

Abstract
PDZ-binding kinase (PBK) plays a major role in proliferation and in safeguarding mitotic fidelity in cancer cells. Frequently upregulated in many cancers, PBK drives tumorigenesis and metastasis. PBK has been shown to be phosphorylated in mitosis by cyclin-dependent kinase 1 (CDK1)/cyclin B, however, no studies have been done examining PBK mitotic phosphorylation in oncogenesis. Additionally to the previously identified Threonine-9 phosphorylation, we found that Threonine-24, Serine-32, and Serine-59 of PBK are also phosphorylated. PBK is phosphorylated in vitro and in cells by CDK1 during antimitotic drug-induced mitotic arrest and in normal mitosis. We demonstrated that mitotic phosphorylation of Threonine-9 is involved in cytokinesis. The non-phosphorylatable mutant PBK-T9A augments tumorigenesis to a greater extent than wild type PBK in breast cancer cells, suggesting that PBK mitotic phosphorylation inhibits its tumor promoting activity. The PBK-T9A mutant also transforms and increases the proliferation of immortalized breast epithelial cells. Collectively, this study reveals that CDK1-mediated mitotic phosphorylation of PBK is involved in cytokinesis and inhibits its oncogenic activity.
Keywords: PBK, mitotic phosphorylation, CDK1, cytokinesis, oncogenesis
1. Introduction
PDZ-binding kinase (PBK) also named Lymphokine-Activated Killer T-Cell-Originated Protein Kinase (TOPK) is a MAPKK-like serine/threonine kinase. PBK was originally isolated due to its increased expression in IL-2 activated killer T-cells and was found to interact with the human homologue of the Drosophila discs large tumor suppressor (hDlg) [1,2]. Normally only having detectible expression in testicular germ cells and some fetal tissues, PBK is overexpressed in a wide array of cancers including pancreatic, breast, ovarian, colorectal, lung, bladder, gastric, and oral cancer [3–11]. In addition, PBK overexpression is sufficient to transform mouse epidermal cells [11] and is strongly associated with patient survival and tumor grade [3,9,12]. Several studies have shown that PBK knockdown decreases cell viability and proliferation [7,11,13–16] as well as suppressing the cologenic potential of cancer cells [7,10,11,15].
PBK has been shown to be upregulated during mitosis and that PBK binding and phosphorylation by CDK1/cyclin B is required for its mitotic activity [17]. Other studies have described a key role for PBK in mitosis, as cells treated with PBK inhibitors or siRNA targeting PBK develop cytokinetic defects [7,18,19]. Threonine-9 of PBK has been described previously as the principle mitotic phosphorylation target of CDK1/cyclin B and that its phosphorylation is required for proper binding of PBK to its effector proteins as well as for PBK’s mitotic activity [2,17,20]. However, no functional studies have been carried out to examine the role of PBK mitotic phosphorylation in cytokinesis and cancer related properties. Interestingly, another study has revealed that phosphorylated bands were still present for HA-tagged PBK-T9A mutant protein in mitotically arrested cells, indicating the possibility of other phosphorylation sites [21].
A recent report uncovered that PBK expression is regulated by Yes-associated protein (YAP), a transcriptional co-activator of the Hippo pathway [22,23], and is responsive to geranylgeranyl signaling in ER- breast cancer cells [24]. In this report, we identified additional CDK1/cyclin B phosphorylation sites on PBK and elucidate the role of the CDK1/PBK axis in breast cancer. We found that CDK1-mediated mitotic phosphorylation of PBK is involved in cytokinesis and inhibits its oncogenic activity.
2. Materials and methods
2.1. Cell culture and transfection
HEK293T, HeLa, U2OS, MCF10A, and T47D cell lines were purchased from American Type Culture Collection (ATCC) and cultured as ATCC instructed. The cell lines were authenticated at ATCC and were used at low (<25) passages. Attractene (Qiagen) was used for transient overexpression of proteins in HEK293T cells following the manufacturer’s instructions. SiRNA transfections were done with HiPerfect (Qiagen). The MISSION siRNA for PBK was purchased from Sigma. The target sequences are: 5′-GACCATAGTTTCTTGTTAA-3′ and 5′-CTGGATGAATCATACCAGA-3′. Lentivirus packaging, infection, and subsequent selection were done as we have described previously [25]. Nocodazole (100 ng/ml for 16 h) and taxol (100 nM for 16 h) (Selleck Chemicals) were used to arrest cells in mitosis unless otherwise indicated. Kinase inhibitors were purchased from Selleck Chemicals (VX680, BI2536, Purvalanol A, SP600125, SB216736, and MK2206), ENZO life Sciences (RO3306 and Roscovitine), or LC Laboratory (U0126 and SB203580). All other chemicals were from either from Sigma or Thermo Fisher.
2.2. Expression constructs
The pWZL-Neo-Myr-Flag-PBK vector was a gift from William Hahn & Jean Zhao (Addgene plasmid # 20558). To make the Lentiviral PBK expression constructs, the above vector was used to clone full-length PBK cDNA into the pSIN4-Flag-IRES-puro vector. The pSIN4-Flag-IRES-puro vector was made by inserting an N-terminal Flag-tag with multiple-cloning-site sequences into the empty pSIN4-EF1α-IRES-Puro vector, which was originally obtained from Addgene [26]. Point mutations were generated by the QuikChange Site-Directed PCR mutagenesis kit (Stratagene) and verified by sequencing.
2.3. EGFP-expressing all-in-one CRISPR construct
To establish the CRISPR vector which expresses EGFP, we modified the pX330A_D10A-1×2 all-in-one vector, from the Takashi Yamamoto laboratory [27]. We removed the small sequence between the FseI and NotI sites and added a T2A-EGFP sequence from pSpCas9(BB)-2A-GFP (PX458) in its place [28].
To construct the all-in-one CRISPR/Cas9n plasmid targeting PBK, the sense and anti-sense oligonucleotides from Table 1 were synthesized, annealed, and Golden Gate assembled into the pX330A_D10A-1×2-EGFP and pX330S-2 vectors as described previously [27]. After the two vectors were generated, a final Golden Gate assembly was performed to generate the all-in-one vector as described previously [27]. The resulting pX330A_D10A-1×2-EGFP-PBK-AB construct was transfected into cells and GFP-positive clones were selected by flow cytometry-based cell sorting.
Table 1.
PBK Guide Sequences
| PBK-A-Fwd | caccGAGGCCGGGATATTTATAGT |
| PBK-A-Rev | aaacACTATAAATATCCCGGCCTC |
| PBK-B-Fwd | caccGCAGAAGCTTGGCTTTGGTAC |
| PBK-B-Rev | aaacGTACCAAAGCCAAGCTTCTGC |
2.4. Recombinant protein purification and in vitro kinase assay
GST-tagged PBK and PBK-4A were cloned in pGEX-5X-1 and proteins were bacterially expressed and purified on GSTrap FF affinity columns (GE Healthcare) following the manufacturer’s instructions. GST-PBK (0.5–1 μg) was incubated with 10 U recombinant CDK1/cyclin B complex (New England Biolabs) or 100 ng CDK1/cyclin B (SignalChem) in kinase buffer (New England Biolabs) in the presence of 5 μCi γ-32P-ATP (3000 Ci/mmol, PerkinElmer). Phosphorylation (32P incorporation) was visualized by autoradiography followed by Western blotting or detected by phospho-specific antibodies.
2.5. Antibodies
The PBK and phospho-PBK (pT9) antibodies were purchased from Cell Signaling Technology (4942 and 4941). Rabbit polyclonal phospho-specific antibodies against PBK T24, S32, and S59 were generated and purified by AbMart. The peptides used for immunizing rabbits were SVLCS-pT-PTINI (T24), INIPA-pS-PFMQK (S32) and RGLSH-pS-PWAVK (S59). The corresponding non-phosphorylated peptides were also synthesized and used for antibody purification and blocking assays. Anti-Flag antibody was from Sigma. Anti-PLK1 was from BioLegend. Anti-β-actin, anti-Mps1/TTK, anti-cyclin E1, and anti-cyclin B antibodies were from Santa Cruz Biotechnology. Anti-aurora-A, anti-CDK2, anti-glutathione S-transferase (GST), anti-BUB1, and anti-BubR1 antibodies were from Bethyl Laboratories. Anti-phospho-S10 H3, anti-YAP, anti-phospho-S127 YAP, anti-phospho-S397 YAP, anti-vimentin, anti-E-cadherin, anti-N-cadherin, anti-CDC25C, anti-CDK4, anti-CDK5, anti-CDK6, anti-Cyclin A2, anti-Cyclin E2, anti-MAD2, anti-phospho-S795 Rb, anti-Wee1, and anti-phospho-S642 Wee1 antibodies were from Cell Signaling Technology. Anti-β-tubulin (Sigma) antibodies were used for immunofluorescence staining.
2.6. Phos-tag and Western blot analysis
Phos-tag™ was obtained from Wako Pure Chemical Industries, Ltd. (cat#: 304-93521) and used at 10–20 μM (with 100 μM MnCl2) in 8% SDS-acrylamide gels as described [29]. Western blotting and lambda phosphatase treatment assays were done as previously described [25].
2.7. Immunofluorescence staining and confocal microscopy
Cell fixation, permeabilization, immunofluorescence staining, and microscopy were done as previously described [30]. For peptide blocking, a protocol from the Abcam website was used, as we previously described [31].
2.8. Proliferation, migration and invasion assays
Proliferation of cells was determined by MTT assay. Briefly, the cell lines MCF10A and T47D were seeded into 12-well plates, in triplicate, at a density of 5×104 and 1×105 cells/well respectively. Next, culture media with 0.5 μg/mL MTT was added to the cells which were incubated in the dark at 37°C for 1 h. After incubation, the media was removed and the reduced MTT was liberated from the cells and dissolved by addition of 500 μL of DMSO. Then 150 μL of the released dye was transferred, in triplicate, to 96-well plates. The absorbance was determined at a wavelength of 570 nm. Wound healing assays were utilized for measuring migratory activity as previously described [32]. In vitro cell invasion assays were performed using the BioCoat invasion system (BD BioSciences) according to the manufacturer’s instructions. The outer section was filled with 750 μL of a culture medium. Cell suspensions in 500 μL of serum free medium were seeded in the inner chambers. After incubation for 24 h, the cells on the upper surface of the chamber were removed with a cotton swab, and the cells that invaded to the lower surface of the chamber were fixed with methanol and stained using ProLong Gold Antifade Reagent with DAPI [31].
2.9. Statistical analysis
Statistical significance was analyzed using a two-tailed, unpaired Student’s t-test.
3. Results and Discussion
3.1 Antimitotic drugs trigger PBK phosphorylation at mitosis of cell cycle and can be blocked by CDK1 inhibitors
To further explore PBK’s involvement in mitosis, we treated HeLa cells with taxol or nocodazole to arrest the cells in mitosis and as shown in Figure 1A, PBK protein was dramatically up-shifted on a Phos-tag gel (Fig. 1A). Lambda phosphatase treatment largely abolished the PBK mobility shift, suggesting that PBK is phosphorylated during mitotic arrest (Fig. 1B). We used various kinase inhibitors to identify the candidate kinase for PBK phosphorylation. Inhibition of PLK1 (with BI2536), Aurora-A, -B, -C (with VX680), Akt (with MK2206), p38 kinase (with SB203580), JNK1/2 (with SP600125), MEK/ERK (with U0126), or GSK3 (with SB216763) failed to alter the phosphorylation level and resultant mobility shift of PBK during G2/M arrest (Fig. 1C, lanes 5–11). These inhibitors are effective under the conditions used [31,33]. Treatments with RO3306 (CDK1-specific inhibitor) or Purvalanol A (CDK1/2/5 inhibitor) virtually blocked the PBK mobility shift/phosphorylation entirely (Fig. 1C, lanes 3–4). These data suggest that PBK is likely phosphorylated by CDK1 during mitotic arrest induced by taxol or nocodazole treatment.
Figure 1. Phosphorylation of PBK by CDK1 during mitotic arrest.
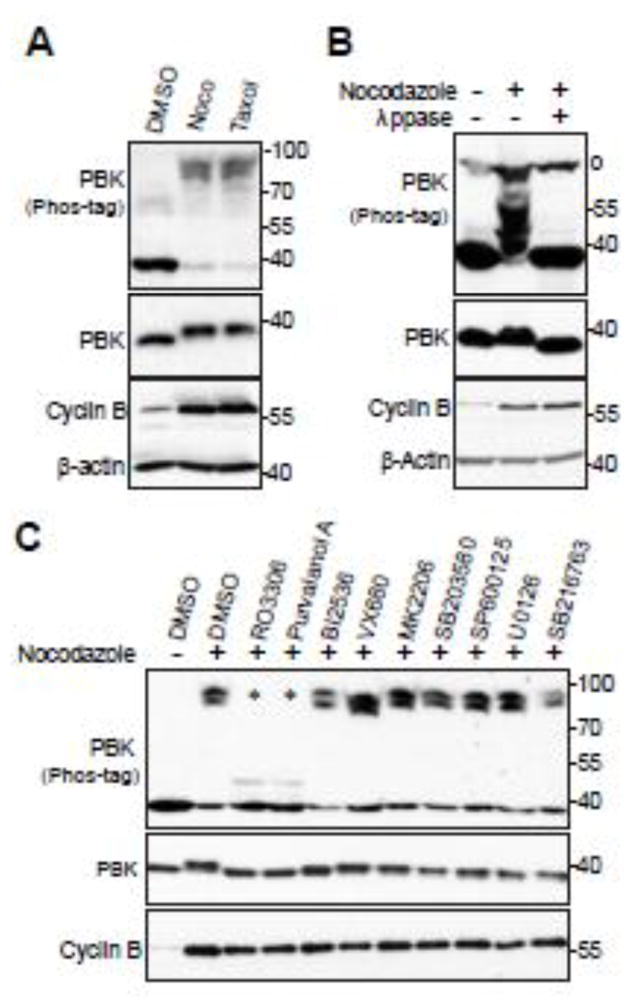
A, HeLa cells were treated with DMSO, taxol (100 nM for 16 h) or nocodazole (100 ng/ml for 16 h). Total cell lysates were probed with the indicated antibodies on Phos-tag or regular SDS-polyacrylamide gels. B, HeLa cells were treated with nocodazole as indicated and cell lysates were further treated with (+) or without (−) λ-phosphatase (ppase). Total cell lysates were probed with anti-PBK antibody. o marks a non-specific band. C, HeLa cells were treated with nocodazole together with or without various kinase inhibitors as indicated. RO3306 (5 μM), Purvalanol A (10 μM), BI2536 (100 nM), VX680 (2 μM), MK2206 (10 μM), SB203580 (10 μM), SB203580 (20 μM), SP600125 (20 μM), U0126 (20 μM), and SB216763 (10 μM) were used. Inhibitors were added 1.5 h before harvesting the cells (with MG132 to prevent cyclin B degradation and consequent mitotic exit). Total cell lysates were subjected to Western blotting with the indicated antibodies. * marks the two lanes in which the PBK mobility-shift was inhibited.
3.2 CDK1 phosphorylates PBK in vitro
Next, with bacterially purified PBK proteins as substrates, we performed in vitro kinase assays to determine whether CDK1 can directly phosphorylate PBK. Figure 2A shows that purified CDK1/cyclin B kinase complex strongly phosphorylated GST-PBK proteins in vitro and that the CDK1 inhibitor RO3306 greatly reduced the phosphorylation of GST-PBK (Fig. 2A). There are a total of four conserved sites on the N-terminus of PBK that fit the proline-directed consensus sequence of CDK1-phosphorylation sites [34] (Fig. 2B). Intriguingly, these four sites (Threonine-9, Threonine-24, Serine-32, and Serine-59) have been identified as mitotic phosphorylation sites from large scale proteomic studies [35,36]. Mutating these four sites to non-phosphorylatable alanines (PBK-4A) completely eliminated the 32P incorporation onto PBK, indicating that T9, T24, S32 and S59 are the main CDK1 phosphorylation sites (Fig. 2C). We generated phospho-specific antibodies against T24, S32 and S59. Using these antibodies, as well as a commercial phospho-specific T9 antibody, we demonstrated that CDK1 phosphorylates PBK at T9, T24, S32 and S59 in vitro and that addition of RO3306 abolished this phosphorylation (Fig. 2D).
Figure 2. CDK1 phosphorylates PBK in vitro.

A, in vitro kinase assays with purified CDK1/cyclin B complex and recombinant GST-PBK. RO3306 (5 μM) was used to inhibit CDK1 kinase activity. B, Conservation of PBK’s mitotic phosphorylation sites. C, in vitro kinase assays with purified CDK1/cyclin B complex to phosphorylate recombinant GST-PBK or GST-PBK-4A. D, In vitro kinase assays were done as in (A) except anti-phospho-PBK T9, T24, S32 and S59 antibodies were used.
3.3 CDK1 phosphorylates PBK at multiple sites in cells
Next, we sought to examine if the four PBK mitotic phosphorylations occur in cells. Taxol and nocodazole treatment significantly increased the phosphorylation of T24 on endogenous PBK in HeLa cells (Fig. 3A). Phospho-antibody incubation with phospho-peptide, but not the regular peptide, completely blocked the phospho-signal, suggesting that this antibody specifically detects the phosphorylated form of PBK (Fig. 3A). Likewise, taxol and nocodazole treatment significantly increased the phosphorylation of T9, S32, and S59 on endogenous PBK (Fig. 3B). The phospho-signal of PBK T9, T24, S32, and S59 during taxol treatment was considerably reduced in PBK knockdown cells, further validating the specificity of these phospho-antibodies (Fig. 3C). Additionally, we further demonstrated that the phosphorylation of PBK is CDK1-dependent by using CDK1 inhibitors (Fig. 3D, lanes 3 and 4). Taken together, these observations indicate that PBK is phosphorylated by CDK1 at T9, T24, S32, and S59 in cells during antimitotic drug-induced mitotic arrest.
Figure 3. CDK1 phosphorylates PBK in cells.

A, HeLa cells were treated with nocodazole or taxol for 16 hours. The T24 phospho-antibody was incubated for 16 hours at 4°C with either PBS (no peptide), peptide, or phospo-peptide used for immunizing rabbits, before Western blotting. B, HeLa cells were treated with nocodazole or taxol for 16 hours before Western blotting was performed with the indicated antibodies. C, HeLa cells were transfected with scrambled siRNA (control) or siRNA against PBK for 48 h and were further treated with (+) or without (−) taxol for 16 hours. The total cell lysates were subjected to Western blotting with the indicated antibodies. * marks a non-specific band. D, HeLa cells were nocodazole together with or without various kinase inhibitors as indicated. Inhibitors were added with MG132 (to halt cyclin B degradation and prevent mitotic exit) for 1.5 h before harvesting the cells. Total cell lysates were subjected to Western blotting with the indicated antibodies.
3.4 CDK1 mediates PBK phosphorylation during mitotic arrest and in normal mitosis
We next performed immunofluorescence microscopy with these phospho-specific antibodies in HeLa cells. The antibody against p-T24 detected a strong signal in nocodazole-arrested prometaphase cells (Fig. 4A, white arrows). The signal was scarcely detectable in interphase cells (Fig. 4A, yellow arrows). Again, pre-incubation with the phospho-peptide, but not the control regular peptide, predominantly blocked the signal, suggesting that these antibodies specifically detect PBK only when it is phosphorylated (Fig. 4A, middle panels). Addition of the CDK1 inhibitor RO3306 largely reduced the signal detected by the PBK p-T24 antibody in mitotically arrested cells, once again demonstrating that this phosphorylation of PBK is CDK1-dependent (Fig. 4A, lower panels).
Figure 4. CDK1 phosphorylates PBK in normal mitosis and during mitotic arrest.

A, HeLa cells were treated with nocodazole or taxol for 16 hours and then fixed. Before the cells were immunostained with the T24 phospho-antibody, the antibody was incubated for 16 hours at 4°C with either PBS (no peptide), peptide, or phospo-peptide. White and yellow arrows mark some of the metaphase cells and the interphase cells, respectively. B, HeLa cells were synchronized by the double thymidine block and release method. Cells were stained with antibodies against p-PBK T24, β-tubulin, and with DAPI. C, A low power (40X objective) lens was used to view various phases of the cells in a field from (B). D, HeLa cells were synchronized by a double thymidine block and release method. Total cell lysates were harvested at the specified time points after release and subjected to Western blotting analysis with the indicated antibodies.
Subsequently, we utilized a double thymidine block and release method [30] to further investigate the spatial and temporal dynamics of PBK phosphorylation in HeLa cells during unperturbed, normal mitosis. This way, we could more accurately determine the phospho-status of PBK during the different mitotic phases. We found that the p-PBK T24 signal was readily detectable in prometaphase and peaked in metaphase. The signal deteriorated in anaphase and was further reduced in telophase and cytokinesis (Fig. 4B, C). We observed phosphorylation patterns for p-PBK T9, S32, and S59 which were similar to p-PBK T24 when probing lysates taken at 0, 8, 9, 13, and 24 hours after double thymidine block and release (Fig. 4D). Collectively, these data strongly indicate a dynamic, mitotic phosphorylation of PBK in cells.
3.5 CRISPR-induced PBK knockout leads to cytokinesis failure and tetraploid
To characterize the impact of PBK on mitotic processes, we generated PBK knockout (KO) U2OS cells using CRISPR/Cas9-nickase (Cas9n) (Fig. 5A). We then performed immunofluorescence staining, so as to observe which types of mitotic defects may appear. We found that the PBK KO U2OS displayed a marked increase in cells that did not fully complete cytokinesis when compared to parental U2OS (Fig. 5B, C).
Figure 5. Generation and characterization of PBK knockout (KO) U2OS cell line.

A, Western blot of parental and PBK knockout U2OS cell lines. Equal amount of U2OS control and PBK knockout cell lysates were probed with anti-PBK antibody. B, U2OS control and PBK knockout cells were used to quantify the percentage of cells in cytokinesis. C, Immunofluorescence confocal images showing the difference in the amount of cytokinesis between the U2OS control and PBK knockout cell lines. β-tubulin is shown in green and nuclei is stained with DAPI (blue). The arrows point to cytokinetic bridges. D, U2OS KO cells were stably transduced with vector, PBK, or PBK-T9A. Total cell lysates from U2OS cell lines which were treated with taxol and probed with the indicated antibodies. E, The indicated U2OS cell lines were used to quantify the percentage of cells in cytokinesis as in (B). F, The specified U2OS cell lines were labeled with PI and flow cytometry analysis was performed to determine the percentage of tetra-ploidy cells in the whole population. G, U2OS control and PBK knockout cell whole lysates were probed with the indicated YAP antibodies. Numbers below the p-S397 YAP blot indicate the quantification of relative band intensity from three independent experiments. H, U2OS control and PBK knockout cell whole lysates were probed with the indicated cell cycle-related antibodies. Numbers below the Cyclin E1 blot indicate the quantification of relative band intensity from three independent experiments. **:p<0.01;***:p<0.001 (t-test).
We next sought to elucidate the roles the mitotic phosphorylation sites engage in to regulate cytokinesis. To do this, we re-expressed exogenous wild type (WT) PBK or the PBK T9A mutant in PBK KO U2OS cells (Fig. 5D). It must be noted that we attempted multiple times to generate both a 3A (T24A/S32A/S59A) and a 4A (T9A/T24A/S32A/S59A) mutant stable cell lines but were unable to get sufficient expression of either protein, so we proceeded with characterizing the T9A mutant. As shown earlier, PBK is phosphorylated at T9, T24, S32 and S59 in parental U2OS when treated with taxol and, as expected, CRISPR-mediated PBK KO fully ablates each of these phospho-antibody signals (Fig. 5D, lanes 1–4). Reconstitution of WT PBK in PBK-null U2OS rescues each of these taxol-induced phosphorylations (Fig. 5D, lanes 5 and 6). In contrast, p-T9 PBK signal cannot be detected in PBK-T9A reconstitution cells, but has no significant effect on phosphorylation at the other additional three sites (Fig. 5D, lanes 7 and 8). To examine the effects of WT and T9A PBK reconstitution on the perturbed cytokinesis seen in PBK KO cells, we again performed immunofluorescence staining. Reconstitution with WT PBK fully rescued the cytokinesis failure, whereas expression of T9A PBK mutant failed to alter this phenotype found in PBK-null U2OS cells (Fig. 5E), suggesting that mitotic phosphorylation on T9 of PBK is involved in proper cytokinesis in U2OS cells. Since mitotic defects classically lead to genomic instability, we looked to quantify the ploidy changes resulting from knocking out PBK. Utilizing flow cytometry, we discovered that the number of tetraploid cells significantly increased in PBK KO U2OS (Fig. 5F). Although WT PBK reconstitution cells had percent-tetraploid cells in similar levels to parental U2OS, T9A PBK reconstitution cells maintained the dramatically high ratio of tetraploid to diploid cells found in PBK-null U2OS cells (Fig. 5F).
A previous study identified PBK as a downstream target of Hippo/YAP signaling [24], so we examined total and phospho-YAP levels in parental and PBK KO U2OS cells to determine if loss of PBK would affect the status of YAP. We found no noticeable changes to the total YAP protein level, nor did we see any changes to phosphorylation levels at S127 (Fig. 5G). However, we observed a twofold increase of p-S397 of YAP (Fig. 5G). Next, we screened numerous mitotic and cell-cycle regulators in parental and PBK KO U2OS cells to attempt to identify proteins with altered expression or phosphorylation levels between the two (Fig. 5H). Of the proteins assayed, none were altered, except cyclin E1. Cyclin E1 expression was found, repeatedly, to be reduced (about 60%) (Fig. 5H). These findings suggest that the mitotic phosphorylation of PBK at T9 is required for proper mitotic division and for maintaining genomic stability.
3.6 PBK overexpression leads to epithelial-mesenchymal transition in normal cells
PBK has been identified as an oncogene in many types of cancer, including breast cancer [7]. Therefore, to determine the biological significance of mitotic phosphorylation of PBK, we used breast cell lines as a model system. We assessed PBK protein levels in MCF10A (an immortalized breast epithelial cell line) and various breast cancer cell lines. PBK expression was very high in most of the cancer cell lines and was barely detectible in MCF10A cells (Fig. 6A). Next, we stably expressed PBK-WT or PBK-T9A in MCF10A cells. Neither the overexpression of PBK-WT or PBK-T9A led to changes in either total YAP protein level or p-YAP S127 (Fig. 6B). Interestingly, PBK-WT and -T9A overexpression in MCF10A led to changes in epithelial-mesenchymal transition (EMT) markers, with E-cadherin downregulation and N-cadherin upregulation (Fig. 6C). Accompanying this molecular change, PBK-WT and PBK-T9A overexpressed MCF10A cells displayed a spindle-shaped morphology and grew in a more loosely arranged manner compared to the cuboidal shape of vector control MCF10A cells which grew in organized, compact islets (Fig. 6D).
Figure 6. Overexpression of PBK elicits EMT in normal cells.

A, PBK protein expression in various normal and breast cancer cell lines. B, MCF10A (an immortalized breast epithelial cell line) cells transduced with vector control, PBK, or PBK-T9A and probed with the indicated YAP antibodies. C, MCF10A cells transduced with vector control, PBK, or PBK-T9A and probed with the indicated EMT marker antibodies. D, 10x microscope images displaying the morphology differences between MCF10A vector control, PBK, and PBK-T9A. E,F Cell migration (wound healing) assays with cell lines established in (B). G, Cell proliferation assays with cell lines established in (B). H, The cell lines established in (B) were used to quantify the percentage of cells in anaphase/telophase. **:p<0.01;***:p<0.001 (t-test).
Both PBK-WT and PBK-T9A MCF10As had significantly increased rates of cell migration in wound healing assays when compared to vector control, although, no difference was observed between the two (Fig. 6E, F). Interestingly, overexpression of PBK-WT in MCF10A led to significantly higher proliferation in comparison to vector control and surprisingly, PBK-T9A expressing MCF10A cells grew even faster than the PBK-WT expressing cells (Fig. 6G). Since PBK-null U2OS cells which were reconstituted with PBK-T9A displayed high levels of cytokinesis failure, we examined the effect of overexpressing PBK-T9A on MCF10A mitotic division. Similarly, immunofluorescence microscopy revealed that MCF10A cells overexpressing PBK-T9A had a significantly higher percentage of mitotic cells in anaphase and telophase than did either vector control or PBK-WT overexpression (Fig. 6H). Taken together, these findings suggest that mitotic phosphorylation of PBK T9 partially inhibits its oncogenic activity.
3.7 Mitotic phosphorylation of PBK inhibits its oncogenic activity in cells
We further determined the impact of mitotic phosphorylation of PBK in cancer cells by attempting to generate a full knockout cell line, but unfortunately were unable to do so, possibly because a complete loss of PBK activity is lethal in breast cancer; something that has been shown previously using PBK inhibitors [18]. We did however generate a PBK mono-allelic knockout (maKO) T47D cell line and used it to overexpress either WT or T9A PBK (Fig. 7A). With these cell lines we examined PBK’s role in invasion using Matrigel. Invasion of PBK maKO T47D cells was dramatically reduced when compared to parental T47D, whereas ectopic expression of PBK-WT rescued this drop in invasiveness (Fig. 7B, C). Expression of PBK-T9A further increased the invading activity when compared to PBK-WT (Fig. 7B, C). T47D PBK maKO cells, having a reduced PBK expression level, possess much lower migratory activity than do parental T47D cells (Fig. 7D, E). Overexpression of PBK-T9A increased cell migration to a greater extent when compared to PBK-WT overexpression (Fig. 7D, E). Interestingly, overexpression of PBK-T9A significantly increased cell proliferation over that of control and PBK-WT-overexpressing cells, suggesting that mitotic phosphorylation of PBK can constrain proliferation (Fig. 7F). As expected, cells partially lacking PBK expression (PBK maKO) proliferated at a rate significantly slower when compared to control cells (Fig. 7F). Together, our data suggest that mitotic phosphorylation of PBK inhibits its oncogenic activity in breast cancer cells.
Figure 7. Mitotic phosphorylation of PBK inhibits its oncogenic activity in cells.

A, Western blot of PBK mono-allelic knockout T47D cell line and transduced PBK and PBK-T9A cell lines. B, C, Invasion assays with cell lines established in (A). D, E, Cell migration (wound healing) assays with cell lines established in (A). F, Cell proliferation assays with cell lines established in (A). *:p<0.05; **:p<0.01; ***:p<0.001 (t-test).
Several groups have described that mitotic phosphorylation at T9 on PBK is required for PBK’s proper mitotic functionality [2,17,20]. However, little is known regarding the role of PBK mitotic phosphorylation in cancer related properties. Our studies revealed that mitotic phosphorylation on T9 is inhibitory on PBK’s oncogenic activity (Figs. 6, 7). Here we identified three novel mitotic phosphorylation sites on PBK which are regulated by CDK1/cyclin B, extricating another layer mitotic phospho-regulation of PBK. However, the function of the other three CDK1 phosphorylation sites (T24, S32, S59) has not been defined. We discovered that the PBK-3A (T24A/S32A/S59A) and -4A (T9A/3A) mutants cannot be stably expressed. Utilizing a proteasomal inhibitor, we found that both of these proteins are being expressed, but are in fact being quickly degraded (data not shown). Further investigation using individual mutants is needed to determine if one or more of these sites are important for proper protein folding or if their phosphorylation is required to block targeting of PBK for proteasomal turnover.
Overexpression of PBK has been associated with increased tumorigenicity and clinical lethality of various cancers [3,9,10,12,37]. Future experiments are also needed to further investigate the relationship between the levels of PBK protein and PBK mitotic phosphorylation in cancer patient samples. Equally important is to identify PBK’s protein substrate(s) that are essential for cytokinesis and cancer cell survival and proliferation. Addressing these questions will not only help with understanding the cellular function of PBK in mitosis, but also provide insights into the underlying mechanisms of PBK overexpression in cancer development.
4. Conclusions
In the current study, we demonstrated that the mitotic phosphorylation of PBK on T9 is involved in proper cytokinesis and inhibits its oncogenic activity in breast cancer cells. We identified novel phosphorylation sites (T24, S32, S59) on PBK by CDK1 during mitosis.
Highlights.
PBK kinase is phosphorylated during mitosis
CDK1 phosphorylates PBK T9, T24, S32, and S59 in vitro and in vivo
Mitotic phosphorylation is involved in proper cytokinesis
Mitotic phosphorylation inhibits PBK oncogenic activity in breast cancer cells
Acknowledgments
All fluorescence images were acquired by Zeiss LSM 710 confocal microscope at the Advanced Microscopy Core at the University of Nebraska Medical Center. The core is supported in part by grant P30 GM106397 from the National Institutes of Health (NIH). Research in the Dong laboratory is supported by Fred & Pamela Buffett Cancer Center Support Grant (P30 CA036727), grants P30 GM106397 and R01 GM109066 from the NIH, and W81XWH-14-1-0150 from the Department of Defense Health Program. Seth Stauffer is supported by a Graduate Studies Office fellowship from the University of Nebraska Medical Center.
Abbreviations
- CDK1
Cyclin-dependent kinase 1
- EMT
Epithelial-mesenchymal transition
- PBK
PDZ-binding kinase
- YAP
Yes-associated protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abe Y, Matsumoto S, Kito K, Ueda N. J Biol Chem. 2000;275:21525–21531. doi: 10.1074/jbc.M909629199. [DOI] [PubMed] [Google Scholar]
- 2.Gaudet S, Branton D, Lue RA. Proc Natl Acad Sci U S A. 2000;97:5167–5172. doi: 10.1073/pnas.090102397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang CF, Chen SL, Sung WW, Hsieh MJ, Hsu HT, Chen LH, Chen MK, Ko JL, Chen CJ, Chou MC. Int J Mol Sci. 2016;17 doi: 10.3390/ijms17071007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fujibuchi T, Abe Y, Takeuchi T, Ueda N, Shigemoto K, Yamamoto H, Kito K. Dev Growth Differ. 2005;47:637–644. doi: 10.1111/j.1440-169X.2005.00834.x. [DOI] [PubMed] [Google Scholar]
- 5.Ikeda Y, Park JH, Miyamoto T, Takamatsu N, Kato T, Iwasa A, Okabe S, Imai Y, Fujiwara K, Nakamura Y, Hasegawa K. Clin Cancer Res. 2016;22:6110–6117. doi: 10.1158/1078-0432.CCR-16-0207. [DOI] [PubMed] [Google Scholar]
- 6.Long J, Liu Z, Wu X, Xu Y, Ge C. Mol Med Rep. 2016;13:3913–3919. doi: 10.3892/mmr.2016.5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park JH, Lin ML, Nishidate T, Nakamura Y, Katagiri T. Cancer Res. 2006;66:9186–9195. doi: 10.1158/0008-5472.CAN-06-1601. [DOI] [PubMed] [Google Scholar]
- 8.Ohashi T, Komatsu S, Ichikawa D, Miyamae M, Okajima W, Imamura T, Kiuchi J, Kosuga T, Konishi H, Shiozaki A, Fujiwara H, Okamoto K, Tsuda H, Otsuji E. Br J Cancer. 2017;116:218–226. doi: 10.1038/bjc.2016.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh PK, Srivastava AK, Dalela D, Rath SK, Goel MM, Bhatt ML. Immunobiology. 2014;219:469–474. doi: 10.1016/j.imbio.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Wei DC, Yeh YC, Hung JJ, Chou TY, Wu YC, Lu PJ, Cheng HC, Hsu YL, Kuo YL, Chen KY, Lai JM. Cancer Sci. 2012;103:731–738. doi: 10.1111/j.1349-7006.2011.02197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu F, Zykova TA, Kang BS, Wang Z, Ebeling MC, Abe Y, Ma WY, Bode AM, Dong Z. Gastroenterology. 2007;133:219–231. doi: 10.1053/j.gastro.2007.04.048. [DOI] [PubMed] [Google Scholar]
- 12.Wang MY, Lin ZR, Cao Y, Zheng LS, Peng LX, Sun R, Meng DF, Xie P, Yang JP, Cao L, Xu L, Huang BJ, Qian CN. Oncotarget. 2016;7:26604–26616. doi: 10.18632/oncotarget.8445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Liu H, Cao H, Song B, Zhang W, Zhang W. Oncol Rep. 2015;34:3288–3296. doi: 10.3892/or.2015.4308. [DOI] [PubMed] [Google Scholar]
- 14.Hu F, Gartenhaus RB, Eichberg D, Liu Z, Fang HB, Rapoport AP. Oncogene. 2010;29:5464–5474. doi: 10.1038/onc.2010.275. [DOI] [PubMed] [Google Scholar]
- 15.Ayllon V, O’connor R. Oncogene. 2007;26:3451–3461. doi: 10.1038/sj.onc.1210142. [DOI] [PubMed] [Google Scholar]
- 16.Rizkallah R, Batsomboon P, Dudley GB, Hurt MM. Oncotarget. 2015;6:1446–1461. doi: 10.18632/oncotarget.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abe Y, Takeuchi T, Kagawa-Miki L, Ueda N, Shigemoto K, Yasukawa M, Kito K. J Mol Biol. 2007;370:231–245. doi: 10.1016/j.jmb.2007.04.067. [DOI] [PubMed] [Google Scholar]
- 18.Matsuo Y, Park JH, Miyamoto T, Yamamoto S, Hisada S, Alachkar H, Nakamura Y. Sci Transl Med. 2014;6:259ra145. doi: 10.1126/scitranslmed.3010277. [DOI] [PubMed] [Google Scholar]
- 19.Park JH, Inoue H, Kato T, Zewde M, Miyamoto T, Matsuo Y, Salgia R, Nakamura Y. Cancer Sci. 2017;108:488–496. doi: 10.1111/cas.13160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsumoto S, Abe Y, Fujibuchi T, Takeuchi T, Kito K, Ueda N, Shigemoto K, Gyo K. Biochem Biophys Res Commun. 2004;325:997–1004. doi: 10.1016/j.bbrc.2004.10.133. [DOI] [PubMed] [Google Scholar]
- 21.Park JH, Nishidate T, Nakamura Y, Katagiri T. Cancer Sci. 2010;101:403–411. doi: 10.1111/j.1349-7006.2009.01400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan D. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu FX, Zhao B, Guan KL. Cell. 2015;163:811–828. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dou X, Wei J, Sun A, Shao G, Childress C, Yang W, Lin Q. Cancer Cell Int. 2015;15 doi: 10.1186/s12935-015-0178-0. 27-015-0178-0. eCollection 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao L, Chen Y, Ji M, Dong J. J Biol Chem. 2011;286:7788–7796. doi: 10.1074/jbc.M110.173468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elcheva I, Brok-Volchanskaya V, Kumar A, Liu P, Lee JH, Tong L, Vodyanik M, Swanson S, Stewart R, Kyba M, Yakubov E, Cooke J, Thomson JA, Slukvin I. Nat Commun. 2014;5:4372. doi: 10.1038/ncomms5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakuma T, Nishikawa A, Kume S, Chayama K, Yamamoto T. Sci Rep. 2014;4:5400. doi: 10.1038/srep05400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X, Stauffer S, Chen Y, Dong J. J Biol Chem. 2016;291:14761–14772. doi: 10.1074/jbc.M116.722751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang L, Iyer J, Chowdhury A, Ji M, Xiao L, Yang S, Chen Y, Tsai MY, Dong J. J Biol Chem. 2012;287:34069–34077. doi: 10.1074/jbc.M112.385518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang S, Zhang L, Liu M, Chong R, Ding SJ, Chen Y, Dong J. Cancer Res. 2013;73:6722–6733. doi: 10.1158/0008-5472.CAN-13-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jube S, Rivera ZS, Bianchi ME, Powers A, Wang E, Pagano I, Pass HI, Gaudino G, Carbone M, Yang H. Cancer Res. 2012;72:3290–3301. doi: 10.1158/0008-5472.CAN-11-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim H, Vick P, Hedtke J, Ploper D, De Robertis EM. Cell Rep. 2015;11:1151–1159. doi: 10.1016/j.celrep.2015.04.048. [DOI] [PubMed] [Google Scholar]
- 34.Nigg EA. Trends Cell Biol. 1993;3:296–301. doi: 10.1016/0962-8924(93)90011-o. [DOI] [PubMed] [Google Scholar]
- 35.Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M. Sci Signal. 2010;3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- 36.Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. Proc Natl Acad Sci U S A. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leary PCO, Penny SA, Dolan RT, Kelly CM, Madden SF, Rexhepaj E, Brennan DJ, McCann AH, Ponten F, Uhlen M, Zagozdzon R, Duffy MJ, Kell MR, Jirstrom K, Gallagher WM. BMC Cancer. 2013;13 doi: 10.1186/1471-2407-13-175. 175-2407-13-175. [DOI] [PMC free article] [PubMed] [Google Scholar]