPCR experiment used 5ng of DNA along with TaqMan probes and the TaqMan Universal Master Mix II (Life Technologies). Two biological replicates were tested per cell line. Each qPCR assay was performed in triplicate. The L1 copy numbers in extrachromosomal DNA were quantified through qPCR using TaqMan probes designed by Life Technologies. The L1 probes amplify within the following 100bp sequences.
ORF1 – ATGGGGAAAAAACAGAACAGAAAAACTGGAAACTCTAAAACGCAGAGCGCCTCTCC TCCTCCAAAGGAACGCAGTTCCTC
ORF2 – GCTCATGGGTAGGAAGAATCAATATCGTGAAAATGGCCATACTGCCCAAGGTAATT TACAGATTCAATGCCATCCCCATC
ORF2–3′UTR – TGGAAACCATCATTCTCAGTAAACTATCGCAAGAACAAAAAACCAAACACCGCATAT TCTCACTCATAGGTGGGAATTGA
The relative quantities of AluS and AluY in extrachromosomal DNA were quantified through qPCR by RealTime qPCR probes from IDT and primers described previously (Macia et al., 2011).
Primers and probe for quantifying AluS:
For—GCCCAGGCGGGCGGATCACC
Rev—GCCTCCCGAGTAGCTGGGAT
Probe—CAGCCTGGCCAACATGGTGAAAC
Primers and probe for quantifying AluY:
For—AGATCGAGACCATCCTGGCT
Rev—CCGCCTCCCGGGTTCACGCC
Probe—AAATTAGCCGGGCGTGGTGG
In silico Extrachromosomal DNA characterization
Raw sequenced extrachromosomal DNA data were filtered to recover high quality (phred-scaled quality score higher than 22) sequencing reads using IlluQC software (Patel and Jain, 2012). High quality reads of each individual library were mapped to human reference genome (build Hg19) using BWA-MEM software (Li and Durbin, 2009) with default parameters over SAM mapping files. Next, we used SAMTOOLS software (Li et al.) over SAM mappings for generation of sorted mappings in compressed BAM files. For correlation of mapped reads with annotated molecules in Hg19, we created a non-redundant GTF annotation file of annotated transcripts (UCSC genes) (Kent et al., 2002) and repetitive elements for the databases RepBase (Kapitonov and Jurka, 2008), DbRIP (Wang et al., 2006) and RepeatMasker (Chen, 2004). GTF annotation file and sorted BAM mappings were compared using HTSeq software package (Anders et al., 2015) to count for each annotated molecule in the Hg19 genome how many sequenced cytosolic reads map to it. Finally, the per sample counted reads were subjected to RPKM (reads per kilobase per million) normalization (Brouha et al., 2002).
Retrotransposition assay
WT63 or V63fs NPCs were pre-treated with 10μM Ri for 1 hour prior to dissociation with Accutase (Invitrogen). Using the Rat Neuronal Stem Cell Nucleofection kit and the A31 program of Amaxa Nucleofector (Lonza), 5×106 single cells were transfected with 10μg p99-GFP-LRE3-Cherry. This construct contains a full-length retrotransposition-competent L1 element (LRE3) tagged with a mEGFP1 retrotransposition indicator cassette (Ostertag et al., 2000) in a modified version of pCEP4 (Invitrogen) that lacks a CMV promoter and contains an mCherry cassette in place of the puromycin resistance marker. After transfection, NPCs were plated on poly-L-ornithine/laminin coated plates and fed daily during the course of the retrotransposition assay.
The percentages of eGFP-positive cells were determined 9 days post-transfection by FACS. Cells were collected using Accutase, centrifuged at 1200rpm for 5 minutes, resuspended in FACS buffer (PBS, 2.5mM EDTA, 25nM HEPES, 5%FBS). A fraction of transfected cells was treated with 500nM histone deacetylase inhibitor Trichostatin A (TSA) for 16h prior to FACS analyses to determine whether L1 retrotransposition events were subjected to epigenetic silencing (Coufal et al., 2009; Garcia-Perez et al., 2010). To calculate the adjusted retrotransposition rate, the mean percentage of eGFP-positive cells was normalized by the transfection efficiency, determined by the mCherry reporter, and by live cell number (Cell Viability Solution, BD Biosciences).
shRNA knockdown of L1
Commercially available lentiviral vectors expressing short hairpin RNAs (shRNAs) against L1ORF1 under the control of the U6 promoter (VectorBuilder, Cyagen) were engineering, containing GFP as a reporter. The shRNA sequence was selected according to a previous publication (Philippe et al., 2016). 3×105 cells were seeded and transduced using MOI=1. After expansion, NPCs were used for ssDNA puncta quantification and further differentiation into neurons. Neurons were differentiated for 2 weeks prior to assessment of ssDNA and CC3.
Cleaved Caspase 3 and TUNEL analysis
Neurons were differentiated, purified, and plated as described supra. Plated neurons were fixed and stained 48 hours after purification with 4% paraformaldehyde, permeabilized with 0.25% Triton X-100 for 15 minutes, blocked with 3% bovine serum album for 60 minutes and stained for Cleaved Caspase-3 or TUNEL (Click-iT TUNEL assay kit from Life Technologies). Cells were incubated in primary antibodies overnight at 4°C and stained with secondary antibodies and DAPI the following day prior to mounting. Images were blindly collected and blindly quantified. For H9 neurons treated with astrocyte-conditioned media, the control H9 neurons were differentiated, purified, and plated as described supra. Concurrently, Neurobasal media with B27 was overlaid onto astrocytes for 48 hours, and then the fresh astrocyte-conditioned media (ACM) was overlaid onto freshly purified neurons with or without 5μg/ml neutralizing antibody against the human interferon alpha/beta receptor chain 2 (MMHAR-2). The H9 neurons were fixed and stained 48 hours after purification and exposure to ACM as described supra. Images were blindly collected and blindly quantified.
ssDNA puncta, Cleaved Caspase 3 and TUNEL quantification
ssDNA puncta were blindly quantified by manually counting puncta per nucleus, excluding puncta farther than 5μm away from the cell outline, indicated by Nestin, MAP2, and GFAP for NPCs, neurons, and astrocytes, respectively. Cell areas for normalization were measured by tracing cell outlines using ImageJ. For Cleaved Caspase 3 and TUNEL analysis, images were also blindly collected and blindly quantified.
Western Blotting
Protein was collected using RIPA Lysis and Extraction buffer (ThermoScientific) with PhosSTOP phosphatase inhibitor (Roche) and cOmplete ULTRA mini protease inhibitor (Roche). 20μg of protein was separated on a 4–12% Bis-Tris protein gel (Novex), transferred using the iBlot2 Gel Transfer device (ThermoScientific) onto a nitrocellulose membrane (Novex) gel, blocked with Rockland Blocking Buffer (Rockland) and incubated overnight at 4°C with primary antibodies anti-TREX1 (Abcam, ab185228, 1:1000), anti-IRF3 (Cell Signaling, 4302S, 1:1000), anti-phospho-IRF3 (Cell Signaling, 4947S, 1:1000), anti-LINE-1ORF1p (Millipore, MABC1152, 1:1000), and anti- β-Actin (Abcam, ab8226, 1:7000), diluted in 3% BSA. The following day, the cells were incubated with the secondary antibodies for two hours at room temperature and imaged and quantified using the Odyssey CLx imaging system (Licor).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Statistical analyses were performed using GraphPad Prism. The reported values are means ± SD or SEM, noted in the figure legend for each panel. Unless otherwise noted in the individual figure legend, n = 3 cell lines for genotypes WT (H9, WT63, WT83), ΔTREX1 (V201D, V63fs, E83fs), ΔTREX1+RTi (V201D+RTi, V63fs+RTi, E83fs+RTi), and n = 1 for ΔTREX1+NVP (V63fs+RTi). Statistical significance was determined using Student’s t-tests.
DATA AND SOFTWARE AVAILABILITY
Exome sequencing data
The exome sequencing data (used in Tables S1, S2 and Figure S1C) have been deposited in the NCBI Bioproject database under ID code PRJNA393747 and can be accessed at http://www.ncbi.nlm.nih.gov/bioproject/393747.
Supplementary Material
The gRNA sequences used to create the V63fs and E83fs lines were entered into the CRISPR design website. The CRISPR design website returned fifteen and seventeen putative off-target lesion sites, respectively. Each putative off-target lesion site was analyzed in the exome sequencing results. The exome sequencing data indicated that no mutagenesis occurred at these sites.
The exome sequencing data were analyzed to find insertions and deletions within coding regions of genes. Using the parental H9 as a reference, several genes contained frame-shift mutations. However, none of the loci containing an indel matched the gRNA, except the expected TREX1 locus. Thus, it is likely these indels likely existed within the genomes of the H9 population and arose due to clonal expansion.
Complete list of oligonucleotide sequences used for CRISPR gRNA construction and as PCR primers/probes in this study.
Highlights.
TREX1-deficient human neural cells accumulate extranuclear L1 ssDNA
Accumulation of extranuclear DNA induces toxicity in human neurons
TREX1-deficient human astrocytes secrete neurotoxic type-I interferons
Blocking L1 reverse transcription or type-I interferon signaling rescues toxicity
Acknowledgments
This work was supported by grants from the National Institutes of Health through the R01MH108528, R01MH094753, R01MH109885, R01MH100175, R21MH107771, R56MH109587, U19MH107367 and the Ruth L. Kirschstein NRSA Fellowship (1F31NS076198). The work was also supported by the California Institute for Regenerative Medicine (CIRM) award DISC1-08825, a UCSD CTRI pilot grant and a NARSAD Independent Investigator Grant to Dr. Muotri. Y.J.C. acknowledges the European Research Council (GA 309449), ERA-NET Neuron (MR/M501803/1) and a state subsidy managed by the National Research Agency (France) under the “Investments for the Future” program bearing the reference ANR-10-IAHU-01. Cl.A.T. was supported by Roche Postdoc Fellowship Program (F. Hoffmann-La Roche Ltd).
Footnotes
Author Contributions:
Ch.A.T. and A.R.M. conceived and designed this study. P.M. and L.T generated and characterized astrocytes. P.M. and P.D.N performed astrocyte IFN/ISG qPCR experiments. Cl.A.T. performed mouse experiments and helped with cell line generation. P.D.N. and Cl.A.T. generated organoids and performed all related experiments. R.H.H. performed all bioinformatics. A.M. performed retrotransposition assays. Ch.A.T. and L.T. performed and analyzed all other experiments. Y.J.C. provided patient fibroblasts, patient data, and critical review of work. Ch.A.T., L.T. and A.R.M. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akwa Y, Hassett DE, Eloranta ML, Sandberg K, Masliah E, Powell H, Whitton JL, Bloom FE, Campbell IL. Transgenic expression of IFN-alpha in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol. 1998;161:5016–5026. [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arad U. Modified Hirt procedure for rapid purification of extrachromosomal DNA from mammalian cells. Biotechniques. 1998;24:760–762. doi: 10.2144/98245bm14. [DOI] [PubMed] [Google Scholar]
- Bloom GS. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014;71:505–508. doi: 10.1001/jamaneurol.2013.5847. [DOI] [PubMed] [Google Scholar]
- Brouha B, Meischl C, Ostertag E, de Boer M, Zhang Y, Neijens H, Roos D, Kazazian HH., Jr Evidence consistent with human L1 retrotransposition in maternal meiosis I. Am J Hum Genet. 2002;71:327–336. doi: 10.1086/341722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chailangkarn T, Trujillo CA, Freitas BC, Hrvoj-Mihic B, Herai RH, Yu DX, Brown TT, Marchetto MC, Bardy C, McHenry L, et al. A human neurodevelopmental model for Williams syndrome. Nature. 2016;536:338–343. doi: 10.1038/nature19067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics/editoral board, Andreas D Baxevanis. 2004;Chapter 4(Unit 4):10. doi: 10.1002/0471250953.bi0410s05. [DOI] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10:691–703. doi: 10.1038/nrg2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cost GJ, Feng Q, Jacquier A, Boeke JD. Human L1 element target-primed reverse transcription in vitro. EMBO J. 2002;21:5899–5910. doi: 10.1093/emboj/cdf592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O’Shea KS, Moran JV, Gage FH. L1 retrotransposition in human neural progenitor cells. Nature. 2009;460:1127–1131. doi: 10.1038/nature08248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A. 2015;167A:296–312. doi: 10.1002/ajmg.a.36887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, et al. Mutations in the gene encoding the 3 ′-5 ′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nature Genetics. 2006a;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Jackson AP, Roberts E, van Beusekom E, Barth P, Corry P, Ferrie CD, Hamel BC, Jayatunga R, Karbani G, et al. Aicardi-Goutieres syndrome displays genetic heterogeneity with one locus (AGS1) on chromosome 3p21. Am J Hum Genet. 2000;67:213–221. doi: 10.1086/302955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006b;38:910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Manel N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15:429–440. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. 2009;18:R130–136. doi: 10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JL, Guimaraes KP, Benazzato C, Almeida N, Pignatari GC, Romero S, et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature. 2016;534:267–271. doi: 10.1038/nature18296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L, Huang Q, Boeke JD. Effect of reverse transcriptase inhibitors on LINE-1 and Ty1 reverse transcriptase activities and on LINE-1 retrotransposition. BMC Biochem. 2011;12:18. doi: 10.1186/1471-2091-12-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Silva U, Choudhury S, Bailey SL, Harvey S, Perrino FW, Hollis T. The crystal structure of TREX1 explains the 3′ nucleotide specificity and reveals a polyproline II helix for protein partnering. J Biol Chem. 2007;282:10537–10543. doi: 10.1074/jbc.M700039200. [DOI] [PubMed] [Google Scholar]
- Doucet AJ, Hulme AE, Sahinovic E, Kulpa DA, Moldovan JB, Kopera HC, Athanikar JN, Hasnaoui M, Bucheton A, Moran JV, et al. Characterization of LINE-1 ribonucleoprotein particles. PLoS Genet. 2010:6. doi: 10.1371/journal.pgen.1001150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Fowler BJ, Gelfand BD, Kim Y, Kerur N, Tarallo V, Hirano Y, Amarnath S, Fowler DH, Radwan M, Young MT, et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science. 2014;346:1000–1003. doi: 10.1126/science.1261754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall A, Treuting P, Elkon KB, Loo YM, Gale M, Jr, Barber GN, Stetson DB. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. 2012;36:120–131. doi: 10.1016/j.immuni.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Perez JL, Morell M, Scheys JO, Kulpa DA, Morell S, Carter CC, Hammer GD, Collins KL, O’Shea KS, Menendez P, et al. Epigenetic silencing of engineered L1 retrotransposition events in human embryonic carcinoma cells. Nature. 2010;466:769–773. doi: 10.1038/nature09209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL. Restricting retrotransposons: a review. Mob DNA. 2016;7:16. doi: 10.1186/s13100-016-0070-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesi-Oliveira K, Acab A, Gupta AR, Sunaga DY, Chailangkarn T, Nicol X, Nunez Y, Walker MF, Murdoch JD, Sanders SJ, et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry. 2015;20:1350–1365. doi: 10.1038/mp.2014.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M, Koch J, Rakheja D, Pattnaik AK, Brugarolas J, Dozmorov I, Levine B, Wakeland EK, Lee-Kirsch MA, Yan N. Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat Immunol. 2013;14:61–71. doi: 10.1038/ni.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heras SR, Macias S, Caceres JF, Garcia-Perez JL. Control of mammalian retrotransposons by cellular RNA processing activities. Mob Genet Elements. 2014;4:e28439. doi: 10.4161/mge.28439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzner AM, Hagmann CA, Goldeck M, Wolter S, Kubler K, Wittmann S, Gramberg T, Andreeva L, Hopfner KP, Mertens C, et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat Immunol. 2015;16:1025–1033. doi: 10.1038/ni.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Zhang F. CRISPR Design. 2013. [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RB, Garrison KE, Wong JC, Duan EH, Nixon DF, Ostrowski MA. Nucleoside analogue reverse transcriptase inhibitors differentially inhibit human LINE-1 retrotransposition. PLoS One. 2008;3:e1547. doi: 10.1371/journal.pone.0001547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat Rev Genet. 2008;9:411–412. doi: 10.1038/nrg2165-c1. author reply 414. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Research. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzi G, D’Arrigo S, Drumbl G, Uggetti C, Fazzi E. Aicardi-Goutieres syndrome: differential diagnosis and aetiopathogenesis. Funct Neurol. 2003;18:71–75. [PubMed] [Google Scholar]
- Lebon P, Badoual J, Ponsot G, Goutières F, Hémeury-Cukier F, Aicardi J. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. Journal of the Neurological Sciences. 1988;84:201–208. doi: 10.1016/0022-510x(88)90125-6. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Du J, Goodier JL, Hou J, Kang J, Kazazian HH, Jr, Zhao K, Yu XF. Aicardi-Goutieres syndrome protein TREX1 suppresses L1 and maintains genome integrity through exonuclease-independent ORF1p depletion. Nucleic Acids Res. 2017 doi: 10.1093/nar/gkx178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston JH, Crow YJ. Neurologic Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutieres Syndrome and Beyond. Neuropediatrics. 2016;47:355–360. doi: 10.1055/s-0036-1592307. [DOI] [PubMed] [Google Scholar]
- Macia A, Munoz-Lopez M, Cortes JL, Hastings RK, Morell S, Lucena-Aguilar G, Marchal JA, Badge RM, Garcia-Perez JL. Epigenetic control of retrotransposon expression in human embryonic stem cells. Mol Cell Biol. 2011;31:300–316. doi: 10.1128/MCB.00561-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia A, Widmann TJ, Heras SR, Ayllon V, Sanchez L, Benkaddour-Boumzaouad M, Munoz-Lopez M, Rubio A, Amador-Cubero S, Blanco-Jimenez E, et al. Engineered LINE-1 retrotransposition in nondividing human neurons. Genome Res. 2017;27:335–348. doi: 10.1101/gr.206805.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MCN, Carromeu C, Acab A, Yu D, Yeo GW, Mu YL, Chen G, Gage FH, Muotri AR. A Model for Neural Development and Treatment of Rett Syndrome Using Human Induced Pluripotent Stem Cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur DJ, Perrino FW. Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3 ′-> 5 ′ exonucleases. Journal of Biological Chemistry. 1999;274:19655–19660. doi: 10.1074/jbc.274.28.19655. [DOI] [PubMed] [Google Scholar]
- Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435:903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- Muotri AR, Marchetto MCN, Coufal NG, Oefner R, Yeo G, Nakashima K, Gage FH. L1 retrotransposition in neurons is modulated by MeCP2. Nature. 2010;468:443–446. doi: 10.1038/nature09544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Prak ET, DeBerardinis RJ, Moran JV, Kazazian HH., Jr Determination of L1 retrotransposition kinetics in cultured cells. Nucleic Acids Res. 2000;28:1418–1423. doi: 10.1093/nar/28.6.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, Kim CH, Park JY, O’Rourke NA, Nguyen KD, et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods. 2015;12:671–678. doi: 10.1038/nmeth.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7:e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrat PN, DasGupta S, Wang J, Theurkauf W, Weng Z, Rosbash M, Waddell S. Transposition-driven genomic heterogeneity in the Drosophila brain. Science. 2013;340:91–95. doi: 10.1126/science.1231965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe C, Vargas-Landin DB, Doucet AJ, van Essen D, Vera-Otarola J, Kuciak M, Corbin A, Nigumann P, Cristofari G. Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. Elife. 2016:5. doi: 10.7554/eLife.13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, Ackroyd S, Allcock R, Bailey KM, Balottin U, et al. Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 2013;12:1159–1169. doi: 10.1016/S1474-4422(13)70258-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards A, van den Maagdenberg AMJM, Jen JC, Kavanagh D, Bertram P, Spitzer D, Liszewski MK, Barilla-LaBarca ML, Terwindt GM, Kasai Y, et al. C-terminal truncations in human 3 ′-5 ′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nature Genetics. 2007;39:1068–1070. doi: 10.1038/ng2082. [DOI] [PubMed] [Google Scholar]
- Smith C, Gore A, Yan W, Abalde-Atristain L, Li Z, He C, Wang Y, Brodsky RA, Zhang K, Cheng L, et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 2014;15:12–13. doi: 10.1016/j.stem.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Veres A, Gosis BS, Ding Q, Collins R, Ragavendran A, Brand H, Erdin S, Cowan CA, Talkowski ME, Musunuru K. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 2014;15:27–30. doi: 10.1016/j.stem.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Song L, Grover D, Azrak S, Batzer MA, Liang P. dbRIP: a highly integrated database of retrotransposon insertion polymorphisms in humans. Hum Mutat. 2006;27:323–329. doi: 10.1002/humu.20307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Krainc D. alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23:1–13. doi: 10.1038/nm.4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Witherspoon DJ, Jorde LB. Mobile element biology: new possibilities with high-throughput sequencing. Trends Genet. 2013;29:280–289. doi: 10.1016/j.tig.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell. 2007;131:873–886. doi: 10.1016/j.cell.2007.10.017. [DOI] [PubMed] [Google Scholar]
- Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, Thomson JA. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan SH, Martin J, Elia J, Flippin J, Paramban RI, Hefferan MP, Vidal JG, Mu Y, Killian RL, Israel MA, et al. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One. 2011;6:e17540. doi: 10.1371/journal.pone.0017540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Sun XL, Qian YM, LaDuca JP, Maquat LE. At least one intron is required for the nonsense-mediated decay of triosephosphate isomerase mRNA: a possible link between nuclear splicing and cytoplasmic translation. Molecular and Cellular Biology. 1998;18:5272–5283. doi: 10.1128/mcb.18.9.5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Du J, Han X, Goodier JL, Li P, Zhou X, Wei W, Evans SL, Li L, Zhang W, et al. Modulation of LINE-1 and Alu/SVA retrotransposition by Aicardi-Goutieres syndrome-related SAMHD1. Cell Rep. 2013;4:1108–1115. doi: 10.1016/j.celrep.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The gRNA sequences used to create the V63fs and E83fs lines were entered into the CRISPR design website. The CRISPR design website returned fifteen and seventeen putative off-target lesion sites, respectively. Each putative off-target lesion site was analyzed in the exome sequencing results. The exome sequencing data indicated that no mutagenesis occurred at these sites.
The exome sequencing data were analyzed to find insertions and deletions within coding regions of genes. Using the parental H9 as a reference, several genes contained frame-shift mutations. However, none of the loci containing an indel matched the gRNA, except the expected TREX1 locus. Thus, it is likely these indels likely existed within the genomes of the H9 population and arose due to clonal expansion.
Complete list of oligonucleotide sequences used for CRISPR gRNA construction and as PCR primers/probes in this study.