

Abstract
Although Chk1 kinase inhibitors are currently under clinical investigation as effective cancer cell sensitizers to the cytotoxic effects of numerous chemotherapeutics, there is still a considerable uncertainty regarding their role in modulation of anticancer potential of platinum-based drugs. Here we newly demonstrate the ability of one of the most specific Chk1 inhibitors, SCH900776 (MK-8776), to enhance human colon cancer cell sensitivity to the cytotoxic effects of platinum(II) cisplatin and platinum(IV)- LA-12 complexes. The combined treatment with SCH900776 and cisplatin or LA-12 results in apparent increase in G1/S phase–related apoptosis, stimulation of mitotic slippage, and senescence of HCT116 cells. We further show that the cancer cell response to the drug combinations is significantly affected by the p21, p53, and PTEN status. In contrast to their wt counterparts, the p53- or p21-deficient cells treated with SCH900776 and cisplatin or LA-12 enter mitosis and become polyploid, and the senescence phenotype is strongly suppressed. While the cell death induced by SCH900776 and cisplatin or LA-12 is significantly delayed in the absence of p53, the anticancer action of the drug combinations is significantly accelerated in p21-deficient cells, which is associated with stimulation of apoptosis beyond G2/M cell cycle phase. We also show that cooperative killing action of the drug combinations in HCT116 cells is facilitated in the absence of PTEN. Our results indicate that SCH900776 may act as an important modulator of cytotoxic response triggered by platinum-based drugs in colon cancer cells.
Abbreviations: CDDP, cisplatin; Chk1, checkpoint kinase 1; Chk2, checkpoint kinase 2; γ-H2AX, phosphorylated histone H2AX at Ser 139; LA-12, (OC-6-43)-bis(acetato)(1-adamantylamine)amminedichloroplatinum(IV); PARP, poly (ADP-ribose) polymerase; PBS, phosphate-buffered saline; PTEN, phosphatase and tensin homolog; SEM, standard error of the mean; siRNA, small interfering RNA
Introduction
Cisplatin is a highly active platinum(II) complex used for treatment of a wide variety of human cancers. Unfortunately, its therapeutic efficacy is often hampered due to intrinsic or acquired cancer cell resistance or undesired side effects to the normal tissues. Although great efforts have been made in last decades to characterize anticancer potential of cisplatin, there are still many unresolved issues regarding precise molecular mechanisms of its action and the crucial determinants of the cell response to the drug [1]. Among other targets, cisplatin is known to bind DNA and create inter- and intrastrand cross-links, which block DNA replication and mobilize signaling pathways including those responsible for activation of checkpoint kinase 1 (Chk1). However, the exact role of Chk1 and its specific inhibition in the cytotoxic/cytostatic effects of cisplatin still remain not fully explained.
Chk1, an evolutionary conserved serine/threonine kinase, is a key component of DNA damage response, a network of processes crucial for the preservation of genomic integrity and regulation of tumorigenesis [2]. Following various types of DNA damage, Chk1 is mainly activated by ATR-mediated phosphorylation on serine-317 and serine-345, which is followed by autophosphorylation on serine-296, a marker of its activity [3]. Chk1 then phosphorylates a number of substrates such as p53, cell division control 25 (cdc25), histone H3, or Rad 51, and is responsible for coordinating pleiotropic cellular response including the cell cycle arrest or delay, DNA replication and repair, cell death, senescence, homologous recombination, and maintenance of somatic cell viability [4].
Small-molecule inhibitors of Chk1 kinase have been synthesized and intensively investigated as single agents or in combination with various DNA-damaging chemotherapeutic drugs and showed to significantly enhance the chemosensitivity of numerous tumor types [5], [6], [7]. One of the most specific Chk1 inhibitors, SCH900776 (also known as MK-8776), a pyrazolo[1,5-a]pyrimidine derivative, identified using high-content cell-based functional screen for induction of H2AX phosphorylation (γ-H2AX) [8], is currently profiled in phase II clinical trials. SCH900776 has been shown to interact synergistically with DNA antimetabolite agents such as hydroxyurea or gemcitabine in vitro and in vivo to induce DNA damage and death of pancreatic and ovarian cancer cells [8]. It also significantly potentiated the cytotoxic response induced by fludarabine, cytarabine, or gemcitabine in various tumor types [9], [10], [11], [12], [13], [14].
While many Chk1 inhibitors often mediate robust sensitization to cytotoxic effects of antimetabolites in numerous cancer models, less is known about their cooperative anticancer action with cisplatin, and currently available studies report remarkably inconsistent results with varying degrees of success. A significant UCN-01–mediated enhancement of cisplatin cytotoxicity has been shown in Chinese hamster ovary cells [15] or cisplatin-resistant HCT116 cell clones [16] but not in MDA-MB-231 or MCF10A breast cancer cell lines [10]. Potentiation of cisplatin cytotoxicity has been observed using V158411 in p53 mutated HT-29 but not p53 wt HCT116 colon [17] or in SKOV-3 ovarian [18] cancer cells, by LY2603618 in several osteosarcoma cell lines [19], or by PF477736 in HT-29 cells [20]. AZD7762 enhanced the cytotoxic effects of cisplatin in p53-mutant or -deficient head and neck squamous cell carcinoma [21] or clear cell carcinoma of the ovary [22]. In contrast, AZD7762 did not affect the clonogenic survival of cisplatin-treated HeLa cells, although it sensitized them to gemcitabine [23]. Furthermore, no sensitization to cisplatin was achieved with SCH900776 in MDA-MB-231 and MCF10A breast [10] or OVCAR-8 and SKOV3 ovarian [24] cancer cells.
Compared to cisplatin, there are even fewer studies focused on the role of Chk1 in the cytotoxic/cytostatic action of other platinum-based drugs, including novel candidates with improved anticancer properties. LA-12 represents a recently introduced platinum(IV) complex [25] with favorable cytotoxic potential against various cancer cell types including colon in vitro [26], [27], [28], [29], [30] and in vivo [31]. LA-12 also exerted potent killing effects in cisplatin-resistant cancer cell lines [32], [33]. To date, no relevant study documents the functional role of Chk1 in anticancer action of LA-12, and the effects of Chk1 inhibitors on cancer cell response to LA-12 remain completely unexplored. Therefore, further research is needed to uncover whether and how the particular Chk1 inhibitors could potentiate the cancer cell killing induced by standard-of-care or new candidate platinum-based drugs, and to define the unique molecular determinants of their action.
Herein, we newly demonstrate the ability of SCH900776 to significantly enhance the human colon cancer cell sensitivity to the cytotoxic effects of cisplatin or LA-12, and describe investigation of the involved mechanisms especially at the level of cell cycle regulation, DNA damage, cell death, and senescence. The particular attention is paid to the role of p53, p21, and PTEN in cooperative anticancer action of SCH900776 and cisplatin/LA-12.
Materials and Methods
Cell Culture and Treatments
Human colon adenocarcinoma cell lines HCT116 wt, p53−/−, p21−/−, Chk2−/− (obtained from Prof. Bert Vogelstein, John Hopkins University, Baltimore, MD) [34], HCT116 PTEN+/+, and PTEN−/− (from Prof. Todd Waldman, Georgetown University School of Medicine, Washington, DC) [35] were maintained in McCoy's 5A medium (Gibco, Thermo Fisher Scientific, USA) supplemented with penicillin (100 U/ml), streptomycin (0.1 mg/ml) (both Duchefa Biochemie B. V., Haarlem, the Netherlands), and 10% heat-inactivated fetal bovine serum (FBS, Gibco, Thermo Fisher Scientific). The cells were cultivated in TPP (TPP Techno Plastic Products AG, Trasadingen, Switzerland) cultivation dishes, flasks, or plates in a humidified incubator at 37°C in atmosphere of 5% CO2 and passaged twice a week after exposure to EDTA/PBS and trypsin solutions. Numbers of cells were determined using a CASY Model TT Cell Counter and Analyzer (Roche Diagnostics GmbH, Germany).
Twenty-four hours after seeding, the cells were treated with the following drugs as specified in figure legends: SCH900776 (synthesized as described in [13]), LA-12 (OC-6-43)-bis(acetato)(1-adamantylamine) amine dichloroplatinum(IV) (Platinum Pharmaceuticals, a.s., Brno, Czech Republic), cisplatin cis-diamminedichloroplatinum(II) (Sigma-Aldrich, Prague, Czech Republic), or pan-caspase inhibitor z-VAD-fmk (BD Bioscience Pharmingen, San Diego, CA). The stock solutions were diluted in dimethylsulfoxide (DMSO, Sigma-Aldrich). Appropriate vehicles were always added to the controls.
Cell Transfection and RNA Interference
The cells were seeded into McCoy's medium with 10% FBS, without antibiotics, and incubated overnight. Before transfection, medium was changed for Opti-MEM I Reduced Serum Medium (Gibco, Thermo Fischer Scientific). The cell transfections were performed using Lipofectamine 2000 Transfection Reagent or Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, Thermo Fisher Scientific) according to the manufacturer's recommendations. The cells were transfected with 100 nM siRNA duplexes directed against Chk1 (sense: GAAGCAGUCGCAGUGAAGA[dT][dT], anti-sense: UCUUCACUGCGACUGCUUC[dT][dT]) [36] or control siRNA against GL2 luciferase (sense: CGUACGCGGAAUACUUCGA[dT][dT], anti-sense: UCGAAGUAUUCCGCGUACG[dT][dT]) [37], both from Sigma-Aldrich. After 4 hours of incubation with transfection complexes, medium was changed for McCoy's medium with 10% FBS and antibiotics. Twenty-four hours after transfection, the cells were treated with the drugs studied.
Immunoblotting Analysis
The cells were harvested, whole cell lysates were prepared and separated by SDS-PAGE, and Western blot analysis was performed as described previously [27]. Immunodetection was carried out using the following primary antibodies: monoclonal mouse anti-p53 (1:1000, #126), monoclonal mouse anti-Chk1(1:1000, #8408), polyclonal rabbit anti-cyclin A (1:1000, #751), monoclonal mouse anti-cyclin B1 (1:1000, #245), monoclonal mouse anti-cyclin D1 (1:1000, #20044), monoclonal mouse anti-PTEN (1:500, #7974), polyclonal rabbit anti-p21 (1:1000, #397), polyclonal rabbit anti-p27 (1:1000, #528) (all from Santa Cruz Biotechnology), polyclonal rabbit anti-phospho-p53 (Ser15) antibody (1:1000, #9284), monoclonal rabbit anti-phospho-Aurora A/B/C (Thr288/232/198) (1:500, #2914), polyclonal rabbit anti-Aurora B (1:500, #3094), polyclonal rabbit anti-phospho-Chk1 (Ser296) (1:500, #2349), monoclonal mouse anti-Chk2 (1:1000, #3440), polyclonal rabbit anti-cleaved caspase-3 (1:500, #9661), polyclonal rabbit anti-cleaved caspase-9 (1:500, #9505), monoclonal rabbit anti-phospho-H2AX (Ser139, γ-H2AX) (1:500, #9718), polyclonal rabbit anti-phospho-histone H3 (Ser10) (1:500, #9701), monoclonal rabbit anti-histone H3 (1:1000, #4499), polyclonal rabbit anti-cleaved PARP (1:1000, #9541), monoclonal rabbit anti-survivin (1:1000, #2808), polyclonal rabbit anti-phospho-Rb (Ser807/811) (1:1000, #9308) (all from Cell Signaling Technology, Danvers, MA), and monoclonal rabbit anti-Cdc25C (1:1000, #1302-1, Epitomics). The proteins were recognized using horseradish peroxidase–labeled secondary antibodies: anti-mouse IgG (1:3000, #NA931) and anti-rabbit IgG (1:3000, #NA934) antibody (both from Amersham Biosciences, Bucks, UK). Detection of the antibody complexes was performed with Immobilon Western Chemiluminescent HRP Substrate (Millipore Corp., Darmstadt, Germany). An equal loading was verified by anti–β-actin antibody (1:5000, #A5441, Sigma-Aldrich).
Annexin V/Propidium Iodide Assay
For detection of phosphatidyl serine externalization as a marker of apoptosis, annexin V-FITC/PI assay was used as described previously [27]. Fluorescence was then analyzed using flow cytometer (FACSVerse, Becton Dickinson, USA) and BD FACSuite software version 1.0.5 (Becton Dickinson). The results were expressed as the percentage of annexin V+/PI− (apoptotic), annexin V+/PI+ (secondary necrotic), or annexin V−/PI+ plus annexin V+/PI+ (total dead) cells.
Cell Cycle Analysis
The cells were fixed in 70% ethanol, washed with PBS, and resuspended in Vindelov solution [propidium iodide (20 μg/ml), RNase A (DNase free, 5 U/ml, Sigma-Aldrich), 1 M Tris (pH 8.0), 0.1% Triton X-100, 20 mM NaCl, in MQ water] at 37°C for 30 minutes and analyzed by flow cytometry (FACSVerse; Becton Dickinson) and ModFit LT version 3.1 software (Verity Software House, Topsham, ME). The results were expressed as the percentage of the cells in G1, S, and G2/M phases.
Active Caspase-3 Detection
The cells were harvested, washed in PBS, fixed, and stained using FITC active caspase-3 apoptosis kit (#550480, BD Pharmingen) according to the manufacturer's protocol. The percentage of cells with cleaved caspase-3 was detected by flow cytometry (FACSVerse, Becton Dickinson) and analyzed using BD FACSuite software version 1.0.5 (Becton Dickinson).
Simultaneous Detection of Active Caspase-3 and the Cell Cycle Progression
The cells were harvested, washed in PBS, fixed, and stained using FITC active caspase-3 apoptosis kit (#550480, BD Pharmingen) according to the manufacturer's protocol. Subsequently, the cells were washed and stained with Vindelov solution as described above and analyzed by flow cytometry (FACSVerse, Becton Dickinson) and BD FACSuite software version 1.0.5 (Becton Dickinson). Results were expressed as percentage of cells with active caspase-3 related to selected cell cycle phases.
Detection of Senescence-Associated β-Galactosidase (SA-β-gal) Activity
The cells were stained for SA-β-gal activity using X-gal substrate (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside; #15243, SERVA, Heidelberg, Germany) at pH 6.0 as previously described [38]. The percentages of cells positive for SA-β-gal staining were determined microscopically from at least total of 200 cells per sample.
Statistical Analysis
Data were expressed as means ± SEM of three independent experiments and statistically analyzed by one-way ANOVA followed by Fisher's Least Significant Difference (LSD) test using STATISTICA 12 software (StatSoft, Inc., Tulsa, OK). For all flow cytometry analyses, a minimum of 10 (annexin V/PI assay) or 20 (all others) thousand events was collected per sample excluding doublets and debris.
Results
SCH900776 or Chk1 siRNA Sensitizes Human Colon Cancer Cells to LA-12/Cisplatin-Induced Apoptosis and DNA Damage
We newly showed that SCH900776 effectively stimulated LA-12– or cisplatin-induced apoptosis of HCT116 human colon adenocarcinoma cells, as demonstrated by significant increase in phosphatidyl serine externalization (Figure 1A) and specific cleavage of PARP and caspase-9 (Figure 1B) upon 48-hour treatment. Pretreatment with a pan-caspase inhibitor z-VAD-fmk significantly blocked these events (Figure 1, A and B). The combination of SCH900776 and LA-12/cisplatin induced an enhanced caspase-independent DNA damage detected by phosphorylation (Ser139) of histone H2AX (γ-H2AX) as compared to the agents used alone (Figure 1B, Supplementary Figure 1A). The drug combination–induced potentiation of apoptosis and DNA damage was also apparent upon 72-hour treatment (Supplementary Figure 1, B and C).
Figure 1.

SCH900776 or Chk1 siRNA mediated sensitization of HCT116 cells to LA-12/cisplatin-induced apoptosis. (A) Percentage of annexin V+/PI− (flow cytometry) HCT116 wt cells and (B) cleavage (cl.) of PARP, caspase-9, level of phosphorylated (Ser139) H2AX (γ-H2AX), phosphorylated (Ser296), and total Chk1 (Western blotting) following pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent treatment (48 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM) in the absence (DMSO) or presence of z-VAD-fmk (10 μM). (C) Cleavage of PARP, caspase-9, caspase-3, level of γ-H2AX and Chk1 (Western blotting), and (D) cleavage of caspase-3 (flow cytometry) in HCT116 wt cells transfected (24 hours) with control or Chk1 siRNA, pretreated (2 hours) with SCH900776 (SCH, 3 μM) and subsequently treated (48 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). Results are means ± SEM or representatives of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH, Ο versus LA-12 or CDDP and Δ for with versus without z-VAD-fmk or control versus Chk1 siRNA.
Supplementary Figure 1.

(A) Percentage of HCT116 wt cells with phosphorylated (Ser139) histone H2AX (γ-H2AX) (flow cytometry) following pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent treatment (48 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). (B) Percentage of annexin V+/PI− or annexin V+/PI+ (flow cytometry) HCT116 wt cells and (C) cleavage of PARP, caspase-9, level of γ-H2AX, and phosphorylated (Ser296) and total Chk1 (Western blotting) in HCT116 wt cells pretreated with SCH900776 (SCH, 3 μM) and subsequently treated (72 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). Results are means ± SEM or representatives of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH, Ο versus LA-12 or CDDP.
The concentration of SCH900776 used in the study was selected based on our investigation of its time- and dose-dependent effects on Chk1 phosphorylation (Ser296) and cell viability. The 3-μM dose of SCH900776 effectively blocked the platinum drug–induced Chk1 phosphorylation during the whole course (2 days) of the HCT116 cell treatment, and when applied individually, it triggered neither apoptosis nor DNA damage (γ-H2AX) (Figure 1, A and B, and not shown). Following the combined treatment (48 hours) with SCH900776 and LA-12/cisplatin, a decrease in total Chk1 protein level in HCT116 cells was observed (Figure 1B).
Similarly to the effects of SCH900776, a specific siRNA-based silencing of Chk1 resulted in a significant stimulation of the cytotoxic response induced by LA-12/cisplatin in HCT116 cells. This was demonstrated by enhanced cleavage of PARP and caspase-9 and -3 (Figure 1, C and D) and phosphorylation of H2AX (Figure 1C) in LA-12/cisplatin-treated (48 hours) HCT116 cells transfected with Chk1 siRNA compared to their control siRNA-transfected counterparts. The Chk1 siRNA-mediated enhancement of LA-12/cisplatin-induced apoptosis in HCT116 cells was further apparent upon 72-hour treatment (Supplementary Figure 2, A and B).
Supplementary Figure 2.

(A) Cleavage of PARP, caspase-9, caspase-3, and Chk1 level (Western blotting), and (B) cleavage of caspase-3 (flow cytometry) in HCT116 wt cells transfected (24 hours) with control or Chk1 siRNA pretreated (2 hours) with SCH900776 (SCH, 3 μM) and subsequently treated (72 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). Results are means ± SEM or representatives of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH, Ο versus LA-12 or CDDP and Δ for control versus Chk1 siRNA.
In order to examine the potential involvement of Chk2 in the above-mentioned effects of SCH900776, we performed analogous experiments also in the absence of this kinase. As in the case of HCT116 wt cells, a significant stimulation of the cytotoxic response induced by the combination of SCH900776 and LA-12/cisplatin was observed in their Chk2-deficient counterparts, which was demonstrated by enhancement of phosphatidyl serine externalization (Supplementary Figure 3A), cleavage of PARP and caspase-9, and phosphorylation of H2AX (Supplementary Figure3B). In addition, siRNA-mediated silencing of Chk1 in Chk2-deficient HCT116 cells augmented their cytotoxic response to LA-12/cisplatin to a similar extent as compared to their Chk1 siRNA-transfected HCT116 wt counterparts (Supplementary Figure3, C and D).
Supplementary Figure 3.

(A) Percentage of dead (annexin V+/PI− plus annexin V+/PI+) wt and Chk2−/− HCT116 cells following pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent treatment (48 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). (B) Cleavage of PARP, caspase-9, level of γ-H2AX, and phosphorylated (Ser296) and total Chk1 and Chk2 (Western blotting) in wt and Chk2−/− HCT116 cells treated as in A. (C) Percentage of Chk2−/− HCT116 cells with cleaved caspase-3 (flow cytometry) following their transfection (24 hours) with control or Chk1 siRNA, pretreatment (2 hours) with SCH900776 (SCH, 3 μM), and subsequent treatment (48 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). (D) Cleavage of PARP, caspase-9, caspase-3, level of γ-H2AX, Chk1, and Chk2 (Western blotting) in Chk2−/− HCT116 cells transfected and treated as in C. In D, HCT116 wt cells were used as a positive control for total Chk2 protein detection. Results are means ± SEM or representatives of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH, Ο versus LA-12 or CDDP and Δ for wt versus Chk2−/− or control versus Chk1 siRNA.
The ability of SCH900776 to stimulate apoptosis (phosphatidyl serine externalization, PARP, and caspase-9 cleavage) and DNA damage (γ-H2AX) induced by LA-12 or cisplatin was also demonstrated in other human colon adenocarcinoma cell lines HT-29 (Supplementary Figure 4, A and B) and RKO (Supplementary Figure 4, C and D).
Supplementary Figure 4.

(A) Percentage of annexin V+/PI− (flow cytometry) HT-29 cells and (B) cleavage of PARP, caspase-9, level of γ-H2AX, and phosphorylated (Ser296) and total Chk1 (Western blotting) following their pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent treatment (48 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). (C) Percentage of annexin V+/PI− (flow cytometry) RKO cells and (D) cleavage of PARP, caspase-9, level of γ-H2AX, and phosphorylated (Ser296) and total Chk1 (Western blotting) following the same treatments as specified in (A and B). Results are means ± SEM or representative of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH and Ο versus LA-12 or CDDP.
SCH900776-Mediated Enhancement of LA-12/Cisplatin-Induced Cytotoxicity Is Accelerated in the Absence of p21
The treatment of HCT116 wt cells with LA-12/cisplatin or their combinations with SCH900776 resulted in apparent upregulation of p53 level and its augmented phosphorylation at Ser15, which was further enhanced in the absence of p21. In HCT116 wt cells, a strong p53-dependent upregulation of p21 level was also detected (Figure 2, A and B). Therefore, the role of p53 and p21 in the cooperative cytotoxic effects of SCH900776 and LA-12/cisplatin was further examined. In the absence of p53, a partial suppression of PARP (Figure 2A) and caspase-3 (Supplementary Figure 5A) cleavage and a significant decrease in the percentage of the drug combination–treated HCT116 cells in annexin V+/PI− (apoptotic) subpopulation (Figure 2C) were observed compared to the wt counterparts. On the other hand, in the absence of p21, apoptosis induced by combination of SCH900776 and LA-12/cisplatin was remarkably accelerated, as documented by a significant increase in the percentage of the drug combination–treated p21−/− versus wt cells in annexin V+/PI+ (secondary necrotic) subpopulation (Figure 2D). An apparent increase in caspase-3 cleavage (Supplementary Figure 5B) and phosphorylation of H2AX (Figure 2B) was also detected in HCT116 p21−/− versus wt cells treated with the SCH900776 and LA-12/cisplatin. The drug combination–induced stimulation of apoptosis but not DNA damage in p21−/− cells was largely caspase dependent, as confirmed by z-VAD-fmk (Supplementary Figure 6, A-C).
Figure 2.

The role of p53 and p21 in SCH900776-mediated sensitization of HCT116 cells to LA-12/cisplatin-induced cytotoxicity. (A, B) Total and phosphorylated (Ser15) p53, total p21, cleaved PARP, γ-H2AX, phosphorylated (Ser296), and total Chk1 level (Western blotting) in wt versus p53−/− or wt versus p21−/− HCT116 cells pretreated (2 hours) with SCH900776 (SCH, 3 μM) and subsequently treated (48 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). (C) Percentage of annexin V+/PI− or (D) annexin V+/PI+ (flow cytometry) wt, p53−/−, and p21−/− HCT116 cells treated as in A and B. Results are means ± SEM or representatives of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH, Ο versus LA-12 or CDDP and Δ for wt versus p21−/− or wt versus p53−/− cells.
Supplementary Figure 5.

Caspase-3 cleavage (flow cytometry) in (A) wt versus p53−/− or (B) wt versus p21−/− HCT116 cells pretreated (2 hours) with SCH900776 (SCH, 3 μM) and subsequently treated (48 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). Results are means ± SEM or representatives of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH, Ο versus LA-12 or CDDP and Δ for wt versus p21−/− or wt versus p53−/− cells.
Supplementary Figure 6.

(A) Percentage of annexin V+/PI− and (B) annexin V+/PI+ HCT116 p21−/− cells (flow cytometry), and (C) cleavage of PARP, caspase-9, caspase-3, level of γ-H2AX, and phosphorylated (Ser296) and total Chk1 (Western blotting) following their pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent treatment (48 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM) in the absence (DMSO) or presence of z-VAD-fmk (10 μM). Results are means ± SEM or representative of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH, Ο versus LA-12 or CDDP and Δ for with versus without z-VAD-fmk.
Loss of p21 Stimulates SCH900776- and LA-12/Cisplatin Combination-Induced Apoptosis Beyond G2 Cell Cycle Phase
To uncover the relationship between apoptosis induction and the cell cycle position in HCT116 cells treated with SCH900776 and LA-12/cisplatin, a simultaneous flow cytometry analysis of caspase-3 cleavage and DNA content was performed. While apoptosis in HCT116 wt cells induced by the examined doses of LA-12/cisplatin occurred predominantly in G1 phase, the combined treatment with SCH900776 apparently increased caspase-3 cleavage in G1 and especially S phase, with lower effects in the subsequent cell cycle phases (Figure 3). The ablation of p21 in the cells treated with individual platinum drugs resulted in increased frequency of G1 and S phase–related apoptosis. Importantly, following their combined treatment with SCH900776 and LA-12/cisplatin, further increase in caspase-3 cleavage was detected during the whole course of the cell cycle progression including G2/M (Figure 3). In p53−/− cells, less apparent drug combination–induced changes in particular cell cycle phase–related apoptosis were detected compared to wt or p21−/− cells (Supplementary Figure 7).
Figure 3.

The relationship between cycle progression and apoptosis induced by combination of SCH900776 and LA-12/cisplatin in HCT116 cells. (A) Percentage of wt and p21−/− HCT116 cells with caspase-3 cleavage related to the cell cycle progression (propidium iodide, flow cytometry) following their pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent treatment (48 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). Results are means ± SEM or representative of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH and Ο versus LA-12 or CDDP.
Supplementary Figure 7.
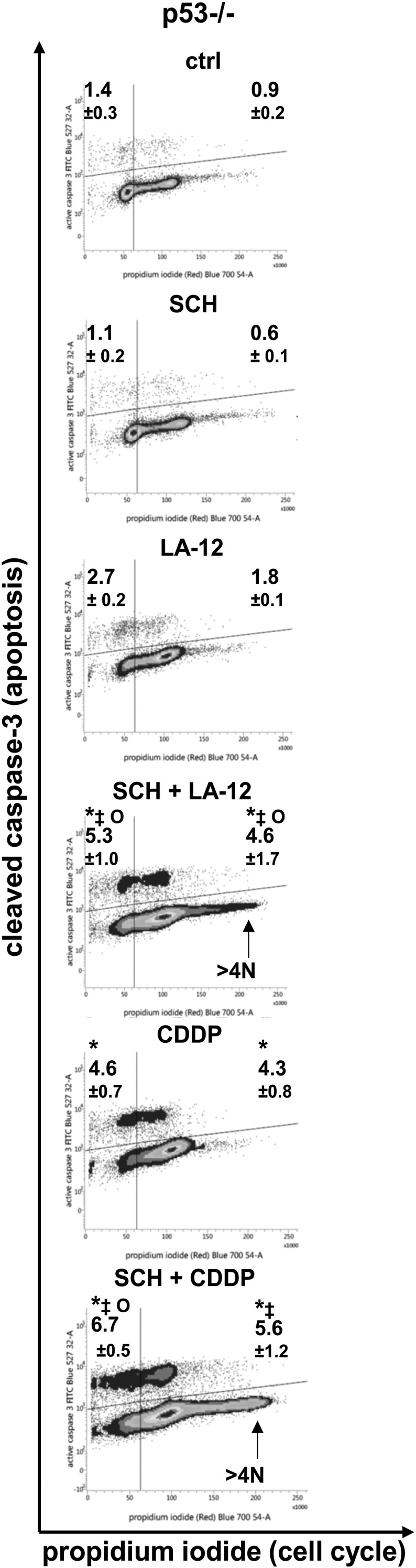
Percentage of HCT116 p53−/− cells with caspase-3 cleavage related to the cell cycle progression (propidium iodide, flow cytometry) following their pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent treatment (48 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). Results are means ± SEM or representative of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH and Ο versus LA-12 or CDDP.
While only a slight (less than 5%) increase in polyploidy was observed in SCH900776- and LA-12/cisplatin-treated HCT116 wt cells, a significant drug combination–induced increase of this parameter (about 10%-15% polyploid cells, depending on the platinum drug and the time of the treatment) was detected in their p21−/− or p53−/− counterparts (Supplementary Figure 8, A-D). Within these polyploid populations, only a minor part of the cells was apoptotic, i.e., positive for caspase-3 cleavage (Supplementary Figure 8, A-D).
Supplementary Figure 8.

(A, B) Percentage of polyploid (>4 N) wt or p21−/− HCT116 cells with (c3+) or without (c3−) active caspase-3 following pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent 48-hour (A) or 72-hour (B) treatment with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). (C, D) Percentage of polyploid (>4 N) wt or p53−/− HCT116 cells with (c3+) or without (c3−) active caspase-3, treated as in A and B. Results are means ± SEM or representative of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH, Ο versus LA-12 or CDDP, and Δ for wt versus p21−/− or wt versus p53−/− cells.
Combination of SCH900776 and LA-12/Cisplatin Induces p21- and p53-Dependent Alterations of the Cell Cycle Progression
The cell cycle analysis of HCT116 wt, p21−/−, and p53−/− cells revealed that, compared to appropriate controls, treatment (48 or 72 hours) with both LA-12 or cisplatin induced a significant increase in their percentage in G2/M phase, associated with decrease in G1 phase (Figure 4, A-D, Supplementary Figure 9, A and B). A significant LA-12/cisplatin-induced decrease in percentage of wt cells in S phase was observed upon 48-hour treatment, and these trends were still present until 72 hours (Figure 4, A and B). This was accompanied by a dramatic suppression of DNA synthesis, as demonstrated by flow cytometric detection of decreased incorporation of modified thymidine analogue EdU (Supplementary Figure 10, A and B).
Figure 4.

The cell cycle phase distribution in wt and p21−/− HCT116 cells treated with SCH900776 and LA-12/cisplatin. Percentage of wt (A and B) and p21−/− (C and D) HCT116 cells in individual cell cycle phases (flow cytometry) following their pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent 48-hour (A and C) or 72-hour (B and D) treatment with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). Results are means ± SEM of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH and Ο versus LA-12 or CDDP.
Supplementary Figure 9.

(A, B) Percentage of HCT116 p53−/− cells in individual cell cycle phases (flow cytometry) following their pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent 48-hour (A) or 72-hour (B) treatment with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). Results are means ± SEM of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH and Ο versus LA-12 or CDDP.
Supplementary Figure 10.

Simultaneous analysis of 5-ethynyl-2′-deoxyuridine (EdU) incorporation and cell cycle (DAPI, flow cytometry) in HCT116 wt cells following their pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent treatment (48 hours) with (A) LA-12 (0.75 μM) or (B) cisplatin (CDDP, 20 μM). Results show percentages of the cells with incorporated EdU and are means ± SEM or representatives of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH and Ο versus LA-12.
SCH900776 alone essentially did not affect the cell cycle distribution in HCT116 wt, p21−/−, or p53−/− cells in the time intervals studied (Figure 4, A-D, Supplementary Figure 9). However, it was responsible for significant suppression of cisplatin- (48 hours) or LA-12–(72 hours) induced increase of percentage of G2/M phase wt cells and a slight increase in S phase (Figure 4, A and B), preceded (48 hours) by a suppression of DNA synthesis (Supplementary Figure 10, A and B). Similar changes in G2/M or S phase distribution were also confirmed following siRNA-mediated silencing of Chk1 and subsequent treatment of HCT116 wt cells with LA-12 or cisplatin (data not shown). The above-mentioned changes in the cell cycle distribution were further much more pronounced in p21−/− cells, where a strong decrease in percentage of the cells in G2/M phase and a dramatic increase in S phase were detected following the combined SCH900776 and LA-12/cisplatin treatment (especially 72 hours) compared to the individual drugs (Figure 4, C and D). The transient changes in G2/M phase representation were also apparent in p53−/− cells treated with combination of SCH900776 and LA-12/cisplatin compared to individual platinum drugs (Supplementary Figure 9, A and B).
The alterations of the cell cycle progression induced by SCH900776 and LA-12/cisplatin were further correlated with the expression of the relevant regulatory proteins. In addition to the already described changes of the level of p53 and p21 (Figure 2, A and B), the wt cells treated (48 hours) with the drug combination largely showed a slight increase in cyclin D1 and p27 level compared to individual platinum drugs, and a dramatic loss of phosphorylated Rb protein, cyclin A, cdc25C, cyclin B1, and survivin (Figure 5). An apparent decrease in cyclin B1 and survivin level was already detected following 24-hour treatment with combination of SCH900776 and LA-12/cisplatin (Supplementary Figure 11). In the wt cells treated with the individual platinum drugs or their combination with SCH900776, loss of phosphorylated (Ser10) but not total histone H3 was detected (Figure 5). The described loss of phosphorylated H3 was already apparent after 8 and 24 (cisplatin) or 24 (LA-12) hours of the treatments (Supplementary Figure 11). The incubation (48 hours) with the platinum drugs also resulted in loss of phosphorylated Aurora A/B/C (Thr288/232/198) level, which was accompanied by decrease in total Aurora B levels upon combined treatments (Figure 5).
Figure 5.

The levels of selected cell cycle regulatory proteins in wt, p21−/−, and p53−/− HCT116 cells treated with SCH900776 and LA-12/cisplatin. The total level of cyclin D1, p27, phosphorylated (Ser 807/811) Rb, cyclin A, cdc25C, cyclin B1, survivin, phosphorylated (Ser10) and total histone 3 (H3), phosphorylated Aurora A/B/C (Thr288/232/198), and Aurora B (Western blotting) in wt, p21−/−, and p53−/− HCT116 cells following their pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent treatment (48 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). Results are representatives of three independent experiments.
Supplementary Figure 11.

The level of γ-H2AX, phosphorylated (Ser296) and total Chk1, cyclin B1, survivin, and phosphorylated (Ser10) histone 3 (p-H3) in HCT116 wt cells (Western blotting) following their pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent treatment (8 or 24 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). Results are representatives of three independent experiments.
Compared to HCT116 wt cells, their p21−/− and p53−/− counterparts treated with the combination of SCH900776 and LA-12/cisplatin showed a reduced cyclin D1 and p27 level and a weaker loss of phosphorylated Rb protein, cyclin A, cdc25C, cyclin B1, or survivin level (Figure 5). The p53−/− or p21−/− cells treated with the individual platinum drugs or their combination with SCH900776 also mostly did not show the loss of phosphorylated histone H3 (Ser10) and Aurora A/B/C (Thr288/232/198) level observed in their wt counterparts (Figure 5). Very similar trends in phosphorylation of histone H3 induced by SCH900776 and LA-12/cisplatin were also confirmed by flow cytometry analysis in wt versus p21−/− or p53−/− cells (Supplementary Figure 12). As in the case of wt cells, the level of total histone H3 remained unchanged following all types of treatment in p21−/− and p53−/− lines. Less dramatic drug combination– versus single agent–induced decrease of total Aurora B level was mostly detected in p21−/− and p53−/− cells compared to the wt cell line (Figure 5).
Supplementary Figure 12.

Percentage of HCT116 wt, p53−/−, and 21−/− cells with phosphorylation (Ser10) of histone 3 (p-H3) (flow cytometry) following their pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent treatment (48 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). Results are means ± SEM of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH, Ο versus LA-12 or CDDP and Δ for wt versus p53−/− or wt versus p21−/−
SCH900776 Triggers p53/p21-Dependent Modulations of LA-12/Cisplatin-Induced Cellular Senescence
We demonstrated that prolonged treatment with LA-12 or cisplatin (72-hour incubation with the drugs and subsequent 72-hour cultivation in drug-free medium) induced a strong increase in percentage of HCT116 wt cells with senescence-like phenotype, manifested by a significant increase in β-galactosidase activity and enlarged, flattened cell morphology (Figure 6A, Supplementary Figure 13A), which was associated with increase in p53, p21, cyclin D1 level, and loss of phosphorylated Rb, cyclin B1, or survivin level compared to control (Figure 6B). The platinum drug–induced β-galactosidase activity in wt cells was slightly enhanced by SCH900776 (Figure 6A, Supplementary Figure 13A), while we could not observe any respective differences in the level of the above-mentioned proteins (Figure 6B). Compared to their wt counterparts, the platinum drug–induced senescence-associated β-galactosidase activity was significantly lower in p53−/− and p21−/− cells and further significantly suppressed by SCH900776 (Supplementary Figure 13, A and B).
Figure 6.

Senescence-associated changes in HCT116 wt cells treated with SCH900776 and LA-12/cisplatin. (A) Percentage of HCT116 wt cells with β-galactosidase activity after pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent treatment (72 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM) followed by 72-hour cultivation in drug-free medium. (B) The level of p53, p21, cyclin D1, phosphorylated (Ser807/811) Rb, cyclin B1, and survivin (Western blotting) in HCT116 wt cells treated as in A. Results are means ± SEM or representatives of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH and Ο versus LA-12 or CDDP.
Supplementary Figure 13.

(A) Representative microscopy pictures of the wt, p53−/−, and p21−/− HCT116 cells stained for β-galactosidase activity and quantified in Figure 6A after their pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent treatment (72 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM) followed by 72-hour cultivation in drug-free medium. (B) Percentage of p53−/− and p21−/− HCT116 cells with β-galactosidase activity treated as in A. Cell images were made by bright field microscopy with 10× magnification. Results are means ± SEM or representatives of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH and Ο versus LA-12 or CDDP.
PTEN Deficiency Facilitates SCH900776-Mediated Enhancement of LA-12/Cisplatin-Induced Apoptosis
Combined treatment of HCT116 PTEN+/+ cells with SCH900776 and LA-12/cisplatin induced a remarkable decrease in PTEN level compared to the action of individual drugs (Figure 7A). We further showed that HCT116 PTEN−/− cells were more sensitive to the killing effects of LA-12/cisplatin alone or their combination with SCH900776, as demonstrated by enhanced PARP cleavage (Figure 7A) and phosphatidyl serine externalization (Figure 7B, Supplementary Figure 14) compared to their PTEN+/+ counterparts. No apparent differences in the level of total/phosphorylated p53, p21, or γ-H2AX induced by platinum drugs or their combination with SCH900776 were detected between PTEN+/+ and PTEN−/− cells (Figure 7A).
Figure 7.

Role of PTEN in SCH900776-mediated sensitization of HCT116 cells to LA-12/cisplatin-induced apoptosis. (A) The level of PTEN, cleaved PARP, phosphorylated (Ser15) and total p53, p21, γ-H2AX, and phosphorylated (Ser296) and total Chk1 (Western blotting) in PTEN+/+ and PTEN−/− HCT116 cells following their pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent treatment (48 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). (B) Percentage of annexin V+/PI− PTEN+/+ and PTEN−/− HCT116 cells treated as in A. Results are means ± SEM or representatives of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH, Ο versus LA-12 or CDDP and Δ for PTEN+/+ versus PTEN −/− cells.
Supplementary Figure 14.

Percentage of annexin V+/PI+ HCT116 PTEN+/+ and PTEN−/− cells following pretreatment (2 hours) with SCH900776 (SCH, 3 μM) and subsequent treatment (48 hours) with LA-12 (0.75 μM) or cisplatin (CDDP, 20 μM). Results are means ± SEM or representatives of three independent experiments. Statistical significance: P < .05, * versus control, ‡ versus SCH, Ο versus LA-12 or CDDP and Δ for PTEN+/+ versus PTEN −/− cells.
Discussion
Although Chk1 kinase inhibitors are currently under clinical investigation as effective sensitizers of numerous cancer cell types to the cytotoxic effects of various chemotherapeutics, there is still a considerable uncertainty over their role in modulation of anticancer potential of platinum-based drugs. While some studies reported enhancement of cisplatin toxicity mediated by various Chk1 inhibitors [15], [16], [17], [18], [19], [20], [21], [22], others found no therapeutic benefit associated with their combined treatment [10], [23], [24]. Here we newly demonstrate the potent ability of one of the most specific Chk1 inhibitors, SCH900776, to enhance the human colon cancer cell sensitivity to the cytotoxic effects of platinum-based drugs cisplatin or LA-12. So far, only a few studies investigating combined cytotoxic action of cisplatin and SCH900776 have been reported, and in contrast to our observations, they showed no further sensitization in breast [10] or ovarian [24] cancer cells. To our best knowledge, we also provide the first demonstration of cooperative anticancer effects of LA-12 and SCH900776.
Similarly to pharmacological inhibition by SCH900776, we clearly confirmed that specific siRNA-mediated depletion of Chk1 rendered the HCT116 cells more sensitive to platinum drug–induced cell death. In agreement with our findings, previous reports also showed the sensitization to cisplatin-induced toxicity mediated by Chk1 knockdown in SKOV3, RMG-I and KK ovarian [22], [39], and HCT116 colon [40] cancer cells. In contrast, no such effects were observed in cisplatin-resistant HCT116 clones [16], Hela cervical [23], HN31 head and neck [21], or OVCAR-8 ovarian [24] cancer cells. The contradictory information calls for more detailed defining of the main determinants of response to cisplatin-induced damage in cells with abrogated function of Chk1. Concerning LA-12, according to our knowledge, we provide the first demonstration of Chk1 siRNA-mediated stimulation of LA-12–induced apoptosis in human cancer cells.
Although many Chk1 inhibitors effectively block Chk1, they may also exert varying degrees of activity against Chk2 [41]. As SCH900776 was shown to be much more selective for Chk1 compared to Chk2 [8], we hypothesized that it would not substantially interfere with the Chk2 activity and its potential involvement in platinum drug–induced cell death. In support of this, we observed that the Chk2 status did not affect the ability of SCH900776 to increase LA-12–induced HCT116 cell killing. This is in agreement with Guzi et al. who reported that knockdown of Chk2 did not contribute to cell death and DNA damage induced by ablation of Chk1 function during antimetabolite exposure [8]. We further demonstrated that silencing of Chk2 resulted in slight decrease in SCH900776 and cisplatin combination-induced HCT116 cell death. However, as the similar effect was also apparent in Chk2−/− cells treated with cisplatin alone, it did not seem to be related to the action of SCH900776. Our results correspond to those obtained by Pabla et al. who showed that Chk2−/− HCT116 cells were less sensitive to cisplatin-induced apoptosis then their wt counterparts [42]. Therefore, we concluded that it is rather Chk1 than Chk2 that is involved in SCH900776-mediated stimulation of cisplatin/LA-12–induced cytotoxicity.
It has been shown that Chk1 inhibitor-mediated potentiation of anticancer effects of some chemotherapeutic drugs including cisplatin occurs preferentially in p53-mutated or -deficient background [13], [17], [21], [43], but other studies contradicted this paradigm [44], [45]. Although we demonstrated the ability of SCH900776 to enhance cisplatin/LA-12–induced cell death in colon cancer cells expressing wt (HCT116, RKO) or mutated (HT-29) p53 and also in those lacking this protein, our results clearly show that the presence of functional p53 may significantly facilitate the drug combination–induced cytotoxicity. It seems that in order to understand the differences in results reporting the anticancer activity of Chk1 inhibitors in relation to p53 status, one should also consider additional factors such as distinct p53 mutations and their functional consequences, selectivity of inhibitor or the type of the chemotherapeutic agent and resulting DNA damage, and possible defects in downstream p53 signaling.
Compared to p53, much less is known about the role of its transcriptional target p21 in modulation of cytotoxic effects of chemotherapeutic drugs in cancer cells with abrogated Chk1 function. Loss of p21 enhanced the excess of thymidine-induced killing in Chk1-depleted HCT116 cells, showing that Chk1 and p21 cooperate to prevent cancer cell death induced by DNA damage [46]. The protective role of p21 from the lethal effects of the combination of AZD7762 and irinotecan was also demonstrated in normal epithelial cells and colorectal tumors [47]. A siRNA-mediated silencing of Chk1 in HCT116 p21−/− cells caused a greater sensitization to cisplatin toxicity compared to parental cell line with the wt protein [40]. In support of these results, we newly demonstrated that apoptosis induced by combination of SCH900776 and LA-12/cisplatin was significantly accelerated in HCT116 p21−/− cells compared to their wt counterparts. Our findings suggest that p21 status is an important determinant of cancer cell response to combined action of platinum-based drugs and Chk1 inhibitors.
One of the most prominent cell responses to cisplatin treatment is arrest or delay in S and G2 phase of the cell cycle, which can be mediated by Chk1 kinase activated by the drug [48]. Inhibition of this kinase may then enable the cells to proceed through the cell cycle despite the presence of unrepaired damaged DNA, with fatal consequences [49]. UCN-01 abrogated cisplatin-induced G2 arrest followed by enhanced killing of cisplatin-resistant HCT116 clones or CHO cells [15], [16]. Combined treatment of AZD7762 and cisplatin abrogated G1 and S phase arrest, followed by clear cell carcinoma cell death [22]. We demonstrated that HCT116 wt cells treated with the combination of SCH900776 and LA-12/cisplatin showed a remarkable loss of important G2/M regulators cyclin A, B1, Cdc25C, survivin, or Aurora B, and undetectable H3 (Ser10) phosphorylation, whereas the observed changes in pRb, p53, and p21 levels accounted for G1 arrest. This indicates that the cells might have undergone mitotic slippage, a premature exit from M to G1 phase without cell division [50]. Our results showing a relatively high percentage of drug combination–treated cells with 4 N DNA content, while preferentially displaying G1 markers, are supportive to this hypothesis [51].
In contrast to their wt counterparts, p53−/− or p21−/− cells treated with SCH900776 and platinum drugs did not so dramatically suppress the crucial mitotic regulators, which indicates their mitotic entry and progression, further supported by their relatively high pH3 (Ser10) level. A significant increase in the histone H3 phosphorylation associated with enhanced apoptosis was observed in p53 null and p53 null/p21 knockdown HCT116 cells treated with the combination of AZD7762 and irinotecan compared to the similarly treated parental cells [47]. An increase in number of mitotic HCT116 p53−/− cells was also demonstrated upon their combined treatment with Chk1 inhibitor Chir-124 and irradiation, whereas no such effect was apparent in their wt counterparts [52]. Overriding Chk1-mediated G2 arrest resulted in premature mitotic entry and mitotic cell death in H2O2-treated HCT116 cells deficient for p53/p21 [50]. Importantly, we newly showed that loss of p21 in p53 wt cells stimulated SCH900776- and LA-12/cisplatin combination-induced apoptosis in G2/M cell cycle phase, which was not so evident in wt or p53−/− cells.
The combined treatment with SCH900776 and platinum drugs also resulted in an enhanced fraction of polyploid p21−/− and p53−/− cells, while the wt cells remained mostly unaffected. Similarly, an increase in polyploidy was also demonstrated in HCT116 p21−/− and p53−/− but not wt cells treated with Chir-214 and irradiation [52] or in p53-mutant or -deficient HNSCC cells exposed to cisplatin and AZD7762 [21]. Finally, in the absence of p53 and especially p21, the ability of platinum drugs to induce senescence-associated phenotype was significantly diminished, and co-treatment with SCH900776 further supported this effect. The senescence-like response induced by chemotherapeutic drugs has been previously reported as strongly decreased but not abolished in p53- or p21-deficient HCT116 colon carcinoma cell lines [53], and the link between senescence and Chk1 signaling has also been suggested in this cancer cell type [50], further supporting our results.
We and others previously demonstrated that the cancer cell sensitivity to the cytotoxic effects of LA-12 or cisplatin could be affected by PTEN status [27], [54]. Importantly, PTEN is known to play a unique role in DNA damage response and cell cycle regulation and can also functionally interact with Chk1 pathway [55]. Depletion of Chk1 resulted in decreased PTEN level in hydroxyurea-treated human osteosarcoma U2OS cells [56]. We also observed that the combined treatment with SCH900776 and LA-12/cisplatin induced a loss in PTEN level in HCT116 cells compared to the action of individual drugs. Moreover, we showed that the cooperative killing action of SCH900776 and LA-12/cisplatin is facilitated in the absence of PTEN.
Taken together, we newly demonstrated that SCH900776 can significantly affect the platinum drug–induced cytotoxicity in human colon cancer cells, and their p53 or p21 status further decides about the preferential type and kinetics of the cell response. In HCT116 wt cells, the combined treatment with SCH900776 and cisplatin/LA-12 resulted in stimulation of mitotic slippage, senescence, or apparent G1/S phase–related apoptosis. However, in the absence of p53 or p21, the cells treated with SCH900776 and LA-12/cisplatin entered mitosis and became polyploid, and the senescence phenotype was strongly suppressed. While the cell death induced by SCH900776 and cisplatin or LA-12 was significantly delayed in the absence of p53, the anticancer action of the drug combinations was significantly accelerated in p21-deficient cells that underwent apoptosis during the whole cell cycle progression including G2/M phase. We also showed that cooperative killing action of the drug combinations in HCT116 cells was facilitated in the absence of PTEN. Further investigation of the anticancer effects of clinical candidate SCH900776 or other Chk1 inhibitors in combination with conventionally used or novel platinum-based drugs, both in vitro and in vivo cancer models, is needed to further confirm whether this approach can be generally applicable to improve the treatment outcomes, and to determine the useful biomarkers of cancer cell response to the combined action of the drugs.
The following are the supplementary data related to this article.
Supplementary Material and Methods
Acknowledgments
Acknowledgements
The authors thank Profs. B. Vogelstein and T. Waldman for providing cell lines (specified in Material and Methods section); Radek Fedr, M.Sc., for his outstanding technical support in flow cytometry; Zuzana Kahounová, Ph.D., for her advice in senescence analysis; Šárka Šimečková, M.Sc., for her help and advice in some flow cytometry analyses; Zuzana Tylichová, M.Sc., and Ondřej Zapletal, M.Sc., for their help with cell cultures; Tereza Suchánková, Ph.D., for helpful discussion; and Iva Lišková, Kateřina Svobodová, and Martina Urbánková for their superb technical assistance. We apologize to those investigators whose work could not be cited due to space limitations.
This work was supported by the Czech Science Foundation (15-06650S), grant CZ-OPENSCREEN: National Infrastructure for Chemical Biology (LM2015063), HistoPARK (CZ.1.07/2.3.00/ 20.0185), project no. LQ1605 from the National Program of Sustainability II (MEYS CR), and Brno City Municipality (Brno Ph.D. Talent).
Competing Interests
The authors declare no conflict of interests. Petr Sova is an employee of the company owning intellectual property rights regarding LA-12 and a coauthor of patents in the field of LA-12.
References
- 1.Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel-Bellan A, Castedo M, Kroemer G. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257. doi: 10.1038/cddis.2013.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y, Hunter T. Roles of Chk1 in cell biology and cancer therapy. Int J Cancer. 2014;134:1013–1023. doi: 10.1002/ijc.28226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okita N, Minato S, Ohmi E, Tanuma S, Higami Y. DNA damage-induced CHK1 autophosphorylation at Ser296 is regulated by an intramolecular mechanism. FEBS Lett. 2012;586:3974–3979. doi: 10.1016/j.febslet.2012.09.048. [DOI] [PubMed] [Google Scholar]
- 4.Tapia-Alveal C, Calonge TM, O'Connell MJ. Regulation of chk1. Cell Div. 2009;4:8. doi: 10.1186/1747-1028-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson R, Eastman A. The cancer therapeutic potential of Chk1 inhibitors: how mechanistic studies impact on clinical trial design. Br J Clin Pharmacol. 2013;76:358–369. doi: 10.1111/bcp.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manic G, Obrist F, Sistigu A, Vitale I. Trial watch: targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol Cell Oncol. 2015;2:e1012976. doi: 10.1080/23723556.2015.1012976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McNeely S, Beckmann R, Bence Lin AK. CHEK again: revisiting the development of CHK1 inhibitors for cancer therapy. Pharmacol Ther. 2014;142:1–10. doi: 10.1016/j.pharmthera.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Guzi TJ, Paruch K, Dwyer MP, Labroli M, Shanahan F, Davis N, Taricani L, Wiswell D, Seghezzi W, Penaflor E. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol Cancer Ther. 2011;10:591–602. doi: 10.1158/1535-7163.MCT-10-0928. [DOI] [PubMed] [Google Scholar]
- 9.Karp JE, Thomas BM, Greer JM, Sorge C, Gore SD, Pratz KW, Smith BD, Flatten KS, Peterson K, Schneider P. Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor Sch 900776 in refractory acute leukemias. Clin Cancer Res. 2012;18:6723–6731. doi: 10.1158/1078-0432.CCR-12-2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montano R, Chung I, Garner KM, Parry D, Eastman A. Preclinical development of the novel Chk1 inhibitor SCH900776 in combination with DNA-damaging agents and antimetabolites. Mol Cancer Ther. 2012;11:427–438. doi: 10.1158/1535-7163.MCT-11-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Montano R, Thompson R, Chung I, Hou H, Khan N, Eastman A. Sensitization of human cancer cells to gemcitabine by the Chk1 inhibitor MK-8776: cell cycle perturbation and impact of administration schedule in vitro and in vivo. BMC Cancer. 2013;13:604. doi: 10.1186/1471-2407-13-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schenk EL, Koh BD, Flatten KS, Peterson KL, Parry D, Hess AD, Smith BD, Karp JE, Karnitz LM, Kaufmann SH. Effects of selective checkpoint kinase 1 inhibition on cytarabine cytotoxicity in acute myelogenous leukemia cells in vitro. Clin Cancer Res. 2012;18:5364–5373. doi: 10.1158/1078-0432.CCR-12-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zemanova J, Hylse O, Collakova J, Vesely P, Oltova A, Borsky M, Zaprazna K, Kasparkova M, Janovska P, Verner J. Chk1 inhibition significantly potentiates activity of nucleoside analogs in TP53-mutated B-lymphoid cells. Oncotarget. 2016;7:62091–62106. doi: 10.18632/oncotarget.11388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Venkatesha VA, Parsels LA, Parsels JD, Zhao L, Zabludoff SD, Simeone DM, Maybaum J, Lawrence TS, Morgan MA. Sensitization of pancreatic cancer stem cells to gemcitabine by Chk1 inhibition. Neoplasia. 2012;14:519–525. doi: 10.1593/neo.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bunch RT, Eastman A. Enhancement of cisplatin-induced cytotoxicity by 7-hydroxystaurosporine (UCN-01), a new G2-checkpoint inhibitor. Clin Cancer Res. 1996;2:791–797. [PubMed] [Google Scholar]
- 16.Shen H, Perez RE, Davaadelger B, Maki CG. Two 4N cell-cycle arrests contribute to cisplatin-resistance. PLoS One. 2013;8:e59848. doi: 10.1371/journal.pone.0059848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rawlinson R, Massey AJ. gammaH2AX and Chk1 phosphorylation as predictive pharmacodynamic biomarkers of Chk1 inhibitor-chemotherapy combination treatments. BMC Cancer. 2014;14:483. doi: 10.1186/1471-2407-14-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bryant C, Rawlinson R, Massey AJ. Chk1 inhibition as a novel therapeutic strategy for treating triple-negative breast and ovarian cancers. BMC Cancer. 2014;14:570. doi: 10.1186/1471-2407-14-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duan L, Perez RE, Hansen M, Gitelis S, Maki CG. Increasing cisplatin sensitivity by schedule-dependent inhibition of AKT and Chk1. Cancer Biol Ther. 2014;15:1600–1612. doi: 10.4161/15384047.2014.961876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Massey AJ, Borgognoni J, Bentley C, Foloppe N, Fiumana A, Walmsley L. Context-dependent cell cycle checkpoint abrogation by a novel kinase inhibitor. PLoS One. 2010;5:e13123. doi: 10.1371/journal.pone.0013123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gadhikar MA, Sciuto MR, Alves MV, Pickering CR, Osman AA, Neskey DM, Zhao M, Fitzgerald AL, Myers JN, Frederick MJ. Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53. Mol Cancer Ther. 2013;12:1860–1873. doi: 10.1158/1535-7163.MCT-13-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Itamochi H, Nishimura M, Oumi N, Kato M, Oishi T, Shimada M, Sato S, Naniwa J, Kudoh A, Kigawa J. Checkpoint kinase inhibitor AZD7762 overcomes cisplatin resistance in clear cell carcinoma of the ovary. Int J Gynecol Cancer. 2014;24:61–69. doi: 10.1097/IGC.0000000000000014. [DOI] [PubMed] [Google Scholar]
- 23.Wagner JM, Karnitz LM. Cisplatin-induced DNA damage activates replication checkpoint signaling components that differentially affect tumor cell survival. Mol Pharmacol. 2009;76:208–214. doi: 10.1124/mol.109.055178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huntoon CJ, Flatten KS, Wahner Hendrickson AE, Huehls AM, Sutor SL, Kaufmann SH, Karnitz LM. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013;73:3683–3691. doi: 10.1158/0008-5472.CAN-13-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zak F, Turanek J, Kroutil A, Sova P, Mistr A, Poulova A, Mikolin P, Zak Z, Kasna A, Zaluska D. Platinum(IV) complex with adamantylamine as nonleaving amine group: synthesis, characterization, and in vitro antitumor activity against a panel of cisplatin-resistant cancer cell lines. J Med Chem. 2004;47:761–763. doi: 10.1021/jm030858+. [DOI] [PubMed] [Google Scholar]
- 26.Jelinkova I, Safarikova B, Vondalova Blanarova O, Skender B, Hofmanova J, Sova P, Moyer MP, Kozubik A, Kolar Z, Ehrmann J. Platinum(IV) complex LA-12 exerts higher ability than cisplatin to enhance TRAIL-induced cancer cell apoptosis via stimulation of mitochondrial pathway. Biochem Pharmacol. 2014;92:415–424. doi: 10.1016/j.bcp.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 27.Laukova J, Kozubik A, Hofmanova J, Nekvindova J, Sova P, Moyer MP, Ehrmann J, Hyrslova Vaculova A. Loss of PTEN facilitates rosiglitazone-mediated enhancement of platinum(IV) complex LA-12–induced apoptosis in colon cancer cells. PLoS One. 2015;10:e0141020. doi: 10.1371/journal.pone.0141020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roubalova E, Kvardova V, Hrstka R, Borilova S, Michalova E, Dubska L, Muller P, Sova P, Vojtesek B. The effect of cellular environment and p53 status on the mode of action of the platinum derivative LA-12. Invest New Drugs. 2010;28:445–453. doi: 10.1007/s10637-009-9270-4. [DOI] [PubMed] [Google Scholar]
- 29.Svihalkova-Sindlerova L, Foltinova V, Vaculova A, Horvath V, Soucek K, Sova P, Hofmanova J, Kozubik A. LA-12 overcomes confluence-dependent resistance of HT-29 colon cancer cells to Pt (II) compounds. Anticancer Res. 2010;30:1183–1188. [PubMed] [Google Scholar]
- 30.Vondalova Blanarova O, Jelinkova I, Hyrslova Vaculova A, Sova P, Hofmanova J, Kozubik A. Higher anti-tumour efficacy of platinum(IV) complex LA-12 is associated with its ability to bypass M-phase entry block induced in oxaliplatin-treated human colon cancer cells. Cell Prolif. 2013;46:665–676. doi: 10.1111/cpr.12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sova P, Mistr A, Kroutil A, Zak F, Pouckova P, Zadinova M. Comparative anti-tumor efficacy of two orally administered platinum(IV) drugs in nude mice bearing human tumor xenografts. Anticancer Drugs. 2006;17:201–206. doi: 10.1097/00001813-200602000-00012. [DOI] [PubMed] [Google Scholar]
- 32.Horvath V, Blanarova O, Svihalkova-Sindlerova L, Soucek K, Hofmanova J, Sova P, Kroutil A, Fedorocko P, Kozubik A. Platinum(IV) complex with adamantylamine overcomes intrinsic resistance to cisplatin in ovarian cancer cells. Gynecol Oncol. 2006;102:32–40. doi: 10.1016/j.ygyno.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 33.Kozubik A, Horvath V, Svihalkova-Sindlerova L, Soucek K, Hofmanova J, Sova P, Kroutil A, Zak F, Mistr A, Turanek J. High effectiveness of platinum(IV) complex with adamantylamine in overcoming resistance to cisplatin and suppressing proliferation of ovarian cancer cells in vitro. Biochem Pharmacol. 2005;69:373–383. doi: 10.1016/j.bcp.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 34.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 35.Lee C, Kim JS, Waldman T. PTEN gene targeting reveals a radiation-induced size checkpoint in human cancer cells. Cancer Res. 2004;64:6906–6914. doi: 10.1158/0008-5472.CAN-04-1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho SH, Toouli CD, Fujii GH, Crain C, Parry D. Chk1 is essential for tumor cell viability following activation of the replication checkpoint. Cell Cycle. 2005;4:131–139. doi: 10.4161/cc.4.1.1299. [DOI] [PubMed] [Google Scholar]
- 37.Tuschl T. Cotransfection of luciferase reporter plasmids with siRNA duplexes. CSH Protoc. 2006:2006. doi: 10.1101/pdb.prot4342. [DOI] [PubMed] [Google Scholar]
- 38.Pernicova Z, Slabakova E, Kharaishvili G, Bouchal J, Kral M, Kunicka Z, Machala M, Kozubik A, Soucek K. Androgen depletion induces senescence in prostate cancer cells through down-regulation of Skp2. Neoplasia. 2011;13:526–536. doi: 10.1593/neo.11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arora S, Bisanz KM, Peralta LA, Basu GD, Choudhary A, Tibes R, Azorsa DO. RNAi screening of the kinome identifies modulators of cisplatin response in ovarian cancer cells. Gynecol Oncol. 2010;118:220–227. doi: 10.1016/j.ygyno.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 40.Carrassa L, Broggini M, Erba E, Damia G. Chk1, but not Chk2, is involved in the cellular response to DNA damaging agents: differential activity in cells expressing or not p53. Cell Cycle. 2004;3:1177–1181. [PubMed] [Google Scholar]
- 41.Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med. 2011;17:88–96. doi: 10.1016/j.molmed.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pabla N, Huang S, Mi QS, Daniel R, Dong Z. ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J Biol Chem. 2008;283:6572–6583. doi: 10.1074/jbc.M707568200. [DOI] [PubMed] [Google Scholar]
- 43.Wang Q, Fan S, Eastman A, Worland PJ, Sausville EA, O'Connor PM. UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J Natl Cancer Inst. 1996;88:956–965. doi: 10.1093/jnci/88.14.956. [DOI] [PubMed] [Google Scholar]
- 44.Husain A, Yan XJ, Rosales N, Aghajanian C, Schwartz GK, Spriggs DR. UCN-01 in ovary cancer cells: effective as a single agent and in combination with cis-diamminedichloroplatinum(II) independent of p53 status. Clin Cancer Res. 1997;3:2089–2097. [PubMed] [Google Scholar]
- 45.Zenvirt S, Kravchenko-Balasha N, Levitzki A. Status of p53 in human cancer cells does not predict efficacy of CHK1 kinase inhibitors combined with chemotherapeutic agents. Oncogene. 2010;29:6149–6159. doi: 10.1038/onc.2010.343. [DOI] [PubMed] [Google Scholar]
- 46.Rodriguez R, Meuth M. Chk1 and p21 cooperate to prevent apoptosis during DNA replication fork stress. Mol Biol Cell. 2006;17:402–412. doi: 10.1091/mbc.E05-07-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Origanti S, Cai SR, Munir AZ, White LS, Piwnica-Worms H. Synthetic lethality of Chk1 inhibition combined with p53 and/or p21 loss during a DNA damage response in normal and tumor cells. Oncogene. 2012;32:577–588. doi: 10.1038/onc.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He G, Kuang J, Khokhar AR, Siddik ZH. The impact of S- and G2-checkpoint response on the fidelity of G1-arrest by cisplatin and its comparison to a non-cross-resistant platinum(IV) analog. Gynecol Oncol. 2011;122:402–409. doi: 10.1016/j.ygyno.2011.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luo Y, Rockow-Magnone SK, Kroeger PE, Frost L, Chen Z, Han EK, Ng SC, Simmer RL, Giranda VL. Blocking Chk1 expression induces apoptosis and abrogates the G2 checkpoint mechanism. Neoplasia. 2001;3:411–419. doi: 10.1038/sj.neo.7900175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poehlmann A, Habold C, Walluscheck D, Reissig K, Bajbouj K, Ullrich O, Hartig R, Gali-Muhtasib H, Diestel A, Roessner A. Cutting edge: Chk1 directs senescence and mitotic catastrophe in recovery from G(2) checkpoint arrest. J Cell Mol Med. 2011;15:1528–1541. doi: 10.1111/j.1582-4934.2010.01143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Restall IJ, Parolin DA, Daneshmand M, Hanson JE, Simard MA, Fitzpatrick ME, Kumar R, Lavictoire SJ, Lorimer IA. PKCiota depletion initiates mitotic slippage-induced senescence in glioblastoma. Cell Cycle. 2015;14:2938–2948. doi: 10.1080/15384101.2015.1071744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tao Y, Leteur C, Yang C, Zhang P, Castedo M, Pierre A, Golsteyn RM, Bourhis J, Kroemer G, Deutsch E. Radiosensitization by Chir-124, a selective CHK1 inhibitor: effects of p53 and cell cycle checkpoints. Cell Cycle. 2009;8:1196–1205. doi: 10.4161/cc.8.8.8203. [DOI] [PubMed] [Google Scholar]
- 53.Chang BD, Xuan Y, Broude EV, Zhu H, Schott B, Fang J, Roninson IB. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene. 1999;18:4808–4818. doi: 10.1038/sj.onc.1203078. [DOI] [PubMed] [Google Scholar]
- 54.Wu H, Cao Y, Weng D, Xing H, Song X, Zhou J, Xu G, Lu Y, Wang S, Ma D. Effect of tumor suppressor gene PTEN on the resistance to cisplatin in human ovarian cancer cell lines and related mechanisms. Cancer Lett. 2008;271:260–271. doi: 10.1016/j.canlet.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 55.Ming M, He YY. PTEN in DNA damage repair. Cancer Lett. 2012;319:125–129. doi: 10.1016/j.canlet.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martin SA, Ouchi T. Cellular commitment to reentry into the cell cycle after stalled DNA is determined by site-specific phosphorylation of Chk1 and PTEN. Mol Cancer Ther. 2008;7:2509–2516. doi: 10.1158/1535-7163.MCT-08-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material and Methods