

Abstract
Among the 22 FKBP domains in the human genome, FKBP12.6 and the first FKBP domains (FK1) of FKBP51 and FKBP52 are evolutionarily and structurally most similar to the archetypical FKBP12. As such, the development of inhibitors with selectivity among these four FKBP domains poses a significant challenge for structure-based design. The pleiotropic effects of these FKBP domains in a range of signaling processes such as the regulation of ryanodine receptor calcium channels by FKBP12 and FKBP12.6 and steroid receptor regulation by the FK1 domains of FKBP51 and FKBP52 amply justify the efforts to develop selective therapies. In contrast to their close structural similarities, these four FKBP domains exhibit a substantial diversity in their conformational flexibility. A number of distinct conformational transitions have been characterized for FKBP12 spanning timeframes from 20 s to 10 ns and in each case these dynamics have been shown to markedly differ from the conformational behavior for one or more of the other three FKBP domains. Protein flexibility-based inhibitor design could draw upon the transitions that are significantly populated in only one of the targeted proteins. Both the similarities and differences among these four proteins valuably inform the understanding of how dynamical effects propagate across the FKBP domains as well as potentially how such intramolecular transitions might couple to the larger scale transitions that are central to the signaling complexes in which these FKBP domains function.
Keywords: FKBP domain, conformational dynamics, NMR, crystallography, allostery
Graphical abstract
Alternate orientations of the Val 90 sidechain in two crystal forms of FKBP12.6 at the tip of the β4–β5 loop where it packs against residues of the β2 and β3a strands.
INTRODUCTION
The archetypical FKBP domain protein FKBP12 is best known for forming binary complexes with the immunosuppressants FK506 and rapamycin which, in turn, form inhibitory ternary complexes with the protein phosphatase calcineurin [1] and the protein kinase mTOR [2], respectively. Although these FKBP12-mediated immunosuppressant interactions have proven to be highly useful in pharmacological therapies, there is little evidence to indicate that analogous endogenous interactions play a significant role in physiological signal processing. Despite more than two decades of research [3], no endogenous small molecule ligand has been shown to mediate protein-protein interactions for any of the 22 FKBP domains within the human genome [4]. In addition to the closely homologous FKBP12.6 and the more evolutionarily divergent FKBP13, there are 19 different FKBP domains that are modules within larger proteins. Some, but not all, of these FKBP domains catalyze the cis-trans prolyl isomerization reaction. The relative significance of this catalytic activity in the overall biological function for many of these FKBP proteins remains an open question, although the prolyl isomerization activity of FKBP12 has been proposed to contribute to the interactions of this protein with the misfolded forms of α-synuclein [5], tau [6] and amyloid precursor protein [7] while prolyl isomerization has also been proposed to have a role in the activities of larger FKBP domain proteins such as in the regulation of NF-κB by FKBP52 [8]. Due to the challenges posed by the particularly close structural similarity of FKBP12 and FKBP12.6 to the first FKBP domains of FKBP51 and FKBP52, as also reflected in their ungapped sequence alignments (Fig. 1), these four FKBP domains have been the primary focus of the conformational dynamics studied considered herein.
Fig. 1. Sequence alignment for FKBP12/12.6 and the FK1 domains of FKBP51/52.

To preserve similarity in the structural description, the initial β strand in the FK1 domains of FKBP51 and FKBP52 is denoted as βo.
Although crystal structures are available for a number of the human FKBP domains, the only reported structures of a physiological protein-protein complex involving an FKBP domain is for FKBP12 bound to the TGF β1 receptor and the closely related activin and bone morphogenetic protein receptors [9, 10]. In these structures, the site of catalytic isomerization in FKBP12 is occupied by a Leu-Leu dipeptide from the receptor. It appears that for these complexes the catalytic cleft of FKBP12 is being utilized for protein recognition interactions without any participation of peptide isomerization, indicating that catalytic site mutations which disrupt signaling function need not imply a role for isomerization.
Genetic and cell biology studies indicate that the physiological roles for various FKBP proteins primarily involve protein-protein recognition interactions that contribute to the regulation of various signaling pathways as typified by the FKBP12/FKBP12.6 regulation of ryanodine receptor calcium channels [11–14] and the FKBP51/FKBP52 regulation of steroid receptor complexes [15–18]. FK506 and rapamycin bind to many of the FKBP domains resulting in disruption of function. Given the wide range of processes in which the FKBP proteins participate in regulation, selective inhibition among the various FKBP domains is an important pharmacological goal. Clinical observations on the effects of non-immunosuppressant FK506 derivatives developed for neurological therapies demonstrate excellent tolerance to broad range inhibition among the FKBP domain proteins [19, 20], indicating the potential for selective FKBP inhibitors exhibiting good clinical tolerance [21].
In part stimulated by the considerable challenges to standard structure-based design of selectivity that is posed by FKBP12 and FKBP12.6 and the FK1 domains of FKBP51 and FKBP52, NMR and crystallographic studies of these four domains are being pursued to examine the degree to which transient conformations that are sampled by each of these domains might offer a degree of structural diversity among the four domains that could prove useful for selectivity design. Similarly of interest for these studies is the question of whether characterizing the conformational transitions for these four domains might provide valuable insights into their physiological functions within the large signaling complexes in which they are found. As large scale conformational transitions play a central role in the signaling process of both the ryanodine receptor and the steroid receptor, it is of considerable interest to determine whether conformational transition within the FKBP domains enable these domains to have direct allosteric coupling to the overall signaling transition of the receptor as a key aspect of their role in regulating these receptors.
FKBP12 preferentially binds to the ryanodine receptor isoform RyR1 found in skeletal muscle [11], while FKBP12.6 preferentially binds to the RyR2 isoform present in cardiac muscle [12, 22]. Correspondingly, most research on the ryanodine receptors has been interpreted in terms of distinct skeletal muscle regulation by FKBP12 and cardiac muscle regulation by FKBP12.6, although some studies have argued for a more interdependent pattern of regulatory interactions [14, 23]. Phosphorylation of the cardiac RyR by protein kinase A during myocardial infarction has been argued to cause FKBP12.6 to dissociate from the receptor [24]. Drugs designed to block this dissociation have been proposed to inhibit the progression of heart failure pathologies in mice [25], although the validity of this approach is controversial [26, 27].
In addition to its presence in cardiac muscle, the FKBP12.6-sensitive RyR2 isoform is predominant in pancreatic islet cells where it plays a role in Ca2+-dependent insulin secretion [28–30]. RyR2 is also quite abundant in the central nervous system. Antisense oligonucleotide knockdown experiments in mice have shown impairment of memory processes, while analogous experiments on the RyR1 channels showed no such effect [31]. Recent evidence suggests that release of FKBP12.6 from neuronal RyR2 channels may play a crucial role in stress-induced cognitive dysfunctions such as posttraumatic stress disorder [32]. Following on the logic of earlier cardiac studies, it has been proposed that drugs which selectively inhibit the dissociation of FKBP12.6 may provide a useful approach for neurological therapy [32].
Recent advances in cryo-electron microscopy have enabled the determination of the closed state of the RyR1 receptor at a resolution near 4 Å [33, 34], while structural analysis of the open state at 8 Å resolution has facilitated the characterization of the large changes in conformation that accompany this transition [35]. Although tantalizing, it is still not feasible to gain detailed structural insight into how FKBP12/FKBP12.6 differentially interacts with the two conformational states of the receptor, in part, because the quality of the cryo-EM maps diminish as one moves away from the central channel to the more distal regions of the complex.
FKBP51 and FKBP52 are two closely homologous proteins each containing a pair of FK506-binding domains (FK1 and FK2) followed by a tetratricopeptide (TPR) repeat domain which mediates their interactions with Hsp90. FKBP52 was first characterized as a cochaperone of Hsp90 in the activated hormone-bound steroid receptor complexes that are formed with the progesterone, androgen or glucocorticoid receptor proteins [16]. In contrast, FKBP51 serves as the predominant Hsp90 cochaperone in the unliganded state for these steroid receptors [17, 18]. Mediated by the FK1 domain [36], steroid binding to the FKBP51-bound receptor is believed to induce the exchange for FKBP52 [37]. While FKBP51 reduces the hormone binding affinity of the receptor, FKBP52 substitution increases that affinity. Substitution with FKBP52 also enhances binding to dynein via its FK1 domain [38], thus facilitating microtubular transport of the receptor from the cytosol to the nucleus [39, 40].
FKBP51 and FKBP52 play qualitatively similar antagonistic roles in regulating the transport from cytoplasm to nucleus for the NF-κB transcription factors [41–43] with the FK1 domain of FKBP52 directly participating in stimulating transcriptional activation [8]. Also mediated by the FK1 domains, FKBP51 and FKBP52 act as antagonists in regulating the phosphorylation state of the tau protein and its proper recycling [44, 45]. In addition to normal regulation of microtubule polymerization, excessive tau phosphorylation contributes to the neurofibrillary tangles that are characteristic of various tauopathies. FKBP51 serves to mediate the regulation of the protein kinase Akt/PKB [46–49]. In addition to its more widely studied role in the control of cell survival and apoptosis, Akt/PKB reciprocally regulates the glucocorticoid receptor via phosphorylation.
Despite considerable effort, it has not been possible to reconstitute the activity of FKBP51 or FKBP52 with Hsp90, p23 and the receptor protein to form a biochemically defined steroid hormone receptor system [21, 50]. On the other hand, a low resolution (38 Å) cryo-EM reconstruction has recently been reported for the complex formed by Hsp90, Hsp70, Hop and the ligand binding domain of the glucocorticoid receptor protein (GR-LBD) [51],which corresponds to an obligatory intermediate in the ATP-dependent assembly process for the mature steroid receptor. Several aspects of the interactions of Hsp70 reported in this study bear an interesting analogy to those of FKBP51 with the mature glucocorticoid receptor complex. The isolated apo-GR-LBD binds hormone with high affinity, and that binding is markedly inhibited upon the binding of Hsp70 which induces a conformational transition affecting residues neighboring the ligand binding pocket of the GR-LBD. Hsp70 not only inhibits the binding of hormone by the apo-GR-LBD, it also stimulates the release of hormone from the liganded receptor. The subsequent binding of Hsp90 to this complex is insufficient to induce an increased hormone binding affinity until the Hsp90 hydrolyzes its bound ATP molecule which, in turn, induces the release of Hsp70. Reflecting the fact that the hormone affinity of the glucocorticoid receptor is modulated in the course of this cycle, Agard and colleagues [51] have proposed that the resultant sensitivity to the intracellular hormone concentration could provide the basis for an ATP-dependent regulatory mechanism for the receptor.
As noted above, the unliganded glucocorticoid receptor complex predominantly incorporates FKBP51 as its TPR cochaperone [17]. The FKBP51-bound form has a lower binding affinity for glucocorticoids than does the FKBP52-bound form [52]. Sanchez and colleagues have provided strong evidence that an initial effect of glucocorticoid binding to the FKBP51-bound receptor is to induce the release of FKBP51 and the subsequent binding of FKBP52 [37]. Using yeast heterologous expression systems for the human glucocorticoid and androgen receptors, Riggs et al. [36] demonstrated that a L119P mutation near the tip of the β4–β5 loop in the FK1 domain of human FKBP51 yielded a 3.5-fold increase in reporter gene expression, approaching the transcriptional level for the FKBP52-bound receptor complex. The complementary P119L mutation in FKBP52 yielded a 2-fold decrease in reporter gene expression. Similar results were also obtained for these FKBP51 and FKBP52 variants in an embryonic fibroblast cell line derived from FKBP52 knockout mice [36], indicating that the differing effects of these two cochaperones on the transcriptional activity of the steroid hormone receptor can be substantially reversed by a single point mutation.
As discussed below, the β4–β5 loop in the FK1 domain of human FKBP51 undergoes an extensive conformational transition that occurs in the submillisecond timeframe, while no such transition is apparent in FKBP52. Introduction of the FKBP52-like L119P mutation into FKBP51 suppresses this transition, while the complementary P119L substitution induces an analogous conformational transition in FKBP52. The reported interactions of Hsp70 discussed above provide a useful framework for considering how this conformational transition in the β4–β5 loop of FKBP51 might function in the regulation of the steroid receptor complex. Both Hsp70 and FKBP51 bind to the activated and inactivated hormone binding states of the glucocorticoid receptor in a differential manner as indicated by their effect of lowering the hormone binding affinity of the receptor. By replacing FKBP51 and stabilizing the activated state of the receptor, FKBP52 can then serve a role analogous to the Hsp90-catalyzed hydrolysis of ATP which leads to the release of Hsp70. If the conformational transition observed in the isolated FK1 domain of FKBP51 were to correspond to its differing conformation when bound to the activated and inactivated hormone binding states of the receptor, no such transition would be required for the FKBP52 interaction. Such a mechanism could potentially provide a means for establishing a hormone concentration-dependent regulation of the activity of the steroid receptor complex analogous to that proposed for the Hsp70 interactions [51] which would function without the requirement of a coupled ATP hydrolysis cycle.
Crystal structures have been obtained for the ligand binding domains from several steroid receptor proteins in various ligand-bound states. These studies demonstrate that the ligand-induced transitions for steroid antagonists generally differ from those induced by steroid agonists, and these distinct conformations can, in turn, differentially interact with co-regulators [53]. A concurrent conformational transition in the Hsp90 subunits has been proposed as a component of a larger scale allosteric response [54] providing further justification for consideration of a potential allosteric coupling to the FKBP domain.
If such allosteric processes are utilized by FKBP domains in mediating their protein-protein interactions within various signaling pathways, it is reasonable to expect that the underlying conformational transitions may often exploit the dynamical behavior that is intrinsic to the isolated FKBP domain. Combined with the further expectation that diversity among the various FKBP domains in the range of significantly populated conformations may provide a basis for the design of selective inhibitors, NMR and crystallographic characterization of such transitions have been actively pursued for the wild type and engineered variants of FKBP12/12.6 and the FK1 domains of FKBP51/52.
NMR CHARACTERIZATION OF CONFORMATIONAL TRANSITIONS IN FKBP DOMAINS
NMR enables the characterization of protein conformational dynamics in several time regimes. Conformational transitions that occur in a timeframe τ slower than ~10–100 ms will generally give rise to distinct resonances for each state. If the timeframe for that transition is shorter than a few seconds (i.e., similar to the T1 relaxation time of the observed nucleus), the rate of the transition can be quantified. As the rate of a conformational transition increases, it approaches the rate defined by the difference in frequency between the resonances arising from the exchanging states. For increasing exchange rates across this so-called conformational exchange linebroadening regime (τ ~ms-µs), the distinct resonances observed for a slow limit transition broaden and migrate together into a single resonance which then progressively sharpens to a fast exchange limit linewidth. The third readily accessible time regime (τ ~ns-ps) monitors conformational transitions that occur more rapidly than the rate of global molecular tumbling (τc = 6 ns for FKBP12 at 25°C [55]). Most commonly, a combination of 15N T1, T2 and heteronuclear NOE experiments are used to characterize the amplitude and timeframe of transitions in the protein backbone that cause a more rapid increase in the orientational disorder of the 1H-15N bond vector than does the overall tumbling of the protein molecule.
Generally most problematic to characterize by NMR are conformational transitions that occur within the time regime which spans ~3 orders of magnitude between the fast exchange limit of conformational exchange linebroadening and the global molecular tumbling time. In this time regime, conformational transitions are usually identified from structure-based analysis that indicates the presence of motional averaging for observed NMR signals such as spin coupling constants [56], residual dipolar couplings [57] or homonuclear NOE intensities as further discussed below.
Conformational transitions of FKBP12 have been observed in each of these time regimes which in each case have been shown to differ from the dynamical behavior of at least one of the FKBP domains: FKBP12.6 or the FK1 domains of FKBP51 and FKBP52. Dynamical processes from each of these time regimes are discussed and the current understanding of their structural basis is considered.
RESONANCE DOUBLING EXCHANGE DYNAMICS IN FKBP12 AND FKBP12.6
During the course of amide hydrogen exchange experiments on FKBP12 [58], a substantial set of minor peaks were noted to be approximately 7-fold weaker than the intensities for the major amide resonances in the standard 1H-15N 2D correlation spectrum (Fig. 2) [55]. Numerous previous publications had presented 1H-15N 2D correlation spectra of unliganded wild type FKBP12 with no such minor peaks being indicated [59–64]. Two other publications had presented such correlation plots at apparently lower contour levels that indicated a pattern of minor peaks [65, 66], although no discussion of this heterogeneity was offered. Mass spectral analysis of the FKBP12 samples indicated a single chemical species, and backbone resonance assignment experiments on a U-13C,15N labeled sample provided the sequential connectivities for these minor crosspeaks [55].
Fig. 2. 1H-15N 2D NMR correlation spectrum of U-2H,15N enriched wild type FKBP12.

Residues exhibiting resolved resonances for the minor slow exchange conformation are indicated in black while all other residues are indicated in gray. Ala 84 and Gly 89 exhibit the most rapid amide hydrogen exchange in the protein [58], resulting in severe broadening in the 1H dimension at pH 6.5. These resonances are readily observed at pH 5.5. The severely broadened resonance for the major conformation of Tyr 82 is visible at a lower contour level. The hydrogen exchange rate for this residue is too slow to contribute to linebroadening. Folded sidechain resonances are indicated with ‘x’. Illustration as modified from research originally published [55], the Biochemical Society copyright holder.
In sum, 31 of the 99 backbone amides and the indole sidechain of Trp 59 at the base of the catalytic cleft were observed to give rise to both major and minor resonances. Excluding significantly overlapped and severely broadened resonances, the mean peak volume ratio between the corresponding minor and major peaks was 14% with an rmsd of 2%, indicating that 12% of the protein adopts this minor conformational state. The largest chemical shift differences for the doubled resonances are seen in the 80’s loop (or β4–β5 loop) which extends from the 310 turn at the end of the β4 strand to the start of the final β5 strand (Fig. 3). The resonance doubling extends beyond the 80’s loop into the underlying β sheet, including residues in the contiguous β2 and β3a, and β5 strands. The doubling also extends backwards along the chain into the 310 turn which forms a hydrogen bonded junction with the N-terminus of the central α-helix that bears the Trp 59 residue. The resonance doubling further extends throughout the α-helix and into residues of the 50’s loop (or β3b-α loop) that surrounds the C-terminus of the helix at the far side of the protein.
Fig. 3. Structural distribution of residues exhibiting amide resonance doubling due to slow conformational exchange in FKBP12.

Residues that yield doublings of their amide resonances are indicated in black. Aromatic sidechains to be further discussed are also indicated. Illustration as modified from research originally published [55], the Biochemical Society copyright holder.
Given that the largest differential chemical shifts for these resonance doublings are concentrated around the tip of the β4–β5 loop, the most straightforward interpretation is a slow local conformational transition centered in this region which has structural effects that propagate across much of the protein structure. Strong support for that interpretation is provided by the H87V mutation which suppresses the resonance doublings to below the population level detection limit (~0.2%) [55]. Given the 12% population of the minor state in wild type FKBP12, the H87V substitution shifts the equilibrium of the slow exchange transition by at least 60-fold or by more than 10 kJ/mol. On the other hand, the 1.7 Å resolution crystal structure of the H87V variant [55, 67] reveals strikingly little structural change (0.25 Å Cα rmsd) with respect to the 0.92 Å crystal structure of the wild type protein [68]. This conclusion is reinforced by the high similarity between the 1H-15N 2D correlation spectra for the H87V variant and for the resonances of the major slow exchange state in the wild type protein spectra [55]. Minimal structural changes were observed near the site of mutation (Fig. 4) with the newly introduced Cγ2 of Val 87 being pressed against the phenyl ring of the absolutely conserved Tyr 82 [69]. Although the aromatic ring of Tyr 82 is not significantly displaced by the interaction with the Val 87 sidechain, its crystallographic B-factor is markedly decreased, relative to the other aromatic rings of the protein. The minimal displacement of the Tyr 82 aromatic ring following the H87V substitution presumably reflects the fact that this ring is tightly packed against the sidechain of Ile 56 which, in turn, is packed against the indole ring of Trp 59 at the base of the catalytic cleft. These data are consistent with the structural constraints imposed by the interactions between the Tyr 82 and Val 87 sidechains serving to block the slow conformational transition which gives rise to the set of minor crosspeaks.
Fig. 4. Superposition of region surrounding residue 87 from the 1.70 Å resolution structure of the H87V variant of FKBP12 and the 0.92 Å resolution wild type structure.

The carbon atoms of the H87V variant are indicated in dark gray (blue) while those of the wild type protein (including dual conformers for the His 87 sidechain) are indicated in light gray (green). The Cγ2 atom of Val 87 is 3.7 Å from both the Cγ and Cδ1 atoms of Tyr 82.
A structural mechanism for propagating dynamical effects away from the tip of the β4–β5 loop might utilize the set of sidechain interactions spanning from near the tip of the β4–β5 loop to the indole ring of Trp 59 in the α-helix as illustrated in Fig. 4. A number of groups have observed that a shifting in the position of the Trp 59 sidechain appears to be coupled to a corresponding lateral shift in the α-helix itself [68, 70, 71]. An additional set of predominantly backbone-mediated interactions may also significantly contribute to propagating these dynamical effects from the tip of the β4–β5 loop to the α-helix and beyond. Central to this set of interactions is the junction between the 310 turn at the beginning of the β4–β5 loop and the N-terminus of the central α-helix mentioned above. In addition to van der Waals packing interactions, there is only one direct mainchain-mainchain hydrogen bond spanning this junction (Gly 58 HN – Tyr 80 O). Either one or both sets of these interactions may provide the basis for the spatial selectivity in the propagation of conformational dynamics as evidenced by the distribution of resonance doublings.
The FK1 domains of FKBP51 and FKBP52 do not exhibit an analogous pattern of amide resonance doublings [72]. In contrast, the more similar FKBP12.6 does show doublings of its amide resonances in the β4–β5 loop and in the neighboring segments of the β2, β3a and β5 strands [72]. On the other hand, no resonance doublings were observed for residues within the α-helix or in the other more distal regions of the FKBP12.6 structure. The interactions which enable the conformational coupling to propagate across the FKBP12 molecule are apparently disrupted in FKBP12.6. In this regard, the most obvious difference in sequence between FKBP12 and FKBP12.6 is the substitution of Trp 59 with Phe 59 at the base of the catalytic cleft. Indeed, FKBP12.6 is the only human FKBP domain protein having a phenylalanine at this position [73]. The crystal structure of the W59F variant of FKBP12 does not exhibit the shifted position in the α-helix seen in the wild type protein that has been ascribed, in part, to the steric interaction of the larger indole ring with the aromatic ring of Phe 99 [71]. With regards to potentially altered interactions at the junction between the 310 turn and the N-terminus of the α-helix, it may be noted that the hydrogen bond acceptor residue Tyr 80 of FKBP12 is replaced with Val 80 in FKBP12.6.
The single chemical species identified from mass spectral analysis of FKBP12 and the H87V mutational studies indicate that the resonance doubling of FKBP12 and FKBP12.6 presumably arises from a slow conformational exchange. To determine whether this conformational exchange could be directly detected, a series of zz-exchange experiments [74] were carried out. This experiment monitors the chemical shift of a given nucleus at both the beginning and the end of a specified mixing period. For a two-state transition, two diagonal peaks will arise from magnetization of the 1H nuclei that remain in the same conformational state at both the beginning and the end of the exchange mixing period, while the nuclei that change conformational state during that mixing period will give rise to two off-diagonal crosspeaks. If the rate of conformational exchange is comparable to the 1H longitudinal relaxation rate R1 or faster, the transition rate for a two-state process can be determined by fitting the diagonal and crosspeak intensities as a function of the length of the mixing period. Sufficiently rapid transition rates were only observed at elevated temperatures. During the moderately long collection times required, wild type FKBP12 samples began to precipitate, despite the presence of both dithiothreitol and tris(2-carboxyethyl)phosphine reducing agents, thus necessitating the use of the cysteine-free evolutionarily conservative C22V variant.
As illustrated for the indole sidechain of Trp 59 in a U-2H,15N enriched sample of the C22V variant (Fig. 5A), the conformational transition of this sidechain is sufficiently rapid so as to yield observable off-diagonal crosspeaks at 43°C. When the intensities of these peaks were analyzed as a function of the exchange mixing period, an exchange lifetime of 3.0 s was obtained (Fig. 5B). A conformational transition lifetime measurement of 1.8 s at 48°C implied an activation energy of 70 kJ/mol. Using this activation energy to extrapolate the exchange rate to 25°C yields a predicted conformational transition lifetime of 20 s. This conformational exchange rate is 102-fold slower than the global folding reaction of FKBP12 under similar conditions which occurs with a rate of 4 s−1 at 25°C [75]. A similar analysis of cysteine-free FKBP12.6 yielded an exchange lifetime of ~2.4 s at 43°C [76].
Fig. 5. Kinetics of the slow conformational exchange in the C22V variant of FKBP12 at 43°C.

(A) zz-exchange diagonal and crosspeaks of the Trp 59 indole HNε1 in the major and minor conformational states at a mix time of 2.2 s. (B) Time course for the normalized peak intensities for the AA and BB diagonal peaks and for the AB crosspeak. The modest deviations in the predictions of the AA and BB diagonal peaks for early timepoints likely reflect a weak violation of equal R1 values for all states that is assumed in the model analysis which has a minimal effect upon the derived conformational exchange constant. Illustration from research originally published [55], the Biochemical Society copyright holder.
Protein conformational transitions in this timeframe most commonly arise from either cis-trans isomerization at proline residues or disulfide bond transitions. As no disulfides are present in these FKBP domains, the conformational state for the seven proline residues of FKBP12 were characterized utilizing the dependence of the Cβ and Cγ chemical shifts on the equilibrium of the proline ring pucker distribution which, in turn, depends upon the cis-trans equilibrium of the peptide linkage [77]. Across a large number of proteins of known structure, the difference between the Cβ and Cγ chemical shifts for trans prolines averaged 4.51 (± 1.17) ppm while the corresponding value for cis prolines was 9.64 (±1.27) ppm [78]. Each of the seven proline residues of FKBP12 adopts a trans peptide linkage in both the major and minor conformations, indicating that this slow transition is not the result of prolyl isomerization [70].
The first direct indication of the site within the tip of the β4–β5 loop of FKBP12 which undergoes a slow conformational transition was drawn from the 1H,13C and 15N backbone chemical shifts for the minor state resonances. These chemical shifts are sensitive to the neighboring ϕ and ψ backbone torsion angles and various algorithms have been developed for empirically predicting the backbone conformation from chemical shift values. For the chemical shifts of both the major and minor states across the β4–β5 loop, the TALOS+ algorithm [79] predicted the largest change at Gly 89 with a ϕ torsion angle of +88° in the major slow exchange state (similar to the X-ray structure [68]), while a ϕ torsion angle of −59° was predicted for the minor slow exchange state. These results suggest that the switch from a positive to a negative ϕ angle for Gly 89 constitutes a major aspect of the structural transition underlying the resonance doubling behavior of FKBP12. This observation offers a straightforward rationalization for why the FK1 domains of FKBP51 and FKBP52 do not exhibit a similar resonance doubling behavior. For both domains the homologous residue is Pro 120 which is covalently constrained to a negative ϕ torsion angle.
Since the chemical shift analysis indicated that the minor slow exchange state of FKBP12 has Gly 89 transitioned to a negative ϕ conformation, the G89P variant was generated as a means of enforcing a negative ϕ torsion angle at that position. The proline substitution eliminates peak doubling for all of the amide resonances. The similarity in chemical shift behavior for the G89P variant and the minor slow exchange conformation of the wild type protein strongly indicates a corresponding similarity in structure [70]. The superposition of these two 2D 1H-15N HSQC spectra indicate appreciable differences only in the absence of an amide resonance for the G89P variant and for the Ile 90 crosspeak in which the 15N of the G89P variant is shifted downfield as anticipated from the inductive effects resulting from the sidechain substitution for the preceding residue [80, 81].
The β4–β5 loop conformation in the G89P crystal structure at 1.50 Å resolution [70] differed from wild type FKBP12 primarily at residues 88, 89 and 90, and it closely resembled that reported for the PDB code 1N1A crystal structure of FKBP52 [82] which has a cis-peptide conformation at Pro 120 analogous to the cis-peptide linkage that was observed at Pro 89 in the FKBP12 variant (Fig. 6). If the transition of the Gly 89 peptide linkage to a cis configuration is responsible for the minor slow exchange state of FKBP12, the G89P crystal structure provides an explanation for how the H87V substitution for FKBP12 might suppress this transition. The Cγ2 atom of that valine is 3.7 Å from the Cδ1 atom of the evolutionarily invariant Tyr 82 sidechain in the H87V crystal structure (Fig. 4) [55]. Modeling the His 87 sidechain of the G89P crystal structure as a valine results in the valine Cγ2 atom being only 2.9 Å from the Cδ1 atom of Tyr 82, suggestive of a substantial steric hindrance to such a conformational transition.
Fig. 6. Superposition of region surrounding the Pro 88–Pro 89 peptide bond in the crystal structure of the G89P variant (light gray/green) as compared to the homologous segment from the first FKBP domain of FKBP52 (dark gray/purple).

The structural changes induced by the cis-peptide linkage in the G89P variant yields a backbone conformation that closely follows that of the first FKBP domain of FKBP52 (PDB code 1N1A). The His 87 sidechain is truncated at Cβ to facilitate visualization of the backbone conformations. Illustration as modified from research originally published [70], the Biochemical Society copyright holder.
The prediction that the peptide linkage of Gly 89 adopts a cis conformation in the minor slow exchange state of FKBP12 is exceptional as non-proline cis-peptide linkages occur in protein crystal structures at a frequency of only 0.03% [83]. Cis-peptide linkages result in a short range distance between the sequential Hα atoms. In the more common trans peptide linkage, the sequential Hα atoms are separated by a longer distance around 4.5 Å that is nearly independent of the intervening ψi,ϕi+1 torsion angles [84]. Based on the G89P crystal structure, the Pro 88 Hα would be separated from the two Gly 89 Hα atoms by 2.0 Å and 3.4 Å.
3D 13C-13C-1H NOESY measurements were carried out to determine whether the Hα atoms of Pro 88 and Gly 89 exhibit a strong NOE interaction in the minor conformation. The intensities of the sequential NOE connectivities between the 1Hα resonances of Gly 89 and the 1Hα of Pro 88 for the minor slow exchange state conformation of wild type FKBP12 are more intense than the intraresidue NOE crosspeaks between Pro 88 1Hα and its own 1Hβ resonances [70]. On the other hand, the sequential NOE connectivities between the 1Hα resonances of Gly 89 and the 1Hα of Pro 88 are much weaker than the intraresidue crosspeaks from the 1Hβ resonances for the major slow exchange state conformation. A similar pattern of NOE crosspeak intensities were observed for transfer in the reverse direction from the 1Hα of Pro 88 to the 1Hα resonances of Gly 89 for both the major and minor slow exchange states. These results indicate that the peptide linkage at Gly 89 in the minor slow exchange state of FKBP12 is in a cis configuration.
Although the 1N1A crystal structure of FKBP52 mentioned above adopts a cis-peptide linkage at Pro 120 as does the 4LAV [85] crystal structure, it should be noted that the 1Q1C [86] and 4LAW [85] crystal structures of FKBP52 have a trans peptide linkage at this residue. [78]. To determine the peptide linkage conformation of this residue in solution, a 2D CT-HSQC spectrum resolved the 1Hδ-13Cδ resonances for all seven prolines of FKBP52. With the proline 1Hδ-13Cδ resonances of FKBP52 exhibiting an average S/N ratio above 50, no evidence was detected of peak doublings arising from prolyl isomerization. The large 10.0 ppm difference in Cβ and Cγ chemical shifts for Pro 120 indicates a cis-peptide linkage for that residue in solution [70].
CONFORMATIONAL EXCHANGE LINEBROADENING IN FKBP12
Conformational exchange linebroadening in 13C or 15N relaxation experiments arises from transitions between states that differ in their 13C or 15N chemical shift values, respectively. The magnitude of the linebroadening effect depends upon the difference in the resonance frequencies, the relative population of the states and the rate of the conformational transition. As germane to the FKBP domains, for dynamical processes having rates near the fast exchange limit, the R2 relaxation data or relaxation dispersion data used to measure the exchange linebroadening effect are often insufficient to separately quantify the values for the underlying chemical shift differences, populations and transition rate(s).
13C relaxation dispersion measurements on FKBP12 identified 12 exchange-broadened methyl resonances which the authors interpreted as representing a single global conformational exchange process with time constant of ~130 µs [87]. Subsequently, Brath and Akke [88] carried out an analogous 15N relaxation dispersion study to characterize conformational dynamics in the backbone of FKBP12. They identified 23 amides that exhibited conformational exchange which when combined with their earlier 13C measurements were fitted to a single global time constant of 120 µs. Brath and Akke [88] as well as Sapienza, Mauldin and Lee [89] observed that the binding of FK506 to FKBP12 quenches the conformational exchange broadening of the backbone resonances. Noting that many of the exchange-broadened residues line the catalytic cleft, it was proposed this apparently collective conformational transition directly participates in the catalytic process for prolyl isomerization [87, 88].
Subsequent 15N relaxation measurements on the H87V variant indicated additional complexity to the conformational exchange linebroadening dynamics of FKBP12. The R1 and NOE values for the H87V variant are quite similar to those for the major species of the wild type protein [55], indicating that both proteins exhibit a similar magnitude of conformational dynamics in the timeframe more rapid than global molecular tumbling. However, the R2 values for the H87V variant differ markedly from those of the major slow exchange state of wild type FKBP12. The conformational broadening for the residues in the 80’s loop (β4–β5 loop) was completely suppressed, while the broadened resonances in the 40’s loop (β3a–β3b loop) were seemingly unaffected. For residues in the 50’s loop (β3b-α loop), the conformational broadening contribution is ~35% smaller in the H87V variant as compared to the wild type protein.
A similarly selective effect was observed when valine was introduced at residue 44. The 1H-15N 2D correlation spectrum of the K44V variant closely follows the resonances for the wild type protein, indicating how precisely the structure is preserved upon this mutation. Conformational exchange linebroadening for residues 39, 42 and 44 in the 40’s loop is reduced 3-fold by the K44V substitution, while the other exchange broadened residues of FKBP12 appear to be unaffected by this mutation [55]. Interestingly, none of the resonance doublings exhibited by the wild type protein were affected by the K44V mutation, including that for the immediately adjacent Ser 38.
The H87V substitution does not alter the peptidyl proline isomerase activity of FKBP12 [67] thus implying that the exchange linebroadening behavior that is observed for much of the active site in the wild type protein does not correspond to a catalytically relevant conformational transition. The H87V and K44V mutations significantly decouple the conformational broadening dynamics among three different regions of the protein. The implied localization of these conformational exchange linebroadening processes clearly indicates that the apparent global character of the motions in this timeframe proposed in previous relaxation studies [87–89] is coincidental.
Many of the residues for which the H87V substitution causes a suppression of exchange broadening also exhibit resonance doublings that are suppressed in the H87V variant. In addition, the largest of these exchange linebroadening effects also occur near the tip of the long β4–β5 loop. This correspondence is intriguing, given that the timeframe for the slow conformational exchange differs by five orders of magnitude from that for the linebroadening transition. On the other hand, the structural details must clearly differ between these two conformational transitions as indicated by the fact that the differences in 15N chemical shift between peaks from the major and minor slow exchange states do not closely correlate with the magnitude of the magnetic field-dependent 15N R2 linebroadening as would be expected if the minor state conformations for both the resonance doubling transition and linebroadening transition were structurally similar [70].
Particularly informative is the fact that for the residues which exhibit exchange linebroadening and resonance doubling that are both suppressed by the H87V substitution, only the major slow exchange resonance exhibits conformational linebroadening [55]. Since the minor slow exchange state corresponds to the cis-peptide conformation for Gly 89, this peptide isomerization must either suppress the conformational transition that underlies the exchange broadening, or alternatively, it must markedly accelerate that transition so as to shift the rate into the fast exchange limit.
The G89A mutation was introduced so as to shift the conformational equilibrium toward a negative ϕ value for this residue. The 2D 1H,15N HSQC spectrum of the G89A variant yielded a pattern of amide resonance doublings with chemical shift values quite similar to that observed for the wild type protein [55]. On the other hand, the set of crosspeaks that correspond to the two distinct slow exchange conformations have markedly different relative intensities for these two proteins. The minor slow exchange state represents only 12% of the total population in the wild type spectrum, while the analogous set of crosspeaks yield 67% of the total protein signal for the G89A variant. This 15-fold change in relative population, induced by the alanine substitution, corresponds to a 6.7 kJ/mol difference in the relative stability of the two slow exchange conformations.
Interestingly, the G89A substitution also markedly enhances the conformational linebroadening for resonances of the 80’s loop which arise from the trans peptide state of residue 89. Indeed for many of the residues near the tip of this loop the crosspeak intensities are too weak to allow for satisfactory relaxation measurements or even too weak for detection. On the other hand, for the resonances corresponding to the cis peptide conformation of the Ala 89 residue, no conformational exchange linebroadening was observed, analogous to the results obtained for wild type FKBP12 [70].
These dynamics data are consistent with a statistically coupled pair of transitions (Fig. 7). First is an exchange linebroadening transition (τ ~100 µs) in which ϕ89 switches from positive to a negative value along with a corresponding shift in ψ88 torsion angle. This second state then undergoes a much slower (τ ~20 s) trans-to-cis peptide transition for the Gly linkage. The fourth conformational state of this cycle (cis peptide with a positive ϕ89) is absent to reflect the fact that no exchange linebroadening is observed for the cis peptide state suggesting that the faster transition for ϕ89 is suppressed in this conformation so that the cis-trans peptide transition for Gly 89 predominantly occurs only when the ϕ89 torsion angle is negative.
Fig. 7. Modeling the conformational transitions at the Pro 88 – Gly 89 peptide bond.

At the lower left is displayed the Pro 88 – Gly 89 crystal structure conformation for wild type FKBP12 (PDB code 2PPN [68]). At the top is the backbone for this dipeptide derived from the FKBP52 structure (PDB code 4LAW [85]) which illustrates the result of a concerted (ψ88,ϕ89) transition. At the lower right, the corresponding Pro-Gly model conformation derived from the crystal structure for the G89P variant of FKBP12 (PDB code 4N19 [70]) illustrates the cis state of this peptide linkage with a negative ϕ89 torsion angle value.
Since the magnitude of the exchange linebroadening effect for a given residue depends upon the difference in chemical shift for the exchanging conformations, structure-based predictions of chemical shifts provides a means of testing the potential validity of models for those exchanging conformations. Complementary to the chemical shiftbased predictions of protein backbone conformation (e.g., TALOS [79]) discussed above, empirical library-based algorithms have been introduced for predicting backbone chemical shifts from a given protein structure. One such algorithm SPARTA+ [90] was used to calculate backbone chemical shifts from models of the β4–β5 loop of FKBP12 mimicking the proposed transient conformation having a negative ϕ89 torsion angle and a trans peptide linkage. As noted above, two of the published crystal structures of FKBP52 exhibit the analogous Pro 120 peptide linkage in a trans configuration FKBP52 (PDB codes 1Q1C [86] and 4LAW molecules A and B [85]). The ϕ angle for this residue is covalently forced to be negative. To enable a differential SPARTA+ analysis with respect to the FKBP12 crystal structure (PDB code 2PPN [68]), the sidechains of the β4–β5 loop for these FKBP52 structures were computationally mutated to a sequence mimicking FKBP12 as illustrated by the dipeptide model above (Fig. 7).
The strongest correlation between the predicted differences in 15N chemical shift and the experimentally observed 15N linebroadening effects was obtained by averaging the predictions over the three FKBP52-derived structures (Fig. 8) [55]. Given the challenges that typically confound quantitative structure-based predictions of conformational exchange linebroadening effects, the degree of correlation that was obtained between the predicted and observed linebroadening for the 80’s loop of FKBP12 strongly suggests that the observed exchange linebroadening does arise from Gly 89 transiently adopting a negative ϕ torsion angle when that residue is in a trans peptide configuration.
Fig. 8. Correlation between the 15N conformational exchange linebroadening (Δex) in the 80’s loop of FKBP12 (black) and the differential chemical shift predictions derived from two crystal forms of FKBP52 (gray).

Model-free [104] analysis of the R1, R2 and NOE 15N relaxation measurements was used to estimate conformational exchange linebroadening [55]. The differences in 15 N chemical shift were predicted with SPARTA+ [90] applied to FKBP12 (PDB code 2PPN [68]) and the first FKBP domain of FKBP52 (PDB code 1Q1C [86] and non-equivalent monomers of PDB code 4LAW [85]). An asterisk indicates no statistically significant conformational exchange linebroadening (< ~0.4 Hz). The amide resonances for both Ala 84 and Gly 89 are severely broadened due to rapid hydrogen exchange [55], precluding a reliable determination of conformational exchange linebroadening, and the predicted chemical shift differences for these two residues are indicated as open bars. Illustration reproduced from [70] Illustration from research originally published [70], the Biochemical Society copyright holder.
CONFORMATIONAL EXCHANGE LINEBROADENING IN FKBP51 AND FKBP52
With respect to motions in the µs-ms timeframe, the FK1 domains of FKBP51 and FKBP52 exhibit strikingly different patterns of exchange linebroadening despite their strong sequence homology. As evidenced by the occurrence of elevated R2 values, only one region of the FK1 domain of FKBP52 exhibits a small amount of the resonance linebroadening from this timescale of motion (Fig. 9). The residues most affected lie within the β3a strand and extend up to the start of the β3α-β3b loop. It should be noted that such linebroadening transitions within the strands of a β-sheet are relatively uncommon. In contrast, strongly elevated R2 values are observed for residues of FKBP51 in the β3α-β3b loop and for many of the residues throughout the long β4–β5 loop (Fig. 9). Residues Ser 70, Arg 73 and Glu 75 in the β3α-β3b loop of FKBP51 exhibit the large linebroadening effect that are closely similar to those observed for the homologous residues in FKBP12 [55].
Fig. 9. 15 N Transverse relaxation measurements for the backbone amide resonances in the FK1 domain of FKBP52 and FKBP51 at 25°C.

Transverse (R2) relaxation rates at 600 (gray) and 900 (black) MHz 1H are shown for FKBP52 (upper panel) and FKBP51 (lower panel). In addition to proline residues, relaxation data is not reported for overlapped resonances and for the severely broadened resonances. Illustration as modified from research originally published [72], the Biochemical Society copyright holder.
The 15N R2 relaxation values for residues in the β3a strand, the β3α-β3b loop, and in the β4–β5 loop of FKBP51 exhibit magnetic field-dependent increases in conformational linebroadening that are approximately proportional to the square of the magnetic field. This implies that the conformational transition rate(s) is substantially faster than the strength of the spinlock field used in the relaxation experiments (1245 Hz at 600 MHz and 1085 Hz at 900 MHz). A similar frequency range for the conformational exchange linebroadening of FKBP12 has been reported [87–89].
Following the mutagenesis studies on steroid receptor transcription activity by Riggs et al. [36], the introduction of the FKBP52-like L119P mutation at the tip of the β4–β5 loop in FKBP51 completely suppressed the exchange linebroadening in this loop while partially suppressing the linebroadening in the neighboring β2 and β3a strands (Fig. 10). Introducing the complementary mutation P119L into FKBP52 induces exchange linebroadening dynamics into this loop, although the magnitude of the effects was ~5-fold smaller than that observed for FKBP51. NMR relaxation analysis of the FKBP51-like P119L, P124S double mutant of FKBP52 yielded a linebroadening effect in the β4–β5 loop that was 60% of that observed for FKBP51. For both the P119L and the P119L,P124S variants of FKBP52, the pattern of differential linebroadening for residues in the β4–β5 loop was quite similar to that for FKBP51. Since the magnitude of the exchange linebroadening depends upon the difference in 15N chemical shift (hence backbone conformation) for the exchanging states as well as their populations and rate of exchange, a proportionality in the linebroadening effects across the residues of the loop strongly indicates that a similar conformational transition is being monitored for each mutant with a differing rate or population applying in each case. The similarity between this pattern of mutational alteration in the conformational dynamics in the β4–β5 loop and the corresponding pattern of transcriptional activity levels for this same set of mutations is striking. Regarding the issue of potential physiological relevance for this conformational transition, it should be noted that only residues 119 and 124 differ between FKBP51 and FKBP52 within the long β4–β5 loop sequence extending from Cys 107 to Leu 128. In contrast, there is only a 63% sequence identity for the rest of the 120 residue FK1 domains analyzed in these studies.
Fig. 10. Differential 15 N transverse relaxation measurements for the wild type and L119P variant of FKBP51 at 25°C.

The differential transverse relaxation rates at 600 MHz 1H are shown in the upper panel, while those for 900 MHz 1H are shown in the lower panel. The data for the two fields are plotted on the same vertical scale to illustrate the approximate 2.25-fold increase for the 900 MHz data indicative of conformational transitions occurring near the fast exchange time limit. As a result, the ΔR2 values for residues 117, 119 and 122 at 900 MHz are truncated. At each field, the median R2 values for the two data sets are scaled to correct for small variations in the global molecular correlation times. Outside of the regions exhibiting significant differential linebroadening (i.e., residues 57–77 and 108–128), the rmsd for the ΔR2 values were 0.15 and 0.18 s−1 for 600 MHz and 900 MHz, respectively, corresponding to 1.5% of the median R2 values in each case. The ΔR2 value for Leu 119 in the wild type protein is given relative to the median R2 value and at 900 MHz this ΔR2 value is too large for reliable quantitation (gray). Due to decreased statistical reliability for the more severely attenuated resonances, the residues in which the R2 value is > 18 s−1 for both wild type and the L119P variant were excluded (Ser 70, Arg 73 and Glu 75). Illustration as modified from research originally published [72], the Biochemical Society copyright holder.
Although introduction of the P119L and P124S substitutions into FKBP52 resulted in a pattern of conformational exchange linebroadening quite similar to that observed for wild type FKBP51, there was no corresponding enhancement of the linebroadening effects within the β2 and β3a strands as had been observed in the comparison between the wild type and L119P variant of FKBP51. These results suggest that linebroadening observed within the β2 and β3a strands for both wild type FKBP51 and FKBP52 arise from a distinct conformational transition which exhibits a different degree of interaction with the conformational transition centered in the β4–β5 loop for the variants of these two proteins. In this regard, it was noted [72] that the largest differential linebroadening effects within the β2 and β3a strands were centered around the bifurcated hydrogen bond formed between the amides of Phe 67 and Asp 68 of the β3a strand with the carbonyl oxygen of Gly 59 in the β2 strand, disrupting the canonical antiparallel hydrogen bonding pattern (Fig. 11). It was proposed that the rearrangement of this bifurcated interaction could serve as a component of the conformational transition underlying the linebroadening behavior [72].
Fig. 11. Structural distribution of residues in the β2 and β3a strands of FKBP51 that exhibit elevated R2 values.

Residues for which the 15N R2 value decreases by more than 0.5 Hz at 900 MHz 1H in the L119P variant are indicated in dark gray (purple). There are no other differences in R2 greater than 0.5 Hz outside of the β4–β5 loop. A kink in the β3a strand occurs at residues Phe 67 and Asp 68 where the amide hydrogen of Asp 68 is slightly too far from the carbonyl oxygen of Gly 59 to form a canonical antiparallel β sheet hydrogen bonding interaction. This kink occurs at the site of direct contact with the tip of the β4–β5 loop as indicated by Lys 121. Illustration as modified from research originally published [72], the Biochemical Society copyright holder.
Subsequently, the supposition of such a conformational transition was strongly vindicated by the recent report of the crystal structure for an altered conformation of FKBP51 when bound to a selective inhibitor [91]. Previous studies of FKBP12 had demonstrated that the F36V mutation introduces a hydrophobic pocket along the side of the packing interface between the β4–β5 loop and the underlying β3a strand which provided a structural basis for designing selective inhibitors [92, 93]. Hausch and colleagues [91] introduced the analogous F67V mutation into FKBP51 and used the earlier reported selective F36V-FKBP12 inhibitor Shield1 [94] as the initial scaffold for developing a FKBP51-selective inhibitor. In contrast to the earlier FKBP12 studies, they found that for their so-called iFit variants of the Shield1 ligand were bound to a conformation of wild type FKBP51 in which the sidechain of Phe 67 is flipped out toward the solvent phase from its typical position packed underneath the tip of the β4–β5 loop (Fig. 12). In this alternate conformation, a canonical antiparallel β-sheet hydrogen bonding pattern is established that extends from the carbonyl oxygen of Tyr 57 to the amide of Leu 61 in the β2 strand and from the carbonyl oxygen of Lys 66 to the amide of Ser 70 in the β3a strand.
Fig. 12. Structural distribution of residues in the β2 and β3a strands of FKBP51 that exhibit large changes in backbone dihedral angles upon binding either the selective inhibitor iFit4 or the nonselective inhibitor iFit1.

As displayed upon the crystal structure of iFit4-inhibited FKBP51 (PDB code 4TW7 [91]), four of the residues in the β2 and β3a strands, indicated in dark gray (purple) exhibit (Δψi−1, Δϕi) dihedral angle values greater than 35° in both the iFit1 and iFit4-inhibited structures, relative to the reference FK506-inhibited structure [95]. The sidechain of Lys 65 was truncated for clarity.
Differences in the backbone ϕ and ψ dihedral angles of the iFit-inhibited FKBP51 crystal structures provide an initial indication that a transition involving a similar transient conformational state of the unliganded FKBP51 domain may be responsible for the observed exchange linebroadening in the β2 and β3a strands. The 15N exchange linebroadening effect depends upon the difference in 15N chemical shift for the exchanging conformational states which, in turn, strongly depends upon the adjacent ψi−1 and ϕi dihedral angles. The sum of the absolute differences for these two dihedral angles were calculated for all of the residues in the selective iFit4-inhibited (PDB code 4TW7 [91]) and the structurally similar nonselective iFit1-inhibited (PDB code 4TW6 [91]) as compared to the reference FK506-inhibited (PDB code 3O5R [95]) crystal structures. With the exception of the substantially disordered Arg 73, the only residues having (ψi−1,ϕi) differences greater than 35° for both the iFit1 and iFit4 structures occur in the β2 and β3a strands (Fig. 12) immediately surrounding the site of the bifurcated hydrogen bonding in the reference structure. Note that the amide of the highlighted Lys 60 is covalently conjugated to the Gly 59 carbonyl oxygen which is the hydrogen bond acceptor at the site of bifurcation.
Hausch and colleagues [91] have interpreted their crystal structures of the iFit inhibitor-bound FK1 domain of FKBP51 in terms of a process of induced fit. However, the similar structural distribution of the residues exhibiting 15N exchange linebroadening in the β2 and β3a strands suggests that a closely related conformational transition is significantly populated in the unliganded FK1 domain, a result that is more readily identified with a population selection mechanism of binding interaction.
Various lines of evidence indicate that the conformational transition which gives rise to the extensive exchange linebroadening in the β4–β5 loop of FKBP51 is structurally distinct from the transition that gives rise to the iFit-inhibited crystal structures. None of the residues in this long loop exhibit (Δψi−1, Δ ϕi) values above 30° in both the iFit1 and iFit4 structures, despite the fact that the tip of the β4–β5 loop exhibits substantial conformational variability among the 24 crystal structures of the unliganded FK1 domain with resolution limits of 2.0 Å or better that have been reported to date [95–97]. On the other hand, the conformational variability illustrated among these 24 crystal structures would also seem unlikely to provide a useful model for the comparatively slow exchange linebroadening transition of the β4–β5 loop, in large part due to the fact that most of that conformational variability can be accounted for by a single mode of flexibility involving the concerted rotations of the ψ angle of Ser 118 and the ϕ angle of Lys 121 [76] which does not appear to have a substantial kinetic barrier to rationalize the slow dynamics of the linebroadening transition. Furthermore, while the β4–β5 loop of FKBP52 exhibits no conformational exchange linebroadening, Hausch and colleagues did find that the FK1 domain of FKBP52 undergoes a qualitatively similar rotation of the Phe 67 sidechain out toward the solvent phase upon binding a structural related iFit-FL ligand (PDB code 4TW8 [91]).
Given the implication that the interactions between the β3a strand and the β4–β5 loop are crucial to steroid receptor function and that the conformational dynamics effects of mutations at residues 119 and 124 in FKBP51 and FKBP52 exhibit an intriguing parallel with the effects of these mutations on steroid receptor transcriptional activity, it seems warranted to consider whether physically plausible conformational transitions might provide a useful model for interpreting the differing roles of FKBP51 and FKBP52 in steroid receptor regulation. In further effort to characterize the structural basis of the exchange linebroadening in the β4–β5 loop in FKBP51, it should be noted that the pattern of conformational linebroadening observed for this loop in FKBP12 is qualitatively similar. However, as discussed above, a large transition in the ϕ torsion angle of Gly 89 lies at the center of the exchange linebroadening dynamics of the β4–β5 loop in FKBP12. Since the analogous ϕ angle of Pro 120 in FKBP51 is covalently constrained, the linebroadening transition in that β4–β5 loop must be mechanistically different.
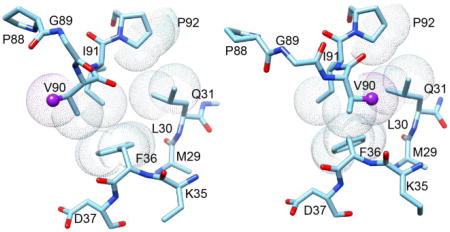
One plausible qualitative model for the relevant structural transition in the β4–β5 loop of FKBP51 can be drawn from the P21 crystal form of FKBP12.6 in the unliganded state (PDB code 4IQ2 [76]). In contrast to the P3121 crystal form of unliganded FKBP12.6 (PDB code 4IQC [76]) and the more than thirty crystal structures of FKBP12, the P21 crystal form of FKBP12.6 exhibits a substantial rearrangement at the tip of the β4–β5 loop. Transitions in the ψ89 and ϕ90 torsion angles of 130° and 60°, respectively, cause the peptide unit linking Gly 89 and Val 90 to be flipped. This transition results in the sidechain of Val 90 switching from an orientation pointed toward the catalytic cleft to one in which that sidechain is pointed in nearly the opposite direction (Fig. 13).
Fig. 13. Superposition of region surrounding the Gly 89–Val 90 peptide bond from the two crystal forms of FKBP12.6.

This region of the P3121 crystal form structure of cysteine-free FKBP12.6 is illustrated on the left. The Cγ of Val 90 (sphere) is pointed toward the catalytic cleft as is typical of analogous crystal structures for FKBP12. On the right is displayed this region from the P21 crystal form structure in which the Val 90 Cγ is pointed away from the catalytic cleft. The difference in conformation primarily arises from the peptide linkage between Gly 89 and Val 90 being flipped in the P21 crystal form relative to its position in the P3121 crystal form. Van der Waals surfaces are illustrated for the evolutionarily conserved hydrophobic sidechain interactions between the β2 and β3a strands and the tip of the β4–β5 loop.
Lys 121 is one of the residues most characteristic of the FKBP51/52 family of FK1 domains as is the homologous Ile/Val 90 for the FKBP12/12.6 family of FKBP domains. Due to the cis-peptide linkage of Pro 120 in FKBP51, the neighboring backbone torsion angles significantly differ from the homologous positions in the FKBP12.6 structures so as to preclude a direct structural equivalence to the two FKBP12.6 conformations. However, a qualitatively similar transition which reorients the Lys 121 sidechain between either side of the β3a strand - β4–β5 loop interface in FKBP51, while FKBP52 adopts only one such orientation, could provide a dynamical basis for their differential interactions within the steroid receptor complex.
CONFORMATIONAL DYNAMICS OF FKBP DOMAINS BEYOND THE FAST EXCHANGE LIMIT
15N relaxation measurements on FKBP51 and FKBP52 [72] and to a lesser extent FKBP12 [55] indicate a small degree of internal motion in the ps-ns timeframe for the backbone residues near the tip of the β4–β5 loop, evidenced by modest decreases in both the longitudinal relaxation rate R1 and the heteronuclear NOE value. Unlike FKBP12, FKBP51 and FKBP52 also exhibit a small degree of internal motion in this timeframe for residues in the β3a–β3b loop (Fig. 14), a region which also shows substantial conformational exchange linebroadening in FKBP51 and FKBP12 but not in FKBP52 as discussed above.
Fig. 14. Structural distribution for residues of the FK1 domain of FKBP52 that exhibit conformational dynamics in either the ps-ns or µs-ms timeframe.

The mainchain conformational schematic of the FK1 domain as viewed from the back side of the β sheet. Discounting the termini, residues that exhibit order parameter values of S2 < 0.78 are indicated in black, while the residues between Asp 63 and Ser 70 exhibiting conformational exchange broadening above 0.5 Hz at 600 MHz 1H and above 1.0 Hz at 900 MHz 1H are indicated in gray. Illustration as modified from research originally published [72], the Biochemical Society copyright holder.
By far the most extensive internal dynamics in the ps-ns timeframe for both FKBP51 and FKBP52 occurs for the β1–β2 loop which corresponds to the outer strand of a topological crossing of two loops that connect β-strands of the central sheet (Fig. 14). No comparable dynamics are apparent for the qualitatively similar backbone geometry of FKBP12. Earlier crystal structure studies of FKBP12 [98] have called attention to the rarity of this type of topology for the linking of strands in antiparallel β-sheets [99]. In the structurally related FKBP13, this loop crossing is stabilized by a disulfide bridge linking the two loops [100].
A closely integrated analysis of crystal structure studies and NMR data have provided further insight into conformational transitions of the FKBP domains that are too rapid to yield exchange linebroadening effects, particularly for a set of interactions surrounding the catalytic cleft. The P3121 crystal form of unliganded FKBP12.6 has two nonequivalent monomers in the asymmetric unit which exhibit a backbone rmsd of only 0.35 Å [76]. In molecule B the aromatic ring of Phe 59 at the base of the catalytic cleft superimposes upon that seen in the rapamycin-inhibited FKBP12.6 [101] and is also closely similar to the orientation of the Trp 59 indole ring in the large number of X-ray structures reported for the wild type FKBP12 protein. In contrast, the electron density map of molecule A clearly indicates a perpendicular orientation of the Phe 59 ring (Fig. 15). This reorientation of the Phe 59 ring leaves the rest of the catalytic cleft largely unperturbed, as illustrated for the other aromatic rings which line the walls of this cavity.
Fig. 15. Superposition of region surrounding the aromatic ring of Phe 59 from the two non-equivalent monomers in the 1.90 Å resolution structure of the P3121 crystal form for the cysteine-free variant of FKBP12.6.

The heavy atoms from molecules A and B are superimposed with the electron density grid (2mFo-DFc at a contour level of 0.0114 e/Å3 = 1σ) for molecule A also illustrated, The aromatic ring of Phe 59 from this molecule is oriented perpendicular to that from molecule B. In the latter case, the plane of the ring forms the base of the catalytic cleft as is seen in previously reported crystal structures of FKBP proteins. Illustration as modified from [76]. Reproduced with permission of the International Union of Crystallography.
The highly similar backbone conformations for the nonequivalent monomers and the absence of any interactions with the neighboring lattice molecule that directly involve this phenyl ring suggest that lattice interactions do not contribute significantly to the difference in free energy between the parallel and perpendicular orientation of the Phe 59 ring. This, in turn, suggests only a modest difference in free energy for the two Phe 59 ring orientations in solution.
Among the 22 FKBP domains in the human genome, only FKBP12.6 contains a phenylalanine residue at the base of the catalytic site [102]. FKBP12 and six other FKBP domains have a tryptophan residue at this position, while the other 14 FKBP domains contain a leucine, isoleucine or methionine at this site. The plasticity of the Phe 59 phenyl ring orientation in FKBP12.6 offers the potential for an energetically accessible catalytic site stereochemistry that could enable the design of binding selectivity with respect to the other FKBP domains.
Crystallographic evidence for a larger scale transition in the catalytic site is provided by the structure for the G89P variant of FKBP12. Despite the 21 Å between the Cα atoms of residues 59 and 89 and seeming absence of appreciable structural changes for the residues lying between these two sites, the indole ring of Trp 59 is oriented perpendicular to the conformation observed in each of the more than thirty crystal structures of wild type FKBP12. In contrast to the crystal structure of unliganded FKBP12.6, the aromatic rings of Phe 48 and Phe 99 in the G89P structure appear to move slightly inward toward the reoriented indole ring, relative to the orientations observed in the wild type FKBP12 crystals. A small cavity lies beneath the indole ring in the wild type structure. Rotation of the indole ring leaves the sidechains surrounding this cavity are essentially unperturbed [70].
This perpendicular reorientation of the Trp 59 aromatic ring in the G89P crystal structure is strikingly similar to that recently reported for the E60Q variant of FKBP12 (PDB code 2PPP [68]). In that earlier study, the indole ring reorientation was proposed to be linked to disrupting the interactions between the residue 60 sidechain and the backbone atoms of the 50’s loop. However, those interactions remain largely intact in the G89P structure. These observations on the G89P crystal structure stimulated a more detailed analysis of the degree to which the reorientation of the Trp 59 indole ring occurs in wild type FKBP12.
The 3D 13C-15N-1H NOESY spectrum on U-13C,15N labeled FKBP12 revealed substantial crosspeaks from the indole HNε1 of Trp 59 to both methyl groups for Val 24 and Val 63 as well as to the Cγ1 methyl of Val 101. This observation was surprising given the fact that these methyls lie ~7 Å from the indole HNε1 in the crystal structure of the wild type protein [70]. To more accurately quantify these NOE interactions, a sample of FKBP12 was prepared with 13C enrichment for the methyl groups of valine, leucine and isoleucine residues with uniform 15N and perdeuteration at all other carbon bound sites as previously described by Kay and colleagues [103]. This labeling pattern largely eliminates the effects of dipolar relaxation interactions involving more than two nonequivalent protons (i.e., spin diffusion) so that effective interproton distances can be estimated with reasonable accuracy. In turn, these data can provide a useful test for whether the observed NOE cross relaxation rates can be robustly explained in terms of an admixture of the two crystallographically observed perpendicular orientations of the Trp 59 indole ring. Each crystal structure provides a set of interproton distances between the Trp 59 HNε1 and various methyl groups. Generalized order parameter calculations [104] were applied to the two crystal structures to predict the relative NOE volumes for each of these methyl - indole HNε1 interactions. For an admixture of 80% and 20% for the wild type and the G89P conformations, respectively, the observed NOE volumes were fitted with a correlation coefficient r of 0.976 (Fig. 16).
Fig. 16. Predicted vs. observed NOE volumes for the methyl crosspeaks to Trp 59 HNε1.

The experimental NOE volumes were normalized to the maximum value (Val 101 Cγ2) and to the volumes of the corresponding 1H-13C crosspeaks in the 2D HSQC spectrum to compensate for relaxation and differential enrichment effects. Generalized order parameter calculations were performed on the crystal structures of the wild type protein and G89P variants and then weighted at 80% and 20%, respectively. Illustration reproduced from research originally published [70], the Biochemical Society copyright holder.
Given that both indole ring orientations are significantly occupied for wild type FKBP12 in solution, the crystal structure of the G89P variant was re-examined. The 1.50 Å resolution dataset initially used for solving the crystal structure did not provide clear evidence for multiple conformations of the Trp 59 indole ring. To gain further data regarding the potential conformational heterogeneity in the catalytic cleft, a more extensive 1.20 Å resolution dataset was collected. During the later stages of refinement, dual conformers were allowed for both Trp 59 and Glu 60. A second conformer was identified for each of these residues with occupancy levels of 0.29 and 0.34, respectively. Looking down along the plane of the Trp 59 ring in its the major conformer state, the electron density for a perpendicular orientation of the indole ring is readily apparent (Fig. 17). In the minor conformational state of Glu 60, the mainchain carbonyl oxygen is shifted toward the 50’s loop into the position seen in the wild type FKBP12 crystal structure.
Fig. 17. Electron density omit map for Gly 58, Trp 59 and Glu 60 in the active site of the G89P variant of FKBP12 at 1.20 Å resolution.

Viewed along the plane of the indole ring for the major conformer (occupancy of 0.71), electron density for the perpendicular orientation of the minor conformer is readily apparent. The carbonyl oxygen in the minor conformer of Glu 60 (occupancy of 0.34) is shifted away from the canonical α-helical hydrogen bonding geometry seen in the major conformer and to a position close to that observed in the wild type FKBP12 structure [68]. The contour level for the electron density grid is 0.1261 e/Å3 = 2σ. Illustration as modified from research originally published [70], the Biochemical Society copyright holder.
Such a shift of the backbone in the second turn of the central α-helix, centered around Glu 60, was first observed in the crystal structure of the W59F variant of FKBP12 as compared to the wild type structure [71]. This helix is kinked in the wild type protein and the smaller phenylalanine sidechain at the base of the catalytic cleft enables this segment of the helix to shift so as to form canonical hydrogen bonding geometry with the following turn of the helix. Subsequently, Saven and colleagues [68] carried out a 0.92 Å resolution crystallographic analysis of wild type FKBP12 to study the highly conserved solvent inaccessible water molecule which mediates the interaction of the Glu 60 carboxylate with the backbone of the 50’s loop. The E60Q structure [68] not only has the flipped indole ring as discussed above, the peptide unit that links Lys 52 and Gln 53 is flipped with a corresponding change in the coordination interactions for the buried water molecule. In addition, the helical backbone is shifted so as to yield a canonical hydrogen bonding geometry similar to that which was subsequently observed for the G89P variant (Fig. 18) [70].
Fig. 18. Superposition of the wild type (dark gray/purple) and G89P (light gray/green) crystal structures.

The transition of the Glu 60 χ1 rotamer from gauche- to trans facilitates the formation of canonical hydrogen bonding geometry between the Glu 60 O and the Ala 64 HN as well as the reorientation of the indole ring of Trp59. Illustration as modified from [105], Elsevier copyright holder.
In the crystal structure of wild type FKBP12, Glu 60 adopts a gauche− χ1 sidechain torsion angle, while in contrast, the G89P variant exhibits a trans χ1 torsion angle for this residue. This sidechain transition allows the backbone atoms of Trp 59 and Glu 60 to shift toward the active site indole ring without disrupting the hydrogen bonding interactions with the 50’s loop. Contrary to the analysis drawn from the E60Q crystal study [68], both the wild type and G89P structures indicate a similar pattern of interactions among the Glu 60 sidechain, the backbone of the 50’s loop and the buried water molecule. In comparing to the E60Q structure, these results suggest that disruption of these interactions are sufficient but not necessary to enable the shift of the backbone of Trp 59 and Glu 60 into a canonical α-helical hydrogen bonding geometry and enable the reorientation of the indole ring.
The readily accessible perpendicular reorientation of the Trp 59 indole ring in FKBP12 appears to be allosterically coupled to interactions with the backbone of the 50’s loop which, in turn, exhibits an appreciable degree of conformational plasticity. These interactions raise the possibility that intermolecular binding interactions at the 50’s loop might modulate the conformational geometry within the catalytic cleft and vice versa. These observations bring two questions to mind: do similar transitions occur to a significant degree in other FKBP domains and can the presumed allosteric interaction between the indole ring and the 50’s loop be analyzed in more quantitative detail.
Carrying out analogous 3D NOESY experiments on selective 13C methyl-labeled FK1 domains of FKBP51 and FKBP52 provide no evidence of a perpendicular reorientation of the homologous Trp 90 ring at a population sensitivity level of ~0.5% [105]. The absence of detectable reorientation of the indole ring in these proteins was anticipated, primarily due to the fact that the Glu 60 of FKBP12 is replaced by the shorter aspartate sidechain which forms similar interactions with the backbone of the 50’s loop, and it is unclear how these interactions could be preserved while undergoing the shift in the α-helical backbone to allow for an analogous reorientation of the tryptophan ring.
An independent line of evidence consistent with the Trp 90 ring of FKBP51 and FKBP52 being resistant to a reorientation transition can be drawn from the highly anomalous 1H chemical shift of this indole HNε1. Craescu and colleagues first correctly identified this resonance at 5.35 ppm in FKBP52, shifted upfield by ~4.8 ppm compared to standard reference values, which they ascribed to the ring current shielding effect from the Phe 99 aromatic ring as well as possibly bond-polarization effects arising from the tight packing of the indole HNε1 against this ring [106, 107]. The homologous resonance in FKBP12 is less strongly shifted by 0.6 ppm, while that of FKBP51 is more strongly upfield shifted by 0.1 ppm. Although not a quantitative measure, the more weakly shifted indole resonance in FKBP12 is consistent with an averaged chemical shift arising from ~15% population of a reoriented indole ring conformation exhibiting a typical reference HNε1 chemical shift value.
In addition to FKBP12, FKBP51 and FKBP52, crystal structures have been reported for FKBP13 [100] and FKBP25 [108], each of which contain both a tryptophan ring at the base of the catalytic cleft and the shorter aspartate sidechain at the following residue. Indeed, during the evolution of FKBP12 this position is occupied by an aspartate throughout lower eukaryotes. The glutamate substitution first appeared at this position during the evolution of bony fish at approximately the time that FKBP12 and FKBP12.6 began to diverge [69]. A further indication that the reorientation of the active site indole sidechain may be energetically disfavored in these other FKBP domains is that the unoccupied volume beneath the plane of the Trp 59 ring in FKBP12, which accommodates one edge of the indole ring in the reoriented position, appears to be occluded in these other crystal structures. Thus a well-populated conformation of FKBP12 which differs substantially from conformations that are appreciably sampled by any of the other FKBP domain may provide a useful design target for selective inhibition within this family.
These NMR studies of the indole ring flip transition in FKBP12 provide little information regarding the timeframe of this transition beyond the fact that the absence of discernible exchange linebroadening effects for the resonances involved would indicate a timeframe that is shorter than ~10–50 µs. 15N NMR relaxation analysis for the Trp 59 Nε1 resonance yielded an order parameter estimate that was modestly smaller than that expected for the absence of any substantial internal motion although not as small as predicted from a 20% population sampling of a perpendicular orientation [70]. These results tentatively suggest that the indole ring flip is likely to be somewhat slower than the global molecular tumbling time of 6.0 ns so that only a portion of the rotational disorder effect of the ring flip is experimentally detected. Another line of evidence for internal mobility of the Trp 59 indole ring comes from the time-resolved fluorescence anisotropy measurements [109] in which S2 order parameter values of 0.75 and ~1.0 were derived for the unliganded protein and the FK506-bound complex, respectively. The time constant of 5.43 ns for the slowest component of the fluorescence anisotropy decay in the FK506-bound protein was decreased to 4.75 ns for uncomplexed FKBP12. The authors interpreted their data in terms of a change in the slowest component of the signal due to unresolved contributions from a local conformational transition of the indole ring and the global molecular tumbling [109].
The indole ring flip appears likely to occur in the timeframe of ~10 ns while the sidechain rotamer transition of Glu 60 and the shifting of the hydrogen bonding geometry in the central α-helix may occur at least as rapidly. To exploit the relatively rapid timescale anticipated for these transitions, full atom molecular simulations were carried out on the µs timescale to examine whether these three distinct local transitions could be satisfactorily sampled. Such simulations would enable the degree of thermodynamic coupling between these local transitions to be analyzed and the degree to which this allosteric process arises from concerted transitions could be assessed. Interpreting the thermodynamics of protein allosteric coupling in terms of specific mechanistic structural models continues to present a significant challenge for the field [110, 111]. Although models invoking concerted conformational transitions have long been a central aspect of these mechanistic analyses, the kinetics underlying many of the best studied allosteric systems are too slow to allow for unbiased molecular dynamics simulations to provide estimates of the transition rates and equilibria to assess the degree to which concerted transitions contribute to the predicted coupling equilibria.
A set of three parallel 400 ns CHARMM27 molecular simulations were carried out on FKBP12 to examine how the Trp 59 indole ring flip might be energetically coupled to a transition of the Glu 60 sidechain which interacts with the backbone of the 50’s loop located ~12 Å from the indole nitrogen [105]. At 5 ps intervals during the simulation the conformational states for the Trp 59 and Glu 60 sidechains and the Glu 60 O – Ala 64 HN hydrogen bonding interaction were assessed. As illustrated by sampling across one of the three simulations, the conformational state of the Trp 59 sidechain is clearly sensitive to the Glu 60 sidechain torsion angle and vice versa (Fig. 19). Note that the predicted population ratio for the Trp 59 ring reorientation deviates from the experimental value, likely reflecting inaccuracies in the modeling of the interactions of the Glu 60 sidechain with the backbone of the 50’s loop and the bridging structural water molecule [68]. Analogous methyl-indole HNε1 NOESY measurements and CHARMM27 simulations were carried out on a V101I variant of FKBP12 designed to inhibit the rotation of the indole ring and the experimental difference in the free energy of the indole ring flip for the wild type and V101I variant was reasonably accurately predicted [105], indicating an increased robustness in the differential free energy predictions.
Fig. 19. CHARMM27 simulations of the sidechain conformation for Trp 59 and Glu 60 in FKBP12.

Illustrated at 400 ps intervals, the orientation of the Trp 59 indole Nε1 – HNε1 bond vector is plotted relative to that of the initial frame (A). The χ1 sidechain torsion angle of Glu 60 is plotted at the same intervals (B) using a 0° to 360° scale to circumvent the discontinuity at 180°/−180°. Illustration reproduced from [105], Elsevier copyright holder.
The transition matrix formed among the 23 states of trans or gauche− χ1 rotamers for Trp 59 and Glu 60 and the presence or absence of a Glu 60 O – Ala 64 HN hydrogen bond was used to analyze the strength of correlation among these transitions (Fig. 20). Since the indole ring reorientation combines a trans to gauche− χ1 rotamer transition with a smaller shift in the χ2 torsion angle, the larger χ1 transition was used as diagnostic. The diagonal values in the transition matrix correspond to conformations that remain in the same state after a 5 ps sampling interval. The states are ordered within the transition matrix so that the rapidly interchanging hydrogen bond breakage/formation transitions lie within 2×2 submatrices. The much slower transitions between the torsion angle states of Glu 60 and Trp59 lie further from the diagonal. The value of each off-diagonal element is generally similar to that of the symmetrical element, indicating reasonably complete statistical sampling of most transitions in both directions.
Fig. 20. Transition matrix for wild type FKBP12 characterizing the trans-to-gauche− χ1 rotamer transitions for the sidechains of Trp 59 and Glu 60 and the hydrogen bonding status between Glu 60 O and Ala 64 HN (> or < 2.5 Å).

The 1.14 µs of molecular simulation was sampled at 5 ps intervals. A small set of transitions of the χ1 rotamers with residence times less than 15 ps were disregarded. These generally corresponded to conformations that move only slightly beyond the torsional barrier maximum at 240° separating the trans and gauche− rotamers. The 59g− denotes Trp 59 sidechains with a gauche− χ1 rotamer and a Nε1-HNε1 angle (relative to the initial frame) < 50°, while 59t denotes the trans χ1 rotamer with a Nε1-HNε1 angle > 50°. The population average for each state is listed along the bottom. The use of either 2.3 Å or 2.7 Å for the cutoff distance in the hydrogen bonding analysis had minimal effects on the differential free energy predictions. Illustration reproduced from [105], Elsevier copyright holder.
Most strikingly, there are no transitions that occur within the 2×2 blocks that lie along the anti-diagonal. Although the sidechain conformations of Trp 59 and Glu 60 are clearly well correlated (Fig. 19), this result indicates that throughout the entire 1.14 µs of simulation not a single concerted transition occurs for these two sidechains. Nevertheless, the statistical sampling is quite adequate since even the site of the slowest rotamer transition, Trp 59, underwent 57 χ1 transitions during this time period.
The allosteric coupling between the transitions of the Trp 59 and Glu 60 sidechains is determined by how the population ratio for the 59g− and 59t states depends upon the state of the Glu 60 sidechain, this is to say [(59g−/59t)|60g−]/[(59g−/59t)|60t]. By symmetry, this population ratio also equals [(60g−/60t)|59g−]/[(60g−/60t)|59t]. The transition matrix yields a predicted coupling ratio of 63 for wild type FKBP12 which corresponds to a coupling free energy of 10.3 kJ/mol. Since the predicted coupling free energy is larger than the free energy difference between any pair of Trp 59 and Glu 60 sidechain states, the molecular dynamics-derived energy level diagram deviates from the generic diamond pattern typically assumed (Fig. 21). Indeed, such a chevron pattern can be generally anticipated for efficiently coupled allosteric systems in which the coupling free energy is larger than that for the population ratio at either site.
Fig. 21. The free energy diagram (kJ/mol) for the gauche− and trans χ1 rotamer states of Trp 59 and Glu 60 predicted from 1.14 µs of simulation for wild type FKBP12.

Illustration reproduced from [105], Elsevier copyright holder.
CONCLUSIONS
Challenges continue in the efforts to gain a direct structural understanding for the physiological role of the various FKBP domains within the large protein complexes in which they function. Despite the considerable structural similarities within this protein family, individual FKBP domains appear to have evolved quite distinct sets of protein recognition interactions which enable them to serve as cofactors in a diverse range of signaling processes. The difficulties in developing selective inhibitors for specific FKBP domain proteins has contributed to the ongoing challenges in adequately characterizing the physiologically significant interactions for each of the FKBP domains. A large proportion of these efforts have been directed toward FKBP12/12.6 and the FK1 domains of FKBP51/52, each of which participate in a number of processes beyond their best characterized roles in the regulation of ryanodine receptor calcium channels and steroid receptor complexes.
Given the particularly strong similarity among the crystal structures of these four FKBP domains, the range of conformational flexibility exhibited by these domains is being studied to determine whether the transient conformations sampled by each protein might be sufficiently distinct so as to offer an opportunity for selective inhibitor design. It has been shown that the Trp 59 indole ring of FKBP12 undergoes a well-populated transition which dramatically altered the geometry of the catalytic cleft, a transition that does not appear to be significantly sampled by any of the other FKBP domains containing an active site Trp. Furthermore, this transition within the catalytic cleft of FKBP12 is allosterically linked to the spatially remote backbone of the 50’s loop which is known to be conformationally plastic, thus presenting a potential mechanism for protein-protein signaling interactions. In contrast to the homologous FKBP52, the β4–β5 loop of FKBP51 FK1 domain undergoes a conformational transition at the site of its regulatory interaction with the steroid receptor protein. Mutations that largely interchange the transcriptional activity levels controlled by these FKBP proteins likewise interchange the dynamical properties of this loop. An allosterically coupled transition in the β2 and β3a strands of FKBP51 which underlie the tip of the β4–β5 loop has been identified and appears to exhibit structural similarity to the conformations recently reported for iFit inhibitor-bound FK1 domains. Several other conformational transitions have been characterized in one or more of these four FKBP domains. Although the detailed structural understanding of these transitions will require significant future efforts, the diversity in dynamical behavior among these FKBP domains offers promising opportunities both for therapeutic design and for gaining insight into how these small domains might participate in the collective transitions that occur within the signaling complexes in which they function.
Acknowledgments
The authors’ work cited in this review was supported in part by National Institutes of Health [GM 088214].
ABBREVIATIONS
- FKBP
FK506-binding protein
- FK1
first FKBP domain
- rmsd
root mean square deviation
- RyR
ryanodine receptor
- TGF
transforming growth factor
- HSP
heat shock protein
- TPR
tetratricopeptide repeat
- NOE
nuclear Overhauser effect
- NOESY
nuclear Overhauser effect spectroscopy
- R1
longitudinal (spin-lattice) relaxation rate
- R2
transverse (spin-spin) relaxation rate
- CT-HSQC
constant time heteronuclear single quantum coherence
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Liu J, JD Farmer J, Lane WS, Friedman J, Wiessman I, Schreiber SL. Calcineurin is a common target for cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 2.Heitman J, Novva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 3.Galat A. Functional diversity and pharmacological profiles of the FKBPs and their complexes with small natural ligands. Cell. Mol. Life Sci. 2013;70:3243–3275. doi: 10.1007/s00018-012-1206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blackburn EA, Walkinshaw MD. Targeting FKBP isoforms with small-molecule ligands. Curr. Opin. Pharmacol. 2011;11:365–371. doi: 10.1016/j.coph.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Gerard M, Deleersnijder A, Daniëls V, Schreurs S, Munck S, Reumers V, Pottel H, Engelborghs Y, VandenHaute C, Taymans JM, Debyser Z, Baekelandt V. Inhibition of FK506 binding proteins reduces α-synuclein aggregation and Parkinson's disease-like pathology. J. Neuroscience. 2010;30:2454–2463. doi: 10.1523/JNEUROSCI.5983-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugata H, Matsuo K, Nakagawa T, Takahashi M, Mukai H, Ono Y, Maeda K, Akiyama H, Kawamata T. A peptidyl-prolyl isomerase, FKBP12, accumulates in Alheimer neurofibrillary tangles. Neurosci. Lett. 2009;459:96–99. doi: 10.1016/j.neulet.2009.04.062. [DOI] [PubMed] [Google Scholar]
- 7.Liu FL, Liu PH, Shao HW, Kung FL. The intracellular domain of amyloid precursor protein interacts with FKBP12. Biochem. Biophys. Res. Commun. 2006;350:472–477. doi: 10.1016/j.bbrc.2006.09.073. [DOI] [PubMed] [Google Scholar]
- 8.Erlejman AG, DeLeo SA, Mazaira GI, Molinari AM, Camisay MF, Fontana V, Cox MB, Piwien-Pilipuk G, Galigniana MD. NF-kB transcriptional activity is modulated by FK506-binding proteins FKBP51 and FKBP52. J. Biol. Chem. 2014;289:26263–26276. doi: 10.1074/jbc.M114.582882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huse M, Chen YG, Massague J, Kuriyan J. Crystal structure of the cytoplasmic domain of the type I TGF-beta receptor in complex with FKBP12. Cell. 1999;96:425–436. doi: 10.1016/s0092-8674(00)80555-3. [DOI] [PubMed] [Google Scholar]
- 10.Chaikuad A, Alfano I, Kerr G, Sanvitale CE, Boegermann JH, Triffitt JT, vonDelft F, Knapp S, Knaus P, Bullock AN. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J. Biol. Chem. 2012;287:36990–36998. doi: 10.1074/jbc.M112.365932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Timerman AP, Ogunbumni E, Freund E, Wiederrecht G, Marks AR, Fleischer S. The calcium release channel of sacroplasmic reticulum is modulated by FK506-binding protein. J. Biol. Chem. 1993;268:22992–22999. [PubMed] [Google Scholar]
- 12.Timerman AP, Onoue H, Xin HB, Barq S, Copello J, Wiederrecht G, Fleischer S. Selective binding of FKBP12.6 by the cardiac ryanodine receptor. J. Biol. Chem. 1996;271:20385–20391. doi: 10.1074/jbc.271.34.20385. [DOI] [PubMed] [Google Scholar]
- 13.Shou W, Aghdasi B, Armstrong DL, Guo Q, Bao S, Charng MJ, Mathews LM, Schneider MD, Hamilton SL, Matzuk MM. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391:489–492. doi: 10.1038/35146. [DOI] [PubMed] [Google Scholar]
- 14.Maruyama M, Li BY, Chen H, Xu X, Song LS, Guatimosim S, Zhu W, Yong W, Zhang W, Bu G, Lin SF, Fishbein MC, Lederer WJ, Schild JH, Field LJ, Rubart M, Chen PS, Shou W. FKBP12 is a critical regulator of the heart rhythm and the cardiac voltage-gated sodium current in mice. Circ. Res. 2011;108:1042–1052. doi: 10.1161/CIRCRESAHA.110.237867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riggs DL, Roberts PJ, Chirillo SC, Cheung-Flynn J, Prapapanich V, Ratajczak T, Gaber R, Picard D, Smith DF. The Hsp90-binding peptidylprolyl isomerase FKBP52 potentiates glucocorticoid signaling in vivo. EMBO J. 2003;22:1158–1167. doi: 10.1093/emboj/cdg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tai PK, Maeda Y, Nakao K, Wakim NG, Duhring JL, Faber LE. A 59-kilodalton protein associated with progestin, estrogen, androgen and glucocorticoid receptors. Biochemistry. 1986;25:5269–5275. doi: 10.1021/bi00366a043. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez ER. HSP56: a novel heat shock protein associated with untransformed steroid receptor complexes. J. Biol. Chem. 1990;265:22067–22070. [PubMed] [Google Scholar]
- 18.Smith DF, Faber LE, Toft DO. Purification of unactivated progesterone receptor and identification of novel receptor-associated proteins. J. Biol. Chem. 1990;265:3996–4003. [PubMed] [Google Scholar]
- 19.Minematsu T, Lee J, Zha J, Moy S, Kowalski D, Hori K, Ishibashi K, Usui T, Kamimura H. Time-dependent inhibitiory effects of FK1706, a novel nonimmunosuppressive immunophilin ligand, on CYP3A4/5 activity in humans in vivo and in vitro. Drug Metab. Disposition. 2010;38:249–259. doi: 10.1124/dmd.109.029280. [DOI] [PubMed] [Google Scholar]
- 20.Kozany C, Marz A, Kress C, Hausch F. Fluorescent probes to characterize FK506-binding proteins. Chembiochem. 2009;10:1402–1410. doi: 10.1002/cbic.200800806. [DOI] [PubMed] [Google Scholar]
- 21.Schmidt MV, Paez-Pereda M, Holsboer F, Hausch F. The prospect of FKBP51 as a drug target. ChemMedChem. 2012;7:1351–1359. doi: 10.1002/cmdc.201200137. [DOI] [PubMed] [Google Scholar]
- 22.Lam E, Martin MM, Timerman AP, Sabers C, Fleischer S, Lukas T, Abraham RT, O'Keefe SJ, O'Neill EA, Wiederrecht GJ. A novel FK506 protein can mediate the immunosuppressive effects of FK506 and is associated with the cardiac ryanodine receptor. J. Biol. Chem. 1995;270:26511–26522. doi: 10.1074/jbc.270.44.26511. [DOI] [PubMed] [Google Scholar]
- 23.Galfré E, Pitt SJ, Venturi E, Sitsapesan M, Zaccai NR, Tsaneva-Atanasova K, O'Neill S, Sitsapesan R. FKBP12 activates the cardiac ryanodine receptor Ca2+-release channel and is antagonised by FKBP12.6. PLoS ONE. 2012;7:e31956. doi: 10.1371/journal.pone.0031956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wehrens XHT, Lehnart SE, Reiken S, vanderNagel R, Morales R, Sun J, Cheng Z, Deng SX, deWindt LJ, Landry DW, Marks AR. Enhanced calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc. Natl. Acad. Sci. USA. 2005;102:9607–9612. doi: 10.1073/pnas.0500353102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, Dura M, Chen BX, Marks AR. Role of chronic ryanodine receptor phosphorylation in heart failure and β-adrenergic receptor blockage in mice. J. Clin. Invest. 2010;120:4375–4387. doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eschenhagen T. Is ryanodine receptor phosphorylation key to the fight or flight response and heart failure? J. Clin. Invest. 2010;120:4197–4203. doi: 10.1172/JCI45251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bers DM. Ryanodine receptor S2808 Phosphorylation in heart failure: Smoking gun or red herring? Circ. Res. 2012;110:796–799. doi: 10.1161/CIRCRESAHA.112.265579. [DOI] [PubMed] [Google Scholar]
- 28.Okamoto H, Takasawa S. Recent advances in the Okamoto model: the CD38-cyclic ADP-ribose signal system and the regenerating gene protein (Reg)-Reg receptor system in β-cells. Diabetes. 2002;51:S462–S473. doi: 10.2337/diabetes.51.2007.s462. [DOI] [PubMed] [Google Scholar]
- 29.Noguchi N, Takasawa S, Nata K, Tohgo A, Kato I, Ikehata F, Yonekura H, Okamoto H. Cyclic ADP-ribose binds to FK506-binding protein 12.6 to release Ca2+ from islet microsomes. J. Biol. Chem. 1997;272:3133–3136. doi: 10.1074/jbc.272.6.3133. [DOI] [PubMed] [Google Scholar]
- 30.Dixit SS, Wang T, Manzano EJQ, Yoo S, Lee J, Chiang DY, Ryan N, Respress JL, Yechoor VK, Wehrens XHT. Effects of CaMKII-mediated phosphorylation of Ryanodine receptor type 2 on islet calcium handling, insulin secretion, and glucose tolerance. PLOS ONE. 2013;8:e58655. doi: 10.1371/journal.pone.0058655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galeotti N, Quattrone A, Vivoli E, Norcini M, Bartolini A, Ghelardini C. Different involvement of type 1, 2, and 3 ryanodine receptors in memory processes. Learn. Mem. 2008;15:315–323. doi: 10.1101/lm.929008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X, Betzenhauser MJ, Reiken S, Meli AC, Xie W, Chen BX, Arancio O, Marks AR. Role of leaky neuronal ryanodine receptors in stress-induced cognitive dysfunction. Cell. 2012;150:1055–1067. doi: 10.1016/j.cell.2012.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zalk R, Clarke OB, desGeorges A, Grassucci RA, Reiken S, Mancia F, Hendrickson WA, Frank J, Marks AR. Structure of a mammalian ryanodine receptor. Nature. 2015;517:44–49. doi: 10.1038/nature13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan Z, Bai XC, Yan C, Wu J, Li Z, XIe T, Peng W, Yin CC, Li X, Scheres SHW, Shi Y, Yan N. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature. 2015;517:50–55. doi: 10.1038/nature14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Efremov RG, Leitner A, Aebersold R, Raunser S. Architecture and conformational switch mechanism of the ryanodine receptor. Nature. 2015;517:39–43. doi: 10.1038/nature13916. [DOI] [PubMed] [Google Scholar]
- 36.Riggs DL, Cox MB, Tardif HL, Hessling M, Buchner J, Smith DF. Noncatalytic role of the FKBP52 peptidyl-prolyl isomerase domain in the regulation of steroid hormone signaling. Mol. Cell. Biol. 2007;27:8658–8669. doi: 10.1128/MCB.00985-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davies TH, Ning YM, Sanchez ER. A new first step in activation of steroid receptors: Hormone-induced switching of FKBP51 and FKBP52 immunophilins. J. Biol. Chem. 2002;277:4597–4600. doi: 10.1074/jbc.C100531200. [DOI] [PubMed] [Google Scholar]
- 38.Silverstein AM, Galigniana MD, Kanelakis KC, Radanyi C, Renoir JM, Pratt WB. Different regions of the immunophilin FKBP52 determine its association with the glucocorticoid receptor, hsp90, and cytoplasmic dynein. J. Biol. Chem. 1999;274:36980–36986. doi: 10.1074/jbc.274.52.36980. [DOI] [PubMed] [Google Scholar]
- 39.Wocknik GM, Rüegg J, Abel GA, Schmidt U, Holsboer F, Rein T. FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J. Biol. Chem. 2005;280:4609–4616. doi: 10.1074/jbc.M407498200. [DOI] [PubMed] [Google Scholar]
- 40.Galigniana MD, Erlejman AG, Monte M, Gomez-Sanchez C, Piwien-Pilipuk G. The hsp90-FKBP52 complex links the mineralocorticoid receptor to motor proteins and persists bound to the receptor in early nuclear events. Mol. Cell. Biol. 2010;30:1285–1298. doi: 10.1128/MCB.01190-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Avellino R, Romano S, Parasole R, Bisogni R, Lamberti A, Poggi V, Venuta S, Romano MF. Rapamycin stimulates apoptosis of childhood acute lymphoblastic leukemia cells. Blood. 2005;106:1400–1406. doi: 10.1182/blood-2005-03-0929. [DOI] [PubMed] [Google Scholar]
- 42.Romano S, D'Angelillo A, Pacelli R, Staibano S, DeLuna E, Bisogni R, Eskelinen EL, Mascolo M, Cali G, Arra C. Role of FKBP51 in the control of apoptosis of irradiated melanoma cells. Cell Death Differ. 2010;17:145–157. doi: 10.1038/cdd.2009.115. [DOI] [PubMed] [Google Scholar]
- 43.Hinz M, Broemer M, Arslan SC, Otto A, Mueller EC, Dettmer R, Scheidereit C. Signal responsiveness of IκB kinases is determined by Cdc37-assisted transient interaction with Hsp90. J. Biol. Chem. 2007;282:32311–32319. doi: 10.1074/jbc.M705785200. [DOI] [PubMed] [Google Scholar]
- 44.Jinwal UK, J Koren I, Borysov SI, Schmid AB, Abisambra JF, Blair LJ, Johnson AG, Jones JR, Shults CL, JC O'Leary I, Jin Y, Buchner J, Cox MB, Dickey CA. The Hsp90 cochaperone, FKBP51, increases tau stability and polymerizes micotubules. J. Neuroscience. 2010;30:591–599. doi: 10.1523/JNEUROSCI.4815-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chambraud B, Sardin E, Giustiani J, Dounane O, Schumacher M, Goedert M, Baulieu EE. A role for FKBP52 in tau protein function. Proc. Natl. Acad. Sci U. S. A. 2010;107:2658–2663. doi: 10.1073/pnas.0914957107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stechschulte LA, TD Hands J, Ghanem SS, Shou W, Najjar SM, Sanchez ER. FKBP51 reciprocally regulates GRα and PPARγ activation via the AKT-P38 pathway. Mol. Endocrinol. 2014;28:1254–1264. doi: 10.1210/me.2014-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stechschulte LA, TD Hinds J, Khuder SS, Shou W, Najjar SM, Sanchez ER. FKBP51 controls cellular adipogenesis through P38 kinase-mediated phosphorylation of GRα and PPARγ. Mol. Endocrinol. 2014;28:1265–1275. doi: 10.1210/me.2014-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z, Wang L. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16:259–266. doi: 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fabian AK, März A, Neimanis S, Biondi RM, Kozany C, Hausch F. InterAKTions with FKBPs - Mutational and Pharmacological Exploration. PLoS ONE. 2013;8:e57508. doi: 10.1371/journal.pone.0057508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith DF, Toft DO. The intersection of steroid receptors with molecular chaperones: Observations and questions. Molec. Endocrinol. 2008;22:2229–2240. doi: 10.1210/me.2008-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kirschke E, Goswami D, Southworth D, Griffin PR, Agard DA. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell. 2014;157:1685–1697. doi: 10.1016/j.cell.2014.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Denny WB, Valentine DL, Reynolds PD, Smith DF, Scammell JG. Squirrel monkey immunophilin FKBP51 is a potent inhibitor of glucocorticoid receptor binding. Endocrinology. 2000;141:4107–4113. doi: 10.1210/endo.141.11.7785. [DOI] [PubMed] [Google Scholar]
- 53.Pfaff SJ, Fletterick RJ. Hormone binding and co-regulator binding to the glucocorticoid receptor are allosterically coupled. J. Biol. Chem. 2010;285:15256–15267. doi: 10.1074/jbc.M110.108118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trebble PJ, Woolven JM, Saunders KA, Simpson KD, Farrow SN, Matthews LC, Ray DW. A ligand-specific kinetic switch regulates glucocorticoid receptor trafficking and function. J. Cell Sci. 2013;126:3159–3169. doi: 10.1242/jcs.124784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mustafi SM, Chen H, Li H, LeMaster DM, Hernández G. Analyzing the visible conformational substates of the FK506-binding protein FKBP12. Biochem. J. 2013;453:371–380. doi: 10.1042/BJ20130276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vuister GW, Wang AC, Bax A. Measurement of three-bond nitrogen-carbon J-couplings in proteins uniformly enriched in 15N and 13C. J. Am. Chem. Soc. 1993;115:5334–5335. [Google Scholar]
- 57.Skrynnikov NR, Goto NK, Yang D, Choy WY, Tolman JR, Mueller GA, Kay LE. Orienting domains in proteins using dipolar couplings measured by liquid-state NMR: Differences in solution and crystal forms of maltodextrin binding protein loaded with β-cyclodextrin. J. Mol. Biol. 2000;295:1265–1275. doi: 10.1006/jmbi.1999.3430. [DOI] [PubMed] [Google Scholar]
- 58.Hernández G, Anderson JS, LeMaster DM. Polarization and polarizability assessed by protein amide acidity. Biochemistry. 2009;48:6482–6494. doi: 10.1021/bi900526z. [DOI] [PubMed] [Google Scholar]
- 59.Rosen MK, Michnick SW, Karplus M, Schreiber SL. Proton and nitrogen sequential assignments and secondary structure determination of the human FK506 and rapamycin binding protein. Biochemistry. 1991;1991:4774–4789. doi: 10.1021/bi00233a020. [DOI] [PubMed] [Google Scholar]
- 60.Egan DA, Logan TM, Liang H, Matayoshi E, Fesik SW, Holzman TF. Equilibrium denaturation of recombinant human FK binding protein in urea. Biochemistry. 1993;32:1920–1927. doi: 10.1021/bi00059a006. [DOI] [PubMed] [Google Scholar]
- 61.Cheng JW, Lepre CA, Moore JM. 15N NMR relaxation studies of the FK506 binding protein: Dynamic effects of ligand binding and implications for calcineurin recognition. Biochemistry. 1994;33:4093–4100. doi: 10.1021/bi00180a001. [DOI] [PubMed] [Google Scholar]
- 62.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 63.Sich C, Improta S, Cowley DJ, Guenet C, Merly JP, Teufel M, Saudek V. Solution structure of a neurotrophic ligand bound to FKBP12 and its effects on protein dynamics. Eur. J. Biochem. 2000;267:5342–5354. doi: 10.1046/j.1432-1327.2000.01551.x. [DOI] [PubMed] [Google Scholar]
- 64.Inomata K, Ohno A, Tochio H, Isogai S, Tenno T, Nakase I, Takeuchi T, Futaki S, Ito Y, Hiroaki H, Shirakawa M. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature. 2009;458:106–109. doi: 10.1038/nature07839. [DOI] [PubMed] [Google Scholar]
- 65.Liu A, Hu W, Majumdar A, Rosen MK, Patel DJ. Detection of very weak side chain-main chain hydrogen bonding interactions in medium-size 13C/15N-labeled proteins by sensitivity-enhanced NMR spectroscopy. J. Biomolec. NMR. 2000;17:79–82. doi: 10.1023/a:1008373501591. [DOI] [PubMed] [Google Scholar]
- 66.Chatterjee A, Bhavesh NS, Panchal SC, Hosur RV. A novel protocol based on HN(C)N for rapid resonance assignment in (15N,13C) labeled proteins: implications to structural genomics. Biochem. Biophys. Res. Commun. 2002;293:427–432. doi: 10.1016/S0006-291X(02)00240-1. [DOI] [PubMed] [Google Scholar]
- 67.Itoh S, DeCenzo MT, Livingston DJ, Pearlman DA, Navia MA. Conformation of FK506 in X-ray structures of its complexes with human recombinant FKBP12 mutants. Bioorg. Med. Chem. Lett. 1995;5:1983–1988. [Google Scholar]
- 68.Szep S, Park S, Boder ET, VanDuyne GD, Saven JG. Structural coupling between FKBP12 and buried water. Proteins. 2009;74:603–611. doi: 10.1002/prot.22176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Somarelli JA, Herrara RJ. Evolution of the 12 kDa FK506-binding protein gene. Biol. Cell. 2007;99:311–321. doi: 10.1042/BC20060125. [DOI] [PubMed] [Google Scholar]
- 70.Mustafi SM, Brecher M, Zhang J, Li H, LeMaster DM, Hernández G. Structural Basis of Conformational Transitions in the Active Site and 80’s loop in the FK506-binding Protein FKBP12. Biochem. J. 2014;458:525–536. doi: 10.1042/BJ20131429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fulton KF, Jackson SE, Buckle AM. Energetic and structural analysis of the role of tryptophan 59 in FKBP12. Biochemistry. 2003;42:2364–2372. doi: 10.1021/bi020564a. [DOI] [PubMed] [Google Scholar]
- 72.Mustafi SM, LeMaster DM, Hernández G. Differential conformational dynamics in the closely homologous FK506-binding domains of FKBP51 and FKBP52. Biochem. J. 2014;458:525–536. doi: 10.1042/BJ20140232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Galat A. A note on clustering the functionally-related paralogues and orthologues of proteins: A case of the FK506-binding proteins (FKBPs) Comput. Biol. Chem. 2004;28:129–140. doi: 10.1016/j.compbiolchem.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 74.Jeener J, Meier BH, Bachmann P, Ernst RR. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 1979;71:4546–4553. [Google Scholar]
- 75.Main ERG, Fulton KF, Jackson SE. Folding pathway of FKBP12 and characterization of the transition state. J. Mol. Biol. 1999;291:429–444. doi: 10.1006/jmbi.1999.2941. [DOI] [PubMed] [Google Scholar]
- 76.Chen H, Mustafi SM, LeMaster DM, Li Z, Héroux A, Li H, Hernández G. Crystal structure and conformational flexibility of the unligated FK506-binding protein FKBP12.6. Acta Cryst. 2014;D70:636–646. doi: 10.1107/S1399004713032112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Siemon IZ, Wieland T, Pook KH. Influence of the distance of the proline carbonyl from the β and γ carbon on the 13C chemical shifts. Angew. Chem. Int. Ed. 1975;14:702–703. doi: 10.1002/anie.197507021. [DOI] [PubMed] [Google Scholar]
- 78.Schubert M, Labudde D, Oschkinat H, Schmieder P. A software tool for the prediction of Xaa-Pro peptide bond conformations in proteins based on 13C chemical shift statistics. J. Biomol. NMR. 2002;24:149–154. doi: 10.1023/a:1020997118364. [DOI] [PubMed] [Google Scholar]
- 79.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR. 1995;5:67–81. doi: 10.1007/BF00227471. [DOI] [PubMed] [Google Scholar]
- 81.Schwarzinger S, Kroon GJ, Foss TR, Chung J, Wright PE, Dyson HJ. Sequence-dependent correction of random coil NMR chemical shifts. J. Am. Chem. Soc. 2001;123:2970–2978. doi: 10.1021/ja003760i. [DOI] [PubMed] [Google Scholar]
- 82.Li P, Ding Y, Wu B, Shu C, Shen B, Rao Z. Structure of the N-terminal domain of human FKBP52. Acta Cryst. D. 2003;59:16–22. doi: 10.1107/s0907444902017523. [DOI] [PubMed] [Google Scholar]
- 83.Jabs A, Weiss MS, Hilgenfeld R. Non-proline cis peptide bonds in proteins. J. Mol. Biol. 1999;286:291–304. doi: 10.1006/jmbi.1998.2459. [DOI] [PubMed] [Google Scholar]
- 84.Wüthrich K. NMR of Proteins and Nucleic Acids. 1. Vol. 1986. John Wiley & Sons; New York: [Google Scholar]
- 85.Bracher A, Kozany C, Hahle A, Wild P, Zacharias M, Hausch F. Crystal structures of the free and ligand-bound FK1-FK2 domain segment of FKBP52 reveal a flexible inter-domain hinge. J. Mol. Biol. 2013;425:4134–4144. doi: 10.1016/j.jmb.2013.07.041. [DOI] [PubMed] [Google Scholar]
- 86.Wu B, Li P, Liu Y, Ding Y, Shu C, Ye S, Bartlam M, Shen B, Rao Z. 3D structure of human FK506-binding protein 52: Implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex. Proc. Natl. Acad. Sci U. S. A. 2004;101:8348–8353. doi: 10.1073/pnas.0305969101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brath U, Akke M, Yang D, Kay LE, Mulder FA. A. Functional dynamics of human FKBP12 revealed by methyl 13C rotating frame relaxation dispersion NMR spectroscopy. J. Am. Chem. Soc. 2006;128:5718–5727. doi: 10.1021/ja0570279. [DOI] [PubMed] [Google Scholar]
- 88.Brath U, Akke M. Differential responses of the backbone and side-chain conformational dynamics in FKBP12 upon binding the transition-state analog FK506: Implications for transition-state stabilization and target protein recognition. J. Mol. Biol. 2009;387:233–244. doi: 10.1016/j.jmb.2009.01.047. [DOI] [PubMed] [Google Scholar]
- 89.Sapienza PJ, Mauldin RV, Lee AL. Multi-timescale dynamics study of FKBP12 along the rapamycin-mTOR binding coordinate. J. Mol. Biol. 2011;405:378–394. doi: 10.1016/j.jmb.2010.10.037. [DOI] [PubMed] [Google Scholar]
- 90.Shen Y, Bax A. SPARTA+: A modest improvement in empircal NMR chemical shift prediction by means of an artificial neural network. J. Biomol. NMR. 2010;48:13–22. doi: 10.1007/s10858-010-9433-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gaali S, Kirschner A, Cuboni S, Hartmann J, Kozany C, Balsevich G, Namendorf C, Fernandez-Vizarra P, Sippel C, Zannas AS, Draenert R, Binder EB, Almeida OFX, Rühter G, Uhr M, Schmidt MV, Touma C, Bracher A, Hausch F. Selective inhibitors of the FK506-binding protein 51 by induced fit. Nature Chemical Biology. 2015;11:33–37. doi: 10.1038/nchembio.1699. [DOI] [PubMed] [Google Scholar]
- 92.Clackson T, Yang W, Rozamus LW, Hatada M, Amara JF, Rollins CT, Stevenson LF, Magari SR, Wood SA, Courage NL, Lu X, F Cerasoli J, Gilman M, Holt DA. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl. Acad. Sci U. S. A. 1998;95:10437–10442. doi: 10.1073/pnas.95.18.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang W, Rozamus LW, Narula S, Rollins CT, Yuan R, Andrade LJ, Ram MK, Phillips TB, vanSchravendijk MR, Dalgarno D, Clackson T, Holt DA. Investigating protein-ligand interactions with a mutant FKBP possessing a designed specificity pocket. J. Med. Chem. 2000;43:1135–1142. doi: 10.1021/jm9904396. [DOI] [PubMed] [Google Scholar]
- 94.Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AGL, Wandless TJ. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bracher A, Kozany C, Thost AK, Hausch F. Structural characterization of the PPIase domain of FKBP51, a cochaperone of human Hsp90. Acta Cryst. D. 2011;67:549–559. doi: 10.1107/S0907444911013862. [DOI] [PubMed] [Google Scholar]
- 96.Gopalakrishnan R, Kozany C, Wang Y, Schneider S, Hoogeland B, Bracher A, Hausch F. Exploration of pipecolate sulfonamides as binders of the FK506-binding proteins 51 and 52. J. Med. Chem. 2012;55:4123–4131. doi: 10.1021/jm201747c. [DOI] [PubMed] [Google Scholar]
- 97.Marz AM, Fabian AK, Kozany C, Bracher A, Hausch F. Large FK506-binding proteins shape the pharmacology of rapamycin. Mol. Cell. Biol. 2013;33:1357–1367. doi: 10.1128/MCB.00678-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.VanDuyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 99.Richardson JS. β-sheet topology and the relatedness of proteins. Nature. 1977;268:495–500. doi: 10.1038/268495a0. [DOI] [PubMed] [Google Scholar]
- 100.Schultz LW, Martin PK, Liang J, Schreiber SL, Clardy J. Atomic structure of the immunophilin FKBP13-FK506 complex: Insights into the composite binding surface for calcineurin. J. Am. Chem. Soc. 1994;116:3129–3130. [Google Scholar]
- 101.Deivanayagam CC, Carson M, Thotakura A, Narayana SV, Chodavarapu RS. Structure of FKBP12.6 in complex with rapamycin. Acta Cryst. D. 2000;56:266–271. doi: 10.1107/s0907444999016571. [DOI] [PubMed] [Google Scholar]
- 102.Galat A. Functional drift of sequence attributes in the FK506-binding proteins (FKBPs) J. Chem. Inf. Model. 2008;48:1118–1130. doi: 10.1021/ci700429n. [DOI] [PubMed] [Google Scholar]
- 103.Goto NK, Gardner KH, Mueller GA, Willis RC, Kay LE. A robust and cost-effective method for the production of Val, Leu, Ile(δ1) methyl-protonated 15N, 13C, 2H labeled proteins. J. Biomolec. NMR. 1999;13:369–374. doi: 10.1023/a:1008393201236. [DOI] [PubMed] [Google Scholar]
- 104.Lipari G, Szabo A. Model-Free Approach to the Interpretation of Nuclear Magnetic Resonance Relaxation in Macromolecules. 1. Theory and Range of Validity. J. Am. Chem. Soc. 1982;104:4546–4559. [Google Scholar]
- 105.Anderson JS, Mustafi SM, Hernández G, LeMaster DM. Statistical allosteric coupling to the active site indole ring flip equilibria in the FK506-binding domain. Biophys. Chem. 2014;192:41–48. doi: 10.1016/j.bpc.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Craescu CT, Rouviere N, Popescu A, Cerpolini E, Lebeau MC, Baulieu EE, Mispelter J. Three-dimensional structure of the immunophilin-like domain of FKBP59 in solution. Biochemistry. 1996;35:11045–11052. doi: 10.1021/bi960975p. [DOI] [PubMed] [Google Scholar]
- 107.Rouviere-Fourmy N, Craescu CT, Mispelter J, Lebeau MC, Baulieu EE. 1H and 15N assignment of NMR spectrum, secondary structure and global folding of the immunophilin-like domain of the 59-kDa FK506-binding protein. Eur. J. Biochem. 1995;231:761–772. [PubMed] [Google Scholar]
- 108.Liang J, Hung DT, Schreiber SL, Clardy J. Structure of the human 25 kDa FK506 binding protein complexed with rapamycin. J. Am. Chem. Soc. 1996;118:1231–1232. [Google Scholar]
- 109.ND Silva J, Prendergast FG. Tryptophan dynamics of the FK506 binding protein: Time-resolved fluorescence and simulations. Biophys. J. 1996;70:1122–1137. doi: 10.1016/S0006-3495(96)79706-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cui Q, Karplus M. Allostery and cooperativity revisited. Prot. Sci. 2008;17:1295–1307. doi: 10.1110/ps.03259908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Swain JF, Gierasch LM. The changing landscape of protein allostery. Curr. Opin. Struct. Biol. 2006;16:102–108. doi: 10.1016/j.sbi.2006.01.003. [DOI] [PubMed] [Google Scholar]