

Abstract
Thyroid hormones are a critical regulator of mammalian physiology. Much of their action is due to effects in the nucleus where T3 engages thyroid hormone receptor isoforms to mediate its effects. In order to function properly the TR isoforms must be recruited to regulatory sequences within genes that they up-regulate. On these positive regulated target genes the TR can activate or repress depending upon whether the receptor is bound to T3 or not and the type of co-regulatory proteins present in that cell type. In contrast to T3 mediated activation, the mechanism by which the TR represses transcription in the presence of T3 remains unclear. Herein we will review the components of the transcriptional response to T3 within the nucleus and attempt to highlight the outstanding questions in the field.
Introduction
The effects of thyroid hormone signaling on different cell types is likely highly variable and unique. Indeed, circulating thyroid hormone levels are likely only the tip of the iceberg. We now know that both T4 and T3 enter most cells via transporters, including the monocarboxylate transporter 8, whose function can be altered based on its sequence and level of expression. Once inside the cell T4 and T3 can be further metabolized by the deiodinases to produce a unique amount of T3. Finally through a still unclear process T3 enters the nucleus to mediate its genomic actions via its cognate receptor isoforms (thyroid hormone receptor isoforms -TRs) the actions of which are influenced by a milieu of co-factors that can be cell-specific. Additionally, T3 is likely to have non-genomic effects that are initiated in the cytoplasm by the TRs that then result in changes in cellular physiology. In this review we will focus on the nuclear actions of the thyroid hormone receptor isoforms and delineate how the cellular context or output can be altered based on the actions of the TRs.
The Thyroid Hormone Receptor Isoforms
The pioneering work of Tata in the early 1960s first predicted the existence of a nuclear machinery that could respond to T3 by increasing transcription but it was not until 1986 when the laboratories of Bjorn Vennstrom and Ronald Evans first identified the thyroid receptor at the molecular level and determined that it was highly related to the chicken leukemia verb-A oncogene [1–4]. We know now that both rodents and humans possess two thyroid hormone receptor encoding genes termed THRA and THRB (Figure 1). The THRA locus, located on human chromosome 17, expresses two major isoforms TRα1 and TRα2. These two isoforms differ at their C-terminal region due to the presence of an alternative exon. Importantly, TRα2 is unable to bind T3 [5]. The THRB locus, present on chromosome 3, also leads to the expression of two major isoforms TRβ1 and TRβ2 who differ at their amino-termini based upon alternative exon use. Both of the TRβ isoforms bind T3. In addition to the major isoforms produced there is evidence that the THR loci can produce truncated receptor isoforms whose function in vivo is not clear [6, 7].
Figure 1. The thyroid hormone receptor isoforms.
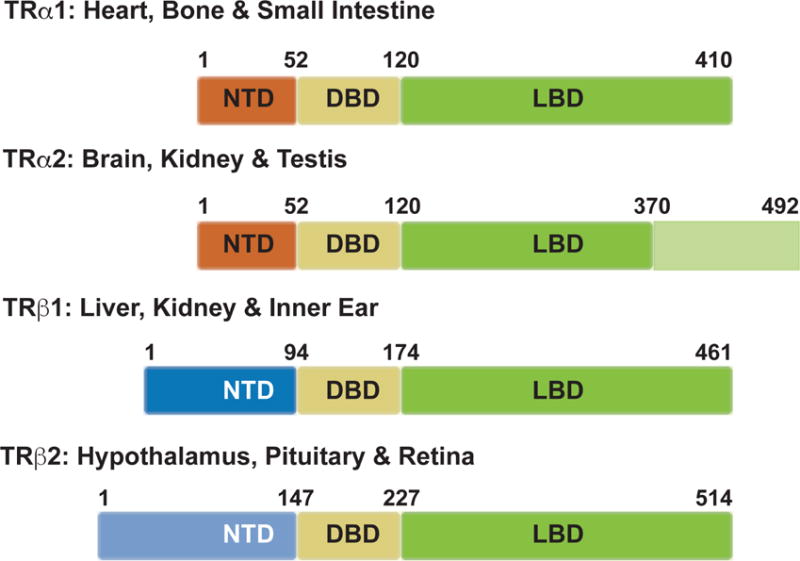
The thyroid hormone receptors (TRs) are encoded by two genes, THRA on chromosome 17 and THRB on chromosome 3. Each gene produces two isoforms, all of which share high sequence homology within their functional domains. Each isoform has an N-terminal domain (NTD, in blue), a DNA binding domain (DBD, in yellow), and a T3 ligand-binding domain (LBD, in green). TRα1 is expressed predominantly in the heart, brain, bone and small intestine. TRα2, which is expressed in the brain, kidney, and testis, has an alternative spliced LBD that prevents binding to T3. TRβ1 is expressed in pituitary, liver, kidney and the inner ear, while TRβ2 is expressed in the hypothalamus, pituitary and retina. Although TRβ1 and TRβ2 are transcribed from the same gene, they have different transcriptional start sites and employ separate promoters.
While encoded for by distinct genes the functions of TRα1 and TRβ are homologous and their separate function is more likely explained by their tissue of expression rather than structural differences that impart unique functions [8]. The molecular structure of the TR isoforms is conserved across species. All TH binding TR-isoforms contains 3 domains that include highly conserved DNA and ligand-binding domains. The most diverse region of the TR isoforms is their amino-terminal or A/B domains. The function of this domain has not been well elucidated though it likely has a function in transcriptional activation through its ability to influence DNA-binding and potentially recruit co-regulatory proteins that then influence the action of the TR. The best-described action of the TR is as a ligand-activated transcription factor. The TR isoforms exist in the nucleus both in the presence and absence of TH and the isoforms have separate ligand-independent and dependent actions that appear to be the result of differential recruitment of co-regulatory protein complexes by the TR. In addition to their basic protein structure the TR isoforms can be modified post-translationally including by sumoylation, which may greatly change their function [9].
The roles of the TR isoforms have been best delineated by: 1. gene knockout studies in mice and 2. by the syndromes of resistance to thyroid hormone in humans which are secondary to mutations in either TRα or TRβ. Initial knockout studies in mice demonstrated a unique role for the TRβ. isoforms in the regulation of TSH production by the pituitary [10, 11]. While early studies delineated a unique role for TRβ2 in TSH regulation more recent work suggests that both TRβ isoforms contribute [12, 13]. These isoforms are felt to exert their effect both in the paraventricular nucleus of the hypothalamus to regulate thyrotropin-releasing hormone (TRH) expression and in the pituitary where TSH subunit expression is regulated. Selective targeted knockout studies of the TRβ isoforms in either the hypothalamus or the pituitary have not been done. However, overexpression of dominant inhibitors of the TR in the pituitary has demonstrated a role for both the pituitary and the hypothalamus in the appropriate regulation of TSH [14]. The role of the TRα in the regulation of TSH is very limited when deleted in isolation. However, when all TR isoforms are deleted a clear role for TRα isoforms is seen [15]. In addition, to the hypothalamus and pituitary, the TRβ2 isoform plays a specialized role in the retina where it allows for the expression of the opsin photopigments in the retina of mice and thus allows for the development of color vision [16]. Interestingly, both TRβ isoforms are important in cochlear development and accordingly hearing development. In adulthood, only the TRβ 1 isoform is required for the maintenance of hearing [10, 13]. The TRβ1 isoform is the principal mediator of TH action in the liver and thus in cholesterol metabolism, while both isoforms have actions in white and brown adipose tissue [17, 18]. Similarly, both isoforms target TH action in the brain, but TRα1 has clear actions on target neurons in the hypothalamus that regulate sympathetic function [19]. Similarly, TRα1 has the majority of actions in the skeleton, heart and intestine but TRβ1 may play a role in certain cell types. Taken together, mouse genetic studies have well delineated the actions of the TR isoforms. However, many of the studies are limited by the fact that global deletions of TR isoforms were studied, which leaves the possibility open that some of the phenotypes seen are due to secondary effects from a nearby cell-type. More recently conditional alleles have become available making tissue-specific deletions of the TR isoforms possible. Indeed study of the TRβ1 isoform in thyroid follicular cells has generated clear evidence that this isoform plays a role in the production of TH by the thyroid gland [20].
While the mouse has been an invaluable tool to elucidate the role of the TR isoforms probably the most powerful model has been the human syndromes of resistance to thyroid hormone (RTH). It is now clear that two RTH syndromes exists due to mutations in the respective TR isoforms (Figure 2). RTHβ was first described in the late 1960s and identified as being secondary to mutations in the TRβ isoforms in the 1980s, thus proving the role of this isoform in the regulation of the hypothalamic-pituitary thyroid (HPT) axis [21–23]. Indeed, the presenting phenotype in patients with RTHβ is inappropriate TSH secretion in the face of elevated thyroid hormone levels. The clinical signs and symptoms of the disorder align with the TR isoform tissue distribution. RTHβ patients present most commonly with a goiter consistent with the resistance present within the HPT axis. While tissues such as the liver and pituitary are resistant to TH, those that express primarily TRα sense the elevated circulating TH levels and are thus hyperthyroid. Thus, there is often evidence of tachycardia and short stature in RTHβ patients, which likely reflects the role of the elevated TH levels on primarily TRα containing tissues including the heart and skeleton. There is also evidence that some of the clinical findings in RTHβ may be the result of a combination of effects of resistant TRβ signaling and activated TRα signaling. This could include the attention deficit hyperactivity syndromes that can occur in RTHβ [24].
Figure 2. Resistance to Thyroid Hormone (RTH).
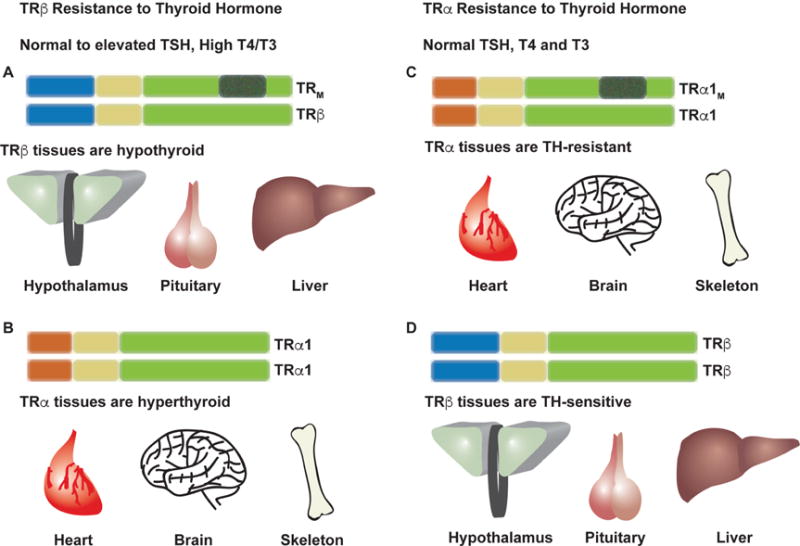
RTH due to mutations in TRβ is characterized by inappropriate TSH secretion despite elevated T4 and T3 levels. Tissues that express either TRβ isoform, which include the hypothalamus, pituitary and liver, are essentially hypothyroid due to TRβ inability to bind T3. Thus positively regulated genes are suppressed and negative T3 targets have elevated expression (A). TRα expressing tissues like the heart, brain and skeleton respond normally to circulating thyroid hormone levels and are therefore hyperthyroid, as circulating T4 and T3 are high (B). RTH due to mutations in TRα is characterized by normal circulating TSH, T4 and T3 levels. Tissues expressing TRα are relatively hypothyroid as TRα can no longer bind T3. Thus positive T3 targets are suppressed and negative T3 targets are elevated in expression (C). Due to normal circulating T4 and T3, TRβ expressing tissues retain normal sensitivity (D).
At the molecular level human TRβ mutations present have helped define its function. All TRβ mutations occur in the ligand-binding domain and result in the inability of the receptor to either bind T3 appropriately or to recruit protein complexes that allow for transcriptional activation or repression. Finally, certain mutations in TRβ demonstrate resistance primarily at the level of the HPT axis but not in peripheral TRβ target tissues suggesting that there are specific structures of the TR that are necessary for the negative regulation of TRH and TSH-subunit genes [25].
Whereas human mutations in the TRβ isoform have been identified for many years, the identification of mutations in the TRα isoform did not occur until 2012 [26]. Indeed, investigators had tried to hasten the identification of such mutations by creating mouse models with analogous mutations to those found in the RTHβ mouse models. Strikingly, the phenotypes present in these TRα mouse models were close to those seen in the index TRα mutant patient described by Bochukova et al [27]. This patient, a young girl identified at age 6, had features consistent with relative hypothyroidism in TRα expressing tissues including a skeletal phenotype, short stature, constipation, bradycardia and neurodevelopmental issues. Of interest, this first patient had slightly decreased T4/T3 ratio with a normal TSH. Subsequently, a number of other patients and families have been identified with TRα mutations present in both the C-terminus of the ligand-binding domain and also more centrally in the ligand-binding domain in a region common to both TRα isoforms. Analysis of all of the TRα mutations cases thus far highlights molecular features similar to mutations found in TRβ. All of the mutations appear to impair T3 binding and lead to the recruitment of a repressive complex to the mutant TR that cannot be released. Furthermore, the degree of resistance in context of T3-binding appears to associate with the severity of the clinical syndrome seen. Thus, patients with more deleterious mutations appear to have more severe cognitive, growth and motor delay compared to those with less deleterious mutations while certain features like macrocephaly and constipation tend to be more uniform. Strikingly, the presence of mutations in regions of TRα that are common to both the TRα1 and TRα2 isoforms has not revealed any unique biochemical or syndromal features, which suggests that TRα2 may not play an important role in TH action. Finally, the small changes in T3 and T4 seen in the first patients with RTHα have not been consistently seen meaning that TH signature akin to that seen in RTHβ is not present in RTHα [28, 29].
Thyroid Receptor Action on Target Genes
While the TR isoforms appear to function in a tissue-specific fashion much remains to be learned about their actions at the molecular level. Since their identification in the 1980s the first 20 years of work on the TRs primarily characterized their actions functionally using knockout studies as described above while their molecular actions were characterized both biochemically and in tissue culture experiments. Seminal findings suggested that TR isoforms functioned through their ability to interact with thyroid hormone response elements (TREs) located in the regulatory regions of target genes. While the TRs contained their own DNA-binding domain their principal mode of action was felt to be through their ability to heterodimerize with the retinoid x receptor (RXR) isoforms on a TRE that in general consisted of two half sites of the common AGGTCA motif either arranged in a: 1. direct repeat with a 4 base pair gap; 2. as a palindrome; or 3. an inverted palindrome with a 6 base pair gap. In the proposed configurations on DNA, the TR binds to the 3′ half site while RXR engages the 5′ half-site [30, 31]. The RXR isoform mainly enhances binding of the TR without an ability to bind its own ligand. While the TR-RXR heterodimer appears to be the favored conformation, a number of studies have demonstrated that the TR-isoforms also possessed the ability to bind DNA as a monomer or as a homodimer. The role of each of these complexes remains to be determined in vivo [32–34].
With the advent of ChlP-sequencing technology there became a significant opportunity to elucidate TR action at the molecular level in vivo. While this technology has greatly advanced our understanding of nuclear receptor signaling in vivo its impact on the TR field has been limited because of the lack of availability of high fidelity antibodies that can recognize and immunoprecipate the TR from in vivo tissues. Still the lessons learned from the studies performed to date have been highly valuable.
In the first published studies on the genomic organization of TR binding sites investigators employed a TRβ1 that was tagged at its amino-terminus with a sequence that can be biotinylated by the BirA enzyme when co-expressed. Thus, this tagged TR isoform could be affinity precipitated very avidly by streptavidin. When performed in the human liver cancer cell line HepG2 the overexpressed tagged TRβ1 was found to bind closely to genes activated by T3 but not repressed by T3. Furthermore, the bound TR was found to enrich around elements that contained the consensus AGGTCA half-site and also co-located in the presence of T3 with histone marks consistent with transcriptional activation ie H3K27ac. While the TR was located close to activated genes the majority of its binding sites were in intergenic regions and thus the function of those sites remains to be determined. This study also revealed that T3 was able to shift the binding of the receptor in some cases. Taken together the work by Ayers et al demonstrated a clear role for TRβ1 in the activation of T3 target genes through proximal or distal TREs that contained a consensus TRE half site usually as multiple copies [35].
Although the HepG2 cell line is a reliable system to study TH action it is not an in vivo system. Therefore, Ramadoss et al used a similar biotinylated TRβ1 approach but in mouse liver [36]. To accomplish this they used BirA transgenic mice that were made hypo or hyperthyroid and were transduced with an adenovirus that harbored the TRβ1 isoform with an amino-terminal sequence that could be biotinylated. Ramadoss et al performed both gene expression analysis and chromatin affinity purification and sequencing on these animals. Like Ayers et al they also found that the majority of TRβ1 binding was not in the proximal promoters of target genes. However, the TR preferred to bind to its consensus half-site arranged in a DR+4 motif especially around targets that were induced by T3. Interestingly, genes that were positively regulated by T3 were more likely to have TR-binding sites located nearby while negatively regulated targets were less likely. This suggests that negative regulation by T3 may not require direct binding by the TR. However, it does not rule out the possibility that the TR may bind further away to mediate its regulation of negative targets. Indeed, Ramadoss et al did determine that two TR half-sites arranged without a gap (DR0) could be found nearby some genes down-regulated by T3. Finally, using this system Ramadoss et al determined that the majority of TRβ1 binding sites that were associated with T3 inducible genes overlapped with RXRα isoform binding sites confirming the importance of the heterodimer in vivo. However, unique T3 inducible targets were also identified where the TR bound nearby as a homodimer only. Finally, many other sites not associated with regulated genes had no evidence of RXR binding. The function of the TR on these sites remains to be determined.
Both the Ayers and Ramadoss studies can be criticized because they used an overexpressed, tagged version of the TR. To get around this Grontved et al used a TR antibody that recognizes both TRβ1 and TRα isoforms on livers derived from mice made hypo or hyperthyroid [37]. They also identified a DR+4 motif as being most common but saw far more ligand-dependent reorganization of binding sites. Furthermore the presence of a TR binding site was also likely to coincide with the presence of a DNAse hypersensitive sensitive site (DHS) implying the existence of an open chromatin structure. Interestingly on negatively regulated T3 targets DHS sites were not as frequent, nor were TR binding sites implying an indirect mechanism of action for negative regulation. Similarly, Grontved et al also demonstrated that the TR-RXR heterodimer was also a key player in genes activated by T3. Finally and most interestingly this work established potentially different mechanisms for ligand-independent repression and ligand-dependent activation on TR-genes. Certainly, the ligand-dependent recruitment of a DHS site and the TR suggests that an interchange of co-regulatory proteins is not always necessary to move from repression to activation on a T3 target gene. While each of these studies has limitations a clearer picture of T3 activation via DR+4 TREs has emerged in vivo. Interestingly, the mechanism by which the TR may mediate negative regulation has remained enigmatic.
Whereas the studies described have addressed the function of TRβ1 to date fewer studies have addressed the molecular actions of TRα1 using genome wide approaches. Chatonnet et al took an innovative approach to compare the actions of TRα1 to TRβ1 in a single neuronal cell line derived from the cerebellum [38]. They expressed equal amounts of either a tagged TRα1 or TRβ1 that could be immunoprecipiated with streptavidin. Furthermore each isoform was co-expressed with GFP so that the transduced cells could be sorted. After the introduction of the isoforms, Chatonnet et al determined the TR isoform cistromes in the presence and absence of T3. Of note, a significant percentage of target genes could only be regulated by one isoform or the other implying that the TR isoforms activate or repress different repertoires of target genes. To determine if isoform-specific regulation was due to selective binding of the isoforms to their target genes they compared the cistromes in both cell lines. Of note the most common binding site in both lines was a DR+4. Importantly, in genes that shared a binding site there was a definite enhancement of a positive T3 response in both cell lines. This was not the case for genes that were negatively regulated. Strikingly, in genes that were differentially regulated by TRα1 versus TRβ1 there was no concordance of TR isoform binding meaning that differential binding could not explain their isoform-specific response to T3. Although TR-binding to a TRE remains critical for activation in a T3-dependent fashion other properties of the TR must dictate whether a binding event leads to activation or not. Some of these properties appear to be isoform-specific.
While genome-wide binding approaches have shed new light on how the TR isoforms engage their target genes they have also identified the presence of other transcription factor binding sites that appear to co-localize with TR-binding sites. These include FOXA sites as well as CTCF sites amongst many others [35, 36, 38]. Thus, it is possible that on certain target genes the co-recruitment of another transcription factor is necessary for T3-mediated activation. Potentially also, other transcription factors may act as pioneer factors which open chromatin that then allow the TR-isoforms to engage their binding site. These complexities are likely to explain differential responses to T3 across individuals based on polymorphisms in binding sites or even in the receptors themselves. Future work in this area will require direct mutational targeting of in vivo TREs to begin to prove their functionality. Additionally, it is likely that T3 analogs in development for disease therapy act differentially based upon their ability to influence TR-isoform binding. Finally, it is also clear that many nuclear receptor binding sites overlap. Indeed, in the liver, the liver x receptor (LXR) binds to many of the same sites as TRβ1. A major question then becomes occupancy time and stability of each nuclear receptor complex on each target gene and how this overall system induces transcription. Future analysis will require a combination of genetic models and genomic technologies to elucidate how T3 and its isoforms selectively regulate target genes.
The Co-Regulators
Considering that T3 can clearly activate or repress transcription via the TR isoforms, the next fundamental question to ask is how do the TR isoforms act to regulate transcription. As discussed previously, the notion for how the TR may do this came from the idea that the TR is a ligand-regulated transcription factor. Indeed, early studies that examined TR binding to DNA using techniques like EMSA demonstrated that the TR structure when bound to a TRE changed in the presence of T3. For example the ability of the TRβ isoform to form homodimers was lost in the presence of T3 [39, 40]. Furthermore, the migration of the very avid RXR/TR heterodimer was also changed in the presence of T3. These observations combined with evidence of a repressive function in the absence of T3 led to the hypothesis that the unliganded and liganded TR isoforms interacted with separate proteins or protein complexes to mediate gene repression or activation. Functional evidence for the presence of such co-factors came from work by Casanova et al who were able to show that a titratable cellular factor was responsible for the unliganded or aporeceptor function of the TR [41]. In contrast evidence for factors that activated the TR in the presence of ligand first came from the experiments of Halachmi et al who showed the existence of a 160 KD protein that was recruited to the estrogen receptor only in the presence of estrogen [42]. These fundamental observations lead to the direct cloning of the major co-regulators of the thyroid hormone receptor isoforms: the corepressors NCoR1 and SMRT and the coactivators SRC-1,2 and 3.
NCoR1 and SMRT
NCOR1 and SMRT were identified in 1997 by a number of groups. Once full-length proteins were identified in became clear that these two separate proteins were large 270KD paralogs with similar functional domains (Figure 3) [43–45]. Both NCoR1 and SMRT interact with the TR isoforms via C-terminal domains first termed CoRNR boxes but more commonly referred to now as nuclear receptor interacting domains (RIDs) [46–48]. Both NCoR1 and SMRT possess 3 RIDs that mediate their interactions with the TR and other nuclear receptors. Importantly the RIDs share a canonical helical domain with a IxxII structure that allow for interaction with the unliganded TR LBD via a region that becomes exposed in the absence of ligand because of the positioning of the 12th helix of the LBD [49, 50]. Remarkably the SMRT and NCoR1 RIDs display specificity such that NCoR1 prefers to interact with the TR via the its more N-terminal RIDs. In contrast, the SMRT RIDs appear to favor RAR isoforms in vitro [51, 52]. A central question in the biology of corepressor activity is underscored by how they choose to act based on the presence of multiple nuclear receptors that they have the potential to interact with at any one time. Based on the structure of the RIDs it is likely that a single NCoR1 molecule interacts with either RXR/TR heterodimer or a TR/TR homodimer.
Figure 3. Representation of the nuclear receptor corepressors.
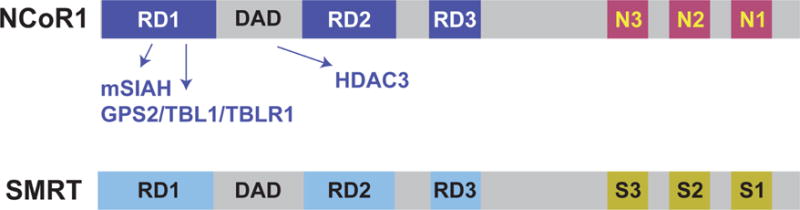
Nuclear receptor corepressor 1 (NCoR1) and silencing mediator for retinoid or thyroid-hormone receptors (SMRT, also known as nuclear receptor corepressor 2, (NCoR2) are transcriptional coregulatory proteins. Both contain three repression domains (RD) and three nuclear receptor interacting domains (N1, N2, and N3 or S1, S2 and S3). RDs work to recruit proteins that are part of the transcriptional repression complex including mSIAH, GPS2, TBL1 and TBLR1. These proteins either regulate corepressor availability (mSIAH) or its interactions with chromatin. Corepressor function requires a deacetylase activation domain (DAD domain) which is required for the enzymatic activity of HDAC3. The nuclear receptor interacting domains bind to nuclear receptors such as the TRs.
While the RIDs are located in the C-terminal region of NCoR1/SMRT the N-terminus and the mid-portion of the proteins contain 3 domains that mediate transcriptional repression. NCoR1 can mediate transcriptional repression by recruiting a multiprotein complex. Included in this complex are histone deactylase 3 (HDAC3), G-protein pathway suppressor 2 (GPS2), transducing β- like (TBL1 and TBLX1) and TBL-related 1 (TBLR1 and TBL1XR1). All of these proteins appear to play essential roles in the stability of the complex and its function in repression [49, 53, 54]. Also present in this region is the DAD or the deacetylase activation domain that is required for the activation of the deacetylase function of HDAC3 [55, 56]. Indeed it is the histone deactylation function of the corepressor complex that is supposed to mediate its ability to repress [57, 58].
While NCoR1 and SMRT are for the most part large 270 KD proteins there is also good evidence that they can undergo alternative splicing especially in context of their RIDs meaning that the stoichiometry of NCoR1/SMRT present in a target cell type can be regulated by expression of both NCoR1 and SMRT and by their modification through alternative splicing [59–62]. While additional corepressors that can be recruited to the TRs have been identified the general feeling in the field is that NCoR1 and SMRT play the most significant roles in TH action.
The Coactivators
After the identification of a 160KD protein that could interact the ER in a ligand-dependent fashion the first coactivator was cloned in 1995 using a yeast-two hybrid system. It was termed steroid receptor coactivator 1 (SRC-1) [42, 63]. Subsequently two highly homologous versions of SRC-1 were identified and were termed SRC-2 and SRC-3. All three of the SRCs share structural homology but appear to have a variety of different functions [64]. The SRCs interact with liganded nuclear receptors including the TR isoforms via a central interacting domain that contains a number of LxxLL motifs. Remarkably, this motif recognizes the liganded-TR as the position of helix 12 changes with the presence of ligand [65]. The C-terminal region of the SRC isoforms contains intrinsic histone acetyl transferase (HAT) activity, which is consistent with the role of coactivators in increasing transcription via histone acetylation. Additionally, the SRC isoforms interact with other chromatin modifying proteins including CBP/p300, which has stronger HAT activity, and co-activator-associated arginine methyltransferase 1 (CARM1) and protein arginine N-methyl transferase 1 (PRMT1), which have alternative enzymatic activity that modifies chromatin [66–68]. These enzymatic activities are likely important for the action of all nuclear receptors including the TRs.
There is some evidence to support the notion that the SRC isoforms can interact with the TRβ2 amino-terminus and that this interaction could be important in TR action [69]. Like the corepressors, members of the SRC family can be differentially expressed in context of a variety of cell types and appear to play non-redundant roles in physiology with SRC-1 having the most significant role in TH action. Also numerous other proteins with coactivator-like activity have been identified and can interact with the TR-isoforms including members of the mediator complex [70]. This complex is essential for the interaction of the TR with the RNA polymerase II basal transcriptional machinery. Additionally, the TR can also recruit additional protein complexes that can remodel chromatin [71]. The roles of other identified coactivators that have been shown to interact with the TR isoforms remain to be determined.
The Co-Regulators and Thyroid Hormone Action
In vitro and cell culture experiments predict a classic model whereby the unliganded TR on positive TREs recruited NCoR1 to mediate repression and the addition of T3 led to the dismissal of NCoR1 and the recruitment of a coactivator complex. However, in vivo experiments do not support this model (Figure 4). The first demonstration of a role for a co-regulator in TH action came from the analysis of SRC-1 KO mice. While SRC-1 KO mice show defects in steroid receptor signaling they also clearly have RTH with inappropriate TSH levels in the presence of elevated circulating TH levels. In contrast, the selective deletion of SRC-2 does not impair the regulation of the HPT axis [72, 73]. While SRC-1 also appears to play a role in T3 signaling in the liver and heart on positively regulated genes its role in the repression of the TRH and TSH subunit genes is paradoxical and against the proposed classic model of coregulatory function in TH action.
Figure 4. Gene transcription is modulated by coregulating proteins.
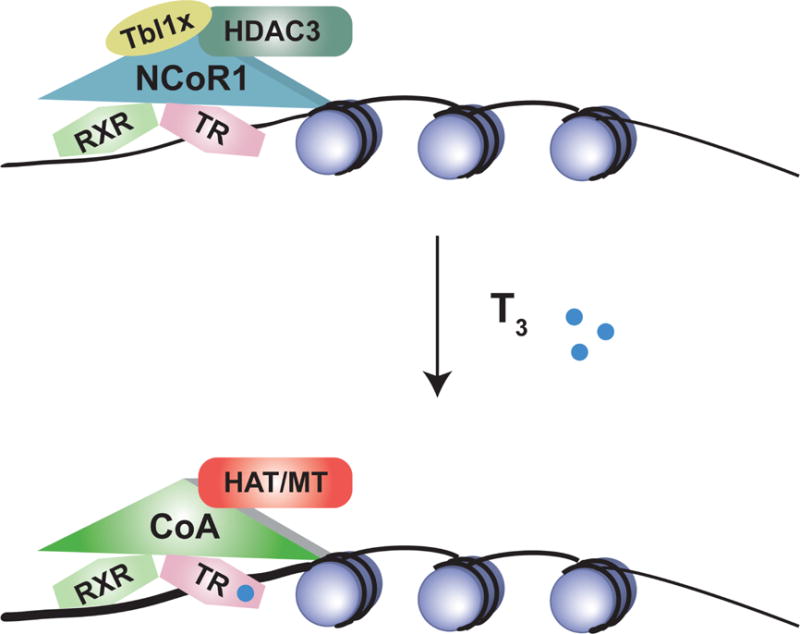
On positive T3 targets, NCoR1 is recruited by thyroid hormone receptors receptors in the absence or presence of T3. NCoR1 then recruits HDAC3 and other inhibitory proteins to alter the histone code. When excess T3 is present, the corepressor complex can be released. Coactivating proteins such as SRC-1 and SRC-2 are recruited by the liganded TR. The RXR/TR/Coactivator complex recruits additional transcriptional machinery including histone acetyltransferase (HAT) and methyl transferase activity to allow for enhanced transcription (see text).
Early attempts at delineating the role of NCoR1 and SMRT in vivo were unsuccessful because deletion of either paralog led to embryonic lethality [74, 75]. To address the role of NCoR1 in TH action Astapova et al developed a mouse model that was able to express a hypomorphic NCoR1 allele (NCoRΔID) that lacked the two principal RIDs that interacted with the TR [76]. Thus, resulting mice would not be able to recruit NCoR1 to the TR. Importantly; this model was developed to express NCoRΔID in either a cell-specific or global manner. When first expressed in hepatocytes alone (L-NCoRΔID mice) a number of TRF31 targets in the liver were unable to be fully repressed in the hypothyroid setting consistent with the classic role predicted for NCoR1. Surprisingly, more targets were up-regulated in the euthyroid setting in L-NCoRΔID mice, suggesting that in the absence of NCoR1 recruitment, TR target genes were more sensitive to the identical amounts of T3 present. Not surprisingly, negative TR target genes were relatively unaffected by the expression of NCoRΔID.
To look at this systemically Astapova et al expressed NCoRΔID globally [77]. Remarkably, these animals had low levels of circulating T4 and T3 with normal TSH levels and normal levels of TRH mRNA in the hypothalamus. Taken together this would suggest central hypothyroidism. However NCoRΔID mice were not small and had evidence of increased energy expenditure. Furthermore, T3 targets in the liver had normal expression. Thus, removal of a functional NCoR1 molecule in vivo appears to increase sensitivity to TH at the level of the HPT axis and the liver. Interestingly, the hearts of NCoRΔID mice did not display increased sensitivity and were in fact bradycardic as they sensed the low TH levels as such. Thus, rather than mediate only ligand-independent repression NCoR1 appears to play an even more essential role in determining sensitivity to T3. In target cell types like the liver or pituitary diminished levels of NCoR1 enhance T3 action while increased levels would be expected to diminish T3 action. Support for this role of the NCoR1 complex has also been found recently in humans where mutations in TBL1X have been found [78]. As discussed TBL1X is part of the NCoR1 complex and plays a role in its interactions with chromatin. Patients with mutations in this protein have thyroid function tests that are identical to those found in NCoRΔID mice suggesting that these patients also have increased sensitivity to thyroid hormone rather than central hypothyroidism.
Given the contrasting roles of RTH in SRC-1 KO mice and increased sensitivity to TH in NCoRΔID mice Vella et al developed a mouse model that combined both of these genetic alterations [79]. As expected deletion of SRC-1 led to RTH at the level of the HPT axis, however when NCoRΔID was introduced on this background, normal sensitivity was re-established. A similar pattern was seen on positively regulated hepatic T3 target genes where the deletion of SRC-1 led to RTH and the introduction of NCoRΔID re-established normal sensitivity. Notably, the normal sensitivity in mice expressing NCoRΔID and lacking SRC-1 was mediated by the recruitment of SRC-2 to the regulatory regions of T3 target genes. Similar to what was seen in NCoRΔID mice, SRC-1 and NCoRΔID together appeared to play little role in other TH responsive tissues such as the heart.
Based on the ability of NCoR1 to potentially be recruited with higher affinity to unliganded TRs, it was also postulated that aberrant corepressor recruitment could be responsible for the defects found in both RTHβ and RTHα. To test this hypothesis Fozzatti et al crossed mouse models with either RTHβ or RTHa with NCoRΔID mice [80, 81]. Consistent with the proposed hypothesis, RTHβ/NCoRΔID mice re-established a normal set point of the HPT axis and improved T3 signaling in the periphery. Similarly RTHα/NCoRΔID mice showed improved growth, enhanced fertility and bone development and rescued impaired adipogenesis. Thus, targeting the NCoR1-mutant TR complex in RTH syndromes could be a reasonable therapeutic option. However, the impaired recruitment of SRC-1 in RTHβ and RTHα is also likely, as disruption of SRC-1 in a RTHβ mouse model worsened the syndrome [82].
Whereas NCoR1 clearly plays a role in TH action, a role for SMRT was not suggested biochemically based on its ability to interact with the TR. However, initial mouse models that mutated the two most C-terminal RIDs in SMRT globally demonstrated an improvement in the hypercholesterolemia present in hypothyroidism. Additionally, these mice showed increased sensitivity to PPARγ in adipocytes consistent with a sensitivity function for SMRT also [83]. Further analysis of these mice on a C57BL/6 background also showed a lethal lung defect leading to respiratory distress syndrome. Importantly, these mice could be rescued by inducing hypothyroidism and rescuing the expression of Klf2, which in wild-type mice is activated through a TR/SMRT pathway and its repression in SMRT mutant mice leads to the lung defect [84].
While the SMRT RID model suggested a role for SMRT in TH action, Shimizu et al chose to compare SMRT to NCoR1 directly by developing mice that lacked SMRT or expressed NCoRΔID in the liver of mice [85]. They also developed mice that both lacked SMRT and expressed NCoRID. In these models SMRT had no role in both TH sensitivity or ligand-independent repression. Consistent with early biochemical experiments the deletion of SMRT was able to enhance signaling on RAR isoform targets. Mice that lacked SMRT but expressed NCoRΔID had evidence of significant triglyceride accumulation in the liver consistent with the role of NCoR1 and SMRT to regulate hepatic lipogenesis and storage via HDAC3 [86]. Finally, Shimizu et al used a post-natal strategy to delete SMRT globally and get around the embryonic lethality seen when SMRT is deleted during embryogenesis. Deletion of SMRT after 6 weeks of life had no impact on mortality in mice. However, unlike the expression of NCoRID in a similar post-natal time period TH levels did not fall in SMRT KO mice. This would again be consistent with a substantially more important role for NCOR1 in TH action than SMRT.
While the in vivo models have clarified the role of co-regulators in TH action many questions remain. Key insight into co-regulator function has really only been established in the HPT axis and in the liver. The role of co-regulators in other TH responsive tissues remains unknown. Additionally, the role of specific co-regulators remains to be better defined. Establishing this will be of utmost importance in beginning to understand why similar levels of TH in humans can exert widely different responses.
Negative Regulation by Thyroid Hormone
While many questions remain in how TH turns on target genes the basic role of the TR isoforms and the co-regulators appear clear. This is in stark contrast to what is known about how TH turns off target genes. This is most importantly seen in the hypothalamus and pituitary where TRH and the TSH subunit genes are negative targets of TH signaling. Furthermore, in the liver there are more negative than positive TH target genes. It is likely that the mechanisms underlying negative regulation are far more complex than positive regulation and may vary widely across target genes.
Data from many studies have suggested negative regulation by T3 occurs through the TR in manner distinctly opposite from positive regulation. On negative targets, in the absence of T3, corepressors are recruited to activate transcription. When T3 is present, coactivators are recruited to suppress transcription. This model is supported by the findings in SRC-1 KO mice and in mice where NCoRΔID is expressed in the pituitary alone [87]. In a unique TSH secreting cell line, the negative regulation of TSHβ appears to be mediated directly by the TRβ isoforms, which interact with regulatory regions of the gene [88]. This direct binding role of TRβ is further supported by a knock-in mutation in mice that impairs DNA binding. In these animals with this mutation there is strong RTH consistent with DNA-binding of the TR being necessary for negative regulation [89]. However, a role for direct binding of the TR isoforms in negative regulation outside of the pituitary appears hard to support. As reviewed above, genome wide binding studies of the TR demonstrate a lack of binding in the vicinity of genes that are negatively regulated thus further work will be required to understand the inherent mechanisms at play here.
While the direct role of the TR in engaging negatively regulated genes remains to be determined it is also clear that there are other mechanisms of negative regulation. These could include the secondary activation of factors or pathways that then mediate negative regulation. For example, T3-induced miRNAs play a significant role in negative regulation in the heart and liver. Indeed, the well-known negatively T3 target in the heart, beta-myosin heavy chain (MYH7), is repressed by a T3-induced miRNA present in an intronic region of the positively regulated T3-target MYH6. This miRNA controls the components of the mediator complex [90]. Thus, when MYH6 is induced by T3 the production of the miRNA leads to the coincidental down-regulation of MYH7. Similarly in the liver, T3 has the ability to induce the miRNA - 181d which in turn down regulates the expression of two targets SOAT and CDX2 [91]. Based on the ability of T3 to regulate miRNAs in a variety of tissues this mechanism is likely to be highly conserved.
Summary
Intra-nuclear TH action is highly dependent upon the levels of T3 that reach the nucleus. However, it is also now clear that the structure and presence of the TR isoforms and their co-regulators can respond in different fashions to a set amount of T3. Positive regulation by T3 appears to be clearly mediated by DNA bound TR isoform that then interacts with families of co-regulators that mediate its tissue-specific effects. While the mechanism appears to be conserved across tissues the co-regulator families involved have not been identified. Furthermore, the mechanism of action of the unliganded TR remains uncertain. Indeed, in the absence of functional NCoR1 or SMRT many TR targets can still be repressed suggesting an alternative pathway for repression. Finally, negative regulation by T3 remains enigmatic and will be a subject of much debate until better models and systems are developed to understand it.
Acknowledgments
ANH is supported by DK105029, DK098525, DK056123
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tata JR. Inhibition of the biological actions of thyroid hormones by actinomycin D and puromycin. Nature. 1963;197:1167–68. doi: 10.1038/1971167a0. [DOI] [PubMed] [Google Scholar]
- 2.Tata JR, Widnell CC. Ribonucleic acid synthesis during the early action of thyroid hormones. Biochem J. 1966;98(2):604–20. doi: 10.1042/bj0980604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sap J, et al. The c-erb-A protein is a high-affinity receptor for thyroid hormone. Nature. 1986;324(6098):635–40. doi: 10.1038/324635a0. [DOI] [PubMed] [Google Scholar]
- 4.Weinberger C, et al. The c-erb-A gene encodes a thyroid hormone receptor. Nature. 1986;324:641–646. doi: 10.1038/324641a0. [DOI] [PubMed] [Google Scholar]
- 5.Lazar MA. Thyroid hormone receptors: multiple forms, multiple possibilities. Endocr Rev. 1993;14(2):184–93. doi: 10.1210/edrv-14-2-184. [DOI] [PubMed] [Google Scholar]
- 6.Chassande O, et al. Identification of transcripts initiated from an internal promoter in the c-erbA alpha locus that encode inhibitors of retinoic acid receptor-alpha and triiodothyronine receptor activities. Mol Endocrinol. 1997;11(9):1278–90. doi: 10.1210/mend.11.9.9972. [DOI] [PubMed] [Google Scholar]
- 7.Williams GR. Cloning and characterization of two novel thyroid hormone receptor beta isoforms. Mol Cell Biol. 2000;20(22):8329–42. doi: 10.1128/mcb.20.22.8329-8342.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brent GA. Mechanisms of thyroid hormone action. J Clin Invest. 2012;122(9):3035–43. doi: 10.1172/JCI60047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu YY, et al. Thyroid hormone receptor sumoylation is required for preadipocyte differentiation and proliferation. J Biol Chem. 2015;290(12):7402–15. doi: 10.1074/jbc.M114.600312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forrest D, et al. Thyroid hormone receptor beta is essential for development of auditory function. Nat Gen. 1996;13:354–357. doi: 10.1038/ng0796-354. [DOI] [PubMed] [Google Scholar]
- 11.Forrest D, Vennstrom B. Functions of thyroid hormone receptors in mice. Thyroid. 2000;10(1):41–52. doi: 10.1089/thy.2000.10.41. [DOI] [PubMed] [Google Scholar]
- 12.Abel ED, et al. Critical role for thyroid hormone receptor beta2 in the regulation of paraventricular thyrotropin-releasing hormone neurons. J Clin Invest. 2001;107(8):1017–23. doi: 10.1172/JCI10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng L, et al. Age-Related Hearing Loss and Degeneration of Cochlear Hair Cells in Mice Lacking Thyroid Hormone Receptor beta1. Endocrinology. 2015;156(10):3853–65. doi: 10.1210/en.2015-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abel ED, et al. Novel insight from transgenic mice into thyroid hormone resistance and the regulation of thyrotropin. J Clin Invest. 1999;103(2):271–9. doi: 10.1172/JCI5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gothe S, et al. Mice devoid of all known thyroid hormone receptors are viable but exhibit disorders of the pituitary-thyroid axis, growth, and bone maturation. Genes Dev. 1999;13(10):1329–41. doi: 10.1101/gad.13.10.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng L, et al. A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat Genet. 2001;27(1):94–8. doi: 10.1038/83829. [DOI] [PubMed] [Google Scholar]
- 17.Gullberg H, et al. Thyroid hormone receptor beta-deficient mice show complete loss of the normal cholesterol 7alpha-hydroxylase (CYP7A) response to thyroid hormone but display enhanced resistance to dietary cholesterol. Mol Endocrinol. 2000;14(11):1739–49. doi: 10.1210/mend.14.11.0548. [DOI] [PubMed] [Google Scholar]
- 18.Gullberg H, et al. Requirement for thyroid hormone receptor beta in T3 regulation of cholesterol metabolism in mice. Mol Endocrinol. 2002;16(8):1767–77. doi: 10.1210/me.2002-0009. [DOI] [PubMed] [Google Scholar]
- 19.Mittag J, et al. Thyroid hormone is required for hypothalamic neurons regulating cardiovascular functions. J Clin Invest. 2013;123(1):509–16. doi: 10.1172/JCI65252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selmi-Ruby S, et al. The targeted inactivation of TRbeta gene in thyroid follicular cells suggests a new mechanism of regulation of thyroid hormone production. Endocrinology. 2014;155(2):635–46. doi: 10.1210/en.2013-1435. [DOI] [PubMed] [Google Scholar]
- 21.Refetoff S, DeWind LT, DeGroot LJ. Familial syndrome combining deaf-mutism, stuppled epiphyses, goiter and abnormally high PBI: possible target organ refractoriness to thyroid hormone. J Clin Endocrinol Metab. 1967;27(2):279–94. doi: 10.1210/jcem-27-2-279. [DOI] [PubMed] [Google Scholar]
- 22.Usala SJ, et al. Tight linkage between the syndrome of generalized thyroid hormone resistance and the human c-erbA gene. Mol Endocrinol. 1988;2:1217–1220. doi: 10.1210/mend-2-12-1217. [DOI] [PubMed] [Google Scholar]
- 23.Usala SJ, et al. A base mutation of the C-erbA beta thyroid hormone receptor in a kindred with generalized thyroid hormone resistance. Molecular heterogeneity in two other kindreds. J Clin Invest. 1990;85(1):93–100. doi: 10.1172/JCI114438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Refetoff S, Weiss RE, Usala SJ. The syndromes of resistance to thyroid hormone. Endocrine Reviews. 1993;14:348–399. doi: 10.1210/edrv-14-3-348. [DOI] [PubMed] [Google Scholar]
- 25.Machado DS, et al. A thyroid hormone receptor mutation that dissociates thyroid hormone regulation of gene expression in vivo. Proc Natl Acad Sci U S A. 2009;106(23):9441–6. doi: 10.1073/pnas.0903227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bochukova E, et al. A mutation in the thyroid hormone receptor alpha gene. N Engl J Med. 2012;366(3):243–9. doi: 10.1056/NEJMoa1110296. [DOI] [PubMed] [Google Scholar]
- 27.Kaneshige M, et al. A targeted dominant negative mutation of the thyroid hormone alpha 1 receptor causes increased mortality, infertility, and dwarfism in mice. Proc Natl Acad Sci U S A. 2001;98(26):15095–100. doi: 10.1073/pnas.261565798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moran C, et al. An adult female with resistance to thyroid hormone mediated by defective thyroid hormone receptor alpha. J Clin Endocrinol Metab. 2013;98(11):4254–61. doi: 10.1210/jc.2013-2215. [DOI] [PubMed] [Google Scholar]
- 29.Moran C, Chatterjee K. Resistance to Thyroid Hormone alpha-Emerging Definition of a Disorder of Thyroid Hormone. Action J Clin Endocrinol Metab. 2016;101(7):2636–9. doi: 10.1210/jc.2016-2317. [DOI] [PubMed] [Google Scholar]
- 30.Kurokawa R, et al. Differential orientations of the DNA-binding domain and carboxy-terminal dimerization interface regulate binding site selection by nuclear receptor heterodimers. Genes Dev. 1993;7:1423–1435. doi: 10.1101/gad.7.7b.1423. [DOI] [PubMed] [Google Scholar]
- 31.Naar AM, et al. The orientation and spacing of core DNA-binding motifs dictate selective transcriptional responses to three nuclear receptors. Cell. 1991;65(7):1267–79. doi: 10.1016/0092-8674(91)90021-p. [DOI] [PubMed] [Google Scholar]
- 32.Katz RW, Koenig RJ. Nonbiased identification of DNA sequences that bind thyroid hormone receptor alpha 1 with high affinity. J Biol Chem. 1993;268(26):19392–7. [PubMed] [Google Scholar]
- 33.Katz RW, Koenig RJ. Specificity and mechanism of thyroid hormone induction from an octamer response element. J Biol Chem. 1994;269(29):18915–20. [PubMed] [Google Scholar]
- 34.Brent GA, et al. Mutations of the rat growth hormone promoter which increase and decrease response to thyroid hormone define a consensus thyroid hormone response element. Mol Endocrinol. 1989;3:1996–2004. doi: 10.1210/mend-3-12-1996. [DOI] [PubMed] [Google Scholar]
- 35.Ayers S, et al. Genome-wide binding patterns of thyroid hormone receptor beta. PLoS One. 2014;9(2):e81186. doi: 10.1371/journal.pone.0081186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramadoss P, et al. Novel Mechanism of Positive versus Negative Regulation by Thyroid Hormone Receptor beta1 (TRbeta1) Identified by Genome-wide Profiling of Binding Sites in Mouse Liver. J Biol Chem. 2014;289(3):1313–28. doi: 10.1074/jbc.M113.521450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grontved L, et al. Transcriptional activation by the thyroid hormone receptor through ligand-dependent receptor recruitment and chromatin remodelling. Nat Commun. 2015;6:7048. doi: 10.1038/ncomms8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chatonnet F, et al. Genome-wide analysis of thyroid hormone receptors shared and specific functions in neural cells. Proc Natl Acad Sci U S A. 2013;110(8):E766–75. doi: 10.1073/pnas.1210626110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yen PM, et al. Triiodothyronine (T3) decreases binding to DNA by T3-receptor homodimers but not receptor-auxiliary protein heterodimers. J Biol Chem. 1992;267(6):3565–8. [PubMed] [Google Scholar]
- 40.Yen PM, et al. Orientation and spacing of half-sites differentially affect T3-receptor (TR) monomer, homodimer and heterodimer binding to thyroid hormone response elements. Endocrine Journal. 1993;1:461–466. [Google Scholar]
- 41.Casanova J, et al. Functional evidence for ligand-dependent dissociation of thyroid hormone and retinoic acid receptors from an inhibitory cellular factor. Mol Cel Bio. 1994;14:5756–5765. doi: 10.1128/mcb.14.9.5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Halachmi S, et al. Estrogen receptor-associated proteins-possible mediators of hormone-induced transcription. Science. 1994;264:1455–1458. doi: 10.1126/science.8197458. [DOI] [PubMed] [Google Scholar]
- 43.Alland L, et al. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature. 1997;387(6628):49–55. doi: 10.1038/387049a0. [DOI] [PubMed] [Google Scholar]
- 44.Heinzel T, et al. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387(6628):43–8. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- 45.Nagy L, et al. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell. 1997;89(3):373–80. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- 46.Hu X, Lazar MA. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature. 1999;402(6757):93–6. doi: 10.1038/47069. [DOI] [PubMed] [Google Scholar]
- 47.Nagy L, et al. Mechanism of corepressor binding and release from nuclear hormone receptors. Genes Dev. 1999;13(24):3209–16. doi: 10.1101/gad.13.24.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perissi V, et al. Molecular determinants of nuclear receptor-corepressor interaction. Genes Dev. 1999;13(24):3198–208. doi: 10.1101/gad.13.24.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Astapova I. Role of co-regulators in metabolic and transcriptional actions of thyroid hormone. J Mol Endocrinol. 2016;56(3):73–97. doi: 10.1530/JME-15-0246. [DOI] [PubMed] [Google Scholar]
- 50.Astapova I, Hollenberg AN. The in vivo role of nuclear receptor corepressors in thyroid hormone action. Biochim Biophys Acta. 2013;1830(7):3876–81. doi: 10.1016/j.bbagen.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cohen RN, et al. The specificity of interactions between nuclear hormone receptors and corepressors is mediated by distinct amino acid sequences within the interacting domains. Mol Endocrinol. 2001;15(7):1049–61. doi: 10.1210/mend.15.7.0669. [DOI] [PubMed] [Google Scholar]
- 52.Cohen RN, et al. The nuclear corepressors recognize distinct nuclear receptor complexes. Mol Endocrinol. 2000;14(6):900–14. doi: 10.1210/mend.14.6.0474. [DOI] [PubMed] [Google Scholar]
- 53.Oberoi J, et al. Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat Struct Mol Biol. 2011;18(2):177–84. doi: 10.1038/nsmb.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang J, et al. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol Cell. 2002;9(3):611–23. doi: 10.1016/s1097-2765(02)00468-9. [DOI] [PubMed] [Google Scholar]
- 55.Guenther MG, et al. A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 2000;14(9):1048–57. [PMC free article] [PubMed] [Google Scholar]
- 56.Li J, et al. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. Embo J. 2000;19(16):4342–50. doi: 10.1093/emboj/19.16.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ishizuka T, Lazar MA. The N-CoR/histone deacetylase 3 complex is required for repression by thyroid hormone receptor. Mol Cell Biol. 2003;23(15):5122–31. doi: 10.1128/MCB.23.15.5122-5131.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.You SH, et al. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol. 2013;20(2):182–7. doi: 10.1038/nsmb.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Faist F, et al. Alternative splicing determines the interaction of SMRT isoforms with nuclear receptor-DNA complexes. Biosci Rep. 2008 doi: 10.1042/BSR20080093. [DOI] [PubMed] [Google Scholar]
- 60.Goodson M, Jonas BA, Privalsky MA. Corepressors: custom tailoring and alterations while you wait. Nucl Recept Signal. 2005;3:e003. doi: 10.1621/nrs.03003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goodson ML, Jonas BA, Privalsky ML. Alternative mRNA splicing of SMRT creates functional diversity by generating corepressor isoforms with different affinities for different nuclear receptors. J Biol Chem. 2005;280(9):7493–503. doi: 10.1074/jbc.M411514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goodson ML, et al. Alteration of NCoR corepressor splicing in mice causes increased body weight and hepatosteatosis without glucose intolerance. Mol Cell Biol. 2014;34(22):4104–14. doi: 10.1128/MCB.00554-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Onate SA, et al. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995;270:1354–1357. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- 64.Lonard DM, Lanz RB, O’Malley BW. Nuclear receptor coregulators and human disease. Endocr Rev. 2007;28(5):575–87. doi: 10.1210/er.2007-0012. [DOI] [PubMed] [Google Scholar]
- 65.McInerney EM, et al. Determinants of coactivator LXXLL motif specificity in nuclear receptor transcriptional activation. Genes Dev. 1998;12(21):3357–68. doi: 10.1101/gad.12.21.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen D, et al. Regulation of transcription by a protein methyltransferase. Science. 1999;284(5423):2174–7. doi: 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- 67.Li H, Chen JD. The receptor-associated coactivator 3 activates transcription through CREB-binding protein recruitment and autoregulation. J Biol Chem. 1998;273:5948–5954. doi: 10.1074/jbc.273.10.5948. [DOI] [PubMed] [Google Scholar]
- 68.Lee YH, Stallcup MR. Minireview: protein arginine methylation of nonhistone proteins in transcriptional regulation. Mol Endocrinol. 2009;23(4):425–33. doi: 10.1210/me.2008-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang Z, Privalsky ML. Isoform-specific transcriptional regulation by thyroid hormone receptors: hormone-independent activation operates through a steroid receptor mode of co-activator interaction. Mol Endocrinol. 2001;15(7):1170–85. doi: 10.1210/mend.15.7.0656. [DOI] [PubMed] [Google Scholar]
- 70.Belakavadi M, Fondell JD. Role of the mediator complex in nuclear hormone receptor signaling. Rev Physiol Biochem Pharmacol. 2006;156:23–43. doi: 10.1007/s10254-005-0002-0. [DOI] [PubMed] [Google Scholar]
- 71.McKenna NJ, et al. Nuclear receptor coactivators: multiple enzymes, multiple complexes, multiple functions. J Steroid Biochem Mol Biol. 1999;69(1–6):3–12. doi: 10.1016/s0960-0760(98)00144-7. [DOI] [PubMed] [Google Scholar]
- 72.Weiss RE, et al. Thyroid function in mice with compound heterozygous and homozygous disruptions of SRC-1 and TIF-2 coactivators: evidence for haploinsufficiency. Endocrinology. 2002;143(4):1554–7. doi: 10.1210/endo.143.4.8828. [DOI] [PubMed] [Google Scholar]
- 73.Weiss RE, et al. Mice deficient in the steroid receptor co-activator 1 (SRC-1) are resistant to thyroid hormone. Embo J. 1999;18(7):1900–4. doi: 10.1093/emboj/18.7.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jepsen K, et al. Cooperative regulation in development by SMRT and FOXP1. Genes Dev. 2008;22(6):740–5. doi: 10.1101/gad.1637108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jepsen K, et al. Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell. 2000;102(6):753–63. doi: 10.1016/s0092-8674(00)00064-7. [DOI] [PubMed] [Google Scholar]
- 76.Astapova I, et al. The nuclear corepressor, NCoR, regulates thyroid hormone action in vivo. Proc Natl Acad Sci U S A. 2008;105(49):19544–9. doi: 10.1073/pnas.0804604105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Astapova I, et al. The nuclear receptor corepressor (NCoR) controls thyroid hormone sensitivity and the set point of the hypothalamic-pituitary-thyroid axis. Mol Endocrinol. 2011;25(2):212–24. doi: 10.1210/me.2010-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Heinen CA, et al. Mutations in TBL1X Are Associated With Central Hypothyroidism. J Clin Endocrinol Metab. 2016;101(12):4564–4573. doi: 10.1210/jc.2016-2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vella KR, et al. Thyroid Hormone Signaling in vivo Requires a Balance Between Coactivators and Corepressors. Mol Cell Biol. 2014 doi: 10.1128/MCB.00129-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fozzatti L, et al. Nuclear receptor corepressor (NCOR1) regulates in vivo actions of a mutated thyroid hormone receptor alpha. Proc Natl Acad Sci U S A. 2013;110(19):7850–5. doi: 10.1073/pnas.1222334110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fozzatti L, et al. Resistance to thyroid hormone is modulated in vivo by the nuclear receptor corepressor (NCOR1) Proc Natl Acad Sci U S A. 2011;108(42):17462–7. doi: 10.1073/pnas.1107474108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kamiya Y, et al. Modulation by steroid receptor coactivator-1 of target-tissue responsiveness in resistance to thyroid hormone. Endocrinology. 2003;144(9):4144–53. doi: 10.1210/en.2003-0239. [DOI] [PubMed] [Google Scholar]
- 83.Nofsinger RR, et al. SMRT repression of nuclear receptors controls the adipogenic set point and metabolic homeostasis. Proc Natl Acad Sci U S A. 2008;105(50):20021–6. doi: 10.1073/pnas.0811012105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pei L, et al. Thyroid hormone receptor repression is linked to type I pneumocyte-associated respiratory distress syndrome. Nat Med. 2011;17(11):1466–72. doi: 10.1038/nm.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shimizu H, et al. NCoR1 and SMRT play unique roles in thyroid hormone action in vivo. Mol Cell Biol. 2015;35(3):555–65. doi: 10.1128/MCB.01208-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sun Z, et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat Med. 2012 doi: 10.1038/nm.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Costa-e-Sousa RH, et al. The thyroid axis is regulated by NCoR1 via its actions in the pituitary. Endocrinology. 2012;153(10):5049–57. doi: 10.1210/en.2012-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chiamolera MI, et al. Fundamentally distinct roles of thyroid hormone receptor isoforms in a thyrotroph cell line are due to differential DNA binding. Mol Endocrinol. 2012;26(6):926–39. doi: 10.1210/me.2011-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shibusawa N, et al. Thyroid hormone action in the absence of thyroid hormone receptor DNA-binding in vivo. J Clin Invest. 2003;112(4):588–97. doi: 10.1172/JCI18377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Grueter CE, et al. A cardiac microRNA governs systemic energy homeostasis by regulation of MED13. Cell. 2012;149(3):671–83. doi: 10.1016/j.cell.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yap CS, et al. Thyroid hormone negatively regulates CDX2 and SOAT2 mRNA expression via induction of miRNA-181d in hepatic cells. Biochem Biophys Res Commun. 2013;440(4):635–9. doi: 10.1016/j.bbrc.2013.09.116. [DOI] [PubMed] [Google Scholar]