


Abstract
Template switching during reverse transcription is crucial for retroviral replication. While strand transfer on the terminal repeated sequence R is essential to achieve reverse transcription, template switching from internal regions of the genome (copy choice) leads to genetic recombination. We have developed an experimental system to study copy-choice recombination in vitro along the HIV-1 genome. We identify here several genomic regions, including the R sequence, where copy choice occurred at high rates. The frequency of copy choice occurring in a given region of template was strongly influenced by the surrounding sequences, an observation that suggests a pivotal role of the folding of template RNA in the process. The sequence R, instead, constituted an exception to this rule since it was a strong hot-spot for copy choice in the different sequence contexts tested. We suggest therefore that the structure of this region has been optimised during viral evolution to ensure efficient template switching independently from the sequences that might surround it.
INTRODUCTION
Recombination is a major source of genetic variability in retroviruses (1). In this family of viruses, recombination is a consequence of template switching by reverse transcriptase (RT) during reverse transcription (2), a process referred to as ‘copy choice’ (3). If the two RNAs are genetically distinct this process leads to genetic recombination. Although the occurrence of recombination in retroviruses was first described in 1971 (4), the burst of human immunodeficiency virus (HIV) genomes sequenced has only recently documented the dramatic impact of recombination on the dynamics of viral populations. For this virus, at least 10% of the infectious strains have been generated by recombination among different viral subtypes (5–7). By allowing the transfer of a whole set of nucleotide changes in a single cycle of replication, recombination accelerates the appearance of new complex variants of the virus, as documented for the generation of resistant strains in response to antiviral treatments (8,9).
Determining the presence of sequences where copy choice preferentially occurs, and locating them on the genome, can reveal the existence of significant biases in the production of specific recombinant forms. Evaluating the rate of generation of individual classes of recombinant genomes regardless of the effect of the selective pressure is essential for understanding the role of recombination in the evolution of retroviruses. Additionally, defining ‘recombination prone’ sequences will provide insight into the molecular mechanism leading to template switching. The identification of such sequences in vivo is a difficult task though, since the analysis of the distribution of the junctions between genomic regions with different phylogenetic histories (breakpoints) is only possible on those genomes that have survived the selective pressure. Each breakpoint identified results, therefore, from a process involving an initial recombination event in that given region of the genome, and the adaptive success of the resulting recombinant virus in the infected organism. Furthermore, only those genomes for which a complete sequence is available allow a systematic analysis of the distribution of breakpoints (10,11). Their number is at present largely insufficient to allow a statistically significant analysis. Other approaches rely on culture cell systems (2,12,13). Through these experimental approaches it has been shown that recombination is frequent along the HIV-1 genome (14). However, the level of resolution at which these results have been obtained (∼0.8 kb) did not allow assessment of whether significant local fluctuations in the frequencies of recombination exist (14).
Reconstituted in vitro systems allow to reach such an accuracy, and the question can then be tackled from a more mechanistic angle. Several factors seem to contribute to the propensity of RTs to switch template during reverse transcription (15). If pausing of DNA synthesis has been invoked as principally responsible (16,17), more recent data suggest that the secondary structures of the RNA templates are determinant in the process (18,19). When copy choice is investigated in vitro, a major role is played by the viral nucleocapsid protein (NC), an RNA chaperone present on the genomic RNA during reverse transcription (20,21). NC enhances strand transfer (22–26), an effect proposed to be exerted, for copy choice, mainly through the interconversion of RNA secondary structures during reverse transcription (19) and possibly by direct interaction of NC with the RT (27).
Transferring DNA synthesis from one region of the genomic RNA to another not only constitutes a stochastic event involved in genetic variability, but is also an obligatory step of reverse transcription in retroviruses. Strand transfer on the terminal repeated sequence R is required at each infectious cycle, a process called minus DNA strong-stop strand transfer. This step allows the synthesis of a full-length minus DNA strand, solves the problem of replicating the terminal sequences of the genomic RNA, and leads to the generation of the long terminal repeated (LTR) sequences of the proviral DNA (28) (Fig. 1). Although according to the prevailing model strand transfer on R is a consequence of the ‘strong stop’ imposed by the 5′ end of the genome (29,30), template switching has also been shown to occur from within R by a copy choice mechanism (31–33). Such observations raise the possibility that R constitutes a site of intense copy choice. For these reasons, while we have studied here copy choice on several regions of the HIV-1 genome, some of which were previously identified as breakpoints in vivo, we have also focused our attention on the R sequence.
MATERIALS AND METHODS
Plasmids and templates for RNA synthesis
All sequences used in this study were isolated from the genome of HIV-1 (LAI strain) by PCR, and verified by sequencing of the cloned region. The primers hybridising to the 3′ end of the retroviral sequence of interest carried, in their non-hybridising tail, either a BamHI or a BspHI site. The primers hybridising to the 5′ end carried an XhoI site. The PCR products were cloned either into a vector containing a part of the malT gene from Escherichia coli (sequence available on request) and the β-lactamase gene, leading to a family of plasmids called pDvir, or into a vector that carries the kanamycin resistance gene and the lacZ’ gene, leading to the family of plasmids called pAvir. These plasmids carry the following retroviral sequences: (i) ΔgagU5RU3a, comprising Δgag, the first 66 nt of the gag gene, U5 and R sequences (84 and 97 nt, respectively) and U3a encompassing the 150 nt at the 3′ end of U3; (ii) The E1 sequence, comprising nucleotides 102–502 of the env gene; (iii) E2, encompassing nucleotides 953–1352 of the env gene; (iv) G1, corresponding to nucleotides 151–550 of the gag gene; (v) G2, nucleotides 551–950 of the gag gene; (vi) G3, nucleotides 951–1350 of the gag gene; (vii) U3r, the first 305 nt from the U3 sequence fused to the 95 nt upstream. U3aU3b spans 400 nt from nucleotide 56 to the end of the U3 sequence. G1aRG1b and G1aE2bG1b are sequences where R and E2b (the 200 nt from the middle of the E2 sequence) have been used to replace the 97 nt sequence located in the middle of G1. E1a-R-E1b represents the sequence where R replaces the 97 nt localised in the middle of E1 (see text for details). pDvir and pAvir were used to synthesise the donor and the acceptor RNAs, respectively. RNA synthesis was carried out by incubating pDvir and pAvir (digested by BspHI) with SP6 as previously described (34).
Nucleocapsid protein
The shorter fragment of the nucleocapsid protein of HIV-1, NC (amino acids 1–55) was prepared by solid phase peptide synthesis and purified as described (35). The peptide was treated in solution by a small excess (2.1 equivalents) of zinc, allowing the folding of the two zinc fingers essential for the biological activity of NC (36). The NC was used at ratio of 1 NC molecule to 8 nt of RNA.
Reverse transcription and cloning of the reverse transcription products
Reverse transcription was performed in the presence of the donor and acceptor RNAs (at a final concentration of 100 nM each), after annealing an oligonucleotide to the donor template to prime reverse transcription (primer FS, Fig. 1B). Annealing was performed at a primer to donor template molar ratio of 10:1 in 50 mM Tris–HCl pH 7.8, 75 mM KCl, 7 mM MgCl2 at 65°C for 5 min followed by a slow cooling to 40°C. For experiments run without NC, DTT (1 mM final concentration), the four dNTPs (1 mM each) and RNasin (100 U; Promega WI) were added after incubation for 5 min on ice. For the experiments with NC, the protein was added at this step and incubated for 10 min at 37°C. Reverse transcription was started by the addition of the RT at a final concentration of 300 nM and carried out for 90 min. After reverse transcription, the samples containing NC were treated for 1 h at 56°C with proteinase K (8 mg/ml), 0.4% (w/v) of SDS, and 50 mM EDTA pH 8.0. The reaction was stopped by extraction with phenol–chloroform followed by RNase treatment, purification of the reverse transcription product, and synthesis of the second DNA strand (using oligo SS, see Fig. 2) as previously described (34). The full-length double-stranded DNA was gel purified, digested by BamHI and PstI and ligated to the vDvir. vDvir was obtained by double digestion of pDvir by BamHI and PstI and double purification of the band containing the origin of replication and the β-lactamase gene on an agarose gel (Fig. 1B). The ligation mixture was used for transformation of XL2-blue MRF’ Epicurian coli cells (Stratagene). As a control, an equivalent amount of vDvir alone was also ligated and used for transformation, providing an estimate of the background of the white colonies resulting from a transformation with circularised vDvir vectors. The background value never exceeded 10% of the white colonies recovered from the recombination samples. The frequency of recombination was computed, as described in the legend to Table 1.
Figure 1.

(A) Outline of the minus DNA strong-stop strand transfer in the strategy of reverse transcription in retroviruses. Reverse transcription begins from a tRNA hybridised to a region of the genome called PBS (top), and proceeds up to the 5′ end of the RNA (middle diagram: RNA, thin line and lower case letters; DNA, thick line, capital letters). The RNaseH activity carried by the RT hydrolyses the RNA template once copied (middle diagram). The nascent DNA is then transferred at the 3′ end of the genomic RNA, allowing the copying of the internal regions of the genome (bottom). (B) The experimental system. The two RNAs present a region of homology constituted by a viral sequence (400 nt in size, grey box) followed, in the sense of reverse transcription, by a genetic marker [lacZ’ for the acceptor RNA (black box) or a truncated and thus non-functional portion of the malT gene from E.coli on the donor template (white box)]. While the 3′ end of the acceptor RNA is constituted by the last nucleotide of the viral sequence studied, the donor RNA also contains at its 3′ end an extension which is used to selectively prime reverse transcription after hybridisation of a complementary oligonucleotide ‘FS’ (black arrow). Processive copying of the donor template will yield lac– genotypes, while template switching during reverse transcription of the retroviral sequence will produce lac+ genotypes. After reverse transcription, the second strand is synthesised by Taq DNA polymerase using a primer ‘SS’ (grey arrow) complementary to the last 18 nt of the single-stranded DNAs resulting from reverse transcription, which will be the same for the parental and the recombinant molecules. We stress that this step does not constitute a polymerase chain reaction. The resulting double-stranded DNAs are restricted with BamHI and PstI and, after ligation to a plasmid vector, used for bacterial transformation. On appropriate media, recombinant DNAs will yield blue colonies distinguishable from the white colonies given by the parental DNAs. The ratio of blue to total colonies allows an estimation of the frequency of recombination.
Figure 2.

(A) Location of the RNA sequences used in the present study on the HIV-1 genome. The viral genome is indicated as black boxes. The individual sequences included in our model templates are shown as open boxes, and their names are given (see text for details). (B) Detail of the retroviral sequences for which the frequency of recombination was determined within for individual sub-regions of the model templates. Left, the sequence referred to in the text as E2 (see Material and Methods) was divided into three regions by the insertion of two point mutations generating the two restriction sites indicated. Right, schematic representation of ΔgagU5-R-U3a (see text for details). The restriction sites were introduced by single base mutations outside R (see also Table 2 legend). (C) Example of generation of a double recombinant molecule. The dotted line traces the path followed by the reverse transcription. The resulting molecule will be positive to a restriction analysis both by NcoI and by ApaLI, but after bacterial transformation this molecule will confer a white phenotype typical of parental molecules. In the example given, the molecule will be recombinant in E2b and in E2c.
Table 1. Recombination rates on the sequences studied.
| Sequence name |
si (nt) |
fi (%) |
ri (×10–4) |
| ΔgagU5RU3a | 397 | 37.9 ± 1.7 | 9.5 ± 0.4 |
| G1 | 400 | <1.0 | <0.3 |
| G2 | 400 | <1.0 | <0.3 |
| G3 | 400 | 35.6 ± 1.8 | 8.9 ± 0.4 |
| E1 | 400 | 30.3 ± 2.2 | 7.5 ± 0.5 |
| E2 | 400 | 52.5 ± 2.7 | 13.1 ± 0.6 |
| U3r | 400 | <1.0 | <0.3 |
| U3aU3b | 400 | 31.3 ± 1.8 | 7.8 ± 0.5 |
| G1aRG1b | 400 | 11.8 ± 1.5 | 3.0 ± 0.3 |
| G1aE2bG1b | 502 | 18.7 ± 0.7 | 3.7 ± 0.2 |
The frequency of recombination (percentage) fi was calculated by dividing the number of blue colonies by number of total colonies obtained (sum of blue and white colonies) (see Fig. 1 legend). The rate of recombination ri was calculated by dividing fi by the size of the sequence, si, in nucleotides: ri = (fi/si) × (1/100). Values were averaged from three independent experiments and the standard deviation was estimated by using the Microsoft Excel program.
Prediction of RNA secondary structure
Folding of the various RNA sequences was predicted according to the Zuker algorithm (37) (also see http://bioinfo.math.rpi.edu/~mfold/rna/form1.cgi). For a given viral insert, we considered not only the most stable secondary structure, but also the set of conformations, i, which presented, with respect to this reference structure, a free energy excess, ΔGi, of <3 kcal/mol. The frequency wi of appearance of a predicted structure i is then
wi = e–ΔGi/RT
where R is the universal gas constant and T is the absolute temperature in degrees Kelvin. Taking wi(o) (the frequency of appearance of the most stable structure) as equal to one, the probability p(j) of finding a given stem–loop motif, j, in this ensemble of structures is then given by:
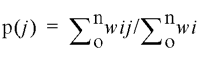
where wij denotes the frequency of appearance of a structure i when the stem–loop j is present (the summation is performed on the i index).
RESULTS
We have developed an experimental system to study in vitro copy choice on retroviral sequences, where a model RNA template is reverse transcribed in the presence of another, partially homologous, RNA. The region of homology between the two RNAs is constituted by a retroviral sequence (Fig. 1B). Template switching during reverse transcription leads to synthesis of DNA coding for the lacZ’ gene (Fig. 1 legend).
We measured the frequency of copy choice on three regions from the gag gene of HIV-1, G1, G2 and G3, each of which is 400 nt long. Altogether, these three regions correspond to 80% of the gag gene. Two other 400 nt long sequences from the env gene, E1 and E2, were also analysed (Fig. 2A and Materials and Methods). An early study localised recurrent intersubtype breakpoints within the regions we call G1, G3, E1 and E2. The locations of the corresponding breakpoints were identified with a resolution of 200 nt (11). With the aim of preserving the local secondary structures of these breakpoints, the 200 nt of interest were embedded in a larger sequence by including 100 nt of flanking genomic RNA. To mimic the physiological context more closely, unless otherwise stated, all experiments were run in the presence of the NC protein. The average recombination rate per nucleotide was found to be 8.9 × 10–4, 7.5 × 10–4 and 13.1 × 10–4 for G3, E1 and E2, respectively, while it was almost undetectable in the case of G1 and G2 (<0.3 × 10–4) (Table 1). The rates for G3, E1 and E2 are above those previously found under the same experimental conditions on non-viral model templates, which ranged from 1 × 10–4 to 6 × 10–4 (19). For the ‘hottest’ sequence (E2), point mutations introduced at the border of the 200 nt breakpoint found in vivo allowed us to discriminate between recombination in the breakpoint itself (E2b, Fig. 2B) or in the flanking sequences (E2a and E2c, Fig. 2B). Most template switches occurred within E2b with a rate reaching 19.1 × 10–4 per nt, while the encompassing sequences (E2a and E2c) yielded rates of 10.1 × 10–4 and 4.3 × 10–4 (Table 2, column 5, lines 10–12). A restriction analysis of plasmids isolated from 21 white colonies demonstrated that four of them corresponded indeed to molecules generated by two template switching events during reverse transcription (double recombinants). In these cases while a first strand transfer brought DNA synthesis from the donor onto the acceptor template, a second template switching brought it back onto the donor template again (Fig. 2C). Therefore we conclude that the growing DNA chain was transferred in at least 38% of the cases during reverse transcription of E2b, a value that rises to ∼50% if the double recombinants are considered (data not shown).
Table 2. Mapping of strand transfer events.
| Sequence name | Part of the sequence | si (nt) | +NC | –NC | ||
| |
|
|
ni/N |
ri (×10–4) |
ni/N |
ri (×10–4) |
| ΔgagU5RU3a | ΔgagU5 | 138 | 37/149 | 6.8 ± 1.1 | 40/166 | 3.0 ± 0.5 |
| R | 110 | 52/149 | 12.0 ± 1.7 | 61/166 | 5.8 ± 0.7 | |
| U3a | 149 | 60/149 | 10.2 ± 1.3 | 65/166 | 4.2 ± 0.5 | |
| G1aRG1b | G1a | 152 | 0/30 | ≤0.2 | 0/30 | ≤0.1 |
| R | 97 | 30/30 | 12.2 ± 2.2 | 30/30 | 5.4 ± 1.0 | |
| G1b | 151 | 0/30 | ≤0.2 | 0/30 | ≤0.1 | |
| E1aRE1b | E1a | 152 | 20/64 | 8.0 ± 2.2 | 26/64 | 7.0 ± 1.6 |
| R | 97 | 20/64 | 13.4 ± 3.5 | 11/64 | 5.4 ± 1.8 | |
| E1b | 151 | 24/64 | 10.4 ± 2.5 | 26/64 | 10.4 ± 2.5 | |
| E2 | E2a | 100 | 14/73 | 10.1 ± 2.7 | 5/54 | 3.2 ± 1.4 |
| E2b | 200 | 53/73 | 19.1 ± 2.6 | 38/54 | 12.3 ± 2.0 | |
| E2c | 100 | 6/73 | 4.3 ± 1.7 | 11/54 | 7.1 ± 2.1 | |
| G1aEbG1b | G1a | 151 | 14/60 | 2.9 ± 0.8 | 9/48 | 2.0 ± 0.7 |
| E2b | 200 | 31/60 | 4.8 ± 0.9 | 16/48 | 2.7 ± 0.7 | |
| G1b | 151 | 15/60 | 3.1 ± 0.8 | 22/48 | 4.9 ± 1.0 | |
| U3aU3b | U3a | 150 | 11/40 | 5.7 ± 1.7 | ND | ND |
| U3b | 250 | 29/40 | 9.1 ± 1.7 | ND | ND |
Recombination resulting from strand transfer was mapped on each sequence studied (in the absence and the presence of NC) by introduction of point mutations creating a restriction site bordering the sequences of interest. The rate of recombination ri was calculated as follows: ri = (ni/N) × fi/(si × 100), where ni is the number of recombinants occurring in the interval i, N is the total number of blue colonies analysed, fi is the frequency of recombination of the whole sequence (see Table 1) and si is the size of the interval i. The standard deviation for ri was estimated as in Table 1. The difference in size of R between ΔgagU5RU3a and either G1aRG1b or E1aRE1b is due to the position of the restriction site used for mapping purposes. In ΔgagU5RU3a these sites were located within U5 and U3a yielding an R interval 13 nt longer than the canonical 97 nt.
ND, not determined
R inserted within a template
The hypothesis that R is a region of frequent copy choice was tested by its insertion into a model template, depicted in Figure 2B, where it is followed by U5 and preceded the proximal part of U3 (U3a, Fig. 2B). Although within the virion R is not followed by U5 and preceded by U3 on the same retroviral RNA molecule, this constituted the best compromise for studying the ability of R to promote internal strand transfer, while preserving its natural sequence context. Restriction sites bordering R either on the donor or on the acceptor RNAs (Fig. 2B) allowed us to distinguish between template switching within R and in the flanking regions. The recombination rates per nucleotide for each portion of template were 6.8 × 10–4 for the U5 region (Δgag-U5), 10.2 × 10–4 for the U3 region (U3a) and 12.0 × 10–4 for R (Table 2, column 5, lines 1–3).
Sequence context and robustness of R
Since recombination had previously been reported to be frequent throughout the whole U3 region (13) we also tested the remaining portion of U3 that was not included in the U3a (sequence named U3r, Fig. 3A). Surprisingly, the average recombination rate on U3r was found to be <0.3 × 10–4 per nt (Table 1 and Fig. 3A). To cross-check these results, we used an overlapping template (U3aU3b, Fig. 3A), spanning part of the U3r sequence (U3b) and U3a. Although the overall rate of recombination on this template (U3aU3b) was equal to 7.8 × 10–4 per nt (see Table 1), the local recombination rate dropped in U3a by approximately a 2-fold factor (Table 2, column 5, lines 3 and 16). In this model template the frequency of strand transfer on U3b was 9.1 × 10–4 per nt (Fig. 3A), a value much higher than the one found when the same sequence was studied as part of U3r (Fig. 3A). Altogether these results indicate the existence of important sequence context effects.
Figure 3.

Effects of the sequence context on copy choice. (A) The terminal regions of the HIV-1 genome that were tested using different individual templates (shown below). The recombination rates (per nucleotide and to be multiplied by 10–4) found for each interval in the presence of NC are also given. (B) Templates generated by replacing 97 central nucleotides of the sequence G1 and E1 with either the 97 nt of the sequence R or the 200 nt of E2b. In all cases R or E2b were surrounded by 151 nt of the exogenous sequences either from G1 or from E1. These surrounding sequences were named E1a or G1a when located upstream of R or E2b (in the sense of reverse transcription), or E1b and G1b when located downstream. The recombination rates found in each interval are given as for (A).
To also test whether the most important hot-spot regions were sensitive to this sequence context effect, the two ‘hottest’ sequences (R and E2b) were independently inserted within the ‘cold’ sequence G1 (Fig. 3B). The insertion of E2b (200 nt) into G1 yielded a global frequency of recombination of 18.7% (RNA named G1aE2bG1b, Tables 1 and 2). The clonal analysis of the recombination products indicated a decrease in the rate of recombination in E2b from 19.1 × 10–4 to 4.8 × 10–4 (Table 2, column 5, lines 11 and 14, and Fig. 3B). At the same time, strand transfer events that were previously undetectable in the surrounding sequences (G1 in Table 1) now took place at a rate of ∼2.9 × 10–4 to 3.1 × 10–4 (Table 2, column 5, lines 13 and 15, and Fig. 3B). Therefore, the hot-spot E2b was also sensitive to the sequence context.
The same approach was used for the R sequence. In this case, R was inserted, as previously done for E2b, within the G1 sequence (Fig. 3B). The overall frequency of recombination was 11.8% (RNA named G1aRG1b, see Tables 1 and 2). All the recombinant molecules analysed (n = 30) originated by template switching during the copy of the R region. A rate of strand transfer of 12.2 × 10–4 per nt was calculated for this interval, a value consistent with that found when R was surrounded by Δgag-U5 and U3a, (Table 2, column 5, lines 2 and 5). This result indicates that R is insensitive to the sequence context. To further confirm this observation, R was also inserted in a second region, this time belonging to the env gene: the E1 sequence. In this case, the rate of strand transfer calculated for R was 13.4 × 10–4 per nt, once again a value consistent with our previous estimates (Table 2, column 5, lines 2, 5 and 8). We therefore define R as a sequence not only efficient but also robust in its ability to promote copy choice.
Recombination on naked RNAs and secondary structure predictions
All the experiments described until now were performed in the presence of the NC. We therefore checked whether this context effect was also found in the absence of NC. We again measured the frequency of recombination of the hottest sequences E2b and R in their different contexts. The results (summarised in Table 2, column 7, lines 2, 5, 8, 11 and 14), indicate that even on naked RNA, R is a hot-spot irrespective of the sequence context. In contrast, E2b remains sensitive to this parameter. As expected, on naked RNA the general trend was a decrease in the frequency of recombination with respect to that observed with NC, in a 2–3-fold range.
We suspected that the different behaviours of E2b and R RNAs could be related to their local secondary structures. To test this idea, we applied the m-fold algorithm (37) to predict the most stable global folding of the five sequences considered in this study, shown in Figure 3B. For a given sequence, among the alternative folding patterns predicted by the program, we retained a set of conformations that had a free energy increase (ΔGi) <3 kcal/mol higher than that of the most stable structure. In all five cases, we examined the predicted stem–loop structures and computed their mean probability of occurrence, p(j) (see Materials and Methods). The results of this simulation are given in Table 3. R encompasses the whole sequence of the transactivation response element (TAR) region, and part of the polyadenylation [poly(A)] hairpin. The base of the stem of this latter hairpin is indeed generated by the hybridisation of part of R to a complementary portion of the U5 region (38). As expected, while both these elements were recurrently predicted in the case of ΔgagU5RU3a, when R was inserted either within G1 or E1, only the hairpin motif of the TAR element was recurrently present, the U5 portion of the poly(A) hairpin being absent in these cases. In the case of the sequence E2, a stable stem–loop motif, named SL, was detected within E2b. Although the stability of SL was close to that of the TAR element (–29.4 kcal/mol versus –30 kcal/mol, respectively), SL was predicted to be erased when E2b was embedded within G1 (Table 3).
Table 3. Probability of appearance, p(j), of the most stable stem–loop motifs found in the predicted RNA secondary structures.
| Sequence name |
Motif name |
P(j) |
| ΔgagU5RU3a | R | 0.85 |
| TAR | 0.86 | |
| pA | 0.99 | |
| G1aRG1b | TAR | 1.0 |
| pA | 0.0 | |
| E1a-R-E1b | TAR | 1.0 |
| pA | 0.0 | |
| E (Ea-Eb-Ec) | SL (Eb) | 0.8 |
| G1aEbG1b | SL (Eb) | 0.0 |
p(j) is calculated as indicated in the Materials and Methods accounting for all the other stable structures predicted using the Zuker program (see Materials and Methods for details). R corresponds to TAR and the pA; TAR is the TAR element within R, pA is the stem–loop of the poly(A) signal structure. SL (E2b) is the stem–loop motif found in the E2b predicted secondary structure.
DISCUSSION
In the present work we have studied template switching generated by HIV-1 RT along various regions of the HIV-1 genome. Globally, 2 900 nt have been analysed, corresponding to approximately one-third of the viral genome. Most regions analysed yielded a high degree of recombination (Fig. 4), in agreement with studies based on cell cultures (14) and with previous in vitro studies performed on non-viral model templates (19). The only exception was a 0.8 kb region at the 5′ end of the gag gene, where the average frequency of recombination was extremely low. Among the sequences tested, the highest recombination frequency was found in a region of the env gene coding for the surface glycoprotein gp120, corresponding to one of the first recurrent breakpoints found in HIV-1 (11). The reverse transcriptase switches template as it copies this region, in at least 38% of the cases. Extrapolated to the physiological context, this observation implies that even when infection is limited to an extremely small population of ‘heterozygous’ virions, mosaic genomes might be generated through template switching in this region. The high frequency of strand transfer observed in the hot-spot described herein suggests that the parts of the gene lying upstream and downstream of this region could segregate almost independently. This hot-spot spans the coding portion of part of the V2 and of the C2 regions of this protein (gp120). Template switching would potentially allow the reshuffling of the V2 domain with respect to the hypervariable V3 domain, among individual viruses. Recombination in this region could participate in the generation of viruses escaping immune response, since the conformational arrangement of V2 and V3 with respect to the CD4 receptor-binding region of gp120 appears to be critical for the recognition by neutralising antibodies (39).
Figure 4.

Recombination rates across the different tested regions from HIV-1 genome. The values are represented as a rate per nucleotide, calculated as described in Tables 1 and 2. The value for the U3 region was calculated as average of the values found in U3a, U3b and U3r (see text for details).
Using long model RNA templates, we provide clear evidence that recombination on a given sequence of RNA occurs at different rates, depending on the context where it is located. This effect was observed, with the exception of R, for all sequences tested in at least two different contexts (Fig. 3), clearly underlining a modulation of strand transfer by the structures of the RNA template. Until now most in vitro studies on retroviral recombination had been performed on short model templates. Under those conditions the influence of the folding of the RNA templates, as well as of the nascent DNA, could hardly be evaluated. A single nucleotide substitution on an RNA molecule can substantially modify the folding of the surrounding regions up to a distance of 50–70 nt (40). Since, as documented in the case of HIV, individual regions of the genome carry multiple nucleotide substitutions in different viral subtypes, they could yield different RNA structures. Therefore the context effect observed here suggests that the ability of individual regions of the genome to promote strand transfer might vary according to the viral subtypes considered.
An exception to the rule of the sequence context was constituted by the repeated sequence R. This region not only was a hot-spot for copy choice, but also proved to be insensitive to the context effect cited above since it yielded the same recombination rate in all the contexts assayed. Therefore R is a robust hot-spot for copy choice along the genome of HIV-1, and it constitutes an autonomous module consistently able to promote efficient strand transfer. In vivo, R is the region where strand transfer takes place during synthesis of the minus DNA strand once for each infectious cycle (strong-stop strand transfer). The potential role of the specific sequence of R in strong-stop strand transfer had been previously addressed both through in vitro and in vivo approaches. Replacing R from the genome of Moloney murine leukaemia virus (MoMLV) with R from HIV-1 or with a non-viral sequence dramatically decreased the efficiency of in vitro strand transfer by MoMLV RT (41). A recent in vivo study using murine leukaemia virus based vectors demonstrated that R can be substituted by a sequence of non-viral origin without abolishing the ability of the virus to replicate in cultured cells, although a 6-fold decrease in the viral titre was observed in this case (42). The robustness of R observed herein could reflect the need for this region to ensure efficient strand transfer regardless of the evolution of the surrounding sequences U5 and U3. This, in turn, suggests the existence of a selective pressure for the conservation of a basal degree of copy choice on R. Furthermore, intensive recombination in R and in U3, also suggested by ex vivo experiments (13), could also be important for the generation of genetic variability in the LTRs. Since individual viral subtypes can display a specific organisation of binding sites for various transcription factors, recombination in these regions could be relevant for the generation of new viruses that differ in the control of gene expression. The highly replicative strain generated by intersubtype recombination between HIV-1 groups M and O (43) provides such an example.
In all three contexts where R was assayed (between U5 and U3, within G1 and within E1) the TAR hairpin was constantly predicted to be present. In contrast, the formation of the other highly stable hairpin, the SL structure present in E2b, was predicted to depend on the sequence context (Table 3). Since high levels of recombination occurred on R at a constant frequency irrespective of the sequence context while on E2b this parameter correlated with the presence of SL, a stable hairpin could be crucial to yield efficient strand transfer on a given primary structure of RNA. Intriguingly the less stable structure constituted by the poly(A) hairpin seems not to play a crucial role for strand transfer along R, since its presence or absence did not affect the frequency of recombination observed along R (Table 3). This conclusion is also supported by recent work on the structural features of R required for efficient strand transfer under strong-stop conditions, which identified such determinants within the TAR hairpin (44). Altogether these results suggest that the determinants for copy choice might be more diverse and complex than suspected. However, signposts for efficient template switching through this process might exist along the genome and, possibly, be conserved during evolution. R is one such an example and might not be unique.
Acknowledgments
ACKNOWLEDGEMENTS
We are much indebted to Torsten Unge for his generous gift of HIV-1 RT, and to Michel Veron for helpful discussion. We are grateful to Anthony Pugsley for critical reading of the manuscript and to Odile Delpech for secretarial assistance. This work was supported by a grant from the Agence Nationale pour la Recherche sur le SIDA to H.B. (ANRS 95004 and 97004). A.M. is a recipient of a fellowship from the ANRS.
References
- 1.Temin H.M. (1993) Retrovirus variation and reverse transcription: abnormal strand transfers result in retrovirus genetic variation. Proc. Natl Acad. Sci. USA, 90, 6900–6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu W.S. and Temin,H.M. (1990) Retroviral recombination and reverse transcription. Science, 250, 1227–1233. [DOI] [PubMed] [Google Scholar]
- 3.Vogt P.K. (1973) The genome of avian RNA tumor viruses: a discussion of four models. In Silvestri,L. (ed.), Meeting “Possible Episomes in Eukaryotes”. North Holland, pp. 35–41.
- 4.Vogt P.K. (1971) Genetically stable reassortment of markers during mixed infection with avian tumor viruses. Virology, 46, 947–952. [DOI] [PubMed] [Google Scholar]
- 5.McCutchan F.E. (2000) Understanding the genetic diversity of HIV-1. AIDS, 14, S31–S44. [PubMed] [Google Scholar]
- 6.Peeters M. and Sharp,P.M. (2000) Genetic diversity of HIV-1: the moving target. AIDS, 14, S129–S140. [PubMed] [Google Scholar]
- 7.Sharp P.M., Bailes,E., Robertson,D.L., Gao,F. and Hahn,B.H. (1999) Origins and evolution of AIDS viruses. Biol. Bull., 196, 338–342. [DOI] [PubMed] [Google Scholar]
- 8.Larder B.A., Kellam,P. and Kemp,S.D. (1993) Convergent combination therapy can select viable multidrug-resistant HIV-1 in vitro. Nature, 365, 451–453. [DOI] [PubMed] [Google Scholar]
- 9.Moutouh L., Corbeil,J. and Richman,D.D. (1996) Recombination leads to the rapid emergence of HIV-1 dually resistant mutants under selective drug pressure. Proc. Natl Acad. Sci. USA, 93, 6106–6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robertson D.L., Gao,F., Hahn,B.H. and Sharp,P.M. (1997) Intersubtype recombinant HIV-1 sequences. Human retroviruses and AIDS. In Korber,B., Foley,B., Kuiken,C., Litner,T., MacCutchan,F., Mellors,J.W. and Hahn,B.H. (eds), Human Retroviruses and AIDS 1997: A Compilation and Analysis of Nucleic Acid and Amino Acid Sequences. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM, pp. III-25–III-30.
- 11.Robertson D.L., Sharp,P.M., McCutchan,F.E. and Hahn,B.H. (1995) Recombination in HIV-1. Nature, 374, 124–126. [DOI] [PubMed] [Google Scholar]
- 12.Stuhlmann H. and Berg,P. (1992)Homologous recombination of copackaged retrovirus RNAs during reverse transcription. J. Virol., 66, 2378–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu H., Jetzt,A.E., Ron,Y., Preston,B.D. and Dougherty,J.P. (1998) The nature of human immunodeficiency virus type 1 strand transfers. J. Biol. Chem., 273, 28384–28391. [DOI] [PubMed] [Google Scholar]
- 14.Jetzt A.E., Yu,H., Klarmann,G.J., Ron,Y., Preston,B.D. and Dougherty,J.P. (2000) High rate of recombination throughout the human immunodeficiency virus type 1 genome. J. Virol., 74, 1234–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Negroni M. and Buc,H. (2001) Retroviral recombination: what drives the switch. Nat. Rev. Mol. Cell. Biol., 2, 151–155. [DOI] [PubMed] [Google Scholar]
- 16.DeStefano J.J., Mallaber,L.M., Rodriguez-Rodriguez,L., Fay,P.J. and Bambara,R.A. (1992) Requirements for strand transfer between internal regions of heteropolymer templates by human immunodeficiency virus reverse transcriptase. J. Virol., 66, 6370–6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu W., Blumberg,B.M., Fay,P.J. and Bambara,R.A. (1995) Strand transfer mediated by human immunodeficiency virus reverse transcriptase in vitro is promoted by pausing and results in misincorporation. J. Biol. Chem., 270, 325–332. [DOI] [PubMed] [Google Scholar]
- 18.Kim J.K., Palaniappan,C., Wu,W., Fay,P.J. and Bambara,R.A. (1997) Evidence for a unique mechanism of strand transfer from the transactivation response region of HIV-1. J. Biol. Chem., 272, 16769–16777. [DOI] [PubMed] [Google Scholar]
- 19.Negroni M. and Buc,H. (2000) Copy-choice recombination by reverse transcriptases: reshuffling of genetic markers mediated by RNA chaperones. Proc. Natl Acad. Sci. USA, 97, 6385–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Darlix J.L., Lapadat-Tapolsky,M., de Rocquigny,H. and Roques,B.P. (1995) First glimpses at structure-function relationships of the nucleocapsid protein of retroviruses. J. Mol. Biol., 254, 523–537. [DOI] [PubMed] [Google Scholar]
- 21.Rein A., Henderson,L.E. and Levin,J.G. (1998) Nucleic-acid-chaperone activity of retroviral nucleocapsid proteins: significance for viral replication. Trends Biochem. Sci., 23, 297–301. [DOI] [PubMed] [Google Scholar]
- 22.Tanchou V., Gabus,C., Rogemond,V. and Darlix,J.-L. (1995) Formation of stable and functional HIV-1 nucleoprotein complexes in vitro. J. Mol. Biol., 252, 563–571. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez-Rodriguez L., Tsuchihashi,Z., Fuentes,G.M., Bambara,R.A. and Fay,P.J. (1995) Influence of human immunodeficiency virus nucleocapsid protein on synthesis and strand transfer by the reverse transcriptase in vitro. J. Biol. Chem., 270, 15005–15011. [DOI] [PubMed] [Google Scholar]
- 24.Negroni M. and Buc,H. (1999) Recombination during reverse transcription: an evaluation of the role of the nucleocapsid protein. J. Mol. Biol., 286, 15–31. [DOI] [PubMed] [Google Scholar]
- 25.Hsu M., Rong,L., de Rocquigny,H., Roques,B.P. and Wainberg,M.A. (2000) The effect of mutations in the HIV-1 nucleocapsid protein on strand transfer in cell-free reverse transcription reactions. Nucleic Acids Res., 28, 1724–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo J., Henderson,L.E., Bess,J., Kane,B. and Levin,J.G. (1997) Human immunodeficiency virus type 1 nucleocapsid protein promotes efficient strand transfer and specific viral DNA synthesis by inhibiting TAR-dependent self-priming from minus-strand strong-stop DNA. J. Virol., 71, 5178–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Druillennec S., Caneparo,A., de Rocquigny,H. and Roques,B.P. (1999) Evidence of interactions between the nucleocapsid protein NCp7 and the reverse transcriptase of HIV-1. J. Biol. Chem., 274, 11283–11288. [DOI] [PubMed] [Google Scholar]
- 28.Gilboa E., Mitra,S.W., Goff,S. and Baltimore,D. (1979) A detailed model of reverse transcription and tests of crucial aspects. Cell, 18, 93–100. [DOI] [PubMed] [Google Scholar]
- 29.Kulpa D., Topping,R. and Telesnitsky,A. (1997) Determination of the site of first strand transfer during Moloney murine leukemia virus reverse transcription and identification of strand transfer-associated reverse transcriptase errors. EMBO J., 16, 856–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Telesnitsky A. and Skalka,A.M. (1997) Reverse transcriptase and the generation of retroviral DNA. In Coffin,J.M., Hughes,S.H. and Varmus,H.E. (eds), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 121–160. [PubMed]
- 31.Lobel L.I. and Goff,S.P. (1985) Reverse transcription of retroviral genomes: mutations in the terminal repeat sequences. J. Virol., 53, 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klaver B. and Berkhout,B. (1994) Premature strand transfer by the HIV-1 reverse transcriptase during strong-stop DNA synthesis. Nucleic Acids Res., 22, 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramsey C.A. and Panganiban,A.T. (1993) Replication of the retroviral terminal repeat sequence during in vivo reverse transcription. J. Virol., 67, 4114–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Negroni M., Ricchetti,M., Nouvel,P. and Buc,H. (1995) Homologous recombination promoted by reverse transcriptase during copying of two distinct RNA templates. Proc. Natl Acad. Sci. USA, 92, 6971–6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Rocquigny H., Gabus,C., Vincent,A., Fournie-Zaluski,M.C., Roques,B. and Darlix,J.L. (1992) Viral RNA annealing activities of human immunodeficiency virus type 1 nucleocapsid protein require only peptide domains outside the zinc fingers. Proc. Natl Acad. Sci. USA, 89, 6472–6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morellet N., Jullian,N., De Rocquigny,H., Maigret,B., Darlix,J.L. and Roques,B.P. (1992) Determination of the structure of the nucleocapsid protein NCp7 from the human immunodeficiency virus type 1 by 1H NMR. EMBO J., 11, 3059–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zuker M., Mathews,D.H. and Turner,D.H. (1999) Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide. In Barciszewski,J. and Clark,B.F.C. (eds), RNA Biochemistry and Biotechnology. NATO ASI Series, Kluwer Academic Publishers, Dordrecht, The Netherlands, pp. 11–43.
- 38.Tang H., Kuhen,K.L. and Wong-Staal,F. (1999) Lentivirus replication and regulation. Annu. Rev. Genet., 33, 133–170. [DOI] [PubMed] [Google Scholar]
- 39.Ye Y., Si,Z.H., Moore,J.P. and Sodroski,J. (2000) Association of structural changes in the V2 and V3 loops of the gp120 envelope glycoprotein with acquisition of neutralization resistance in a simian-human immunodeficiency virus passaged in vivo. J. Virol., 74, 11955–11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen L.X., Basilion,J.P. and Stanton,V.P. (1999) Single-nucleotide polymorphisms can cause different structural folds of mRNA. Proc. Natl Acad. Sci. USA, 96, 7871–7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allain B., Rascle,J.B., de Rocquigny,H., Roques,B. and Darlix,J.L. (1998) CIS elements and trans-acting factors required for minus strand DNA transfer during reverse transcription of the genomic RNA of murine leukemia virus. J. Mol. Biol., 277, 225–235. [DOI] [PubMed] [Google Scholar]
- 42.Cheslock S.R., Anderson,J.A., Hwang,C.K., Pathak,V.K. and Hu,W.S. (2000) Utilization of nonviral sequences for minus-strand DNA transfer and gene reconstitution during retroviral replication. J. Virol., 74, 9571–9579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peeters M., Liegeois,F., Torimiro,N., Bourgeois,A., Mpoudi,E., Vergne,L., Saman,E., Delaporte,E. and Saragosti,S. (1999) Characterization of a highly replicative intergroup M/O human immunodeficiency virus type 1 recombinant isolated from a Cameroonian patient. J. Virol., 73, 7368–7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Berkhout B., Vastenhouw,L.N., Klasens,I.F.B. and Huthoff,H. (2001) Structural features in the HIV-1 repeat region facilitate strand transfer during reverse transcription. RNA, 7, 1097–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]