

Abstract
Prokaryotic proteins must be deformylated before the removal of their first methionine. Peptide deformylase (PDF) is indispensable and guarantees this mechanism. Recent metagenomics studies revealed new idiosyncratic PDF forms as the most abundant family of viral sequences. Little is known regarding these viral PDFs, including the capacity of the corresponding encoded proteins to ensure deformylase activity. We provide here the first evidence that viral PDFs, including the shortest PDF identified to date, Vp16 PDF, display deformylase activity in vivo, despite the absence of the key ribosome-interacting C-terminal region. Moreover, characterization of phage Vp16 PDF underscores unexpected structural and molecular features with the C-terminal Isoleucine residue significantly contributing to deformylase activity both in vitro and in vivo. This residue fully compensates for the absence of the usual long C-domain. Taken together, these data elucidate an unexpected mechanism of enzyme natural evolution and adaptation within viral sequences.
Introduction
Peptide deformylases (PDFs) are enzymes involved in a pervasive and essential process called N-terminal methionine excision (NME). During NME, PDFs catalyze the removal of the formyl moiety from initiator methionine, found at the beginning of all prokaryotic translation products. Deformylation involves 95% of the proteins of a bacterial proteome1.
PDFs belong to the HEXXH-containing metalloprotease super-family, with high conservation of only 3 sets of residues, which build the active site of the enzyme (motif I, GΦGΦΑAXQ, motif II, EGCXS and motif III, HEΦDHXXG, where Φ is a hydrophobic aliphatic amino acid and X is any amino acid; for a review, see ref. 2). Cys from motif II and the two His of motif III, together with a water molecule, coordinate a metal ion involved in catalysis3–5. For decades, PDFs were surmised to be present only in bacteria. However, the surprising discovery of plant PDFs allowed to reveal the presence of these enzymes also in other higher eukaryotes6. Three Types (1, 2 and 3) could be progressively defined according to their overall levels of similarities (for review, see refs 2, 7 and 8). In this early classification of PDFs, Type 1 comprised two subgroups, Types 1B and 1A. Type 1B PDF was exemplified by the E. coli enzyme, showing a globular core and a C-domain involving a 310-helix (referred as η3), common to all known PDF structures, followed by an α-helix (α3)9, 10. Interestingly, the α3 helix of E. coli PDF was shown to be crucial for activity in vivo by allowing tight interaction with the ribosome next to the exit tunnel11. In addition, significant reduction of the activity in vitro was noticed when further trimming the E. coli PDF C-domain beyond the η3 310-helix made of the 4 residues FMDY, which folding correctly orientates the α3 helix back towards the N-terminal domain12. Gram-negative bacteria PDFs all belonged to the Type 1B subgroup. Type 1B PDFs were also found in the organelles of eukaryotes where protein synthesis occurs. Type 1A PDFs, only present in mitochondria, feature a long insertion that partially tethers the active site entrance, contributing to a reduction in the size of the ligand binding pocket, and a C-domain folding in the opposite orientation compared to Type 1B PDFs13, 14. Type 2 PDFs were represented in this early classification by Bacillus stearothermophilus PDF and also included PDFs from Mycoplasma and Gram-positive bacteria. However, when two PDFs occurred in some Gram-positive bacteria, the second was often a Type 1 PDF. Type 2 PDFs were characterized by several insertions and a completely different fold of the C-domain compared to other PDFs7. Finally, Type 3 PDFs clustered the mostly weakly active or inactive PDFs, found in Archaea and in trypanosomatid mitochondria, as they displayed substitutions in one or several crucial amino acids of the conserved motifs6. The C-termini of Type 3 PDFs, for which no structure is known, displayed no conservation in length.
Recent high-throughput screening, sequencing and annotation of thousands of genomes have strongly contributed to revolutionizing our perception of the distribution of PDFs among kingdoms, revealing putative PDFs in almost all organisms, including viruses. In particular, the pioneer marine metagenomics study of viruses within oceanic microbial samples retrieved unpredicted modified PDF genes as the most abundant family in most of these phage genomes15. Sequence comparisons with other known PDFs reveal that viral PDFs are devoid of the key ribosome-interacting C-terminal region. Despite this initial report and the following study uncovering other phage PDFs, no further characterization and functional studies of these intriguing PDFs have been described thus far, particularly in vivo. However, the first sequence alignments of these phage PDFs showing perfect conservation of the motifs of the catalytic residues15, together with the recent characterization of Synechococcus phage S-SSM7 PDF16, prompted the suggestion that all discovered putative phage PDFs might be active enzymes.
To support this trend and enlarge our insufficient knowledge on viral PDFs, we set out to definitively check the deformylase activity of a number of marine viral PDFs, including Vp16 PDF, which is derived from bacteriophages originally isolated from Vibrio parahaemolyticus strain 16 (Vp16)17. We provide the first evidence that some viral PDFs, including the shortest PDF identified to date, Vp16 PDF, display deformylase activity in vivo. Large scale N-terminomics characterization reveals that Vp16 PDF has substrate specificity similar to that of other bacterial PDFs. Surprisingly, biochemical and structural data, combined with site-directed mutagenesis analyses, showed that Vp16 PDF significantly differs from previously characterized PDFs in terms of their properties, which can be related to its few infrequent peculiarities. This uniqueness, not involving the predictable active site, underscored unexpected structural and molecular characteristics of the last rare residue Ile137, which significantly contributes to deformylase activity in vitro and in vivo, fully compensating for the absence of the otherwise indispensable C-terminal domain.
Results
An updated, accurate phylogenetic tree recapitulating the large diversity of PDFs
The two first closely related viral deformylase sequences were identified in 2003 in both Vp16T and Vp16C bacteriophages17. The initial classification in a phylogenetic tree of these two putative PDFs, together with other identified viral PDFs, clustered Vp16T and Vp16C PDFs in a subclass of bacterial PDFs (PDF1B classic), also including E. coli PDF, not embracing the other viral PDFs assembled in other specific subclasses of PDF1B15. A different phylogenetic analysis confirmed the particularity of Vp16T and Vp16C PDFs, as they did not cluster with other phage PDFs16. Given the increasing number of discovered PDF sequences, we built a new accurate phylogenetic tree using sequences selected to represent PDF sequence diversity among the 20,609 entries annotated as “peptide deformylase” available in RefSeq. Only 602 sequences originate from organisms different from bacterial sequences, which represent 97% of the available sequences. Nevertheless, strong redundancy due to metagenomics programs and the many clinical isolates of pathogenic bacteria led us to strongly simplify the sequence diversity and to significantly underweight the bacterial sequence number to obtain a clearer picture of the sequence diversity. We, therefore, extracted PDF sequences from completely sequenced genomes or from genomes for which sequencing was almost complete. A total of 262 sequences recapitulating the sequence diversity were finally aligned (Supplementary information). The four main PDF Types, including 3 subtypes in Type 1 (A/B/C), are clustered and colored in Fig. 1. As previously shown, in this updated phylogenetic tree, both Vp16T and Vp16C PDFs clustered in subtype 1B, as do the majority of bacterial PDFs. Type 1B PDFs are considered both plastid and bacterial PDFs, as this branch also includes all plant, algal and diatom plastid PDFs. Noticeably, with the increasing number of complete sequences now available, rather than the fragmented information from short reads of early metagenomics work, our early classification of marine viral PDF sequences tentatively assigned as Type 115 had to be reconsidered and clustered into a new type - Type 4 - in the updated version of the phylogenetic tree (Fig. 1). Type 4 also includes various sequences from several apicomplexan eukaryotic parasites, such as Toxoplasma gondii, that were previously difficult to cluster. Finally, Type 3 is now enriched with a large variety of PDFs originating from all types of unicellular organisms, including prokaryotes and eukaryotes. As such, Type 3 is the most diverse in terms of organism diversity. This is unlike Type 2 PDFs, which are almost exclusively found in Gram-positive bacteria. Interestingly, Type 1A PDFs, previously believed to be only in eukaryotes, were also found in different bacteria.
Figure 1.

A robust phylogenetic tree of PDFs reveals a new classification for these enzymes. 262 sequences were selected to represent PDF sequence diversity among 20,609 entries. The sequences were aligned with Clustal X33 and the bootstrap tree was constructed with PHYLIP. The random number generator seed was 111 and the number of bootstrap trials was 1,000. The rooted phylogenic tree was constructed with N-J Tree and Drawn with TreeView1.6634. Internal values labelled on each node record the stability of the branch over the 1,000 bootstrap replicates. Four main PDF Types (PDF1, PDF2, PDF3 and PDF4) and 3 PDF1 subtypes (PDF1A, PDF1B, PDF1C) are clustered and shown in color.
Phage Vp16 PDF is the smallest active PDF
Although an increasing number of viral PDF sequences are regularly being identified, no data are available on the capacity of the corresponding proteins to function as PDF in vivo. Most viral PDFs, including marine viral PDFs and Vp16 PDF proteins, show high conservation in the three motifs that build the catalytic site; however, most contain shorter C-terminal extensions compared to the different active PDFs15. Because this C-terminal extension was shown to be important for PDF-ribosome binding and required for the in vivo deformylase activity of E. coli PDF (Fig. 2a and refs 11 and 12), we investigated the in vivo deformylase activity of several C-terminally truncated viral PDFs using the def-conditional strain PAL421Tr-pMAKdef18. Based on their relative distribution in the phylogenetic tree, we assessed (i) the representative members of marine viral PDFs – namely, those referred to as 1906, 1577 and 2750 in ref. 15, all three Type 4 PDFs, and (ii) the representative of the two almost identical Vp16 PDF proteins, which, unlike marine viral PDFs, clustered in Type 1B. The four sequences featured short C-domains of significantly reduced size (Fig. 2a). All viral PDF genes were cloned into the pBAD plasmid19, which features a tightly tunable arabinose-inducible promoter. We observed that both the Vp16 phage PDF and the marine viral 1906 PDF were active in vivo, as these enzymes allowed PAL421Tr growth under non-permissive conditions at the lowest arabinose concentrations (Fig. 2b). In contrast, no complementation was observed with marine viral 1577 and 2750 PDFs (Figs 2b and S1). Detailed analysis of the C-domains of these proteins (Fig. 2a,c) showed that the C-domain of the 1906 PDF is the longest compared to other viral PDFs, embracing the short η3 helix shown to be indispensable to fulfill deformylase activity of E. coli PDF (Fig. 2a and refs 11 and 12). Accordingly, a deletion in the 1906 PDF of the two last C-terminal residues located within the η3 helix, (1906QR), 1577 and 2750, did not lead to complementation (Figs 2a,b and S1). Instead, Vp16 PDF complemented PAL421Tr strain growth at non-permissive temperatures, although it did not contain residues expected to contribute to the crucial η3 helix of the C-domain (Fig. 2a). With only 137 residues vs an average of 165–175 residues for the prokaryotic PDFs, Vp16 PDF is thus unique among other PDFs, as it is the shortest PDF completely devoid of the C-domain (Fig. 2c) while exhibiting deformylase activity in vivo.
Figure 2.

Complementation of strain PAL421Tr by PDFs with various length of their C-domain. pBAD plasmids encoding different PDFs (E. coli, V. parahaemolyticus phage and marine viral PDFs named: 1906 and 1577) were used to transform strain PAL421Tr at 30 °C. Strains were serially diluted and spotted in parallel at 42 °C on LB Petri dishes containing PDF expression inducer arabinose. Control corresponds to the empty cloning vector pBAD. (a) List of all used constructs, highlighting the corresponding C-terminus and results of the complementation. Various variants of E. coli PDF lacking the C-terminal region were previously constructed and activity tested in vitro and in vivo 12. (b) Image of the Petri dishes incubated at 30 °C or 42 °C at different concentration of arabinose. (c) sequence alignment between E. coli and Vp16 PDFs, realized with ESPript (http://endscript.ibcp.fr)35. Identical residues are shown on red boxes and secondary structures (α and 310-helices, β-strands and turns) of E. coli PDF are indicated on top.
Characterization of Vp16 PDF, a protein with high catalytic efficiency in vitro and with substrate specificity comparable to other PDFs
Enzymatic properties of purified Vp16 PDF were further determined in vitro with a formylated tripeptide (Fo-Met-Ala-Ser), which is used as a reference to compare various PDFs20, 21. Vp16 PDF showed a catalytic efficiency and K m values for Fo-Met-Ala-Ser similar to that of other active PDFs (Table 1). However, it could not be excluded that Vp16 PDF has a particular substrate specificity to promote deformylation of either the proteins encoded by the phage genome Vp16T or to disfavor deformylation of some proteins of the host bacterium. Of note, the in vitro characterization of the deformylase activity of the PDF from cyanophage Synechococcus S-SSM7 using various formylated tetrapeptide derivatives of phage protein sequences has suggested a slight difference in the enzyme substrate specificity relative to the host enzyme16. To determine whether Vp16 PDF could specifically deformylate proteins encoded by the phage genome, we tested several formylated tripeptides derived from the N-terminal sequences of proteins encoded by the phage. Based on sequencing data, the genome of the phage Vp16T contains 64 ORFs17. We chose to test peptides Fo-Met-Pro-Ala and Fo-Met-Ser-Asn, corresponding to putative capsid and helicase N-termini, respectively. In addition, we chose peptides Fo-Met-Lys-Leu, Fo-Met-Thr-Thr and Fo-Met-Ala-Lys, corresponding to N-terminal sequences that are represented up to 3 times in the phage genome. The catalytic efficiencies of Vp16PDF and E. coli PDF for these peptides were determined and compared to reference peptide Fo-Met-Ala-Ser, which was set at 100%. The catalytic efficiencies of Vp16 PDF for Fo-Met-Lys-Leu and Fo-Met-Thr-Thr peptides were similar, while those of the Fo-Met-Pro-Ala and Fo-Met-Ser-Asn peptides were decreased (Table S1). Nevertheless, although slight differences appeared from these results, they were kinetically insignificant, and the same trends were obtained with the E. coli PDF (Table S1).
Table 1.
Enzymatic properties of purified Vp16 PDF and comparison with other PDFs.
| PDF enzyme | k cat (s−1)a | K m (mM)a | k cat/K m (M−1.s−1)a |
|---|---|---|---|
| Ni-E. coli PDF1Ba | 34–144 | 0.2–3.8 | 15,355–86,000 |
| Ni-T. thermophilus PDF1Bc | 27 ± 3 | 2.3 ± 0.5 | 11,739 ± 2,500 |
| Synechocystis PCC PDF1Bd | 150–250 | 1–2 | 88–313 |
| Ni-A. thaliana PDF1Bb | 75 ± 15 | 5.6 ± 1.9 | 13,300 ± 1,500 |
| Ni-Vp16 PDF1B | 5.3–27.4 | 1–2.9 | 2,286–9448 |
| Synechococcus phage S-SSM7 PDF4d | 433–800 | 0.3–1.3 | 449–2,204 |
| Zn-A. thaliana PDF1Ab | 22 ± 2 | 0.3 ± 0.1 | 88,000 ± 150 |
| Ni-H. sapiens PDFIAe | 0.26 ± 0.04 | 3.6 ± 0.9 | 72 ± 7 |
| Ni-B. stearotermophilus PDF2c | 1007 ± 191 | 4.1 ± 1.2 | 245,000 ± 20,000 |
| Ni-S. agalactiae PDF2f | 50 ± 3 | 1.2 ± 0.8 | 41,993 |
| Ni-P. falciparum PDF3g | ND | ND | 13,700 ± 1,000 |
| Zn-T. brucei PDF3h | ND | ND | 8 |
aKinetic constants were determined using the coupled assay, as indicated in Materials and Methods, using substrates Fo-Met-Ala-Ser, Fo-Met-Lys-Leu, Fo-Met-Pro-Ala, Fo-Met-Ser-Asn, Fo-Met-Thr-Thr, Fo-Met-Ala-Lys when tested Vp 16 PDF1B and Ni-E. coli PDF1B.
bData for E. coli and Arabidopsis thaliana PDF1Bs were taken from ref. 26.
cData for Thermus thermophilus PDF1B and Bacillus stearothermophilus PDF2 were taken from refs 20, 28 and 37.
dData were taken from ref. 16, substrates used: Fo-Met-Thr-Ser-Ile, Fo-Met-Leu-Ile-Ser, Fo-Met-Thr-Thr-Ala, Fo-Met-Ala-Lys-Lys, Fo-Met-Ala-Arg-Ile, Fo-Met- Ser-Arg-Val.
eData for Homo sapiens PDF1A were taken from ref. 38.
fData for Streptococcus agalactiae PDF2 were taken from ref. 28.
gData for Plasmodium falciparum PDF3 were taken from ref. 27, substrate used: Fo-Met-Leu-p-nitroanilide.
hFor Trypanosoma brucei PDF3 Data were taken from ref. 39.
ND, not determined.
Ni- or Zn indicates Ni2+- or Zn2+-containing PDFs.
Next, a large scale N-termini proteomics characterization from culture cells expressing only E. coli or phage Vp16 PDF (Fig. 3a) revealed a similar distribution of the modifications affecting the N-termini along the growth profile (Fig. 3b). No difference was observed when the sequences of the identified N-termini undergoing deformylation were compared (Fig. 3b,c), further indicating that the two PDFs share an identical cleavage profile in vivo, i.e., show little preference, if any, for amino acids located beyond the amino-terminal formyl-methionine residue, as already described for E. coli PDF1, 21, 22.
Figure 3.
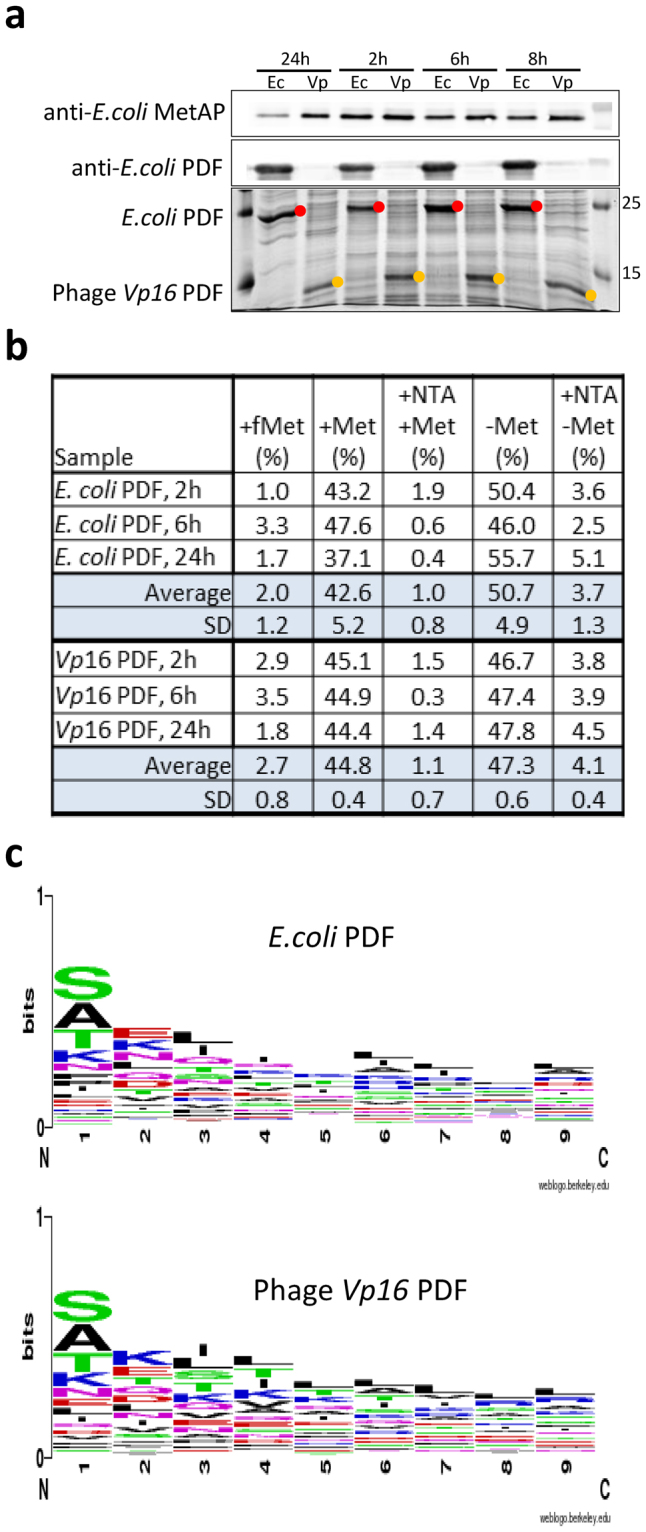
Large scale N-terminomics characterization for in vivo PDF substrate specificity determination. (a) PAL421Tr strain transformed with pBAD plasmids encoding E. coli or Vp16 PDFs were grown in LB liquid medium in the presence of 2% arabinose. At different time course growth rate, cultures were tested for the expression of E. coli MetAP and PDF (upper panels showing proteins detected by western blot with appropriate antibodies) or Vp16 PDF (lower panel showing culture lysates resolved on 12% SDS-PAGE stained with Comassie brilliant blue). Expressed Vp16 PDF protein is marked with yellow dots, expressed E. coli PDF is marked with red dots. The full-lenght blots and gel are presented in Supplementary Figure 3. (b) Samples from A were subjected to SILProNAQ methodology1 and distribution of the characterized N-terminal modifications was analyzed. (c) Protein N-terminus logo obtained for experimentally characterized N-termini from (a).
The crystal structure of Vp16 PDF reveals a classical PDF fold, in addition to a number of peculiarities
To gain insight into the properties of these novel viral PDFs, we determined the X-ray crystal structure of Vp16 PDF. The crystal structure of the protein could be solved by molecular replacement using the structure of Pseudomonas aeruginosa PDF without its C-terminal extremity as a search model. Two crystal forms of Vp16 PDF grown under different crystallization conditions were used. Both diffracted at a high resolution (1.7 to 1.5 Å) in space group P3 221 with nearly identical cell parameters and two molecules per asymmetric unit (Table S2). Superimposition of all chains showed very similar structures (r.m.s.d. of all of Cα < 0.5 Å). Vp16 PDF adopted an overall fold that is consistent with the Type 1B PDF family, and a search for structural homologs by DALI23 identified two Type 1B PDFs, namely, Vibrio cholerae (PDB code 3FWX, unpublished structure) and E. coli PDFs (PDB code 2AI824), as the closest structural neighbors (sequence identity = 27 and 28%, r.m.s.d. = 1.05 Å and 1.08 Å, respectively). Vp16 PDF was found in a super-closed conformation, as evidenced by the delta aperture angle measured between Cys85, His131 and Ile41 (δap = 41°–42°) (δap is defined in ref. 25). Together, the data further validate the classification of Vp16 PDF as a Type 1B PDF (Fig. 1).
The Vp16 PDF structure is composed of two α-helices (α1 and α2), two 310-helices (η1 and η2) and seven β-strands (β1 to β7) organized into two sub-domains between which lies the active site (Fig. 4a). Although Vp16 PDF displays a classical PDF fold, several significant differences could be noted (Fig. 4b). First, Vp16 PDF displays a shorter N-terminus and an α1 helix reduced by one turn at its N-terminal extremity (Fig. 4b). The significance of the absence of one helical turn is currently unknown. Moreover, Vp16 PDF lacks the α3 helix usually found in Type 1B PDFs, as expected from sequence alignments (Fig. 4b,c). Unforeseen, Vp16 PDF is also devoid of the conserved η3 helix (Fig. 4c). Thus, Vp16 PDF lacks the two conserved structural elements of the C-domain. Interestingly, PDF from cyanophage S-SSM7, with a C-terminus slightly longer than that of Vp16 PDF (Fig. 4c), lacks the Type 1B typical α3 helix but displays the typical η3 helix, which is well-preserved in all PDFs determined thus far, including Type 1 and Type 2 (Fig. 4c).
Figure 4.

Crystal structure of Vp16 PDF. (a) Vp16 PDF ribbon diagram with α and 310- helices in pink, β strands in green, showing the two sub-domains encircling the active site. The three consensus motifs (I, II and III) are highlighted in yellow. (b) Superimposition of Vp16 PDF with E. coli PDF structure. The main differences are indicated with arrows. (c) Comparison of C-terminal extremities of Vp16 PDF, E. coli PDF and S-SSM7 PDF was done by aligning sequences and secondary structures with ENDscript/ESPript tool (http://endscript.ibcp.fr)35. Identical residues are shown on red boxes and secondary structures (α and 310-helices, β-strands and turns) are indicated on top. Motifs II and III (m2 and m3, respectively) are indicated. (d) Comparison of C-terminal folding. In contrast to Vp16 PDF, E. coli and S-SSM7 PDFs share the typical η3 310-helix that follows conserved α2 helix. The flexible C-terminal extremity of S-SSM7 PDF is indicated with dotted lines. As in panel a, conserved motif III is highlighted in yellow.
Further study of the C-terminal extremity of Vp16 PDF revealed that it is tightly bound to the globular core of the protein involving (i) a hydrogen bond made between the side chains of the penultimate residue, Thr136, and that of Tyr88 located immediately after motif II (m2, composed of residues 83–87) and (ii) a salt bridge between the C-terminal carboxyl group of the last residue, Ile137, and the side chain of Lys91, with both amino acids sitting in a structured loop between strands β4 and β5 (Fig. 5a). In addition, the side chain extremity of Lys95 is within hydrogen bonding distance of the carboxyl terminal of Ile137. The side chain of Ile137 is clamped by residues coming from β4 and β5 strands and their connecting loop, especially by Tyr88, Lys91, Val93 and Lys95 side chains (Fig. 5a and insert). These residues form a hydrophobic pocket where Ile137 is buried (Fig. 5a insert). Structural inspection of other Type 1B PDFs revealed that the overall network of interactions in the region of Vp16 PDF Ile137 does not occur in other PDFs (Fig. 5a). For instance, Thr136 and Ile137 of Vp16 PDF are usually replaced by two very apolar, bulkier residues in other PDFs regardless of the Type (see below and Fig. 6c), including in E. coli and S-SSM7 PDFs (Fig. 5a). More strikingly, the residues forming the Ile137 pocket of the Vp16 PDF are not conserved in E. coli and S-SSM7 PDFs (Fig. 5a), leading to two main differences in comparison to the Vp16 PDF. First, the hydrogen bond and the salt bridge observed in Vp16 PDF cannot occur in E. coli and S-SSM7 PDFs (Fig. 5a). Second, while the hydrophobic pocket around Vp16 PDF Ile137 appears rather rigid and constrained, the one found in E. coli and S-SSM7 PDFs is more open and agile. Of note, the special interaction network around the C-terminal extremity of Vp16 PDF is located in the vicinity of the three catalytic residues (Fig. 5b), evoking a possible link between the unusual extremity of Vp16 PDF and its biological function.
Figure 5.

Comparison of interactions network at the C-terminal extremity of Vp16 PDF with the E. coli and S-SSM7 PDFs counterparts. The interactions network of Vp16 PDF C-terminal extremity was compared to that of E. coli and S-SSM7 PDFs. (a) The last three residues (V135T136I137) of Vp16 PDF are drawn in stick representation and colored in red. Other residues are shown in grey, with O and N atoms colored in red and blue, respectively. Hydrogen bond and salt bridge are represented by dotted lines. The same region is shown for E. coli and S-SSM7 PDFs. Inset: The hydrophobic pocket where Vp16 PDF Ile137 lies is highlighted through a surface representation of the protein in this region, in a slightly different orientation. The same region is also shown for E. coli and S-SSM7 PDFs. For more clarity, E. coli and S-SSM7 PDFs were truncated after counterpart residues of the last three residues of Vp16 PDF. (b) The proximity of Vp16 PDF C-terminal extremity with the active site (composed of residues His127, His131 and Cys85) is shown, and compared to the equivalent region in E. coli and S-SSM7 PDFs.
Figure 6.

Complementation of strain PAL421Tr by different Vp16 PDF chimeras. pBAD plasmids encoding different PDF chimeras were used to transform strain PAL421Tr at 30 °C. Strains were serially diluted and spotted in parallel at 42 °C on LB Petri dishes containing PDF expression inducer arabinose. (a) Outline of used chimeras. (b) Image of the Petri dishes incubated at 30 °C and 42 °C at different concentration of arabinose. (c) The iceLogo 1.2 was used as the tool to display the data resulting from a collection of 237 distinct proteins36. Alignment starts with conserved motif III and the following 6 residues.
Several metal cations could be identified in the structure. Particularly, a zinc cation was identified within the active site, which was coordinated similarly to other PDF structures, i.e., by Cys of motif II, the two His of motif III and one water molecule.
The crucial role of the last Vp16 PDF residue, Ile137, required for deformylase activity
The major sequence and structural differences between Vp16 PDF and E. coli PDF reside at the level of the C-terminal region, and we have shown that the η3 helix was necessary to preserve the complete deformylase activity of E. coli PDF9 but not Vp16 PDF (Fig. 2a). Therefore, to clearly identify the major determinant of the C-domain contributing to the full hydrolytic activity of Vp16 PDF, even without the η3 and α3 helices, we built a series of chimeras between the two proteins (Fig. 6a). All these chimeras were used to assess their deformylase activity both in vivo and in vitro. First, we generated a chimera made by Vp16 PDF to which we added the two C-terminal η3 and α3 helices of E. coli (Vp16 PDF(KLF)helices, Fig. 6a). As the last residue of Vp16 PDF (Ile137) corresponds to the first residue of the η3 helix of E. coli PDF (Fig. 2c), to ensure that this region would fold well in the form of a 310-helix, we have substituted the last three residues of Vp16 PDF (Val135Thr136I137) with the E. coli counterpart motif Lys135Leu136Phe137. This chimera was unexpectedly found to be unable to complement the conditional PAL421Tr bacteria at non-permissive temperatures (Fig. 6b). In contrast, a chimera where the bacterial motif Lys135Leu136Phe137 was replaced with the phage residues Val135Thr136Ile137 (Vp16 PDF(VTI)helices, Fig. 6a) was able to fully complement the conditional PAL421Tr bacteria at non-permissive temperatures with an effectiveness comparable to E. coli PDF and Vp16 PDF (Fig. 6b). These data led us to a close inspection of the sequence surrounding the η3 helix of other known PDFs. The final Vp16 PDF dipeptides involving Thr136 and Ile137 are rather unusual and correspond generally to Leu or Met and Phe or Tyr residues, respectively, in other PDFs (Fig. 6c). We then constructed a new chimera in which we replaced the last Ile137 residue of Vp16 PDF with the equivalent Phe of E. coli (Vp16 PDF(VTF)helices, Fig. 6a). The only mutation in Ile137 of Vp16 PDF completely abolished the capacity of the protein to guarantee deformylase activity in vivo (Fig. 6b). This was in accordance with the enzymatic activity of the corresponding proteins measured in vitro (Table 2), highlighting the unexpected primary role of Ile137 in the deformylase activity of Vp16 PDF.
Table 2.
Comparative enzymatic constants of Vp16 PDF and chimeras using Fo-Met-Ala-Ser as substrate.
| PDF enzyme | k cat (s−1) | K m (mM) | k cat /K m (M−1.s−1) | Relative k cat/K m (%) |
|---|---|---|---|---|
| Vp16 PDF1B | 20 ± 1 | 2.3 ± 0.9 | 8,478 ± 2276 | 100 |
| Vp16 PDF(KLF)helices | nm | nm | nm | nm |
| Vp16 PDF(VTI)helices | 3.17 | 1 | 7,417 ± 6006 | 92 |
| Vp16 PDF(VTF)helices | 0.4 | 0.5 | 80 | 18 |
| E. coli PDF1B | 144 | 3.8 | 53,846 ± 21,667 | 100 |
| E. coli PDF(KLF)Δhelices | 77 ± 9.4 | 5.9 ± 2.1 | 13,125 ± 4,560 | 66 |
| E. coli PDF(VTI)helices | 26 ± 20 | 1.6 ± 1.3 | 19,316 ± 9,916 | 97 |
| E. coli PDF(VTI)Δhelices | ND | ND | ND | ND |
| E. coli PDF(KLI)helices | 41 ± 0.4 | 3.1 ± 0.11 | 13,189 ± 3,515 | 67 |
nm, not measurable.
ND, not determined.
To complete the above characterization, the constructs of mutant proteins of E. coli PDF were also carried out to mimic the structure of Vp16 PDF and to check the influence of the last three phage amino acids in the E. coli PDF enzyme in the presence and the absence of the η3 and α3 helices (Figure S2a). All these E. coli chimeras were able to complement strain PAL421Tr (Figure S2b). Nonetheless, the E. coli PDF enzyme lacking its η3 and α3 helices (PDF(KLF)Δhelices) was less efficient in complementing the PAL421Tr strain at low concentrations compared to all other constructs, including the corresponding proteins where Phe143 was substituted with the phage Ile137 (see PDF(VTI) helices, PDF(VTI)Δhelices and PDF(KLI) helices in Figure S2b). Taken together, all these data showed the crucial importance of Ile137 for efficient deformylase activity in the absence of the η3 and α3 helices.
Discussion
The updated phylogenetic tree presented in this report, taking in account both the latest released PDF sequences, including those of viruses, and sequence diversity, allowed us to re-classify PDFs into four Types. In this new classification, phage PDFs clustered mainly into two Types: PDF1B and the new class, PDF4. Interestingly, PDF sequences retrieved in the GOS metagenomics database15, including phage S-SSM716, clustered in the new class 4. Unlike previous studies, this phylogenetic tree shows that Vp16 PDF with other phage PDFs, clusters within the classical Type 1B PDF. Given this new classification, aside from the obvious but enthralling issue related to the purpose of why viruses express a PDF, the first query to solve was whether the respective encoded putative viral PDFs do display canonical deformylase activity. Indeed, despite strict conservation of the three motifs building the deformylase catalytic site, all identified viral PDF sequences, independent of the class to which they belong, show shorter C-termini compared to other PDFs15. Previous studies on E. coli PDF revealed that the C-domain can be trimmed, including the complete removal of α3 helix without affecting the activity of the enzyme in vitro or in vivo 12. However, too large of a deletion in the E. coli PDF C-terminus results in a decrease in activity until complete inactivation of the enzyme. Thus, a deletion after residues Leu142 or Met144 located just before or at the beginning of the η3 helix of E. coli PDF (Fig. 2a) causes a dramatic decrease in enzyme activity in vitro. A deletion beyond amino acid Gly140, in turn, contributes to a total loss of E. coli PDF activity11, 12. As the last amino acid of the only in vitro characterized viral PDF16, S-SSM7, corresponds to residue Gln152 in E. coli PDF (Fig. 4c), we could have assumed that despite its short size, S-SSM7 has a sufficient length to display full deformylase activity. However, the low uncommon enzymatic constant values obtained in vitro with both S-SSM7 and Synechocystis PCC PDFs (Table 2 in ref. 16), and particularly when compared to other known PDFs (Table 1), have left open the question regarding the deformylase activity of viral PDFs, independent of their affiliation. Of note, this uncertainty was much more marked for other shorter C-terminal viral PDFs and notably for Vp16 PDF, the last residue of which corresponds to residue Phe143 of E. coli PDF.
In this study, we show that several marine viral Type 4 PDFs and Type 1B Vp16 PDF do display deformylase activity in vivo. Moreover, Vp16 PDF displays in vitro deformylase activity comparable to other known active PDFs26–30. Clearly, with only 137 residues, i.e., 30 less than the average size of bacterial PDFs, Vp16 PDF is the smallest active PDF identified to date. This reduced size might reflect the need to take optimal advantage of the limited sequence information encoded in compact viral genomes, as already observed for other viral proteins (for review see ref. 31). On the other hand, it has been previously suggested that viral PDFs might exhibit specific distinct substrate specificity compared to bacterial PDFs16. Here, we provide evidence, using large-scale N-terminomics analysis, that Vp16 PDF has widespread substrate specificity similar to that of E. coli PDF.
What are the key elements allowing Vp16 PDF to ensure deformylase activity despite its extremely short C-terminus? Comparison of the structures of Vp16 and E. coli PDFs revealed that both PDFs display a classical PDF fold. However, the crystal structure of Vp16 PDF revealed, in addition, a number of differences. The most unexpected peculiarity is the absence of the η3 helix conserved in all characterized active PDFs, including PDF from cyanophage S-SSM716. Interestingly, the two final residues of Vp16 PDF, Thr136 and Ile137, lying very close to the active site, are involved in a network of interactions unseen in any PDF thus far. Interestingly, Phe142 of E. coli PDF is highly conserved among the PDF family, due to the fact that a Phe is found in 76% of all analyzed sequences (it is replaced by a Tyr, which also contains an aromatic ring in less than 17% of the cases), and it is very unusual to find an Ile in this position (less than 2% of the cases). The Val/Ile/Lys-Leu-Phe/Tyr tripeptide appears to be by far the preferred consensus (Fig. 6c).
A close interdependency was observed between the dynamics of Vp16 PDF and its immediate environment (Fig. 5). We noticed indeed that the lateral chain of Ile137 of Vp16 PDF sits into a more tethered space than its counterpart residues in E. coli PDF (Phe142) and S-SSM7 PDF (Phe144). The large space around Phe142 in E. coli PDF and Phe144 in S-SSM7 PDF, compared to Ile137 from Vp16 PDF, confers a significant degree of freedom to the corresponding lateral chain, which might, in turn, contribute to unsettling the catalytic center and promote decreased activity. This mobility is most likely circumvented by the following C-domain residues building the η3 and α3 helices of the corresponding proteins. For instance, α3 helix in E. coli PDF is kept close to the rest of protein by a salt bridge between Asp162 and His7, which might restrain the environment around Phe142 and indirectly influence the optimal shape of the catalytic center. This might explain the low deformylase activity of the mutant E. coli PDF(KLF)Δhelices while pointing out the crucial role of Ile137 in Vp16 PDF. This was strengthened by a number of variants. The addition of the η3 and α3 helices of E. coli PDF at the C-terminus of Vp16 PDF, where the triplet Val135Thr136I137 was also substituted by the E. coli motif Lys135Leu136Phe137 or Val135Thr136Phe137, induces the complete loss of deformylase activity both in vitro and in vivo. In parallel, a simple substitution of the E. coli motif Lys135Leu136Phe137 with the Vp16 PDF triplet Val135Thr136I137 in the chimera E. coli PDF devoid of its C-terminal helices E. coli PDF(VTI)Δhelices improves the deformylase activity compared to the mutant E. coli PDF(KLF)Δhelices, particularly in vivo.
To conclude, characterization of the phage Vp16 PDF allows us to unexpectedly uncover the crucial role of the most unusual final amino acid in fully compensating for the 30 amino acid longer C-domain to ensure full deformylase activity.
Materials and Methods
PDFs cloning into the pBAD and pET16b vectors
Sequences of Vibrio parahaemolyticus phage PDF17 and representative members of marine viral PDFs: 1906, 1577, 275015 were designed on the basis of data deposited in the metagenomics libraries. DNA sequences were synthesized and cloned into pBAD/Myc-HisA plasmid (Invitrogen) using BspHI and PstI cloning sites by GeneArt Company. Proteins expressed from final constructs resulted in amino acids sequences described in Table S3. Sequence of 1906 PDF was further modified by deletion of the last two amino acids Q144 and R145. Vp16 PDF was subcloned from pBAD/Myc-HisA plasmid into pET16b using NcoI and XhoI cloning site.
V. parahaemolyticus phage PDF expression and purification
Vp16 PDF purification has been performed using previous protocols with several modifications (Supplementary information). Identity of purified Vp16 PDF protein was confirmed by MALDI-TOF analysis with 88% of sequence coverage.
Complementation test
The def-conditional strain PAL421Tr-pMAKdef (fms_1, galK,rpsL, recA56, srl-300::Tn10), also known as PAL421Tr, with inactivated fms gene on the chromosome and the wild-type allele on the thermosensitive pMAK vector has been described in ref. 18. pBAD plasmid encoding PDF proteins were transformed into the PAL421Tr E. coli strain at 30 °C. Next day strains were streaked out in parallel at 42 °C on LB dishes containing different concentrations of the PDF inducer arabinose. Negative control corresponded to the empty cloning vector pBAD.
N-terminal proteomic analysis
Proteomic analyses were performed essentially as previously described1. Further details are provided in the Supplementary information.
Crystallization and structure determination
Crystallization conditions of apo Vp16 PDF were screened by a robot using commercial kits (JCSG+ suite and PEGs suite from QIAgen, and Precipitant Synergy screen from Jena Bioscience). Crystallization conditions were obtained and manual optimizations were then carried out at 19 °C with drops of 1:1 mixture of protein (5 mg/mL) and reservoir solution. Best crystals were obtained in 38% MPD, 100 mM CAPS pH 10.5 (form I) and 28% PEG-1,000, 100 mM sodium acetate pH 5.5 (form II). Prior to data collection, manually reproduced crystals were harvested, transferred to a cryoprotectant solution (mother liquor containing 20% of glycerol) and flash frozen in liquid nitrogen. Data collection and processing are described in the Supplementary information.
Electronic supplementary material
Acknowledgements
This work was supported by an ANR grant for the Ribo-Dyn project, which was awarded to CG (ANR-10-Blanc-1510). The team benefits from the support of the Labex Saclay Plant Sciences-SPS (ANR-10-LABX-0040-SPS), the Lidex BIG (Paris-Saclay University) and the I2BC crystallization platform (FRISBI ANR-10-INSB-05-01). This study has benefited from the facilities and expertise of the SICaPS and crystallization platforms of I2BC. JN and FL were supported by a PhD fellowship from University Paris-Sud. The authors acknowledge K. Cherouali for her contribution to initial kinetic analysis of E. coli PDF chimeras.
Author Contributions
C.G. and T.M. designed the research. C.G. supervised the overall project. W.B. performed N-terminomics analysis, F.L. and C.D. cloning and analysis of PDFs chimeras. S.F. X-ray crystal structures resolution. R.G. and J.N. performed all other experiments. C.G. and T.M. wrote the paper. All authors discussed the results and contributed to the final version of the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-11329-3
Accession codes: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository32, with the dataset identifier: PXD001983 and PXD006008 (reviewer account details: Username: reviewer95206@ebi.ac.uk Password: 3d6DUTj8). Coordinates and structure factors of the three Vp16 PDF structures have been deposited in the Protein Data Bank under accession codes 5MTC and 5MTD.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Thierry Meinnel, Email: thierry.meinnel@i2bc-paris-saclay.fr.
Carmela Giglione, Email: carmela.giglione@i2bc.paris-saclay.fr.
References
- 1.Bienvenut WV, Giglione C, Meinnel T. Proteome-wide analysis of the amino terminal status of Escherichia coli proteins at the steady-state and upon deformylation inhibition. Proteomics. 2015;15:2503–2518. doi: 10.1002/pmic.201500027. [DOI] [PubMed] [Google Scholar]
- 2.Giglione C, Boularot A, Meinnel T. Protein N-terminal methionine excision. Cell Mol Life Sci. 2004;61:1455–1474. doi: 10.1007/s00018-004-3466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meinnel T, Lazennec C, Blanquet S. Mapping of the active site zinc ligands of peptide deformylase. J Mol Biol. 1995;254:175–183. doi: 10.1006/jmbi.1995.0609. [DOI] [PubMed] [Google Scholar]
- 4.Dardel F, Ragusa S, Lazennec C, Blanquet S, Meinnel T. Solution structure of nickel-peptide deformylase. J Mol Biol. 1998;280:501–513. doi: 10.1006/jmbi.1998.1882. [DOI] [PubMed] [Google Scholar]
- 5.Becker A, et al. Iron center, substrate recognition and mechanism of peptide deformylase. Nat Struct Biol. 1998;5:1053–1058. doi: 10.1038/4162. [DOI] [PubMed] [Google Scholar]
- 6.Giglione C, Serero A, Pierre M, Boisson B, Meinnel T. Identification of eukaryotic peptide deformylases reveals universality of N-terminal protein processing mechanisms. Embo J. 2000;19:5916–5929. doi: 10.1093/emboj/19.21.5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giglione, C., Fieulaine, S. & Meinnel, T. Cotranslational processing mechanisms: towards a dynamic 3D model. Trends Biochem Sci34, 417–426, doi:S0968-0004(09)00121-2 [pii] 10.1016/j.tibs.2009.04.003 (2009). [DOI] [PubMed]
- 8.Giglione C, Fieulaine S, Meinnel T. N-terminal protein modifications: Bringing back into play the ribosome. Biochimie. 2015;114:134–146. doi: 10.1016/j.biochi.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Meinnel T, Blanquet S, Dardel F. A new subclass of the zinc metalloproteases superfamily revealed by the solution structure of peptide deformylase. J Mol Biol. 1996;262:375–386. doi: 10.1006/jmbi.1996.0521. [DOI] [PubMed] [Google Scholar]
- 10.Chan MK, et al. Crystal structure of the Escherichia coli peptide deformylase. Biochemistry. 1997;36:13904–13909. doi: 10.1021/bi9711543. [DOI] [PubMed] [Google Scholar]
- 11.Bingel-Erlenmeyer R, et al. A peptide deformylase-ribosome complex reveals mechanism of nascent chain processing. Nature. 2008;452:108–111. doi: 10.1038/nature06683. [DOI] [PubMed] [Google Scholar]
- 12.Meinnel T, Lazennec C, Dardel F, Schmitter JM, Blanquet S. The C-terminal domain of peptide deformylase is disordered and dispensable for activity. FEBS Lett. 1996;385:91–95. doi: 10.1016/0014-5793(96)00357-2. [DOI] [PubMed] [Google Scholar]
- 13.Fieulaine S, et al. The crystal structure of mitochondrial (Type 1A) peptide deformylase provides clear guidelines for the design of inhibitors specific for the bacterial forms. J Biol Chem. 2005;280:42315–42324. doi: 10.1074/jbc.M507155200. [DOI] [PubMed] [Google Scholar]
- 14.Escobar-Alvarez S, et al. Structure and activity of human mitochondrial peptide deformylase, a novel cancer target. J Mol Biol. 2009;387:1211–1228. doi: 10.1016/j.jmb.2009.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharon I, et al. Comparative metagenomics of microbial traits within oceanic viral communities. ISME J. 2011;5:1178–1190. doi: 10.1038/ismej.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frank JA, et al. Structure and function of a cyanophage-encoded peptide deformylase. ISME J. 2013;7:1150–1160. doi: 10.1038/ismej.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seguritan V, Feng IW, Rohwer F, Swift M, Segall AM. Genome sequences of two closely related Vibrio parahaemolyticus phages, VP16T and VP16C. J Bacteriol. 2003;185:6434–6447. doi: 10.1128/JB.185.21.6434-6447.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meinnel T, Blanquet S. Characterization of the Thermus thermophilus locus encoding peptide deformylase and methionyl-tRNA(fMet) formyltransferase. J Bacteriol. 1994;176:7387–7390. doi: 10.1128/jb.176.23.7387-7390.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meinnel T, Blanquet S. Enzymatic properties of Escherichia coli peptide deformylase. J Bacteriol. 1995;177:1883–1887. doi: 10.1128/jb.177.7.1883-1887.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ragusa S, Mouchet P, Lazennec C, Dive V, Meinnel T. Substrate recognition and selectivity of peptide deformylase. Similarities and differences with metzincins and thermolysin. J Mol Biol. 1999;289:1445–1457. doi: 10.1006/jmbi.1999.2832. [DOI] [PubMed] [Google Scholar]
- 22.Meinnel T, Patiny L, Ragusa S, Blanquet S. Design and synthesis of substrate analogue inhibitors of peptide deformylase. Biochemistry. 1999;38:4287–4295. doi: 10.1021/bi982622r. [DOI] [PubMed] [Google Scholar]
- 23.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith KJ, et al. Structural variation and inhibitor binding in polypeptide deformylase from four different bacterial species. Protein Sci. 2003;12:349–360. doi: 10.1110/ps.0229303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fieulaine S, et al. Trapping conformational states along ligand-binding dynamics of peptide deformylase: the impact of induced fit on enzyme catalysis. PLoS Biol. 2011;9:e1001066. doi: 10.1371/journal.pbio.1001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serero A, Giglione C, Meinnel T. Distinctive features of the two classes of eukaryotic peptide deformylases. J Mol Biol. 2001;314:695–708. doi: 10.1006/jmbi.2001.5175. [DOI] [PubMed] [Google Scholar]
- 27.Bracchi-Ricard V, et al. Characterization of an eukaryotic peptide deformylase from Plasmodium falciparum. Arch Biochem Biophys. 2001;396:162–170. doi: 10.1006/abbi.2001.2631. [DOI] [PubMed] [Google Scholar]
- 28.Fieulaine S, et al. A unique peptide deformylase platform to rationally design and challenge novel active compounds. Scientific reports. 2016;6:35429. doi: 10.1038/srep35429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang J, et al. Phylogenomic and biochemical characterization of three Legionella pneumophila polypeptide deformylases. J Bacteriol. 2006;188:5249–5257. doi: 10.1128/JB.00866-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin P, et al. Characterization of peptide deformylase homologues from Staphylococcus epidermidis. Microbiology. 2010;156:3194–3202. doi: 10.1099/mic.0.038174-0. [DOI] [PubMed] [Google Scholar]
- 31.Opella SJ. Relating structure and function of viral membrane-spanning miniproteins. Curr Opin Virol. 2015;12:121–125. doi: 10.1016/j.coviro.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vizcaino JA, et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41:D1063–1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ. Multiple sequence alignment with Clustal X. Trends Biochem Sci. 1998;23:403–405. doi: 10.1016/S0968-0004(98)01285-7. [DOI] [PubMed] [Google Scholar]
- 34.Page RDM. TREEVIEW: An application to display phylogenetic trees on personal computers. Comput Appl Biosci12, 357–358 (1996). [DOI] [PubMed]
- 35.Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colaert N, Helsens K, Martens L, Vandekerckhove J, Gevaert K. Improved visualization of protein consensus sequences by iceLogo. Nature methods. 2009;6:786–787. doi: 10.1038/nmeth1109-786. [DOI] [PubMed] [Google Scholar]
- 37.Ragusa S, Blanquet S, Meinnel T. Control of peptide deformylase activity by metal cations. J Mol Biol. 1998;280:515–523. doi: 10.1006/jmbi.1998.1883. [DOI] [PubMed] [Google Scholar]
- 38.Serero A, Giglione C, Sardini A, Martinez Sanz J, Meinnel T. An unusual peptide deformylase features in the human mitochondrial N-terminal methionine excision pathway. J Biol Chem. 2003;278:52953–52963. doi: 10.1074/jbc.M309770200. [DOI] [PubMed] [Google Scholar]
- 39.Bouzaidi-Tiali N, et al. Type 3 peptide deformylases are required for oxidative phosphorylation in Trypanosoma brucei. Mol Microbiol. 2007;65:1218–1228. doi: 10.1111/j.1365-2958.2007.05867.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.