

Abstract
A new codon property, codon directional asymmetry in nucleotide content (CDA), reveals a biologically meaningful genetic code dimension: palindromic codons (first and last nucleotides identical, codon structure XZX) are symmetric (CDA = 0), codons with structures ZXX/XXZ are 5′/3′ asymmetric (CDA = − 1/1; CDA = − 0.5/0.5 if Z and X are both purines or both pyrimidines, assigning negative/positive (−/+) signs is an arbitrary convention). Negative/positive CDAs associate with (a) Fujimoto's tetrahedral codon stereo-table; (b) tRNA synthetase class I/II (aminoacylate the 2′/3′ hydroxyl group of the tRNA's last ribose, respectively); and (c) high/low antiparallel (not parallel) betasheet conformation parameters. Preliminary results suggest CDA-whole organism associations (body temperature, developmental stability, lifespan). Presumably, CDA impacts spatial kinetics of codon-anticodon interactions, affecting cotranslational protein folding. Some synonymous codons have opposite CDA sign (alanine, leucine, serine, and valine), putatively explaining how synonymous mutations sometimes affect protein function. Correlations between CDA and tRNA synthetase classes are weaker than between CDA and antiparallel betasheet conformation parameters. This effect is stronger for mitochondrial genetic codes, and potentially drives mitochondrial codon-amino acid reassignments. CDA reveals information ruling nucleotide-protein relations embedded in reversed (not reverse-complement) sequences (5′-ZXX-3′/5′-XXZ-3′).
Keywords: Secondary structure, Codon-amino acid assignment, Mitochondrial genetic code, Synonymous codon, Alpha helix, Beta turn
Graphical Abstract
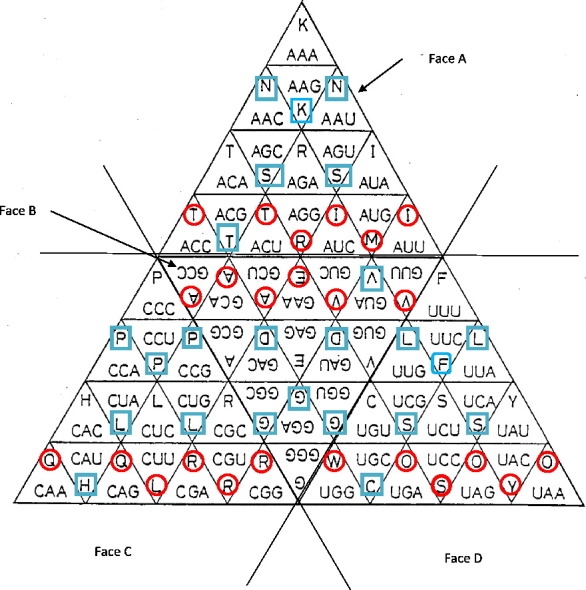
Fujimoto's tetrahedral genetic code representation and codon directional asymmetry, CDA.
Red circles: CDA < 0; blue squares: CDA > 0
Highlights
-
•
Codon directional asymmetry, CDA, is zero for palindromes (XZX), -/+ 1 for ZXX, XXZ.
-
•
CDA fits a tetrahedral genetic code representation.
-
•
tRNA synthetase classes I/II associate with CDA direction.
-
•
Antiparallel betasheet conformational indices of amino acids correlate negatively with mean CDA.
1. Introduction
The genetic code is optimised along several dimensions. Correlations between codon and amino acid properties have frequently been interpreted as resulting from evolutionary optimizations of the genetic code's codon-amino acid assignments. These minimise effects of: replicational/transcriptional nucleotide substitutions on amino acid hydrophobicity [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11] and along multiple properties [12]. The genetic code is also optimised in relation to other processes, such as tRNA misloading with non-cognate amino acids [13], [14], [15], [16]; ribosomal frameshifts [17], [18], [19], [20], [21], [22], [23]; and protein folding kinetics [24], [25], [26].
Another approach assumes that the genetic code coevolved with codon/amino acid metabolic pathways [27], [28], [29], [30], [31]. It remains unclear whether genetic code optimizations are circumstantial byproducts of the metabolic coevolution hypothesis [32], [33], [34], [35], [36], or whether some combination of both processes produced the genetic code [34], [37], [38], [39], [40], [41], [42].
Here we present a previously unknown dimension of the genetic code. Analyses suggest that the genetic code is optimised in relation to this new property. The property reflects differences between nucleotides at first versus second codon positions, as compared to differences between nucleotides at third versus second codon positions. In this context, previous analyses [43] showed that the subtraction of dipole moments of nucleotides at first and second codon positions correlate with hydrophobicities of corresponding amino acids, after accounting for another, previously reported, correlation between codon and amino acid hydrophobicities [44], [45]. Here analyses generalise the principle to all codon positions and nucleotide properties.
2. Codon Directional Asymmetry
The new codon property is derived from comparing two differences in nucleotide contents, the difference between nucleotides at first and second codon positions, and the difference between nucleotides at second and third codon positions. This defines a codon's directional asymmetry in nucleotide content, CDA. CDA reflects semi-quantitatively extents by which a nucleotide at either 5′ or 3′ codon extremity differs from the codon's two remaining nucleotides. Along this principle, palindromic codons with the same nucleotide at 5′ and 3′ extremities (at first and third positions, XZX (including codons with X = Z)) are symmetric, CDA = 0. When the nucleotide at the 5′ extremity belongs to a different nucleotide group (purine/pyrimidine) than the two other positions and the latter are identical (ZXX), CDA = − 1. When the nucleotide at the 3′ extremity differs from other positions (XXZ), CDA = + 1. Signs for 5′-and 3′-dominant CDAs are arbitrary, but necessarily opposite (positive versus negative).
2.1. Purines and Pyrimidines
For codons of types ZXX/XXZ, CDA = − 0.5/+0.5, when both X and Z are purines, or both pyrimidines. This reflects lesser purine-purine and pyrimidine-pyrimidine structural differences than for purine-pyrimidine comparisons. This principle assigns a CDA score also for some codons of type XZW, where all three nucleotides differ, and Z belongs to the same chemical group (purine or pyrimidine) as the nucleotide at either codon extremity. For codons where nucleotides Z and W are both purines/pyrimidines, X is the most different nucleotide (CDA = − 0.5), because chemical structural differences between X and Z are greater than between W and Z. According to that rationale, for codons where nucleotides X and Z are both purines (or both pyrimidines), W is the most different nucleotide (CDA = + 0.5).
2.2. Complementarity Between Nucleotides at Different Codon Positions
For some codons with structure XZW, Z does not belong to the same group (in terms of purines/pyrimidines) as any nucleotide at the other positions. In these cases, an additional rule determines which of the nucleotides among X or W, differs more from the two others. We propose that complementarity between canonical base pairs (C:G and A:T/U) defines that complementary nucleotide pairs are the most different pairs. Hence for codons with structure XZW, CDA = − 0.5 and CDA = + 0.5 when X is the canonical complement of Z, and when W is the complementary of Z, respectively. This rule set defines CDA for all 64 codons (Table 1).
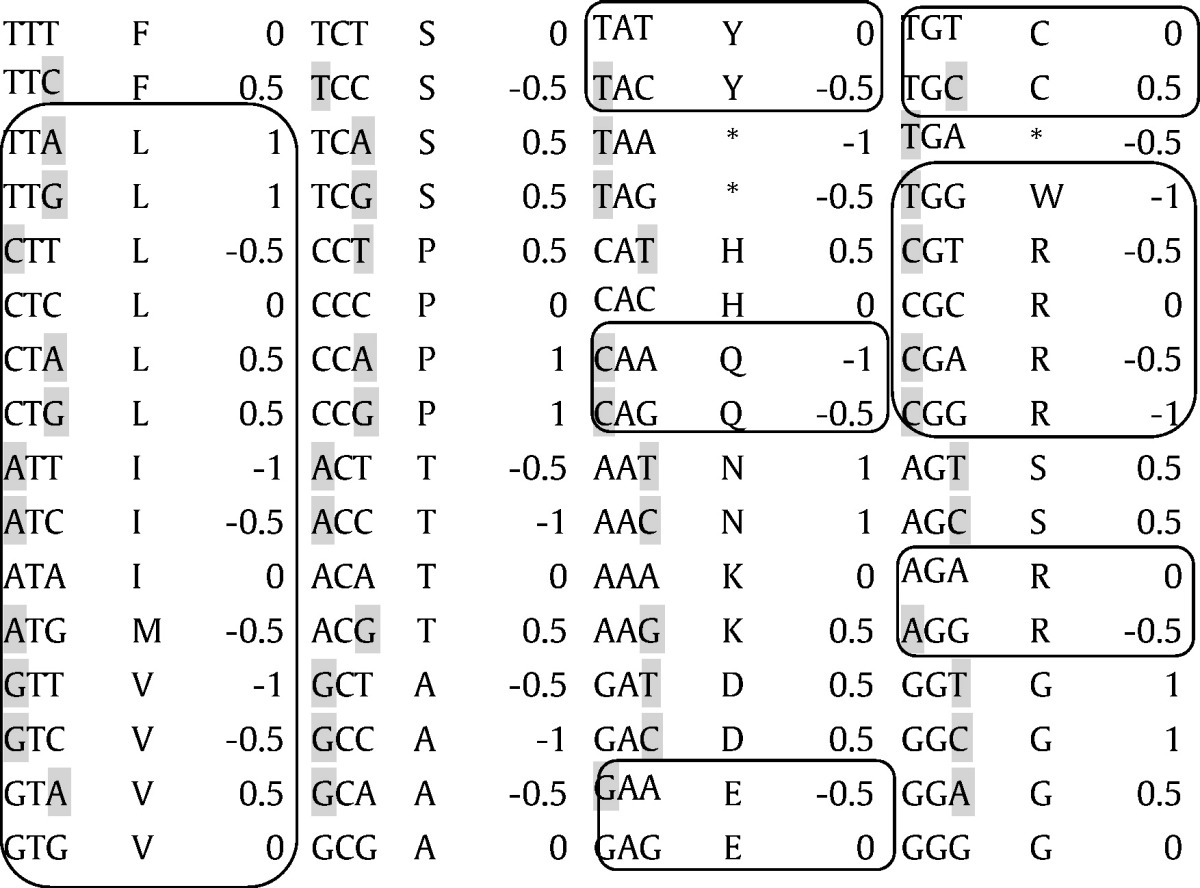
Table 1.
The genetic code's 64 codons and their codon directional asymmetry, CDA. Shaded nucleotides indicate the nucleotide at one of the codon's extremities that is the most different from nucleotides at other positions, along rules described in text, and which determines the dominant side of codon directional asymmetry: negative CDA when the first (5′) codon position has the most different nucleotide, and positive CDA when the third (3′) position has the most different nucleotide. Codons assigned to amino acids aminoacylated by class I tRNA synthetases are framed, remaining amino acids are aminoacylated by class II tRNA synthetases.
3. A New Dimension of the Genetic Code
The distribution of CDA in Table 1 is symmetric. Therefore, the genetic code table could probably be reordered so as to reveal graphically this symmetry, as done for other symmetry properties of the genetic code [46].
To what extent does CDA represent a dimension of the genetic code that is independent of other dimensions? In this respect, we compare Table 1 with the binary representation of the genetic code [47, therein figure 6], a rather complete 6-bit representation of each codon. It assigns to each codon position two binary values, the first representing the purine-pyrimidine divide, the second value represents whether the nucleotide forms two or three hydrogen interactions when in duplex conformation with an inverse-complementary strand. This defines two binary variables for each codon position, hence six binary variables for each codon.
Pearson correlation coefficients r of CDA with any of these six binary codon properties are ‘zero’, indicating that CDA is independent of each of these properties. Correlations with sums and subtractions between any pairs of these six binary values also yield r = 0. Results are identical if one pairs nucleotides according to keto versus amino nucleotides as previously reported [47], [48]. This means that CDA catches a genetic code dimension that differs from classically recognised codon properties.
3.1. Tetrahedral Representations and CDA
The genetic code can also be presented as a tetrahedron, with four equal triangular faces each subdivided into 16 equilateral, smaller triangles, representing the 64 codons. Castro-Chavez [49] reviews these representations, and proposes a tetrahedral representation, placing codons so that hydrophobic amino acids are central to each tetrahedral face, named faces A–D. Applying CDA to Castro-Chavez's tetrahedral representation, faces A and D tend to have CDA < 0, and faces C and B CDA > 0. Within each face, in total 19 triangle vertices (over all 4 faces) with CDA < 0 are common with vertices belonging to triangles with CDA > 0. This is very close to the 18 vertices expected if codons were randomly distributed in relation to CDA (P > 0.5, chi-square test), considering that 24 codons have CDA < 0, 16 have CDA = 0, and 24 have CDA > 0. Eleven among 24 vertices common between triangles from different faces of the tetrahedron are for triangles/codons with opposite CDA. This is slightly more than the 6.75 expected by random CDA distribution (P = 0.054, chi-square test). Hence the tetrahedral representation of Castro-Chavez [49] is random in relation to CDA within tetrahedral faces, and probably also between faces.
Fujimoto's tetrahedral codon stereo-table [50] is much more ordered in relation to CDA's distribution among and within tetrahedral faces (Fig. 1): Faces A–D each have six codons with CDA < 0, six codons with CDA > 0, and four codons with CDA = 0. Within each face, there are exactly two contacts between codons/triangles with opposite CDA. This total of eight contacts between triangles with opposite CDA is significantly less than the expected 18 contacts for randomly distributed CDA within faces of the tetrahedron (P = 0.018, chi-square test). There are no contacts between tetrahedron faces for codons/triangles with opposite CDA (P = 0.0096, chi-square test). Hence Fujimoto's tetrahedral representation is most compatible with the genetic code's symmetries implied by CDA in Table 1.
Fig. 1.

Fujimoto's tetrahedral codon stereo-table, a genetic code's representation that seems non-random in relation to codon directional asymmetry. The tetrahedron has four equal, equilateral faces (A–D), and consist each of 16 equilateral triangles representing each one codon. Red circles: CDA < 0; blue squares: CDA > 0. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Adapted from Fig. 1 at http://www.google.com/patents/US4702704.
The specific examples used here illustrate randomness versus CDA, and close to perfect reorganisation of the genetic code in relation to CDA, respectively. Other representations might reorganise the genetic code more optimally in relation to CDA. However, these representations may not relate to interpretable phenomena in the real world.
3.2. Codon Directional Asymmetry and Codon Participation in Error Correcting Codes
Genetic codes include a subjacent punctuation code called the natural circular code that enables retrieving the ribosomal translation frame [51], [52], [53], [54], [55]. Mechanisms for coding frame retrieval remain unknown, but are probably associated with circular code motifs conserved in tRNAs and ribosomal RNAs [56], [57], [58], [59]. Codon symmetry is particularly informative in relation to frame retrieval, as codons of type XZX (CDA = 0) have maximal capacity for reading frame retrieval [55], [60], [61], and have highest occurrences within various types of error-correcting codes [62]. Absolute values of CDA are lower for codons belonging to the natural circular code than for the remaining codons (P = 0.016, two tailed Mann-Whitney test). This principle is confirmed also when comparisons imply only codons belonging to the natural circular code: their absolute CDA increases with codon-specific reading frame retrieval (r = − 0.615, P = 0.002; rs = 0.44, P = 0.026, one tailed tests). Hence processes determining the near-universal natural circular code probably contributed biological functions to CDA.
4. Codon Directional Asymmetry and tRNA Synthetase Classes
CDA in Table 1 reflects a genetic code symmetry that does not follow the purine-pyrimidine, keto-amino, nor the weak-strong base-pairing patterns. A little known symmetry within the genetic code relates to Rumer's transformation [63], [64], [65], which replaces systematically all adenine (A) with cytosine (C) and vice versa, and also all guanine (G) with thymine (T) and vice versa. It is one among 23 bijective transformations [60], also called systematic nucleotide exchanges [66], [67] or ‘swinger’ transformations [68], [69], [70], [71]. RNA and DNA sequenced by several different methods and published in GenBank by various groups match these transformations. Hence while a priori, transformations such as Rumer's seem theoretical processes, they reflect biological realities, such as actual nucleotide sequences that were presumably produced by replication or transcription that systematically inserts a specific nucleotide instead of another specific nucleotide. This phenomenon of systematic nucleotide exchanges has similarities with isolated nucleotide misinsertions [60], [66], [67].
Rumer's transformation also correlates with a notable biological property, tRNA synthetase classes [72] of amino acids assigned to codons. The tRNA synthetases are enzymes that load amino acids to their cognate tRNA. The twenty tRNA synthetases form two groups of equal size, tRNA synthetase classes I and II based on structural homology [73], [74]. tRNA synthetases class I covalently link cognates to the 2′ hydroxyl group of the tRNA's last ribose, and class II to its 3′ hydroxyl group [75], [76].
The symmetry in the genetic code that correlates with tRNA synthetase classes exchanges nucleotides at the first and third codon positions along rule A ↔ C + G ↔ T (Rumer's transformation), and A ↔ G + C ↔ T at the second codon position. If instead of applying the nucleotide exchange rule A ↔ C + G ↔ T to the third codon position, one applies the exchange rule A ↔ T + C ↔ G, the symmetry between codons whose corresponding tRNA is aminoacylated by tRNA synthetase class I or class II is also recovered [77]. These symmetries by nucleotide exchanges are not mere theoretical considerations. Homologies of some DNA and RNA sequences in GenBank were detected after accounting for systematic nucleotide exchanges for the mitogenome [66], [67], [68], [69], [70], [71], [78], [79], [80]. In addition, the regular human mitogenome includes numerous repeats that can only be detected when assuming systematic exchanges [81], including palindromes [82].
CDA associates with tRNA synthetase classes. On average, codons assigned to amino acids aminoacylated by tRNA synthetases class I have CDA < 0 (15 among 21 codons (stops excluded), P = 0.039, two tailed sign test). For tRNA synthetases class II, the situation is opposite: most codons have CDA > 0 (17 among 24, CDA = 0, P = 0.032, two-tailed sign test). Sign tests are inadequate to handle codons with CDA = 0, therefore codons with CDA = 0 are excluded from these calculations. Mean CDA for tRNA synthetase classes differ significantly (two-tailed P = 0.002 for each t-test and Mann-Whitney test). These comparisons between means include codons with CDA = 0.
CDAs are averaged for codons assigned to specific amino acids. Mean CDA < 0 for 8 among 10 amino acids for class I; and CDA > 0 for 8 among 10 amino acids for class II (P = 0.006, two-tailed sign test for each tRNA synthetase class). Exceptions are Cys and Leu for class I, and Ala, and Thr for class II. Overall, the sign of mean CDA for codons assigned to an amino acid follows expected patterns (class I, CDA < 0; class II, CDA > 0) for 16 among 20 amino acids/tRNA synthetases (P = 0.00296, one tailed sign test).
Note that stop codons have CDA < 0, predicting tRNA synthetase class I. However, the tRNA synthetase of pyrrolysine, which is inserted at some stop codons, belongs to tRNA synthetase class II [83]. Exceptions might reflect historical constraints on the genetic code's genesis [77].
Hence the rationale defining CDA reveals a symmetry that is close to that of the combination of nucleotide exchanges that reveal the genetic code's symmetry in relation to tRNA synthetase classes. However, the rationale behind CDA is simpler and perhaps more amenable to mechanistic reduction.
4.1. Alternative Scores for Codons with CDA = | 0.5 |
Three different types of codons get CDA = | 0.5 |, based on different rationales: (a) codons with structures ZXX/XXZ where both X and Z are purines/pyrimidines; (b) codons with structure XZW where Z belongs to the same nucleotide family (purine/pyrimidine) as either X or W; and (c) codons with structure XZW where Z belongs to a different nucleotide family than X and Z. This scoring is somewhat arbitrary, and might not be optimal to reflect biological properties. Keeping signs, we rescore each of these three codon types with values | 0.25 | and | 0.75 |, resulting in different scoring systems for these three codon groups: alternative CDAs of groups (a, b, c) are (0.5, 0.25, 0.75), (0.5, 0.75, 0.25), (0.25, 0.5, 0.75), (0.25, 0.75, 0.5), (0.75, 0.5, 0.25), and (0.75, 0.25, 0.5). CDA of codons with CDA = 0 and CDA = | 1 | remain unchanged. These different scoring systems do not alter the strength of the CDA-tRNA synthetase class association: according to all these scoring systems, the same 8 among 10 codon families in class I have CDA < 0, and 8 among 10 amino acids in class II have CDA > 0. Excluding palindromic codons (CDA = 0) from calculations does not change results.
This heuristic approach suggests that associations between tRNA synthetase classes (an ancient property of the translational apparatus) and CDA are robust in relation to CDA's semi-quantitative scoring.
5. Translation Kinetics
The tRNA synthetase classes differ in the position of aminoacylation of the amino acid on the tRNA's acceptor stem. This probably affects the spatial kinetics of peptide elongation. We suggest that CDA also affects the spatial kinetics of codon-anticodon interactions in the ribosome's translational core (site P [84]; site A [85]). Hence both tRNA synthetase class and CDA would affect cotranslational protein folding, meaning folding during the process of peptide extension by ribosomal translation [86], [87], [88], [89], [90], [91], [92], [93], [94], [95], [96], [97]. Tentatively, we consider that associations between CDA and tRNA synthetase classes suggest synergistic effects on cotranslational protein folding by each CDA and tRNA synthetase class.
Note that cotranslational protein folding does not occur for all proteins [98]. Cotranslational protein folding frequently increases the yield of proper folds, but is not always an absolute requirement [99], [100], [101], [102], [103]; yet decreases misfolding probabilities [104], [105], [106]. Among others, at least in some cases, cotranslational folding requires complete protein structural subdomains [107], [108]. Cotranslational protein folding following the sense of translation (from the N terminal) predicts more accurately protein structures than when proceeding in the opposite sense (from the C terminal) [109], [110], indicating that cotranslational protein folding is a reality for most proteins. Nevertheless, cell free protein folding shows that cotranslational folding is not always required [111].
mRNA properties affecting translation speed and ribosomal pausing [112], [113], [114], also affect protein folding independently of that protein's amino acid sequence. Synonymous codons associate with different types of protein secondary structures [115], [116], in particular for clusters of rare codons on mRNAs [117], [118], [119]. These associations might explain effects of synonymous single nucleotide polymorphisms on protein function [120], [121], [122], [123] and are in line with selection at amino acid level that affects synonymous codon choice [124], [125].
More specifically, rare codons concentrate in mRNA regions that code for transmembrane helical structures [116]. Optimization of codon usage means that organisms match codon usage frequencies with anticodons of common tRNAs [126], [127], [128], [129], [130], [131], [132], [133], speeding translation, affecting cotranslational protein folding [134]. Lopez and Pazos [135] suggest that proper folding into transmembrane structures requires specific spatial kinetics and particular accuracy in the process. Cotranslational protein folding is most apparent on alpha helices and betasheet secondary structures [136], [137], [138], [139], [140]. Hence one expects associations between CDA and these conformational indices of amino acids. Chemical kinetics of the transfer of the amino acid loaded on the tRNA's acceptor stem to the elongating peptide (kinetic estimates from [141]) also constrain codon-anticodon interactions [43].
Following these rationales, CDA might reflect (a) indirectly tRNA synthetase classes and their effects on amino acid positioning during peptide elongation; and (b) directly the spatial kinetics of codon-anticodon interactions, such as tRNA-mRNA approach angles during codon-anticodon duplex formation in the ribosomal translational core(s). These two components should affect according to the cotranslational protein folding hypothesis folding patterns of elongating peptides. Hence CDA is predicted to correlate with amino acid secondary structure conformational parameters for alpha helices, beta turns and/or betasheets (conformational indices are from [142], [143], [144], [145]). The main candidates are the conformational parameters associated with transmembrane foldings (beta turns, and/or parallel and antiparallel betasheets, from references [146], [147]).
6. Antiparallel Betasheet Formation and Codon Directional Asymmetry
The hypothesis that CDA associates with cotranslational protein folding predicts correlations between CDA and secondary structure conformation parameters. Betasheets are the major secondary structures found in transmembrane proteins, antiparallel betasheets are more frequent than parallel betasheets [147]. Biases in tRNA synthetase amino acid contents correlate with the amino acid's antiparallel betasheet conformation parameter [148]. Hence, we predict correlations between CDA and conformation parameters, and in particular antiparallel betasheet conformation parameters.
Indeed, antiparallel betasheet conformation parameters correlate negatively with mean CDA of codons assigned to the amino acid according to the standard genetic code (Pearson correlation coefficient r = − 0.642, two-tailed P = 0.0023; non-parametric Spearman rank correlation coefficient rs = − 0.564, two-tailed P = 0.01; Fig. 2). In contrast, and functioning as a negative control, the correlation between mean CDA and parallel betasheet conformation parameters is not statistically significant (r = − 0.28, two-tailed P = 0.23, not shown). The presumed effect of CDA is specific for formation of antiparallel, not parallel, betasheets.
Fig. 2.

Antiparallel betasheet conformation parameter of amino acids as a function of the mean codon directional asymmetry (CDA) of codons assigned to that amino acid, for the standard genetic code. Amino acids aminoacylated by tRNA synthetases from class I have open circles, filled circles are for tRNA synthetases from class II.
The variation around the regression line is similar for negative and positive CDA ranges (Fig. 2). Hence the determinism of CDA on conformation is comparable for 5′ versus 3′ CDA dominance: effects are independent of coding importance of codon positions. In other words, the ‘information’ in CDA that is relevant to protein secondary structure is similar for asymmetry at first and third codon positions. Alternative scores (Section 4.1) do not change qualitatively the results (P values for rs remain above 0.05).
The correlation between mean CDA of codons assigned to amino acids and these amino acids' antiparallel betasheet conformational indices might be due to transitivity, due to associations between CDA and tRNA synthetase classes (see above section) and the association between tRNA synthetase class and conformational indices. In order to control for effects of tRNA synthetase classes, we calculate mean CDA and mean antiparallel betasheet index separately for each tRNA synthetase class. These means are subtracted from CDA and conformational indices of each amino acid in that respective class. These values are residual CDA and conformational indices after excluding effects of tRNA synthetase classes. Residual CDA and residual antiparallel betasheet indices correlate negatively (r = − 0.435, P = 0.0275; rs = − 0.461, P = 0.0205, one tailed tests). Hence the correlation between CDA and antiparallel betasheet indices is not indirect, through colinearity with tRNA synthetase classes.
The association between CDA and antiparallel betasheet indices has rs with P < 0.05 for eight among ten alternative scores (as in Section 4.1) after controlling for tRNA synthetase class. The genetic code seems structured so as to enable synergistic effects of CDA and tRNA synthetase classes on antiparallel betasheet formation, presumably by cotranslational protein folding.
Independently of the correlation between CDA and antiparallel betasheet conformation parameters, a weaker correlation exists between CDA and alpha-helix conformation parameters (r = − 0.556, P = 0.011; rs = − 0.499, P = 0.05, two-tailed test, not shown). This further correlation confirms that CDA affects protein folding. To our knowledge, these are the first described correlations between a codon property and secondary structure conformational parameters of assigned amino acids. CDA < 0 associates independently with each alpha and antiparallel beta conformational indices, in line with the literature on cotranslational protein folding [136], [137], [138], [139], [140]. Hence according to the working hypothesis, similar kinetic conditions favor each of these two very different secondary structures. Presumably, factors other than CDA (for example chain polarity) determine whether an alpha helix rather than an antiparallel betasheet is initiated during peptide elongation.
7. Codon Directional Asymmetry and Prediction of Protein Secondary Structure
The correlation between CDA and conformation parameters might have two causes. First, it could be intrinsic to the genesis of the genetic code, but relatively inconsequent to modern organisms. Secondly, CDA still affects protein folding. In the latter case, correlations between CDA and secondary structure conformation parameters could explain that some synonymous mutations perturb protein function. Indeed, several amino acids have some synonymous codons with opposite CDA, such as for alanine, leucine, serine and valine. Putatively, this would indicate that for these amino acids, synonymous codons with CDA < 0 occur preferentially for mRNA regions coding for antiparallel betasheets, and those with CDA > 0 in other mRNA regions.
Codon usage frequencies are adapted to minimise effects of mutations and translation errors [149], [150], [151], [152]. Hence weighing mean CDA for a given amino acid according to observed synonymous codon usages might increase correlations between CDA and conformation parameters. However, this is not the case for the pool of genes encoded by the human nucleus, nor those coded by the human mitogenome: correlations become in both cases weaker (not shown).
CDAs of stop codons are negative, suggesting a bias for amino acids with high tendencies to participate in antiparallel betasheets when amino acids are inserted at stop codons. Indeed, the evolution of mitochondrial genetic codes seems best reconstructed when assuming insertion of amino acids at stops [153], in line with coevolution between predicted suppressor tRNAs [154], [155], [156] and protein alignment analyses [16], [78], [79], [157], [158], [159], [160], [161]. However, frequencies of amino acids inserted at stops [71], [162], [163], [164], [165], [166] do not significantly correlate with antiparallel betasheet conformation parameters.
This does not mean that associations between synonymous codons in modern mRNAs and secondary structures of modern proteins do not exist. However, this suggests that testing these predictions is not as straightforward as it seems. Among others, secondary structure annotations available in GenBank don't indicate whether a betasheet is parallel or antiparallel. Hence these tests will require involvement of more adequately equipped specialised proteomics teams (for example Caudron and Jestin [147]). Until then, the contribution of CDA for improving secondary structure predictions [26], [167], especially such based on optimization of multiple approaches [168], will remain speculative.
8. Mitochondrial Genetic Codes Optimise Codon Directional Asymmetry
Many variant genetic codes are from mitochondria [169]. The reduced mitogenomes almost exclusively encode for mitochondrial transmembrane proteins, which include mainly antiparallel betasheets. In contrast, nuclear genomes encode also for large proportions of cytosolic proteins, which include much fewer betasheets. Hence, we predict that the correlation between CDA and antiparallel betasheet conformation parameters is weaker for genetic codes associated with nucleus-encoded proteomes than for mitochondrial genetic codes. The correlation in Fig. 2 (for the standard genetic code) is calculated for the remaining genetic codes listed by Elzanowski and Ostell [169], after recalculating mean amino acid CDA, considering codon-amino acid reassignments. The correlation's strength for each genetic code is estimated by the Pearson correlation coefficient r.
The correlation between tRNA synthetase classes and CDA is also calculated, by assigning to tRNA synthetase classes I and II values ‘1’ and ‘2’, respectively, and calculating the Pearson correlation coefficients r between this dummy variable representing tRNA synthetase classes and the mean CDA of codons assigned to the corresponding amino acid, for each variant genetic code. The CDA-antiparallel betasheet correlation coefficients are plotted as a function of the CDA-tRNA synthetase class correlation coefficients for the various genetic codes (Fig. 3). The line in Fig. 3 indicates y = x, meaning that both correlations have equal strengths. Note that in context of this particular section, Pearson correlation coefficients are used as quantitative estimates of the strength of a correlation, not as test statistics to infer that a correlation exists.
Fig. 3.

Correlation between antiparallel betasheet conformation parameter of amino acids and mean directional asymmetry (CDA) of codons assigned to that amino acid as a function of the correlation between CDA and the tRNA synthetase class for the corresponding amino acid for different genetic codes. Correlations are Pearson correlation coefficients. Filled/open circles are nuclear/mitochondrial genetic codes, shaded circles are for genetic codes existing in nuclei and mitochondria. The line indicates y = x. Nuclear genetic codes tend to optimise the association between CDA and tRNA synthetase classes, mitochondrial genetic codes tend to optimise the association between CDA and the antiparallel betasheet conformation parameter. Most mitogenome-encoded proteins are transmembrane proteins, hence antiparallel betasheets are particularly frequent in these proteins. Hence genetic code evolution optimises the CDA-antiparallel betasheet association in mitochondria. Open circles: mitochondrial genetic codes; filled circles: nuclear genetic codes; shaded circles: genetic codes used in nuclei and mitochondria.
All eleven mitochondrial genetic codes have stronger correlations between CDA and antiparallel betasheet conformation parameters than between CDA and tRNA synthetase classes. Obtaining this result for all eleven mitochondrial genetic codes has P = 0.00049 (two-tailed sign test). Two additional genetic codes occur in nuclear and mitochondrial genomes: the standard genetic code, and the Mycoplasma/Spiroplasma genetic code that also occurs in mold, protozoan and coelenterate mitochondria. These two genetic codes follow the pattern observed for the eleven genetic codes only found in mitochondria.
Six among eight genetic codes associated only with nuclear genomes are below the line y = x in Fig. 2, indicating that the CDA-tRNA synthetase class correlation is frequently a greater constraint for nuclear genetic codes than mitochondrial ones. This qualitative difference between nuclear and mitochondrial genetic codes has P = 0.001 (two-tailed Fisher exact test). This divide might reflect different constraints on protein folding for populations of mitochondrion-encoded versus nucleus-encoded proteins. This pattern might indicate stronger synergy between effects of CDA and tRNA synthetase class on cotranslational folding for nucleus-encoded proteins translated in the cytosol than mitogenome-encoded ones.
These results indicate that associations between CDA and conformation parameters, and between CDA and tRNA synthetase classes, drive differentially evolutions of mitochondrial versus nuclear genetic codes. Tentatively, amino acid positioning on the tRNA acceptor stem is less relevant for mitochondrial translation than CDA, the opposite is true for cytosolic translations.
9. Whole Organism Properties and Codon Directional Asymmetry
Whole organism properties correlate sometimes with molecular properties [170], [171]: morphological versus molecular rates of evolution [172], [173], [174]; growth rates and genome sizes [175], [176], [177], [178]; and metabolic costs of protein synthesis [179]; body temperatures and predicted expanded codons [180], [181], [182], [183]; developmental stability estimated by lateral differences between bilateral morphological traits and accuracy of various aspects of molecular processes, such as replication [184], ribosomal translation [20], [185], and tRNA loading [14]. CDA might also correlate with whole organism properties.
9.1. Lepidosaurian Body Temperature and Codon Directional Asymmetry
Temperature reflects noise in molecular movements, potentially affecting contranslational protein folding, which indeed depends on optimal temperatures [186]. Hence, formation of antiparallel betasheets might be impeded by high temperatures. Therefore, we expect that negative CDAs promote betasheet formation despite high temperature. Hence when comparing the mean CDA calculated across all 13 membrane-embedded mitogenome-encoded proteins of different organisms, we expect that organisms with high temperatures have low mean CDA for the same homologous genes. Indeed, the mean CDA of lepidosaurian mitochondrion-encoded proteins decreases with their body temperature (ro = − 0.283, one tailed P = 0.018, Fig. 4, temperature data compiled for species with complete mitogenome available in GenBank by Seligmann and Labra [183], therein Table 1).
Fig. 4.

Lepidosaurian body temperature as a function of mean codon directional asymmetry of codons in protein coding genes encoded by complete mitogenomes available in GenBank. Compilation of body temperatures and mitochondrial genomes as in Table 1 of Seligmann and Labra [183]. Agamidae are indicated by triangles, Gekkota by crosses and Lacertidae by filled circles. Species from various other families have open circles.
This correlation is also statistically significant within the family Lacertidae (ro = − 0.842, one-tailed P = 0.001). It is negative for Agamidae (ro = − 0.255, one-tailed P = 0.238), Gekkota (ro = − 0.25, one-tailed P = 0.258), iguanid lizards (ro = − 0.333, one-tailed P = 0.21), Varanidae (ro = − 1.00, one-tailed P = 0.005) and Chamaeleo (ro = − 0.40, one-tailed P = 0.30) and for the pool of remaining isolated species from various families (Heloderma, Shinisaurus, Lepidophyma, Sphenodon (ro = − 0.238, one tailed P = 0.285). The correlation is positive for Amphisbaenia (ro = 0.40, one tailed P = 0.30). Hence seven among eight phylogenetically independent samples yield negative correlations, which is a significant majority according to a sign test (P = 0.0176). Considering the qualitative direction of correlations for phylogenetically independent species samples follows the principle of phylogenetically independent contrasts [187]. This confirms that positive results are not confounded by phylogenetic inertia among species. Results of this sign test are valid independently of P value adjustments for multiple tests.
GC contents could confound this correlation, because G:C base pairs are linked by three hydrogen interactions, while A:T and A:U base pairs by only two hydrogen bridges. Hence GC contents usually increases with temperature, as it confers higher stability to structures formed by nucleotide chains [188], [189], [190]. However, GC codon content does not correlate with body temperature for mitochondria of the above mentioned lepidosaurian species (r = − 0.0425, one-tailed P = 0.379). This is in line with results from various analyses [191], [192], [193]; that didn't detect the expected GC-temperature correlation. This negative control stresses that the association in Fig. 4 is not trivial.
9.2. Developmental Stability and CDA
Molecular noise (in terms of erratic molecular movements) affecting mitochondrial transmembrane protein folding might cause developmental inaccuracies at the whole organism level. Hence, we explore the correlation between mean CDA of mitogenome-encoded proteins and developmental stability of the 4th toe of Lepidosauria, estimated by the Pearson correlation coefficient r between subdigital lamellae counts on left and right sides (data from [194], [195], [196], [197], [198]). Developmental stability/accuracy decreases with mean CDA of mitogenome-encoded proteins (ro = − 0.316, one-tailed P = 0.0235), as expected by the working hypothesis. However, analyzing separately species grouped according to phylogenetic groups (as in previous section) yields negative correlations only in five among eight groups, which is not statistically significant at P < 0.05 according to a one sided sign test. Hence this preliminary result on CDA and developmental stability is at best tentative.
9.3. Lifespan and CDA
Patterns between CDA and temperature, and CDA and developmental stability (Fig. 4, Fig. 5) suggest that CDA < 0 for mitogenome-encoded proteins associates with longevity. For this purpose, we compared codon contents in mitogenomes of 112 semi-supercentenarians and 96 centenarians versus those of 97 healthy young controls [199], [200] (Table 3). Codons with CDA = − 1 are more frequent in supercentenarians than in controls for seven among eight comparisons, which is a significant majority according to a one tailed sign test (P = 0.0176). No tendencies are observed for other CDA values (− 0.5, 0, 0.5, 1), nor for comparisons between centenarians and controls. The result is suggestive that CDA < 0 could contribute to extreme longevity, but the high number of tests and the small differences in codon frequencies stress cautious interpretation.
Fig. 5.

Developmental stability of bilateral counts of subdigital lamellae on 4th toe of Lepidosauria (estimated by Pearson correlation coefficients r between counts on left and right sides) as a function of mean codon directional asymmetry (CDA) of codons in all 13 genes of mitogenome-encoded transmembrane proteins.
Table 3.
Mean codon frequencies (promil) in the 13 mitogenome-encoded genes of three groups of Japanese males: 97 healthy controls, 112 semi-supercentenarians and 96 centenarians from references [199], [200].
| Codon | CDA | Control | Super | Cent | Codon | CDA | Control | Super | Cent |
|---|---|---|---|---|---|---|---|---|---|
| UUU | 0 | 20.34 | 20.32 | 20.34 | UAU | 0.00 | 12.11 | 12.13 | 12.15 |
| UUC | 0.5 | 36.59 | 36.61 | 36.60 | UAC | − 0.50 | 23.39 | 23.35 | 23.35 |
| UUA | 1 | 19.07 | 19.04 | 19.01 | UAA | − 1.00 | 2.09 | 2.09 | 2.10 |
| UUG | 1 | 4.59 | 4.57 | 4.57 | UAG | − 0.50 | 0.81 | 0.81 | 0.80 |
| CUU | − 0.5 | 17.02 | 17.03 | 17.00 | CAU | 0.50 | 4.71 | 4.71 | 4.71 |
| CUC | 0 | 43.89 | 43.88 | 43.89 | CAC | 0.00 | 20.80 | 20.80 | 20.79 |
| CUA | 0.5 | 72.88 | 72.87 | 72.90 | CAA | − 1.00 | 21.61 | 21.61 | 21.59 |
| CUG | 0.5 | 11.74 | 11.78 | 11.79 | CAG | − 0.50 | 2.08 | 2.09 | 2.10 |
| AUU | − 1 | 33.06 | 33.03 | 33.07 | AAU | 1.00 | 8.38 | 8.39 | 8.41 |
| AUC | − 0.5 | 51.34 | 51.30 | 51.25 | AAC | 1.00 | 34.72 | 34.74 | 34.72 |
| AUA | 0 | 43.77 | 43.81 | 43.82 | AAA | 0.00 | 22.37 | 22.40 | 22.39 |
| AUG | − 0.5 | 10.72 | 10.74 | 10.73 | AAG | 0.50 | 2.63 | 2.61 | 2.61 |
| GUU | − 1 | 8.19 | 8.19 | 8.18 | GAU | 0.50 | 3.89 | 3.92 | 3.93 |
| GUC | − 0.5 | 12.61 | 12.64 | 12.68 | GAC | 0.50 | 13.43 | 13.38 | 13.38 |
| GUA | 0.5 | 18.39 | 18.40 | 18.36 | GAA | − 0.50 | 16.87 | 16.85 | 16.86 |
| GUG | 0 | 4.71 | 4.71 | 4.73 | GAG | 0.00 | 6.23 | 6.25 | 6.24 |
| UCU | 0 | 8.42 | 8.42 | 8.41 | UGU | 0.00 | 1.30 | 1.30 | 1.31 |
| UCC | − 0.5 | 25.99 | 25.99 | 26.00 | UGC | 0.50 | 4.50 | 4.50 | 4.50 |
| UCA | 0.5 | 21.81 | 21.81 | 21.80 | UGA | − 0.50 | 24.48 | 24.46 | 24.42 |
| UCG | 0.5 | 1.80 | 1.79 | 1.80 | UGG | − 1.00 | 2.91 | 2.92 | 2.97 |
| CCU | 0.5 | 10.70 | 10.66 | 10.68 | CGU | − 0.50 | 1.80 | 1.80 | 1.80 |
| CCC | 0 | 31.39 | 31.43 | 31.42 | CGC | 0.00 | 6.80 | 6.80 | 6.80 |
| CCA | 1 | 13.71 | 13.71 | 13.72 | CGA | − 0.50 | 7.40 | 7.40 | 7.40 |
| CCG | 1 | 1.78 | 1.78 | 1.78 | CGG | − 1.00 | 0.50 | 0.50 | 0.50 |
| ACU | − 0.5 | 13.41 | 13.39 | 13.43 | AGU | 0.50 | 3.70 | 3.69 | 3.69 |
| ACC | − 1 | 40.44 | 40.45 | 40.36 | AGC | 0.50 | 10.26 | 10.27 | 10.29 |
| ACA | 0 | 34.80 | 34.78 | 34.77 | AGA | 0.00 | 0.30 | 0.29 | 0.30 |
| ACG | 0.5 | 2.57 | 2.58 | 2.58 | AGG | − 0.50 | 0.30 | 0.30 | 0.30 |
| GCU | − 0.5 | 11.73 | 11.73 | 11.74 | GGU | 1.00 | 6.30 | 6.32 | 6.33 |
| GCC | − 1 | 32.55 | 32.56 | 32.60 | GGC | 1.00 | 22.88 | 22.87 | 22.85 |
| GCA | − 0.5 | 21.60 | 21.58 | 21.60 | GGA | 0.50 | 18.00 | 18.02 | 18.03 |
| GCG | 0 | 2.08 | 2.09 | 2.08 | GGG | 0.00 | 8.55 | 8.52 | 8.51 |
Overall, analyses weakly confirm predictions for correlations between CDA and whole organism properties (body temperature, developmental stability, longevity). These suggest that analyses considering additional information, such as residue-specific location in three dimensional protein structures, might yield positive results. More up-to-date methods for including phylogenetic information in relation to evolutionary adaptive optima might also alter conclusions [201].
9.4. Replicational Deamination Gradients
Mitochondrial DNA replication differs from nuclear chromosome replication [202] and is usually strand asymmetric resulting in replicational deamination gradients where C → T and A → G substitutions exceed reversed mutations proportionally to the time spent single stranded during replication [203], [204], [205], [206], [207], [208]. Inverting the direction of the light strand replication origin also inverts the direction of the replicational deamination gradients [209], [210], [211], [212], [213], [214], [215], [216], [217]). These physico-chemical mutation pressures could affect CDA according to gene locations on the mitogenome, independently of protein properties.
Mean CDA of the 13 human mitogenome-encoded proteins does not correlate with time spent single stranded by that gene during replication, assuming light strand replication initiates at the OL, the light strand replication origin (ro = 0.033, P = 0.92, two tailed test). DNA templating for tRNA genes presumably also functions sometimes as replication origins [184], [218], [219]. Integrating the possibility of these multiple replication origins yields gene-wise single-strand durations that converge with transcriptional singlestrandedness [220]. The correlation between transcriptional duration of singlestrandedness and mean gene CDA is also not statistically significant (ro = 0.418, P = 0.156, two tailed test). Hence, we do not detect statistically significant effects of mutation pressures on mean CDA of human mitochondrial genes.
9.5. Adjusting Statistical Significances for Multiple Tests
Analyses that include several tests have to adjust P values according to the number of tests. This is because, when deciding that a result is positive at P < 0.05, when k tests are performed, on average, k × 0.05 tests are false positives. Bonferroni's correction considers that when performing k tests, results are statistically significant at P = 0.05 for any specific test among k tests if P < 0.05/k. This correction is reputedly overconservative [221], [222]. Unadjusted Ps minimise risks of false negative results, Bonferroni's method minimises risks of false positives. The Benjamini-Hochberg adjustment for false discovery rates [223] optimises between these two risks and seems most adequate [224]. This method ranks all k P values from highest to lowest (best), adjusted Ps are the product of P with k divided by the rank i, where i ranges from 1 to k. This means that the ‘best’ (lowest) P is unchanged, and that the ‘worst’ (highest) P value after adjustment follows Bonferroni's adjustment. Ps with intermediate rank are intermediate between these extremes.
Here we consider only P values from non-parametric tests, when also parametric tests were done. For some of the associations described, more than one test was done, but these are then summarised by a test that integrates the previous tests. Adjustments consider in these cases only the latter P value. Along this approach a total of 29 hypothesis tests were done, as detailed in Table 2. Control analyses (such as with GC contents, and mutational gradients, in total 29 tests) are also included in the list of multiple tests. These are not related to the main CDA hypothesis and could arguably be excluded. Excluding controls does not alter qualitatively results of the adjustments of P values.
Table 2.
Benjamini-Hochberg adjustment of P values of non-redundant hypothesis tests. The total number of tests including controls is 58 (Rank 1 and adjusted P1), excluding controls and considering only tests pertaining directly to CDA, there are 29 tests (rank 2 and adjusted P2). Only tests with unadjusted P < 0.05 are shown, all these tests pertain to CDA directly.
| Test | P | Rank 1 | Adj P1 | Rank 2 | Adj P2 |
|---|---|---|---|---|---|
| Number of mitochondrial genetic codes above line in Fig. 3 | 0.00050 | 58 | 0.00050 | 29 | 0.00050 |
| Number of mitochondrial genetic codes above line vs number of nuclear codes below line in Fig. 3 | 0.00100 | 57 | 0.00102 | 28 | 0.00100 |
| tRNA synthetase classes and CDA | 0.00200 | 56 | 0.00207 | 27 | 0.00207 |
| tRNA synthetase classes and mean CDA of codons assigned to amino acids | 0.00295 | 55 | 0.00312 | 26 | 0.00319 |
| Contacts between Fujimoto's tetrahedron faces | 0.00960 | 54 | 0.01031 | 25 | 0.01075 |
| Correlation CDA-antiparallel betasheet indices | 0.01000 | 53 | 0.01094 | 24 | 0.01167 |
| Absolute CDA and circular code | 0.01600 | 52 | 0.01785 | 23 | 0.01948 |
| Temperature and mean CDA of 13 lepidosaurian mitogenome-encoded proteins | 0.01760 | 51 | 0.02002 | 22 | 0.02240 |
| Human lifespan and mitochondrial codon usages-CDA | 0.01760 | 50 | 0.02042 | 21 | 0.02347 |
| Contacts within Fujimoto's tetrahedron faces | 0.01800 | 49 | 0.02131 | 20 | 0.02520 |
| Partial correlation CDA-antiparallel betasheet indices | 0.02050 | 48 | 0.02477 | 19 | 0.03021 |
| CDA-developmental stability | 0.02350 | 47 | 0.02900 | 18 | 0.03656 |
| Absolute CDA-reading frame retrieval capacity | 0.02600 | 46 | 0.03278 | 17 | 0.04435 |
| Alpha helix-CDA | 0.05000 | 45 | 0.06444 | 16 | 0.08750 |
The analysis for codon usage associated with lifespan includes 10 tests (for CDA values − 1, − 0.5, 0, 0.5, and 1, and this for comparisons between controls and centenarians, and between controls and supercentenarians). Among unadjusted P values with P < 0.05, only the adjusted P value for the correlation between mean CDA of codons assigned to amino acids and the amino acids' alpha helix conformational indices is above 0.05. This occurs when considering all 58 tests, and when considering only the 29 tests directly pertaining to the working hypothesis about CDA. Qualitatively, results of P adjustments are robust in relation to numbers of tests included in this analysis: for example, for P with rank 17 to get P > 0.05 after adjustment, one requires k = 89 when including negative controls and k = 33 when excluding negative controls. Hence even if one was to increase numbers of tests included in the analyses, the relevant cutoff property of the distribution of adjusted Ps is relatively robust, so that issues related to multiple tests are unlikely to alter conclusions.
10. A New Directional Codon Dimension
Intuitively, it seems conceivable that CDA, via its plausible effects on codon-anticodon interactions, affects cotranslational protein folding. However, developing a mechanistic scenario that explains why this effect should occur for antiparallel betasheets rather than parallel ones, or for alpha helices, is more difficult. We propose that some (unspecified) conformations depend on translational speed. Other conformations might be favored by random movements of the tRNA's loaded acceptor stem in relation to the elongating peptide, versus more directed movements of that stem, hence some ratio between kinetic noise and direction. Our educated guess (but nothing beyond that) is that CDA relates more to the latter type of mechanisms. We also lack clues on why CDA < 0 promotes antiparallel betasheets, and CDA > 0 prevents them. Alpha helices might be more simple structures that require less order than antiparallel betasheets. A similar rationale might function for parallel and antiparallel betasheets. In addition, the ratio between parallel and antiparallel betasheets is about 1:7 [26]: the genetic code might be optimised towards ‘coding’ for the most frequent protein conformation.
The genetic code can be characterised as a hypercomplex mathematical multidimensional symmetry structure [225]. In other terms, the genetic code reminds spontaneously self-organizing structures such as crystals [226], [227]. Crystals result from specific rules organizing relations between atoms. Similarly, but at a much higher level of molecular complexity, the genetic code organises relations between nucleic and amino acid sequences. The genetic code might be thought as an imaginary polyhedron with 64 triangular faces (64 codons with three nucleotide positions). The geometrical form of this structure remains unknown, but several symmetries implied by RNA/DNA structure and chemistry are known, such as reverse-complementarity (implied by the double helix structure), and the purine-pyrimidine as well as the alpha-keto groupings of nucleic acids. Formulation of a generalised description of this complex structure is a difficult task. It is simplified by projections of the complex structure on specific scales/planes of probable biological interest.
Here learned intuition detects a new symmetry property, based on codon content directionality. Analyses here can be seen as projecting that complex genetic code structure on the CDA scale, enabling to detect some new properties of the genetic code. The details of the scale of CDA scores as presented here is probably inaccurate and will hopefully be amended. CDA implies that a directional dimension that had not been apprehended links codons and amino acids: biologically meaningful information relating to protein structure is embedded in the comparison between codons and their reversed (not reverse-complemented) sequence. This palindrome-minded approach to codons probably reflects error-correcting properties of primitive genetic code(s) [228].
11. Conclusions
A property of codons, codon directional asymmetry (CDA), is defined for the genetic code. Codons are classified into symmetric (CDA = 0), 5′- and 3′-asymmetric (negative and positive CDA). CDA maps non-randomly on Fujimoto's tetrahedral representation of the genetic code. Symmetric codons are the most common codons in frame-error-correcting codes, such as comma-free and circular codes. Most codons assigned to amino acids aminoacylated to cognate tRNAs by tRNA synthetases class I have CDA < 0, those assigned to cognates of tRNA synthetases class II have usually CDA > 0.
Amino acid tendencies to participate in antiparallel betasheets decrease with CDA. Results suggest that CDA and tRNA synthetase class affect spatial kinetics of peptide elongation. These spatial kinetics affect local peptide elongation rates, which determine cotranslational peptide folding during peptide synthesis. Hence CDA, a property of gene sequences, bears useful information to predict protein folding. Some synonymous codons have CDA with opposite signs, potentially explaining how some synonymous mutations alter protein function.
CDA probably played a role in the evolution of genetic codes. Mitochondrial genetic codes optimise associations between CDA and antiparallel betasheet formation, nuclear genetic codes tend to optimise associations between CDA and tRNA synthetase class. This difference might mean that synergistic effects of CDA and tRNA synthetase class on cotranslational protein folding are stronger for nuclear than mitochondrial genetic codes. CDA affects codon-amino acid (re)assignments, hence plays an important role in genetic code evolution.
Preliminary analyses suggest that average CDA of mitochondrion-encoded proteins decreases with body temperature, increases developmental stability and lifespan, but further controlled analyses are required to confirm these potential whole organism effects of codon directional asymmetry (CDA).
Conflicts of Interests
None.
Acknowledgments
Acknowledgments
This study was supported by Méditerranée Infection and the National Research Agency under the program “Investissements d’avenir”, reference ANR-10-IAHU-03 and the A*MIDEX project (no ANR-11-IDEX-0001-02).
References
- 1.Woese C.R. Order in the genetic code. Proc Natl Acad Sci. 1965;54:71–75. doi: 10.1073/pnas.54.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Giulio M. The extension reached by the minimization of the polarity distances during the evolution of the genetic code. J Mol Evol. 1989;29:288–293. doi: 10.1007/BF02103616. [DOI] [PubMed] [Google Scholar]
- 3.Haig D., Hurst L.D. A quantitative measure of error minimization in the genetic-code. J Mol Evol. 1991;33:412–417. doi: 10.1007/BF02103132. [DOI] [PubMed] [Google Scholar]
- 4.Ardell D.H. On error minimization in a sequential origin of the standard genetic code. J Mol Evol. 1998;47:1–13. doi: 10.1007/pl00006356. [DOI] [PubMed] [Google Scholar]
- 5.Freeland S.J., Hurst L.D. Load minimization of the genetic code: history does not explain the pattern. Proc R Soc B Biol Sci. 1998;265:2111–2119. [Google Scholar]
- 6.Freeland S.J., Hurst L.D. The genetic code is one in a million. J Mol Evol. 1998;47:238–248. doi: 10.1007/pl00006381. [DOI] [PubMed] [Google Scholar]
- 7.Ardell D.H., Sella G. On the evolution of redundancy in genetic codes. J Mol Evol. 2001;53:269–281. doi: 10.1007/s002390010217. [DOI] [PubMed] [Google Scholar]
- 8.Freeland S.J., Wu T., Keulmann N. The case for an error minimizing standard genetic code. Origins Life Evol B. 2003;33:457–477. doi: 10.1023/a:1025771327614. [DOI] [PubMed] [Google Scholar]
- 9.Błażej P., Miasojedow B., Grabińska M., Mackiewicz P. Optimization of mutation pressure in relation to properties of protein-coding sequences in bacterial genomes. PLoS One. 2015;10(6) doi: 10.1371/journal.pone.0130411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Błażej P., Mackiewicz D., Grabińska M., Wnętrzak M., Mackiewicz P. Optimization of amino acid replacement costs by mutational pressure in bacterial genomes. Sci Rep. 2017;7(1):1061. doi: 10.1038/s41598-017-01130-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blazej P., Wnetrzak M., Mackiewicz P. The role of crossover operator in evolutionary-based approach to the problem of genetic code optimization. Biosystems. 2016;150:61–72. doi: 10.1016/j.biosystems.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 12.de Oliveira L.L., de Oliveira P.S., Tinos R. A multiobjective approach to the genetic code adaptability problem. BMC Bioinformatics. 2015;16:52. doi: 10.1186/s12859-015-0480-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seligmann H. Do anticodons of misacylated tRNAs preferentially mismatch codons coding for the misloaded amino acid? BMC Mol Biol. 2010;11:41. doi: 10.1186/1471-2199-11-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seligmann H. Error compensation of tRNA misacylation by codon-anticodon mismatch prevents translational amino acid misinsertion. Comput Biol Chem. 2011;35(2):82–95. doi: 10.1016/j.compbiolchem.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 15.Seligmann H. Coding constraints modulate chemically spontaneous mutational replication gradients in mitochondrial genomes. Curr Genomics. 2012;13(1):38–52. doi: 10.2174/138920212799034802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barthélémy R.M., Seligmann H. Cryptic tRNAs in chaetognath mitochondrial genomes. Comput Biol Chem. 2016;62:119–132. doi: 10.1016/j.compbiolchem.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 17.Seligmann H., Pollock D.D. The ambush hypothesis: hidden stop codons prevent off-frame gene reading. DNA Cell Biol. 2004;23(10):701–705. doi: 10.1089/dna.2004.23.701. [DOI] [PubMed] [Google Scholar]
- 18.Itzkovitz S., Alon U. The genetic code is nearly optimal for allowing additional information within protein-coding sequences. Genome Res. 2007;17(4):405–412. doi: 10.1101/gr.5987307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seligmann H. Cost minimization of ribosomal frameshifts. J Theor Biol. 2007;249(1):162–167. doi: 10.1016/j.jtbi.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 20.Seligmann H. The ambush hypothesis at the whole-organism level: off frame, ‘hidden’ stops in vertebrate mitochondrial genes increase developmental stability. Comput Biol Chem. 2010;34(2):80–85. doi: 10.1016/j.compbiolchem.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 21.Singh T.R., Pardasani K.R. Ambush hypothesis revisited: evidences for phylogenetic trands. Comput Biol Chem. 2009;33(3):239–244. doi: 10.1016/j.compbiolchem.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 22.Tse H., Cai J.J., Tsoi H.W., Lam E.P., Yuen K.Y. Natural selection retains overrepresented out-of-frame stop codons against frameshift peptides in prokaryotes. BMC Genomics. 2010;11:491. doi: 10.1186/1471-2164-11-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Křižek M., Křižek P. Why has nature invented three stop codons of DNA and only one start codon? J Theor Biol. 2012;304:183–187. doi: 10.1016/j.jtbi.2012.03.026. [DOI] [PubMed] [Google Scholar]
- 24.Gilis D., Massar S., Cerf N.J., Rooman M. Optimality of the genetic code with respect to protein stability and amino-acid frequencies. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-11-research0049. [RESEARCH0049] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guilloux A., Jestin J.L. The genetic code and its optimization for kinetic energy conservation in polypeptide chains. Biosystems. 2012;109(2):141–144. doi: 10.1016/j.biosystems.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 26.Guilloux A., Caudron B., Jestin J.L. A method to predict edge strands in beta-sheets from protein sequences. Comput Struct Biotechnol J. 2013;7 doi: 10.5936/csbj.201305001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong J.T. A co-evolution theory of the genetic code. Proc Natl Acad Sci U S A. 1975;72:1909–1912. doi: 10.1073/pnas.72.5.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Giulio M. On the origin of the genetic code. J Theor Biol. 1997;187:573–581. doi: 10.1006/jtbi.1996.0390. [DOI] [PubMed] [Google Scholar]
- 29.Di Giulio M. The coevolution theory of the origin of the genetic code. J Mol Evol. 1999;48:253–255. doi: 10.1007/pl00006464. [DOI] [PubMed] [Google Scholar]
- 30.Di Giulio M. An extension of the coevolution theory of the origin of the genetic code. Biol Direct. 2008;3:37. doi: 10.1186/1745-6150-3-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong J.T. The coevolution theory at age thirty. Bioessays. 2005;27(4):416–425. doi: 10.1002/bies.20208. [DOI] [PubMed] [Google Scholar]
- 32.Guimarães R.C. Metabolic basis for the self-referential genetic code. Orig Life Evol Biosph. 2011;41(4):357–371. doi: 10.1007/s11084-010-9226-x. [DOI] [PubMed] [Google Scholar]
- 33.Morgens D.W., Cavalcanti A.R.O. An alternative look at code evolution: using non-canonical codes to evaluate adaptive and historic models for the origin of the genetic code. J Mol Evol. 2013;76:71–80. doi: 10.1007/s00239-013-9542-7. [DOI] [PubMed] [Google Scholar]
- 34.Guimarães R.C. The self-referential genetic code is biologic and includes the error minimization property. Orig Life Evol Biosph. 2015;45:69–75. doi: 10.1007/s11084-015-9417-6. [DOI] [PubMed] [Google Scholar]
- 35.Di Giulio M. The lack of foundation in the mechanism on which are based the physico-chemical theories for the origin of the genetic code is counterposed to the credible and natural mechanism suggested by the coevolution theory. J Theor Biol. 2016;399:134–140. doi: 10.1016/j.jtbi.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 36.Di Giulio M. Some pungent arguments against the physico-chemical theories of the origin of the genetic code and corroborating the coevolution theory. J Theor Biol. 2017;414:1–4. doi: 10.1016/j.jtbi.2016.11.014. [DOI] [PubMed] [Google Scholar]
- 37.Higgs P.G., Pudritz R.E. A thermodynamic basis for prebiotic amino acid synthesis and the nature of the first genetic code. Astrobiology. 2009;9(5):483–490. doi: 10.1089/ast.2008.0280. [DOI] [PubMed] [Google Scholar]
- 38.Novozhilov A.S., Koonin E.V. Exceptional error minimization in putative primordial genetic codes. Biol Direct. 2009;4:44. doi: 10.1186/1745-6150-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santos J., Monteagudo A. Genetic code optimality studied by means of simulated evolution and within the coevolution theory of the canonical code organization. Nat Comput. 2009;8:719. [Google Scholar]
- 40.Tlusty T. A colorful origin for the genetic code: information theory, statistical mechanics and the emergence of molecular codes. Phys Life Rev. 2010;7(3):362–376. doi: 10.1016/j.plrev.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 41.Di Giulio M. The origin of the genetic code: matter of metabolism or physicochemical determinism? J Mol Evol. 2013;77:131–133. doi: 10.1007/s00239-013-9593-9. [DOI] [PubMed] [Google Scholar]
- 42.Banhu A.V., Aggarwal N., Sengupta S. Revisiting the physico-chemical hypothesis of code origin: an analysis based on code-sequence coevolution in a finite population. Orig Life Evol Biosph. 2013;43:465–489. doi: 10.1007/s11084-014-9353-x. [DOI] [PubMed] [Google Scholar]
- 43.Seligmann H., Amzallag G.N. Chemical interactions between amino acid and RNA: multiplicity of the levels of specificity explains origin of the genetic code. Naturwissenschaften. 2002;89(12):542–551. doi: 10.1007/s00114-002-0377-0. [DOI] [PubMed] [Google Scholar]
- 44.Woese C.R., Dugre, Saxinger W.C., Dugre S.A. The molecular basis for the genetic cocde. Proc Natl Acad Sci U S A. 1978;55:966–974. doi: 10.1073/pnas.55.4.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weber A.L., Lacey J.C. Genetic code correlations: amino acids and their anticodon nucleotides. J Mol Evol. 1966;11:199–210. doi: 10.1007/BF01734481. [DOI] [PubMed] [Google Scholar]
- 46.Shu J.J. A new integrated symmetrical table for genetic codes. Biosystems. 2017;151:21–26. doi: 10.1016/j.biosystems.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 47.Nemzer L.R. A binary representation of the genetic code. Biosystems. 2017;155:10–19. doi: 10.1016/j.biosystems.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez D.L., Giannerini S., Rosa R. Strong short-range correlations and dichotomic codon classes in coding DNA sequences. Phys Rev E Stat Nonlin Soft Matter Phys. 2008;78(5 Pt 1):051918. doi: 10.1103/PhysRevE.78.051918. [DOI] [PubMed] [Google Scholar]
- 49.Castro-Chavez F. A tetrahedral representation of the genetic code emphasizing aspects of symmetry. BIOcomplexity. 2012;2012(2):1–6. doi: 10.5048/BIO-C.2012.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fujimoto M. 1987. Tetrahedral codon stereo-table. [4,702,704. U.S. Patent] [http://www.google.com/patents/US4702704] [Google Scholar]
- 51.Arquès D.G., Michel C.J. A complementary circular code in the protein coding genes. J Theor Biol. 1996;182(1):45–58. doi: 10.1006/jtbi.1996.0142. [DOI] [PubMed] [Google Scholar]
- 52.Michel C.J. The maximal C3 self-complementary trinucleotide circular code X in genes of bacteria, eukaryotes, plasmids and viruses. J Theor Biol. 2015;380:156–177. doi: 10.1016/j.jtbi.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 53.Michel C.J. The maximal C(3) self-complementary trinucleotide circular code X in genes of bacteria, eukaryotes, plasmids and viruses. Life. 2017;7(2) doi: 10.3390/life7020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahmed A., Frey G., Michel C.J. Frameshift signals in genes associated with the circular code. In Silico Biol. 2007;7(2):155–168. [PubMed] [Google Scholar]
- 55.Ahmed A., Frey G., Michel C.J. Essential molecular functions associated with the circular code evolution. J Theor Biol. 2010;264(2):613–622. doi: 10.1016/j.jtbi.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 56.Michel C.J. Circular code motifs in transfer and 16S ribosomal RNAs: a possible translation code in genes. Comput Biol Chem. 2012;37:24–37. doi: 10.1016/j.compbiolchem.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 57.Michel C.J. Circular code motifs in transfer RNAs. Comput Biol Chem. 2013;45:17–29. doi: 10.1016/j.compbiolchem.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 58.El Soufi K., Michel C.J. Circular code motifs in the ribosome decoding center. Comput Biol Chem. 2014;52:9–17. doi: 10.1016/j.compbiolchem.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 59.El Soufi K., Michel C.J. Circular code motifs near the ribosome decoding center. Comput Biol Chem. 2015;59(Pt A):158–176. doi: 10.1016/j.compbiolchem.2015.07.015. [DOI] [PubMed] [Google Scholar]
- 60.Michel C.J., Seligmann H. Bijective transformation circular codes and nucleotide exchanging RNA transcription. Biosystems. 2014;118:39–50. doi: 10.1016/j.biosystems.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 61.El Houmami N., Seligmann H. Evolution of nucleotide punctuation marks: from structural to linear signals. Front Genet. 2017;8:36. doi: 10.3389/fgene.2017.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fimmel E., Strüngmann L. Codon distribution in error-detecting circular codes. Life. 2016;6(1) doi: 10.3390/life6010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rumer Y.B. About the codon systematization in the genetic code. Proc Acad Sci USSR. 1966;167:1393–1394. [PubMed] [Google Scholar]
- 64.Shsherbak V.I. Rumer's rule and transformation in the context of the co-operative symmetry of the genetic code. J Theor Biol. 1989;139(2):271–276. doi: 10.1016/s0022-5193(89)80104-3. [DOI] [PubMed] [Google Scholar]
- 65.Gumbel M., Fimmel E., Danielli A., Strüngmann L. On models of the genetic code generated by binary dichotomic algorithms. Biosystems. 2015;128:9–18. doi: 10.1016/j.biosystems.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 66.Seligmann H. Polymerization of non-complementary RNA: systematic symmetric nucleotide exchanges mainly involving uracil produce mitochondrial RNA transcripts coding for cryptic overlapping genes. Biosystems. 2013;111(3):156–174. doi: 10.1016/j.biosystems.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 67.Seligmann H. Systematic asymmetric nucleotide exchanges produce human mitochondrial RNAs cryptically encoding for overlapping protein coding genes. J Theor Biol. 2013;324:1–20. doi: 10.1016/j.jtbi.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 68.Seligmann H. Species radiation by DNA replication that systematically exchanges nucleotides? J Theor Biol. 2014;363:216–222. doi: 10.1016/j.jtbi.2014.08.036. [DOI] [PubMed] [Google Scholar]
- 69.Seligmann H. Mitochondrial swinger replication: DNA replication systematically exchanging nucleotides and short 16S ribosomal DNA swinger inserts. Biosystems. 2014;125:22–31. doi: 10.1016/j.biosystems.2014.09.012. [DOI] [PubMed] [Google Scholar]
- 70.Seligmann H. Sharp switches between regular and swinger mitochondrial replication: 16S rDNA systematically exchanging nucleotides A ↔ T + C ↔ G in the mitogenome of Kamimuria wangi. Mitochondrial DNA A DNA Mapp Seq Anal. 2016;27(4):2440–2446. doi: 10.3109/19401736.2015.1033691. [DOI] [PubMed] [Google Scholar]
- 71.Seligmann H. Translation of mitochondrial swinger RNAs according to tri-, tetra- and pentacodons. Biosystems. 2016;140:36–48. doi: 10.1016/j.biosystems.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 72.Delarue M. An asymmetric underlying rule in the assignment of codons. RNA. 2007;13:161–169. doi: 10.1261/rna.257607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eriani G., Delarue M., Poch O., Gangloff J., Moras D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature. 1990;347:203–206. doi: 10.1038/347203a0. [DOI] [PubMed] [Google Scholar]
- 74.Cusack S. Aminoacyl-tRNA synthetases. Curr Opin Struct Biol. 1997;7:881–889. doi: 10.1016/s0959-440x(97)80161-3. [DOI] [PubMed] [Google Scholar]
- 75.Sprinzl M., Cramer F. Site of aminoacylation of tRNAs from Escherichia coli with respect to the 2′2′- or 3′3′-hydroxyl group of the terminal adenosine. Proc Natl Acad Sci U S A. 1975;72:3049–3053. doi: 10.1073/pnas.72.8.3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Arnez J.G., Moras D. IRL Press; Oxford: 1994. Aminoacyl-tRNA synthetase tRNA recognition; pp. 61–81. [Google Scholar]
- 77.Jestin J.L., Soulé C. Symmetries by base substitutions in the genetic code predict 2′2′ or 3′3′ aminoacylation of tRNAs. J Theor Biol. 2007;247(2):391–394. doi: 10.1016/j.jtbi.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 78.Seligmann H. Overlapping genes coded in the 3′-to-5′-direction in mitochondrial genes and 3′-to-5′ polymerization of non-complementary RNA by an ‘invertase’. J Theor Biol. 2012;315:38–52. doi: 10.1016/j.jtbi.2012.08.044. [DOI] [PubMed] [Google Scholar]
- 79.Seligmann H. Triplex DNA:RNA, 3′-to-5′ inverted RNA and protein coding in mitochondrial genomes. J Comput Biol. 2013;20(9):660–671. doi: 10.1089/cmb.2012.0134. [DOI] [PubMed] [Google Scholar]
- 80.Seligmann H. Systematic exchanges between nucleotides: genomic swinger repeats and swinger transcription in human mitochondria. J Theor Biol. 2015;384:70–77. doi: 10.1016/j.jtbi.2015.07.036. [DOI] [PubMed] [Google Scholar]
- 81.Seligmann H. Swinger RNAs with sharp switches between regular transcription and transcription systematically exchanging ribonucleotides: case studies. Biosystems. 2015;135:1–8. doi: 10.1016/j.biosystems.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 82.Seligmann H. Swinger RNA self-hybridization and mitochondrial non-canonical swinger transcription, transcription systematically exchanging nucleotides. J Theor Biol. 2016;399:84–91. doi: 10.1016/j.jtbi.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 83.Nozawa K., O'donoghue P., Gundllapalli S., Araiso Y., Ishitani R., Umehara T. Pyrrolysyl-tRNA synthetase-tRNAPyl structure reveals the molecular basis of orthogonality. Nature. 2009;457:1163–1167. doi: 10.1038/nature07611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ashraf S.S., Guenther R., Agris P.F. Orientation of the tRNA anticodon in the ribosomal P-site: quantitative footprinting with U33-modified, anticodon stem and loop domains. RNA. 1999;5(9):1191–1199. doi: 10.1017/s1355838299990933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dale T., Fahlman R.P., Olejniczak M., Uhlenbeck O.C. Specificity of the ribosomal A site for aminoacyl-tRNAs. Nucleic Acids Res. 2009;37(4):1202–1210. doi: 10.1093/nar/gkn1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Krasheninnikov I.A., Komar A.A., Adzhubei I.A. Nonuniform size distribution of nascent globin peptides, evidence for pause localization sites, and a contranslational protein-folding model. J Protein Chem. 1991;10(5):445–453. doi: 10.1007/BF01025472. [DOI] [PubMed] [Google Scholar]
- 87.Fedorov A.N., Baldwin T.O. Contribution of cotranslational folding to the rate of formation of native protein structure. Proc Natl Acad Sci U S A. 1995;92(4):1227–1231. doi: 10.1073/pnas.92.4.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kolb V.A., Makeyev E.V., Kommer A., Spirin A.S. Cotranslational folding of proteins. Biochem Cell Biol. 1995;73(11 − 12):1217–1220. doi: 10.1139/o95-131. [DOI] [PubMed] [Google Scholar]
- 89.Gross M. Linguistic analysis of protein folding. FEBS Lett. 1996;390(3):249–252. doi: 10.1016/0014-5793(96)00727-2. [DOI] [PubMed] [Google Scholar]
- 90.Fedorov A.N., Baldwin T.O. Cotranslational protein folding. J Biol Chem. 1997;272(52):32715–32718. doi: 10.1074/jbc.272.52.32715. [DOI] [PubMed] [Google Scholar]
- 91.Kolb V.A. Cotranslational protein folding. Mol Biol. 2001;35:584–590. [PubMed] [Google Scholar]
- 92.Dana A., Tuller T. Determinants of translation elongation speed and ribosomal profiling biases in mouse embryonic stem cells. PLoS Comput Biol. 2012;8(11):e1002755–5. doi: 10.1371/journal.pcbi.1002755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.O'Brien E.P1., Vendruscolo M., Dobson C.M. Prediction of variable translation rate effects on cotranslational protein folding. Nat Commun. 2012;3:868. doi: 10.1038/ncomms1850. [DOI] [PubMed] [Google Scholar]
- 94.Nissley D.A1., O'Brien E.P. Timing is everything: unifying codon translation rates and nascent proteome behavior. J Am Chem Soc. 2014;136(52):17892–17898. doi: 10.1021/ja510082j. [DOI] [PubMed] [Google Scholar]
- 95.O'Brien E.P1., Ciryam P., Vendruscolo M., Dobson C.M. Understanding the influence of codon translation rates on cotranslational protein folding. Acc Chem Res. 2014;47(5):1536–1544. doi: 10.1021/ar5000117. [DOI] [PubMed] [Google Scholar]
- 96.Ray S.K., Baruah V.J., Satapathy S.S., Banerjee R. Cotranslational protein folding reveals the selective use of synonymous codons along the coding sequence of a low expression gene. J Genet. 2014;93(3):613–617. doi: 10.1007/s12041-014-0429-1. [DOI] [PubMed] [Google Scholar]
- 97.Trovato F., O'Brien E.P. Insights into cotranslational nascent protein behavior from computer simulations. Annu Rev Biophys. 2016;45:345–369. doi: 10.1146/annurev-biophys-070915-094153. [DOI] [PubMed] [Google Scholar]
- 98.Lu H.M., Liang J. A model study of protein nascent chain and cotranslational folding using hydrophobic-polar residues. Proteins. 2008;70(2):442–449. doi: 10.1002/prot.21575. [DOI] [PubMed] [Google Scholar]
- 99.Ugrinov K.G1., Clark P.L. Cotranslational folding increases GFP folding yield. Biophys J. 2010;98(7):1312–1320. doi: 10.1016/j.bpj.2009.12.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ciryam P., Morimoto R.I., Vendruscolo M., Dobson C.M., O'Brien E.P. In vivo translation rates can substantially delay the cotranslational folding of the Escherichia coli cytosolic proteome. Proc Natl Acad Sci U S A. 2013;110(2):E132–40. doi: 10.1073/pnas.1213624110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sander I.M1., Chaney J.L., Clark P.L. Expanding Anfinsen's principle: contributions of synonymous codon selection to rational protein design. J Am Chem Soc. 2014;136(3):858–861. doi: 10.1021/ja411302m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Holtkamp W., Kokic G., Jäger M., Mittelstaet J., Komar A.A., Rodnina M.V. Cotranslational protein folding on the ribosome monitored in real time. Science. 2015;350(6264):1104–1107. doi: 10.1126/science.aad0344. [DOI] [PubMed] [Google Scholar]
- 103.Nilsson O.B., Nickson A.A., Hollins J.J., Wickles S., Steward A., Beckmann R. Cotranslational folding of spectrin domains via partially structured states. Nat Struct Mol Biol. 2017;24(3):221–225. doi: 10.1038/nsmb.3355. [DOI] [PubMed] [Google Scholar]
- 104.O'Brien E.P., Vendruscolo M., Dobson C.M. Kinetic modelling indicates that fast-translating codons can coordinate cotranslational protein folding by avoiding misfolded intermediates. Nat Commun. 2014;5:2988. doi: 10.1038/ncomms3988. [DOI] [PubMed] [Google Scholar]
- 105.Cabrita L.D., Cassaignau A.M., Launay H.M., Waudby C.A., Wlodarski T., Camilloni C. A structural ensemble of a ribosome-nascent chain complex during cotranslational protein folding. Nat Struct Mol Biol. 2016;23(4):278–285. doi: 10.1038/nsmb.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Trovato F., O'Brien E.P. Fast protein translation can promote co- and posttranslational folding of misfolding-prone proteins. Biophys J. 2017;112(9):1807–1819. doi: 10.1016/j.bpj.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Eichmann C1., Preissler S., Riek R., Deuerling E. Cotranslational structure acquisition of nascent polypeptides monitored by NMR spectroscopy. Proc Natl Acad Sci U S A. 2010;107(20):9111–9116. doi: 10.1073/pnas.0914300107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Han Y., David A., Liu B., Magadán J.G., Bennink J.R., Yewdell J.W. Monitoring cotranslational protein folding in mammalian cells at codon resolution. Proc Natl Acad Sci U S A. 2012;109(31):12467–12472. doi: 10.1073/pnas.1208138109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ellis J.J1., Huard F.P., Deane C.M., Srivastava S., Wood G.R. Directionality in protein fold prediction. BMC Bioinformatics. 2010;11:172. doi: 10.1186/1471-2105-11-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Srivastava S., Patton Y., Fisher D.W., Wood G.R. Cotranslational protein folding and terminus hydrophobicity. Adv Bioinformatics. 2011;2011:176813. doi: 10.1155/2011/176813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Focke P.J., Hein C., Hoffmann B2., Matulef K., Bernhard F., Dötsch V. Combining in vitro folding with cell free protein synthesis for membrane protein expression. Biochemistry. 2016;55(30):4212–4219. doi: 10.1021/acs.biochem.6b00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li G.-W., Oh E., Weissman J.S. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012;484:538–541. doi: 10.1038/nature10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ta T., Argos P. Protein secondary structural types are differentially coded on messenger RNA. Protein Sci. 1996;5(10):1973–1983. doi: 10.1002/pro.5560051003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Brule C.E., Grayhack E.J. Synonymous codons: choose wisely for expression. Trends Genet. 2017;33(4):283–297. doi: 10.1016/j.tig.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Oresic M., Shalloway D. Specific correlations between relative synonymous codon usage and protein secondary structure. J Mol Biol. 1998;281(1):31–48. doi: 10.1006/jmbi.1998.1921. [DOI] [PubMed] [Google Scholar]
- 116.Saunders R., Deane C.M. Synonymous codon usage influences the local protein structure observed. Nucleic Acids Res. 2010;38(19):6719–6728. doi: 10.1093/nar/gkq495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Phoenix D.A., Korotkov E. Evidence of rare codon clusters within Escherichia coli coding regions. FEMS Microbiol Lett. 1997;155(1):63–66. doi: 10.1111/j.1574-6968.1997.tb12686.x. [DOI] [PubMed] [Google Scholar]
- 118.Clarke T.F., Clark P.L. Rare codons cluster. PLoS One. 2008;3(10):e3412–2. doi: 10.1371/journal.pone.0003412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chartier M., Gaudreault F., Najmanovich R. Large scale analysis of conserved rare codon clusters suggests an involvement in co-translational molecular recognition events. Bioinformatics. 2012;28(11):1438–1445. doi: 10.1093/bioinformatics/bts149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kimchi-Sarfaty C., Oh J.M., Kim I.-W., Sauna Z.E., Calcagno A.M., Ambudkar S.V. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315(5811):525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 121.Komar A.A. Silent SNPs: impact on protein function and phenotype. Pharmacogenomics. 2007;8(8):1075–1080. doi: 10.2217/14622416.8.8.1075. [DOI] [PubMed] [Google Scholar]
- 122.Agashe D., Martinez-Gomez N.C., Drummond D.A., Marx C.J. Good codons, bad transcript: large reductions in gene expression and fitness arising from synonymous mutations in a key enzyme. J Mol Evol. 2012;30(3):549–560. doi: 10.1093/molbev/mss273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fu J., Murphy K.A., Zhou M., Li Y.H., Lam V.H., Tabuloc C.A. Codon usage affects the structure and function of the Drosophila circadian clock protein period. Genes Dev. 2016;30(15):1761–1775. doi: 10.1101/gad.281030.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Morton B.R. Selection at the amino acid level can influence synonymous codon usage: implications for the study of codon adaptation in plastid genes. Genetics. 2001;159:347–358. doi: 10.1093/genetics/159.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Błażej P., Mackiewicz D., Wnętrzak M., Mackiewicz P. The impact of selection at the amino acid level on the usage of synonymous codons. G3 (Bethesda) 2017;7(3):967–981. doi: 10.1534/g3.116.038125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J Mol Biol. 1981;151:389–409. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- 127.Bennetzen J.L., Hall B.D. Codon selection in yeast. J Biol Chem. 1982;257:3026–3031. [PubMed] [Google Scholar]
- 128.Gouy M., Gautier C. Codon-usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 1982;10:7055–7074. doi: 10.1093/nar/10.22.7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol. 1985;2:13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- 130.Kanaya S., Yamada Y., Kudo Y., Ikemura T. Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene. 1999;238:143–155. doi: 10.1016/s0378-1119(99)00225-5. [DOI] [PubMed] [Google Scholar]
- 131.Akashi H. Translational selection and yeast proteome evolution. Genetics. 2003;164:1291–1303. doi: 10.1093/genetics/164.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ghaemmaghami S., Huh W.K., Bower K., Howson R.W., Belle A., Dephoure N. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 133.Goetz R.M., Fuglsang A. Correlation of codon bias measures with mRNA levels: analysis of transcriptome data from Escherichia coli. Biochem Biophys Res Commun. 2005;327:4–7. doi: 10.1016/j.bbrc.2004.11.134. [DOI] [PubMed] [Google Scholar]
- 134.Pechmann S1., Frydman J. Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat Struct Mol Biol. 2013;20(2):237–243. doi: 10.1038/nsmb.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lopez D., Pazos F. Protein functional features are reflected in the patterns of mRNA translation speed. BMC Genomics. 2015;16:513. doi: 10.1186/s12864-015-1734-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Deane C.M., Dong M., Huard F.P., Lance B.K., Wood G.R. Cotranslational protein folding—fact or fiction? Bioinformatics. 2007;23(13):i142–8. doi: 10.1093/bioinformatics/btm175. [DOI] [PubMed] [Google Scholar]
- 137.Evans M.S1., Sander I.M., Clark P.L. Cotranslational folding promotes beta-helix formation and avoids aggregation in vivo. J Mol Biol. 2008;383(3):683–692. doi: 10.1016/j.jmb.2008.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kelkar D.A., Khushoo A., Yang Z., Skach W.R. Kinetic analysis of ribosome-bound fluorescent proteins reveals an early, stable, cotranslational folding intermediate. J Biol Chem. 2012;287(4):2568–2578. doi: 10.1074/jbc.M111.318766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kim S.J., Yoon J.S., Shishido H., Yang Z., Rooney L.A., Barral J.M. Protein folding. Translational tuning optimizes nascent protein folding in cells. Science. 2015;348(6233):444–448. doi: 10.1126/science.aaa3974. [DOI] [PubMed] [Google Scholar]
- 140.Paslawski W., Lillelund O.K., Kristensen J.V., Schafer N.P., Baker R.P., Urban S. Cooperative folding of a polytopic α-helical membrane protein involves a compact N-terminal nucleus and nonnative loops. Proc Natl Acad Sci U S A. 2015;112(26):7978–7983. doi: 10.1073/pnas.1424751112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Siemion I.Z., Stefanowicz P. Periodical changes of amino acid reactivity within the genetic code. Biosystems. 1992;18:297–303. doi: 10.1016/0303-2647(92)90048-4. [DOI] [PubMed] [Google Scholar]
- 142.Chou P.Y., Fasman G.D. Prediction of the secondary structure of proteins from their amino acid sequence. Adv Enzymol Relat Areas Mol Biol. 1978;47:45–147. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]
- 143.Levitt M. Conformational preferences of amino acids in globular proteins. Biochemistry. 1978;17(20):4277–4285. doi: 10.1021/bi00613a026. [DOI] [PubMed] [Google Scholar]
- 144.Deléage G., Roux B. An algorithm for protein secondary structure prediction based on class prediction. Protein Eng. 1987;1(4):289–294. doi: 10.1093/protein/1.4.289. [DOI] [PubMed] [Google Scholar]
- 145.Chen H., Gu F., Huang Z. Improved Chou-Fasman method for protein secondary structure prediction. BMC Bioinformatics. 2006;7(Suppl. 4):S14. doi: 10.1186/1471-2105-7-S4-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lifson S., Sander C. Antiparallel and parallel β-strands differ in amino acid residue preferences. Nature. 1979;282:109–111. doi: 10.1038/282109a0. [DOI] [PubMed] [Google Scholar]
- 147.Caudron B., Jestin J.L. Sequence criteria for the anti-parallel character of protein beta-strands. J Theor Biol. 2012;315:146–149. doi: 10.1016/j.jtbi.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 148.Seligmann H. Positive and negative cognate amino acid bias affects compositions of aminoacyl-tRNA synthetases and reflects functional constraints on protein structure. BIO. 2012;2:11–26. [Google Scholar]
- 149.Zhu C.T., Zeng X.B., Huang W.D. Codon usage decreases the error minimization within the genetic code. J Mol Evol. 2003;57(5):533–537. doi: 10.1007/s00239-003-2505-7. [DOI] [PubMed] [Google Scholar]
- 150.Archetti M. Codon usage bias and mutation constraints reduce the level of error minimization of the genetic code. J Mol Evol. 2004;59(2):258–266. doi: 10.1007/s00239-004-2620-0. [DOI] [PubMed] [Google Scholar]
- 151.Marquez R., Smit S., Knight R. Do universal codon-usage patterns minimize the effects of mutation and translation error? Genome Biol. 2005;6(11):R91. doi: 10.1186/gb-2005-6-11-r91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mackiewicz P., Biecek P., Mackiewicz D., Kiraga J., Baczkowski K., Sobczynski M. Optimisation of asymmetric mutational pressure and selection pressure around the universal genetic code. Comput Sci - ICCS. 2008;Pt 3(5103):100–109. [Google Scholar]
- 153.Seligmann H. Phylogeny of genetic codes and punctuation codes within genetic codes. Biosystems. 2015;129:36–43. doi: 10.1016/j.biosystems.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 154.Seligmann H. Avoidance of antisense, antiterminator tRNA anticodons in vertebrate mitochondria. Biosystems. 2010;101:42–50. doi: 10.1016/j.biosystems.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 155.Seligmann H. Undetected antisense tRNAs in mitochondrial genomes? Biol Direct. 2010;5:39. doi: 10.1186/1745-6150-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Seligmann H. Pathogenic mutations in antisense mitochondrial tRNAs. J Theor Biol. 2011;269(1):287–296. doi: 10.1016/j.jtbi.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 157.Seligmann H. Two genetic codes, one genome: frameshifted primate mitochondrial genes code for additional proteins in presence of antisense antitermination tRNAs. Biosystems. 2011;105(3):271–285. doi: 10.1016/j.biosystems.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 158.Seligmann H. Putative protein-encoding genes within mitochondrial rDNA and the D-loop region. In: Lin Z., Liu W., editors. Ribosomes: molecular structure, role in biological functions and implications for genetic diseases. Nova Science Publishers; 2013. pp. 67–86. [Google Scholar]
- 159.Faure E., Delaye L., Tribolo S., Levasseur A., Seligmann H., Barthélémy R.M. Probable presence of an ubiquitous cryptic mitochondrial gene on the antisense strand of the cytochrome oxidase I gene. Biol Direct. 2011;6:56. doi: 10.1186/1745-6150-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Seligmann H. An overlapping genetic code for frameshifted overlapping genes in Drosophila mitochondria: antisense antitermination tRNAs UAR insert serine. J Theor Biol. 2012;298:51–76. doi: 10.1016/j.jtbi.2011.12.026. [DOI] [PubMed] [Google Scholar]
- 161.Seligmann H. Overlapping genetic codes for overlapping frameshifted genes in Testudines, and Lepidochelys olivacea as special case. Comput Biol Chem. 2012;41:18–34. doi: 10.1016/j.compbiolchem.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 162.Seligmann H. Codon expansion and systematic transcriptional deletions produce tetra-, pentacoded mitochondrial peptides. J Theor Biol. 2015;387:154–165. doi: 10.1016/j.jtbi.2015.09.030. [DOI] [PubMed] [Google Scholar]
- 163.Seligmann H. Chimeric mitochondrial peptides from contiguous regular and swinger RNA. Comput Struct Biotechnol J. 2016;14:283–297. doi: 10.1016/j.csbj.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Seligmann H. Natural chymotrypsin-like-cleaved human mitochondrial peptides confirm tetra-, pentacodon, non-canonical RNA translations. Biosystems. 2016;147:78–93. doi: 10.1016/j.biosystems.2016.07.010. [DOI] [PubMed] [Google Scholar]
- 165.Seligmann H. Unbiased mitoproteome analyses confirm non-canonical RNA, expanded codon translations. Comput Struct Biotechnol J. 2016;14:391–403. doi: 10.1016/j.csbj.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Seligmann H. Natural mitochondrial proteolysis confirms transcription systematically exchanging/deleting nucleotides, peptides coded by expanded codons. J Theor Biol. 2017;414:76–90. doi: 10.1016/j.jtbi.2016.11.021. [DOI] [PubMed] [Google Scholar]
- 167.Srivastava S., Lal S.B., Mishra D.C., Angadi U.B., Chaturvedi K.K., Rai S.N. An efficient algorithm for protein structure comparison using elastic shape analysis. Algorithms Mol Biol. 2016;11:27. doi: 10.1186/s13015-016-0089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Knecht C., Mort M., Junge O., Cooper D.N., Krawczak M., Caliebe A. IMHOTEP—a composite score integrating popular tools for predicting the functional consequences of non-synonymous sequence variants. Nucleic Acids Res. 2017;45(3) doi: 10.1093/nar/gkw886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Elzanowski A., Ostell J. The genetic codes. https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi November 2016 update.
- 170.Moraes E.M., Spressola V.L., Prado P.R.R.R., Costa L.F., Sene F.M. Divergence in wing morphology among sibling species of the Drosophila buzzatii cluster. J Zool Syst Evol Res. 2004;42(2):154–158. [Google Scholar]
- 171.Renaud S., Chevret P., Michaux J. Morphological vs. molecular evolution: exology and phylogeny both shape the mandible of rodents. Zool Scr. 2007;36(5):525–535. [Google Scholar]
- 172.Davies T.J., Savolainen V. Neutral theory, phylogenies, and the relationship between phenotypic change and evolutionary rates. Evolution. 2006;60(3):476–483. [PubMed] [Google Scholar]
- 173.Seligmann H. Positive correlations between molecular and morphological rates of evolution. J Theor Biol. 2010;264(3):799–807. doi: 10.1016/j.jtbi.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 174.Graham B.E. Animal evolution: trilobites on speed. Curr Biol. 2013;23(19):R878–80. doi: 10.1016/j.cub.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 175.Sessions S.K., Larson A. Developmental correlates of genome size in plethodontid salamanders and their implications for genome evolution. Evolution. 1987;41(6):1239–1251. doi: 10.1111/j.1558-5646.1987.tb02463.x. [DOI] [PubMed] [Google Scholar]
- 176.Licht L.E., Lowcock L.A. Genome size and metabolic-rate in salamanders. Comp Biochem Physiol B Biochem Mol Biol. 1991;100(1):83–92. [Google Scholar]
- 177.Roth G., Blanke J., Wake D.B. Cell-size predicts morphological complexity in the brains of frogs and salamanders. Proc Natl Acad Sci U S A. 1994;91(11):4796–4800. doi: 10.1073/pnas.91.11.4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Roth G., Walkowiak W. The influence of genome and cell size on brain morphology in amphibians. Cold Spring Harb Perspect Biol. 2015;7(9):a019075. doi: 10.1101/cshperspect.a019075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Seligmann H. Cost minimization of amino acid usage. J Mol Evol. 2003;56(2):151–161. doi: 10.1007/s00239-002-2388-z. [DOI] [PubMed] [Google Scholar]
- 180.Seligmann H. Putative mitochondrial polypeptides coded by expanded quadruplet codons, decoded by antisense tRNAs with unusual anticodons. Biosystems. 2012;110(2):84–106. doi: 10.1016/j.biosystems.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 181.Seligmann H. Pocketknife tRNA hypothesis: anticodons in mammal mitochondrial tRNA side-arm loops translate proteins? Biosystems. 2013;113(3):165–175. doi: 10.1016/j.biosystems.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 182.Seligmann H. Putative anticodons in mitochondrial tRNA sidearm loops: pocketknife tRNAs? J Theor Biol. 2014;340:155–163. doi: 10.1016/j.jtbi.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 183.Seligmann H., Labra A. Tetracoding increases with body temperature in Lepidosauria. Biosystems. 2013;114(3):155–163. doi: 10.1016/j.biosystems.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 184.Seligmann H., Krishnan N.M. Mitochondrial replication origin stability and propensity of adjacent tRNA genes to form putative replication origins increase developmental stability in lizards. J Exp Zool B Mol Dev Evol. 2006;306:433–449. doi: 10.1002/jez.b.21095. [DOI] [PubMed] [Google Scholar]
- 185.Seligmann H. Error propagation across levels of organization: from chemical stability of ribosomal RNA to developmental stability. J Theor Biol. 2006;242(1):69–80. doi: 10.1016/j.jtbi.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 186.Chwastyk M., Cieplak M. Cotranslational folding of deeply knotted proteins. J Phys Condens Matter. 2015;27(35):354105. doi: 10.1088/0953-8984/27/35/354105. [DOI] [PubMed] [Google Scholar]
- 187.Felsenstein J. Phylogenies and the comparative method. Am Nat. 1985;125:1–15. doi: 10.1086/703055. [DOI] [PubMed] [Google Scholar]
- 188.Sato Y., Fujiwara T., Kimura H. Expression and function of different guanine-plus-cytosine content 16S rRNA genes in Haloarcula hispanica at different temperatures. Front Microbiol. 2017;8:482. doi: 10.3389/fmicb.2017.00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Musto H., Naya H., Zaval A., Romero H., Alvarez-Valin F., Bernardi G. Genomic GC level, optimal growth temperature, and genome size in prokaryotes. Biochem Biophys Res Commun. 2006;347(1):1–3. doi: 10.1016/j.bbrc.2006.06.054. [DOI] [PubMed] [Google Scholar]
- 190.Zheng H., Wu H.W. Gene-centric association analysis for the correlation between the guanine-cytosine content levels and temperature range conditions of prokaryotic species. BMC Bioinformatics. 2010;11:S7. doi: 10.1186/1471-2105-11-S11-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ream R.A., Johns G.C., Somero G.N. Base compositions of genes encoding alpha-actin and lactate dehydrogenase-A from differently adapted vertebrates show no temperature-adaptive variation in G + C content. Mol Biol Evol. 2003;20(1):105–110. doi: 10.1093/molbev/msg008. [DOI] [PubMed] [Google Scholar]
- 192.Marashi S.A., Ghalanbor Z. Correlations between genomic GC levels and optimal growth temperatures are not 'robust'. Biochem Biophys Res Commun. 2004;20(1):381–383. doi: 10.1016/j.bbrc.2004.10.051. [DOI] [PubMed] [Google Scholar]
- 193.Wang H.C., Susko E., Roger A.J. On the correlation between genomic G + C content and optimal growth temperature in prokaryotes: data quality and confounding factors. Biochem Biophys Res Commun. 2006;342:681–684. doi: 10.1016/j.bbrc.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 194.Seligmann H. Evidence that minor directional asymmetry is functional in lizard hindlimbs. J Zool. 1998;248:205–208. [Google Scholar]
- 195.Seligmann H. Evolution and ecology of developmental processes and of the resulting morphology: directional asymmetry in hindlimbs of Agamidae and Lacertidae (Reptilia: Lacertilia) Biol J Linn Soc. 2000;69:461–481. [Google Scholar]
- 196.Seligmann H., Beiles A., Werner Y.L. Avoiding injury or adapting to survive injury? Two coexisting strategies in lizards. Biol J Linn Soc. 2003;78:307–324. [Google Scholar]
- 197.Seligmann H., Beiles A., Werner Y.L. More injuries in left-footed lizards. J Zool. 2003;260:129–144. [Google Scholar]
- 198.Seligmann H., Moravec J., Werner Y.L. Morphological, functional and evolutionary aspects of tail autotomy and regeneration in the ‘living fossil’ Sphenodon (Reptilia: Rhynchocephalia) Biol J Linn Soc. 2008;93(4):721–743. [Google Scholar]
- 199.Bilal E., Rabadan R., Alexe G., Fuku N., Ueno H., Nishigaki Y. Mitochondrial DNA haplogroup D4a is a marker for extreme longevity in Japan. PLoS One. 2008;3(6) doi: 10.1371/journal.pone.0002421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tanak M., Cabrera M., Gonzalez M., Larruga M., Takeyasu T., Fuku N. Mitochondrial genome variation in eastern Asia and the peopling of Japan. Genome Res. 2004;14:1832–1850. doi: 10.1101/gr.2286304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Alexe G., Fuku N., Bilal E., Ueno H., Nishigaki Y., Fujita Y. Enrichment of longevity phenotype in mtDNA haplogroups D4b2b, D4a, and D5 in the Japanese population. Hum Genet. 2007;121:347–356. doi: 10.1007/s00439-007-0330-6. [DOI] [PubMed] [Google Scholar]
- 202.Hansen T.F., Pienaar J., Orzack S.H. A comparative method for studying adaptation to a randomly evolving environment. Evolution. 2007;62:1965–1977. doi: 10.1111/j.1558-5646.2008.00412.x. [DOI] [PubMed] [Google Scholar]
- 203.Bailey L.J., Doherty A.J. Mitochondrial DNA replication: a PrimPol perspective. Biochem Soc Trans. 2017;45(2):513–529. doi: 10.1042/BST20160162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Reyes A., Gissi C., Pesole G., Saccone C. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol Biol Evol. 1998;15(8):957–966. doi: 10.1093/oxfordjournals.molbev.a026011. [DOI] [PubMed] [Google Scholar]
- 205.Krishnan N.M., Seligmann H., Raina S.Z., Pollock D.D. Detecting gradients of asymmetry in site-specific substitutions in mitochondrial genomes. DNA Cell Biol. 2004;23(10):707–714. doi: 10.1089/1044549042476901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Seligmann H., Krishnan N.M., Rao B.J. Possible multiple origins of replication in primate mitochondria: alternative role of tRNA sequences. J Theor Biol. 2006;241(2):321–332. doi: 10.1016/j.jtbi.2005.11.035. [DOI] [PubMed] [Google Scholar]
- 207.Seligmann H., Krishnan N.M., Rao B.J. Mitochondrial tRNA sequences as unusual replication origins: pathogenic implications for Homo sapiens. J Theor Biol. 2006;243(3):375–385. doi: 10.1016/j.jtbi.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 208.Seligmann H. Hybridization between mitochondrial heavy strand tDNA and expressed light strand tRNA modulates the function of heavy strand tDNA as light strand replication origin. J Mol Biol. 2008;379(1):188–199. doi: 10.1016/j.jmb.2008.03.066. [DOI] [PubMed] [Google Scholar]
- 209.Seligmann H. Coding constraints modulate chemically spontaneous mutational replication gradients in mitochondrial genomes. Curr Genomics. 2012;13(1):37–54. doi: 10.2174/138920212799034802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Helfenbein K.G., Brown W.M., Boore J.L. The complete mitochondrial genome of the articulate brachiopod Terebratalia transversa. Mol Biol Evol. 2001;18:1734–1744. doi: 10.1093/oxfordjournals.molbev.a003961. [DOI] [PubMed] [Google Scholar]
- 211.Hassanin A., Leger N., Deutsch J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst Biol. 2005;54(2):277–298. doi: 10.1080/10635150590947843. [DOI] [PubMed] [Google Scholar]
- 212.Fonseca M.M., Froufe E., Harris D.J. Mitochondrial gene rearrangements and partial genome duplications detected by multigene asymmetric compositional bias analysis. J Mol Evol. 2006;63(5):654–661. doi: 10.1007/s00239-005-0242-9. [DOI] [PubMed] [Google Scholar]
- 213.Min X.J., Hickey D.A. DNA asymmetric strand bias affects the amino acid composition of mitochondrial proteins. DNA Res. 2007;14:201–206. doi: 10.1093/dnares/dsm019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Fonseca M.M., Harris D.J. Relationship between mitochondrial gene rearrangements and stability of the origin of light strand replication. Genet Mol Biol. 2008;31(2):566–574. [Google Scholar]
- 215.Fonseca M.M., Posada D., Harris D.J. Inverted replication of vertebrate mitochondria. Mol Biol Evol. 2008;25:805–808. doi: 10.1093/molbev/msn050. [DOI] [PubMed] [Google Scholar]
- 216.Masta S.E., Longhorn S.J., Boore J.L. Arachnid relationships based on mitochondrial genomes: asymmetric nucleotide and amino acid bias affects phylogenetic analyses. Mol Phylogenet Evol. 2009;50:117–128. doi: 10.1016/j.ympev.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 217.Wei S.J., Shi M., Chen X.X., Sharkey M.J., van Achterberg C., Ye G.Y. New views on strand asymmetry in insect mitochondrial genomes. PLoS One. 2010;5(9) doi: 10.1371/journal.pone.0012708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Fonseca M.M., Harris D.J., Posada D. The inversion of the control region in three mitogenomes provides further evidence for an asymmetric model of vertebrate mtDNA replication. PLoS One. 2014;9(9) doi: 10.1371/journal.pone.0106654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Seligmann H. Mitochondrial tRNAs as light strand replication origins: similarity between anticodon loops and the loop of the light strand replication origin predicts initiation of DNA replication. Biosystems. 2010;99(2):85–93. doi: 10.1016/j.biosystems.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 220.Seligmann H., Labra A. The relation between hairpin formation by mitochondrial WANCY tRNAs and the occurrence of the light strand replication origin in Lepidosauria. Gene. 2014;542(2):248–257. doi: 10.1016/j.gene.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 221.Seligmann H. Mutation patterns due to converging mitochondrial replication and transcription increase lifespan and cause growth rate-longevity tradeoffs. In: Seligmann H., editor. DNA replication—current advances. InTech; 2011. Chapter 4. [Google Scholar]
- 222.Perneger T.V. What's wrong with Bonferroni adjustments. Br Med J. 1998;316(7139):1236–1238. doi: 10.1136/bmj.316.7139.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bender R., Lange S. Multiple test procedures other than Bonferroni's deserve wider use. Br Med J. 1999;318(7183):600. doi: 10.1136/bmj.318.7183.600a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57(1):289–300. [Google Scholar]
- 225.Käll L., Storey J.D., MacCoss M.J., Noble W.S. Posterior error probabilities and false discovery rates: two sides of the same coin. J Proteome Res. 2008;7(01):40–44. doi: 10.1021/pr700739d. [DOI] [PubMed] [Google Scholar]
- 226.Petoukhov S.V. Genetic coding and united-hypercomplex systems in the models of algebraic biology. Biosystems. 2017;158:31–46. doi: 10.1016/j.biosystems.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 227.Cairns-Smith A.G., Hartman H. Cambridge University Press; 1986. Clay minerals and the origin of life; p. 197. [Google Scholar]
- 228.Gonzalez D.L., Giannerini S., Rosa R. Available from nature Precedings. 2012. On the origin of the mitochondrial genetic code: Towards a unified mathematical framework for the management of genetic information. [Google Scholar]