

Abstract
Gap junctions (GJs) are important for maintenance of CNS homeostasis. GJ proteins, connexin 43 (Cx43) and connexin 47 (Cx47), play a crucial role in production and maintenance of CNS myelin. Cx43 is mainly expressed by astrocytes in the CNS and forms gap junction intercellular communications between astrocytes-astrocytes (Cx43–Cx43) and between astrocytes-oligodendrocytes (Cx43–Cx47). Mutations of these connexin (Cx) proteins cause dysmyelinating diseases in humans. Previously, it has been shown that Cx43 localization and expression is altered due to mouse hepatitis virus (MHV)-A59 infection both in vivo and in vitro; however, its mechanism and association with loss of myelin protein was not elaborated. Thus, we explored potential mechanisms by which MHV-A59 infection alters Cx43 localization and examined the effects of viral infection on Cx47 expression and its association with loss of the myelin marker proteolipid protein. Immunofluorescence and total internal reflection fluorescence microscopy confirmed that MHV-A59 used microtubules (MTs) as a conduit to reach the cell surface and restricted MT-mediated Cx43 delivery to the cell membrane. Co-immunoprecipitation experiments demonstrated that Cx43–β-tubulin molecular interaction was depleted due to protein–protein interaction between viral particles and MTs. During acute MHV-A59 infection, oligodendrocytic Cx47, which is mainly stabilized by Cx43 in vivo, was down-regulated, and its characteristic staining remained disrupted even at chronic phase. The loss of Cx47 was associated with loss of proteolipid protein at the chronic stage of MHV-A59 infection.
Keywords: astrocyte, connexin, gap junction, hepatitis virus, microtubule, myelin, oligodendrocyte, tubulin
Introduction
Microtubules (MTs)4 are components of the cytoskeleton that consist of polymers of α- and β-tubulins. Highly dynamic structures of MTs spontaneously undergo phases of polymerization and catastrophe and play major roles in various cellular processes. These processes include intracellular transportation of organelles and molecules. The movement and delivery of proteins, vesicular carriers, and organelles from their site of production to specific destinations are crucial for their function. Hence, the trafficking of different membrane proteins like cell junction proteins and signaling molecules to the cell membrane plays an essential role in cellular functioning. A group of cell junction proteins, gap junctions (GJs), form intercellular channels between two neighboring cells only after successful delivery and docking to the cell membrane. GJs are made up of connexin (Cx) proteins, which are synthesized in the endoplasmic reticulum (ER), oligomerized in intercellular organelles like ER or the trans-Golgi network, and finally delivered to the cell surface (1) and a pair of hemichannels from a GJ plaque.
One of the most studied Cxs, Cx43, is expressed in multiple organs, including the CNS, heart, and lungs. In the CNS, Cx43 is mainly expressed by astrocytes (2). Cx43 forms homotypic Cx43–Cx43–mediated channels with other astrocytes and heterotypic Cx43–Cx47–mediated channels with oligodendrocytes (3). These channels are important for maintaining CNS nutrient homeostasis, ionic buffering, and small-molecule (<1 kDa) exchange (4–9). Astrocytes also express Cx30 and Cx26, which form gap junction intercellular communication (GJIC) with other cells in panglial network but are not redundant for Cx43 expression and function (10–12). A number of recent studies examined mechanistic details of Cx43 delivery to the cell surface. Delivery of Cx43 specifically relies on the MT network. Cx43 molecules are delivered in vesicular carriers that traffic along MTs from the Golgi to the plasma membrane (13). The delivery of Cx43-containing vesicles involves intermediate proteins like EB1, p150 (Glued) of dynein–dynactin complexes, and β-catenin (14). Cx43 molecules are also known to bind MTs at the cell surface (15).
It has been shown that viral infections, such as Rous sarcoma virus, Borna disease virus, and human influenza virus, can alter Cx43 expression in vivo or in vitro, but the mechanism of Cx43 alteration remains elusive (16–18). Our recent study showed that a neurotropic demyelinating strain of mouse hepatitis virus (MHV)-A59 causes retention of Cx43 in the endoplasmic reticulum and/or endoplasmic reticulum Golgi intermediate complex (ER and/or ERGIC). The expression of Cx43 is also reduced in protein and RNA level in cultured astrocytes. These phenomena affect functional channel formation between primary astrocytes. Similar alterations of Cx43 expression and localization are also observed in the mouse brain (19), but the molecular mechanism of Cx43 retention in the ER–ERGIC was not described. For efficient infection and cell-to-cell spread, many viruses have developed mechanisms for utilizing cytoskeletal elements. The JHM strain of MHV uses MTs for transneuronal spread within the CNS, and the viral nucleocapsid (N) protein is predicted to have MT interactive properties (20). A recent study shows a demyelinating recombinant strain of MHV, RSA59, uses the MT network as a conduit for cell-to-cell spread (21). Other viruses like Herpes simplex virus 1 utilize the cellular MT network for trafficking of virions and viral glycoproteins to virus release sites (22). Vaccinia virus is reported to use both MT networks and actin filament for egression (23). Adenovirus entry to host cells is dependent on MTs (24), and the virus also uses MT-associated molecular motors for retrograde transport (25). The adeno-associated virus also displays unidirectional movement on MTs toward the nuclei (26). In contrast, there are few studies available delineating which specific molecules, if any, exhibit altered intracellular localization due to MT-mediated viral trafficking. Thus, our current study is designed to understand whether specific utilization of the MT network for virus trafficking by MHV-A59 may be an underlying mechanism by which Cx43 is restricted from localizing to the cell surface.
MHV-A59 causes hepatitis and meningoencephalitis in the acute phase of infection and leads to demyelination and concurrent axonal loss during the chronic phase, serving as a virus-induced chronic progressive model of the human CNS demyelinating disease, multiple sclerosis (27, 28). Cx43-mediated GJICs play a critical role in formation and maintenance of normal myelin. Recent studies indicate that GJIC formation between astrocytic Cx43 and oligodendrocytic Cx47 is crucial for human myelination (29, 30). These GJICs are important in K+ buffering and nutrient homeostasis (31). Upon docking, Cx47 GJ hemichannels are phosphorylated and stabilized as GJ plaques by Cx43 (32). Hence, alteration of Cx43 expression and trafficking is hypothesized to have a critical impact on Cx47 expression in this viral model of neuroinflammation. Altered Cx43–Cx47–mediated heterotypic GJIC formation between astrocytes and oligodendrocytes could be associated with loss of myelin protein. In this context, we extended our study to examine alterations of Cx47 during MHV-A59 infection and its association with myelin marker proteolipid protein (PLP).
In summary, the current study was focused on understanding the molecular mechanisms of virus-induced alteration of Cx43. This study was also extended to understand whether the alteration of Cx43 was also associated with altered oligodendrocytic GJ and myelin protein expression. The prior studies in this field showed that the MT network is important for trafficking of Cx43 as well as viral particles. In this context, we observed that Cx43 and β-tubulin colocalization was reduced due to viral infection. Consistently, viral particles colocalized with the MT network. Upon MHV-A59 infection, the delivery of Cx43 to the cell surface, along MT threads, was reduced. At the molecular level, Cx43–β-tubulin protein–protein interaction was reduced, and it was confirmed that MHV-A59 interacted with MTs. In addition, Cx47 was depleted in MHV-A59–infected mouse brain, and this GJ alteration was associated with loss of myelin protein PLP.
Results
Localization of Cx43 upon colchicine treatment
Primary astrocytes were treated with 100 μm colchicine for 24 h as a representative dose to disrupt the MT network without affecting cell viability. The cell viability of primary astrocytes, upon colchicine treatment, is shown in supplemental Fig. S1. Control cells were maintained in parallel. Cells were coimmunostained with rabbit anti-Cx43 antiserum (red) and mouse anti-β-tubulin antiserum (green). Z-stacking was obtained by a confocal microscope, with signals obtained from the base of the cells (Fig. 1, Plane 1) to the medial part of the cells (Fig. 1, Plane 4), to observe the distribution of Cx43 upon MT disruption. Untreated cells showed the presence of Cx43 at the basal stack (Fig. 1A, thin arrow) and peripheral part of the medial stacks of cells (Fig. 1, B–D). In contrast, at the basal stack of colchicine-treated cells (Fig. 1E, thick arrow), the presence of Cx43 was diminished, whereas the medial stacks showed the continued presence of Cx43 mostly inside the cells (Fig. 1, F–H, thick arrow), suggesting that Cx43 delivery to cell surface was restricted upon MT disruption. This was confirmed by disrupted β-tubulin staining (Fig. 1, F–H). Digitally magnified insets (Fig. 1, I and J) show that Cx43 was present on MT threads with an appearance of “beads-on-a-string” in a single focal plane. Colocalization was evident, specifically where the intensity of Cx43 and β-tubulin was similar (Fig. 1I, ringlike yellow spots, thin arrow). Colchicine-treated cells showed smearlike disruption of β-tubulin signal, whereas Cx43 surface localization and colocalization with MT were restricted (Fig. 1J, thick arrow). For better understanding of Cx43 and tubulin localization, individual channel images of each plane of untreated and colchicine-treated cells are shown in supplemental Fig. S2.
Figure 1.
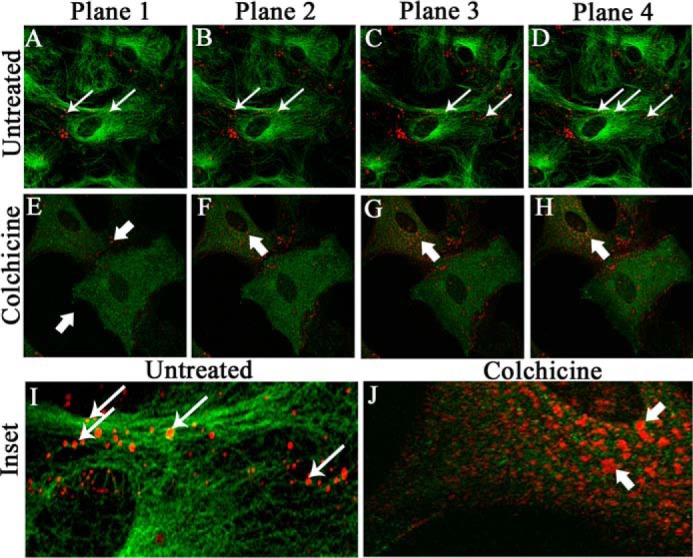
Localization of Cx43 in primary astrocytes upon colchicine treatment. Primary astrocytes were treated with 100 μm colchicine for 24 h. Control cells were maintained in parallel. Cells were subjected to double-label immunofluorescence with rabbit anti-Cx43 antiserum (red) and mouse anti-β-tubulin antiserum (green). Z-stacking was obtained by a confocal microscope from the base of the cells, at the coverslip (Plane 1), to the medial part of the cells (Plane 4), to observe the distribution of Cx43 with microtubule morphology. Untreated cells showed the presence of Cx43 at both the basal (A, thin arrow) and medial parts of cells (B–D). In contrast, at the basal stack of colchicine-treated cells (E, thick arrow), the presence of Cx43 was minimal. The medial stacks showed the presence of Cx43 mostly inside the cells, showing that Cx43 delivery was restricted upon MT disruption, which was confirmed by disrupted β-tubulin staining (F–H, thick arrow). Digitally magnified insets showed that Cx43 was present on MT threads in a single focal plane, and colocalization was evident, specifically where intensities of Cx43 and β-tubulin were similar (I, ringlike yellow spots, thin arrow). Colchicine-treated cells showed smearlike disrupted β-tubulin signal, whereas Cx43 surface localization was restricted (J, thick arrow).
Reduction of Cx43–β-tubulin colocalization due to virus infection and colocalization of viral particles with β-tubulin
Cx43 molecules are reported to directly bind to MTs (15), and the MT network helps direct delivery of Cx43 hemichannels to the cell surface (14). Primary astrocytes were infected with MHV-A59, and mock-infected cells were maintained in parallel. After 24 h postinfection (p.i.), cells were cytofixed and subjected to double-label immunofluorescence with monoclonal anti-β-tubulin (green in Fig. 2, A, E, and I) and polyclonal anti-Cx43 (red in Fig. 2, B, F, and J) antibodies. Cells were counterstained with DAPI (blue). Both mock- and virus-infected cells showed normal MT morphology (Fig. 2, A and E), whereas 100 μm colchicine treatment disrupted the MT network (Fig. 2I). Characteristic punctate staining of Cx43 was observed for mock-infected cells (Fig. 2B). Virus-infected (Fig. 2F) and colchicine-treated cells (Fig. 2J) showed intracellular localization of Cx43. Consistent with prior studies, primary astrocytes showed a proximal association between Cx43 and the MT network (Fig. 2C, thin arrow). When the astrocytes were infected with MHV-A59, Cx43 was localized in a perinuclear compartment, with minimal association with the MT network (Fig. 2G, thick arrow). Colchicine-treated cells showed a disrupted MT network, and Cx43 staining was mainly redistributed in the cytosol (Fig. 2K, thick arrow). The number of points containing colocalization of immunostaining and the intensity of staining was maximal for mock-infected cells (Fig. 2D). Upon virus infection (Fig. 2H) or microtubule disruption with colchicine (Fig. 2L), colocalization was reduced. There was ∼62.8% reduction in colocalized staining within virus-infected cells compared with mock-infected cells (**, p < 0.01; Mann–Whitney U test) and ∼80.3% reduction in colchicine-treated cells compared with mock-infected cells (**, p < 0.01; Mann–Whitney U test). Kruskal–Wallis analysis showed that the differences in a three-way comparison of groups were statistically significant (****, p < 0.0001). Five different images from n = 3 biological replicates were quantified for each experimental group (Fig. 2M). A staining intensity profile was drawn in digitally magnified images, which showed that Cx43 molecules were present along a MT thread (Fig. 2, N and O), whereas Cx43 retained in the intracellular compartment in MHV-A59–infected cells did not show such alignment (Fig. 2, P and Q). Cx43 molecules showed high-intensity peaks on MT threads in mock-infected cells (Fig. 2, R and S), but not in virus-infected cells (Fig. 2, T and U).
Figure 2.

Altered association of Cx43 with microtubule in primary astrocytes upon MHV-A59 infection and colchicine treatment. Primary astrocytes were infected with MHV-A59 at an MOI of 2 or treated with 100 μm colchicine for 24 h. Control cells were maintained in parallel. Cells were subjected to double-label immunofluorescence with rabbit anti-Cx43 antiserum (red) and mouse anti-β-tubulin antiserum (green). Cells were counterstained with DAPI (blue). In control astrocytes, punctate Cx43 (B, thin arrow) was observed to be closely associated with β-tubulin (A), and Cx43 staining was aligned along the typical radial structure of MTs (C, thin arrow). Spots of colocalized signal are shown in D. Upon infection of the cells with MHV-A59, Cx43 was retained in the intracellular compartment (F, thick arrow). Although the MT morphology appeared to be normal (E), intracellular compartment–retained Cx43 had a minimal association with the MT network (G, thick arrow). The intensity and number of colocalization spots were reduced (H). Upon colchicine treatment, the MT network was depolymerized in the primary astrocytes, and diffuse tubulin staining was observed in the cytosol (I). Colchicine treatment showed retention of Cx43 in the intracellular compartment, predominantly in the perinuclear area (J and K, thick arrow). The number of colocalization spots was reduced significantly (L). The number of colocalization points compared between experimental groups showed there was an ∼62.8% reduction in virus-infected cells compared with mock-infected cells (**, p < 0.01; Mann–Whitney U test) and ∼80.3% reduction in colchicine-treated cells compared with mock-infected cells (**, p < 0.01; Mann-Whiney U test). Comparison of all three groups by Kruskal–Wallis ANOVA showed that differences were significant (****, p < 0.0001; M). Five fields were analyzed for each group from n = 3 experiments. Digitally magnified images show that Cx43 molecules were aligned along a MT thread (N and O), whereas intracellular compartment–retained Cx43 did not show such alignment (P and Q). Association of Cx43 molecules on a single MT thread is shown (red line, Cx43; green line, tubulin). Cx43 molecules showed high-intensity peaks on MT threads in mock-infected cells (R and S), but not in virus-infected cells (T and U). Error bars, S.D. AU, arbitrary units.
When mock-infected and virus-infected cells were similarly stained with monoclonal anti-viral N (green in Fig. 3, A and D) and polyclonal anti-β-tubulin (red in Fig. 3, B and E) antibodies, similar tubulin morphology was observed for both. Maximum intensity projection images, obtained from an apotome microscope, showed dispersion of viral staining from the perinuclear compartment to the cell periphery. Viral N signal colocalized with β-tubulin staining, specifically noticeable at the cell surface (Fig. 3F, arrow). As expected, no viral antigen was present in the mock-infected cells (Fig. 3C).
Figure 3.
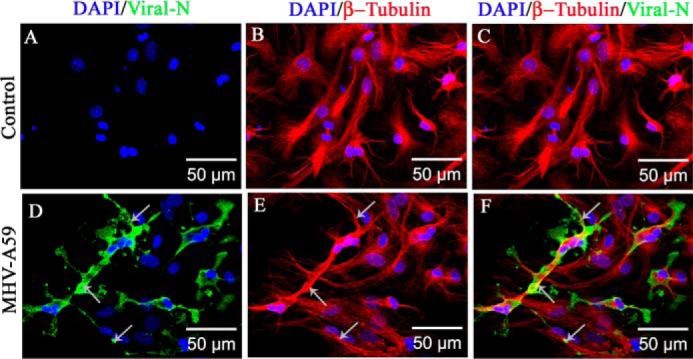
Association of MHV-A59 virus particles with microtubules in primary astrocytes. Primary astrocytes were infected with MHV-A59 at an MOI of 2, and mock-infected cells were maintained in parallel. Cells were subjected to double-label immunofluorescence with rabbit anti-β-tubulin antiserum (red) and mouse anti-N antiserum (green). Cells were counterstained with DAPI (blue), and merged image projection (mip) signals were imaged with an apotome microscope. No viral N–specific signal was observed in mock-infected cells (A), but viral N staining was observed to be dispersed from the perinuclear space to the surface of MHV-A59–infected cells (D, arrow). MT morphology is shown in mock-infected (B) and virus-infected (E, arrow) cells. MHV-A59–infected cells showed the virus-specific signal colocalizing with the MTs. Specifically, near the cell periphery, viral N signal was present on MT threads (F, arrow). As expected, no viral N signal was seen in mock-infected cells (C).
Kinetics of viral spread in primary astrocytes and colocalization with MT network
To examine whether viral particles were spreading along the MT network, primary astrocytes were mock-infected (Fig. 4, A and B) or MHV-A59–infected, and at 6 h (Fig. 4, C and D), 12 h (Fig. 4, E and F), 18 h (Fig. 4, G and H), and 24 h (Fig. 4, I and J) p.i., cells were labeled for β-tubulin (red) and viral N (green). The amount of viral N staining increased from 6 to 24 h p.i. At 6 and 12 h p.i., discrete viral particles were observed to be present on the MT threads (Fig. 4, C and E, arrow and inset), and colocalization points were mainly located at the cell periphery (Fig. 4, D and F). Later, at 18 and 24 h p.i., the anti-N signal was more dispersed throughout the whole cell, and toward the cell border, viral particles were localized on MTs (Fig. 4, G and I, arrow and inset). The number of colocalization points increased visually and were mainly located at the cell periphery and cell-to-cell junctions (Fig. 4, H and J). The number of spots containing colocalization of staining were counted and plotted against increasing time p.i., which showed that the number of colocalization spots increased from 6 h p.i. to 12–18 h p.i. and reached its maximum at 24 h p.i. (**, p < 0.01 for 6, 12, 18, and 24 h p.i. and 24 h p.i. each compared with mock by Mann–Whitney U test). ANOVA was performed by Kruskal–Wallis testing and confirmed that the variations between groups were significant (***, p < 0.001). Five different fields were obtained from each experimental group in n = 3 experiments (Fig. 4K).
Figure 4.

Kinetics of viral particle spread along microtubule threads. To observe viral spread along MT threads, primary astrocytes were mock-infected (A and B) or MHV-A59–infected at 6 h (C and D), 12 h (E and F), 18 h (G and H), and 24 h (I and J) p.i., and cells were labeled for β-tubulin (red) and viral N (green). The amount of anti-N staining increased from 6 to 24 h p.i. At 6 and 12 h p.i., discrete viral particles were observed on MT threads (C and E, arrow and inset), and colocalization points were mainly located at the cell periphery (D and F). At 18 and 24 h p.i., anti-N signal was more dispersed throughout the whole cell, and toward the cell border, viral particles were localized on MTs (G and I, arrow and inset). The number of colocalization points increased visually, and they were mainly located at the cell periphery and cell-to-cell junctions (H and J). Spots containing colocalization of signal were counted and plotted with increasing time p.i. Colocalization spots increased from 6 to 12 to 18 h p.i. and reached a maximum at 24 h p.i. (**, p < 0.01 each for 6, 12, and 18 h p.i. and 24 h p.i. as compared with mock; Mann-Whitney U test). Kruskal–Wallis testing showed that the difference was significant in a five-group comparison (***, p < 0.001). Five to six fields were analyzed for each group from n = 3 experiments (K). Error bars, S.D. AU, arbitrary units.
Confirmation of altered Cx43–β-tubulin colocalization at cell surface was due to viral particle–β-tubulin association
To understand the Cx43 and β-tubulin interaction at the cell membrane, total internal reflection fluorescence (TIRF) microscopy was performed, which specifically magnifies staining of molecules present at or in close proximity (100 nm) to the cell surface. Cells plated on glass coverslips were mock- or virus-infected and subjected to double-label immunofluorescence for Cx43 (green in Fig. 5, A and F) and β-tubulin (red in Fig. 5, B and G), and nuclei were stained with DAPI (blue). Cx43 was subjected to TIRF microscopy, keeping the imaging depth restricted to 100 nm. The whole-cell MT network was captured by epifluorescence microscopy. In mock-infected astrocytes, TIRF images showed the signal of Cx43 exclusively present at or near the cell surface (Fig. 5, A and C). In contrast, MHV-A59–infected cells showed minimal surface expression of Cx43 (Fig. 5, F and H). MT morphology appeared normal in mock-infected and MHV-A59–infected cells (Fig. 5, panels B and C and panels G and H). In mock-infected cells, merged TIRF images showed that Cx43 molecules were closely associated with tubulin threads (Fig. 5C, thin arrow) and indeed aligned along the MT threads (Fig. 5D, inset, thin arrow) or positioned at the tip of the MT threads (Fig. 5E, inset, thin arrow). MHV-A59–infected astrocytes showed an absence of MT-associated Cx43 signal near the cell surface (Fig. 5H). Insets showed that Cx43 molecules were restricted from reaching the cell surface (no TIRF signal was detected; Fig. 5I, thick arrow), and Cx43–β-tubulin association was lost (Fig. 5J, thick arrow).
Figure 5.

TIRF microscopy confirmed association of MT network with cortical Cx43, which was depleted due to MT/MHV-A59 interaction. Primary astrocytes, plated on glass coverslips, were mock-infected or infected with MHV-A59 and, upon staining with β-tubulin (red) and Cx43 (green), subjected to TIRF imaging to specifically capture the immunofluorescent signal of cortical Cx43. The imaging depth was limited to within 100 nm of the coverslip. Simultaneously, the whole-cell MT network was imaged by epifluorescence microscopy. In control astrocytes, Cx43 (A) was observed to be closely associated with positive ends of MTs or the MT network (B and C (thin arrow)). Insets show that Cx43 molecules were either precisely aligned along the MT thread (D, thin arrow) or present at the tip of the MT thread (E, thin arrow). In MHV-A59–infected cells, minimal cell surface-associated Cx43 signal was observed (F), because intracellular compartment–retained Cx43 was restricted from reaching the cell surface. MT morphology appeared normal (G). As expected, in infected cells, Cx43 molecules were not associated with positive ends of MTs (H, I, and J, thick arrow) Similarly, mock- and virus-infected cultures of primary astrocytes were stained for β-tubulin (red) and viral N (green), and TIRF imaging was performed as described before. Mock-infected astrocytes showed no viral N–specific signal (K and M), and MT morphology was normal (L and M). The inset shows a digitally magnified area of the mock-infected cell's periphery (N). MHV-A59–infected astrocyte cultures showed the presence of viral N staining (O and Q), and MT morphology is shown in P. Merged images show a pattern of viral spread at the cell surface in a single infected cell, where viral N staining colocalized with MTs (Q). The inset shows that the viral particles were aligned along the MT thread at the cell surface (R). The arrow shows the alignment of viral spread in O–R.
The loss of Cx43–β-tubulin colocalization was predicted to be due to association between viral particles and the MT network. Hence, cells were double-immunolabeled for viral N (green in Fig. 5, K and O) and β-tubulin (red in Fig. 5, L and P), and nuclei were stained with DAPI (blue). Mock-infected astrocytes showed no specific N-staining (Fig. 5K), whereas MHV-A59–infected cells showed profuse N expression (Fig. 5O, arrow). The MT network appeared normal in both mock- and virus-infected cells (Fig. 5, L and P). High magnification (×100) TIRF images were captured specifically for single infected cells, where viral spread at the surface was evident. In contrast to merged images for mock-infected cells (Fig. 5M), viral particles were observed to be colocalized with MT signal (Fig. 5Q). In digitally magnified insets, viral particles were observed to be aligned along the MT thread at the cell surface, confirming that Cx43–MT association could be replaced by MHV-A59–MT association (Fig. 5R). No viral N staining was observed in the inset (Fig. 5N).
For Cx43 localization, compared with the MT network, TIRF microscopy was used to capture the signal of Cx43 residing near the cell surface (near 100 nm), and parallel epifluorescence images were captured for the same field. Cx43 was observed to be present in profuse amounts as its characteristic punctate stain (Fig. 6, A (thin arrow) and C (merged)). In contrast, MHV-A59–infected astrocytes showed mainly perinuclear localization of Cx43 (Fig. 6, D (thick arrow) and F (merged)), which was not observed by TIRF imaging. MT morphology is shown for mock-infected (Fig. 6B) or MHV-A59–infected cells (Fig. 6E). The distance of Cx43 molecules from the nuclear centroid was measured with the help of ImageJ (Fig. 6G). For mock-infected cells, Cx43 was present ∼25.9 μm away, which was reduced to ∼12.7 μm in MHV-A59–infected cells. Data were obtained from nine different images from n = 3 biological replicates, and average ± S.D. is represented in the graph (****, p < 0.0001; t test).
Figure 6.
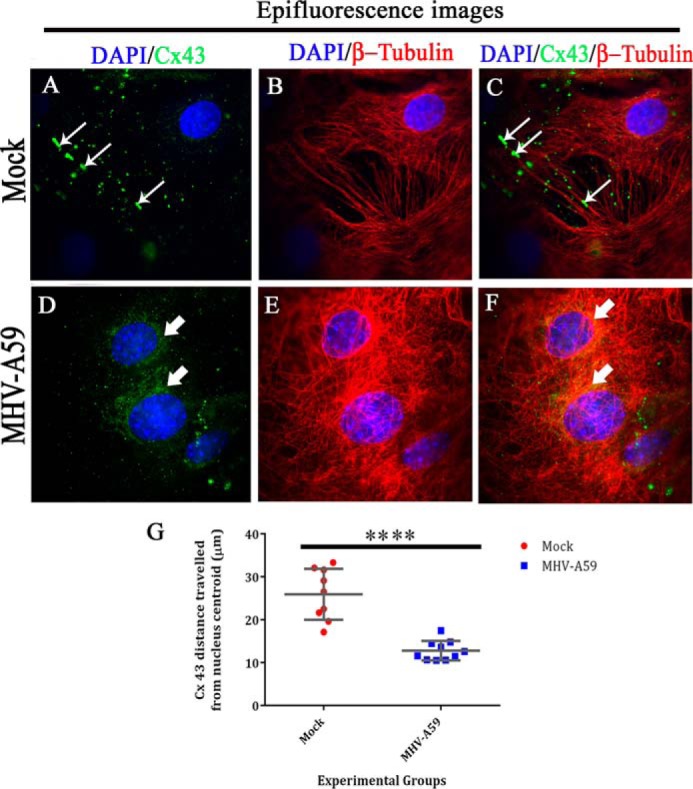
Whole-cell expression of Cx43 and β-tubulin upon MHV-A59 infection. Primary astrocytes immunolabeled for Cx43 and β-tubulin, which was subjected to TIRF microscopy, were simultaneously taken for epifluorescence microscopy to obtain the whole-cell Cx43 expression. Thus, parallel epifluorescence images were captured for the same field. Cx43 was observed to be present in profuse amounts as its characteristic punctate stain of Cx43 (A (thin arrow) and C (merged)). In contrast, MHV-A59–infected astrocytes showed mainly perinuclear localization of Cx43 (D (thick arrow) and F (merged)), which was not observed by TIRF imaging. MT morphology is shown for mock-infected (B) or MHV-A59–infected cells (E). The distance of Cx43 molecules from the nucleus (distance was calculated from nuclear centroid) was measured with the help of ImageJ (G). For mock-infected cells, Cx43 was present ∼25.9 μm away, which was reduced to ∼12.7 μm in MHV-A59–infected cells. Data were obtained from nine different images from n = 3 biological replicates, and average ± S.D. (error bars) is represented (****, p < 0.0001; t test).
Altered protein-level interaction between Cx43 and β-tubulin upon MHV-A59 infection
To investigate the direct interaction between β-tubulin and Cx43, the same number of primary astrocytes were mock-infected or infected with MHV-A59. Cells were lysed in non-denaturing conditions; mouse monoclonal anti-β-tubulin antibody was used to immunoprecipitate β-tubulin, with the help of anti-mouse MagnaBind antibody. The sample was further denatured with Laemmli's buffer and was probed by rabbit polyclonal anti-Cx43 antibody (detectable at nearly 43 kDa). Uninfected primary astrocytes showed that Cx43 was co-immunoprecipitated with β-tubulin, and this interaction was significantly reduced upon MHV-A59 infection. Five percent input of the total extract showed there was a reduction of total Cx43 upon virus infection, but the expression of internal control γ-actin (detectable at nearly 42 kDa) was similar. The beads, without any primary antibody, showed no nonspecific interaction in immunoprecipitation (Fig. 7A). Densitometric analysis showed that Cx43, bound to β-tubulin, was reduced ∼44.25% in MHV-A59–infected cells, compared with the mock-infected cells (Fig. 7B; ****, p < 0.0001; t test; n = 3).
Figure 7.

Reduction in Cx43–β-tubulin interaction in protein level upon virus infection. Primary astrocytes were either mock-infected or infected with MHV-A59 at an MOI of 2. Proteins were extracted, followed by immunoprecipitation with monoclonal anti- β-tubulin antibody and subjected to immunoblot analysis using polyclonal anti-Cx43 antibody (detected at nearly 43 kDa). γ-Actin was used as a loading control (detected at nearly 42 kDa). Inputs showed reduction of total Cx43 upon MHV-A59 infection, where γ-actin expression was not altered. Upon co-IP, substantial reduction in tubulin-associated Cx43 was observed in the MHV-A59–infected cells, compared with the mock-infected cells. Beads, used in preclearing, showed no signal upon probing with anti-Cx43 (A). Densitometric analysis showed that Cx43, associated with β-tubulin, was reduced ∼44.25% in MHV-A59–infected cells, compared with the mock-infected cells (B; ****, p < 0.0001; t test). Similarly, the virus- and mock-infected cells were co-immunoprecipitated with polyclonal anti-Cx43 antibody and probed for β-tubulin using monoclonal anti- β-tubulin antibody (detected at nearly 50 kDa). β-Tubulin was expressed in equal amount in mock– and MHV-A59–infected cells. Cx43-associated β-tubulin signal was down-regulated significantly in virus-infected cells (C). Densitometric analysis showed that β-tubulin, associated with Cx43, was depleted ∼55.61% upon virus infection (D; ****, p < 0.0001; n = 3; t test). Error bars, S.D.
The reduction in Cx43–β-tubulin interaction was verified with the help of reverse co-IP, where proteins extracted in non-denaturing conditions from both mock- and MHV-A59–infected astrocytes were immunoprecipitated with polyclonal anti-Cx43 antibody and probed with monoclonal anti-β-tubulin antibody (detected at nearly 50 kDa). As expected, the expression of β-tubulin was similar between mock- and MHV-A59–infected cells. Loading of the beads showed no nonspecific binding. Upon co-IP, there was a substantial reduction in β-tubulin signal, which was bound to Cx43 in virus-infected cells, compared with control cells (Fig. 7C). There was an approximately 55.61% reduction in β-tubulin–Cx43 interaction, demonstrated by densitometric analysis (Fig. 7D; ****, p < 0.0001; t test; n = 3).
Interaction between viral particles and β-tubulin in MHV-A59–infected primary astrocytes
Primary astrocytes were mock– or MHV-A59–infected at a multiplicity of infection (MOI) of 2, and proteins were extracted in non-denaturing conditions. Polyclonal anti-β-tubulin antibody was used to immunoprecipitate β-tubulin and associated proteins. The samples were denatured and probed for viral N by Western blotting using mouse monoclonal anti-N antibody, which is detectable at nearly 50 kDa. Samples showed similar amounts of γ-actin expression, whereas anti-N was detected only in samples from MHV-A59–infected cells but not in mock-infected ones. As expected, no nonspecific binding with the beads was observed. Anti-N signal was observed upon co-IP of β-tubulin in the infected astrocytes, suggesting that viral particles were interacting with the MT network (Fig. 8).
Figure 8.

Interaction between viral particles and β-tubulin. To understand whether reduced Cx43–β-tubulin interaction was due to interaction between viral particle and MTs, co-IP was performed using polyclonal anti-β-tubulin antibody. The immunoprecipitated samples were probed with monoclonal anti-N antibody. Samples were probed for γ-actin, as an internal control. Viral N protein was selectively detectable near 50 kDa, in the MHV-A59–infected sample only, whereas γ-actin expression was similar between mock- and virus-infected cells. MHV-A59–infected cells also showed that viral N-protein interacted with β-tubulin. The beads that were used in preclearing showed absence of nonspecific signal upon probing with anti-Cx43.
Inhibition of dynein affected Cx43 delivery to cell surface
A small-molecule dynein inhibitor, ciliobrevin D (33), was used to block dynein in primary astrocytes upon treatment at 30, 50, and 100 μm concentration for 24 h, and untreated cells were maintained in parallel. Cells were immunolabeled with β-tubulin (green in Fig. 9, A–D), Cx43 (red in Fig. 9, A–D), and nuclei were counterstained with DAPI (blue). Mock-infected cells showed normal punctate appearance of Cx43 at the cell surface (Fig. 9A, thin arrow). Ciliobrevin D treatment induced clustering of Cx43 inside primary astrocytes (Fig. 9, B–D, thick arrow), depleting the delivery of Cx43 at the cell surface. Dose-dependent treatment of ciliobrevin induced localization of Cx43 aggregates around the nucleus, suggesting that dynein might have played a crucial role in altered MT-mediated Cx43 trafficking to the cell surface.
Figure 9.
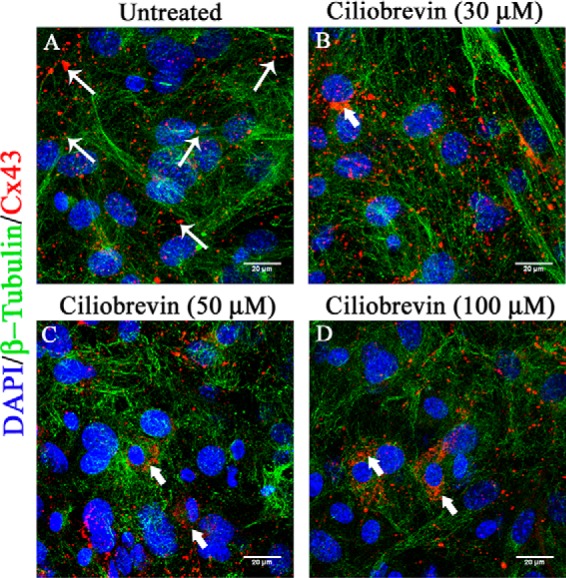
Altered Cx43 localization upon inhibition of cytoplasmic dynein. Primary astrocytes were treated with a cytoplasmic dynein inhibitor, ciliobrevin D, for 24 h, and DMSO (vehicle)-treated control cells were maintained in parallel. Cells were immunolabeled for β-tubulin (green) and Cx43 (red), and nuclei were counterstained with DAPI (blue). Mock-infected cells showed prominent presence of Cx43 at the cell surface (A, thin arrow). Dose-dependent treatment of ciliobrevin treatment induced aggregated localization of Cx43 around the nucleus, showing that inhibition of dynein restricted Cx43 surface localization (B–D, thick arrow).
Persistent loss of oligodendrocytic Cx47 due to MHV-A59 infection in mouse brain
Cx43 is reported to form heterotypic GJ plaques with oligodendrocytic partner Cx47 and also phosphorylates and stabilizes Cx47 in vivo (32). In MHV-A59 infection, whether the coupling partner of Cx43, oligodendrocytic Cx47, was altered or not was examined at the total protein level. C57Bl/6 mice were either mock– or MHV-A59–infected and sacrificed at day 5 p.i. (acute phase) and at day 30 p.i. (chronic phase). Total protein was extracted from mouse brain, and Cx47 expression (detectable at nearly 47 kDa) was compared by Western blotting. γ-Actin (detected at nearly 42 kDa) was probed as an internal control. In whole-brain protein, Cx47 levels were reduced at the peak of inflammation at day 5 p.i., whereas γ-actin expression was similar in both mock– and MHV-A59–infected brains (Fig. 10A). A cross-reactive signal is observed near 51 kDa for Cx47 Western blotting, as it was seen earlier in previous reports (34–36). The densitomeric analysis confirmed reduction in Cx47 expression, showing ∼32.78% depletion of Cx47 signal, upon virus infection (Fig. 10B; ***, p < 0.001; t test). At day 30 p.i. (peak of demyelination), Cx47 levels remained depleted in MHV-A59–infected brains, compared with similar expression of γ-actin in all brains (Fig. 10C). There was a significant ∼35.83% reduction of Cx47, which was sustained until the peak of demyelination (Fig. 10D; ****, p < 0.0001; t test).
Figure 10.

Persistent loss of oligodendrocytic Cx47 in mouse whole-brain protein. C57Bl/6 mice were mock– or MHV-A59–infected, and proteins were extracted from brain at day 5 p.i. (acute phase) and at day 30 p.i. (chronic phase). Whole-brain proteins were probed for Cx47 (detected at nearly 47 kDa) and internal control, γ-actin (detected at nearly 42 kDa). Oligodendrocytic Cx47 was reduced in brain in the acute stage of inflammation, at day 5 p.i. (A). There was ∼32.78% depletion reduction (normalized with γ-actin) of Cx47 in the whole-brain protein (B). The mean ± S.D. (error bars) incidences from three different animals are shown (***, p < 0.001). At the peak of demyelination at day 30 p.i., Cx47 was not replenished back to its normal level (C). Both of the Cx47-immunoprobed blots showed a nonspecific signal at about 51 kDa. A persistent ∼35.83% reduction in Cx47 expression signal was observed upon normalization with internal control γ-actin (D; ****, p < 0.0001). Error bars, S.D.
In situ expression of Cx47 in MHV-A59–infected mouse brain
To verify whether loss of Cx47 measured in protein extracts from MHV-A59–infected brains was also detectible in situ, mock– and MHV-A59–infected mouse brains were subjected to cryosectioning and double-label immunofluorescence for viral N (green in Fig. 11, A, E, and I) and Cx47 (red in Fig. 11, B, F, and J). Nuclei were counterstained with DAPI (blue). At day 5 p.i., MHV-A59 infection was observed in the mouse brain (Fig. 11E), whereas the mock-infected brain (Fig. 11A) as well as the day 30 p.i. MHV-A59–infected brain (Fig. 11I) showed no viral N signal. Cx47 showed a characteristic signal at the perikarya in mock-infected brains (Fig. 11B). Characteristic Cx47 signal was disrupted in the MHV-A59–infected brain (Fig. 11F). This depletion of the Cx47 perikaryonic signal was sustained at day 30 p.i. (Fig. 11J). Merged images show prominent Cx47 puncta at oligodendrocytic somata and proximal processes without infection (Fig. 11C, thin arrow), whereas depletion and degradation of Cx47 were observed around the infected brain regions at day 5 p.i. (Fig. 11G, thick arrow), and this depletion remained noticeable at day 30 p.i., when no productive viral infection was observed (Fig. 11K, thick arrow). Digitally magnified insets for Cx47 show numerous Cx47 puncta distributed in a perikaryonic fashion (Fig. 11D, thin arrow). The number as well as the characteristic staining pattern of Cx47 puncta were depleted at day 5 p.i. (Fig. 11H, thick arrow), and this loss was sustained at day 30 p.i. (Fig. 11L, thick arrow). Images (with an area of 135 × 135 μm) obtained from n = 3 biological replicates were quantified for the presence of complete perikaryonic punctate signal or disrupted signal of Cx47 (Fig. 11M). A reduction of perikaryonic signal was observed for both at day 5 and day 30 p.i. At day 5 p.i., MHV-A59–infected mice showed that ∼7.567 Cx47 perikaryonic plaques were reduced in an area of 135 × 135 μm2 (***, p < 0.001; t test), and at day 30 p.i., this depletion was ∼6.9 intact Cx47 plaques in an area of 135 × 135 μm2 (**, p < 0.01; t test) (Fig. 11M).
Figure 11.

In situ immunofluorescence data on infected brain tissue demonstrated sustained loss of perikaryonic Cx47 signal in MHV-A59–infected mouse brain. Cryosections were obtained from mock– and MHV-A59–infected mouse brains at days 5 and 30 p.i., and double-label immunofluorescence was performed for viral N (green) and Cx47 (red). Nuclei were stained with DAPI (blue). No virus-specific staining was observed for mock-infected brains (A and C), and prominent Cx47 staining was observed around oligodendrocytic somata (B and C (thin arrow); merged image). The characteristic perikaryonic signal of Cx47 was evident (D, inset). At day 5 p.i., MHV-A59–infected brains showed the presence of viral N signal (E and G). Loss of Cx47 signal was observed specifically around the virus-infected area of the brain (F and G (thick arrow)). The inset shows Cx47 immunostaining was disrupted (H, thick arrow). At the peak of demyelination at day 30 p.i., there was no infectious viral particle observed in brain (I and K). In contrast, disrupted Cx47 staining was noticeable in some areas of the brain (J and K (thick arrow)). Normal Cx47-specific signal, visible in oligodendrocytic perikarya, remained depleted at day 30 p.i. (L, inset, thick arrow). Images (with an area of 135 × 135 μm2) obtained from n = 3 biological replicates were quantified for the presence of complete perikaryonic punctate or disrupted signal of Cx47 (M). A reduction of perikaryonic Cx47 plaque count was observed at day 5 as well as at day 30 p.i. At day 5 p.i., ∼7.6 Cx47 plaques were reduced in the MHV-A59 infected mice, in an area of 135 × 135 μm2 (***, p < 0.001; t test). At day 30 p.i., ∼6.9 intact Cx47 plaques were reduced in an area of 135 × 135 μm2 (**, p < 0.01; t test) (M). Error bars, S.D.
Association of disrupted Cx47 signal with myelin marker PLP
Ablation of Cx47 is reported to be associated with loss of myelin. Hence, mouse brain sections were similarly immunostained for Cx47 (green in Fig. 12, A, E, I, M, Q, and U) and myelin marker PLP (red in Fig. 12, B, F, J, N, R, and V), and nuclei were stained with DAPI (blue). In mock-infected brains, Cx47 expression was profuse and perikaryonic. Cx47 was most abundant in and around white-matter regions, including the corpus callosum (Fig. 12A), anterior commissure (Fig. 12I), and deep cerebellar white matter (Fig. 12Q). Prominent PLP expression was observed in these regions of the mock-infected brain (Fig. 12, B, J, and R), where myelinated axon fibers were associated with normal Cx47 staining at oligodendrocyte somata (Fig. 12, C, K, and S). Upon infection with MHV-A59, at day 30 p.i., prominent loss of characteristic Cx47 signal was observed in corpus callosum (Fig. 12E), but in anterior commissure (Fig. 12M) and deep cerebellar white matter (Fig. 12U), only marginal loss in number and disrupted perikaryonic signal of Cx47-positive puncta was observed. In these brains, loss of PLP signal was specifically observed in the corpus callosum (Fig. 12F), and marginal loss of PLP signal was observed in the anterior commissure (Fig. 12N) and cerebellum (Fig. 12V). The prominent loss of Cx47 in corpus callosum was associated with loss of PLP (Fig. 12G), whereas, in other regions, the alteration was marginal (Fig. 12, O and W). Digitally magnified insets show that in mock-infected brains, perikaryonic Cx47 signal was arranged in a beads-on-a-string fashion around myelinated fibers (Fig. 12, D, L, and T, thin arrow). In MHV-A59–infected brains, characteristic Cx47 staining was diminished around the degenerated myelin fibers in corpus callosum (Fig. 12H, thick arrow), and the number of Cx47 puncta were reduced in and around other myelinated regions (Fig. 12, P and X) also.
Figure 12.

Loss of Cx47 staining was associated with loss of PLP staining in chronic phase. Brain sections obtained from mock- and MHV-A59–infected mice were immunolabeled for Cx47 (green) and myelin marker, PLP (red). Nuclei were counterstained with DAPI (blue). In mock-infected mice, Cx47 showed the characteristic stain at oligodendrocytic perikarya, specifically in and around the white-matter regions of brain (e.g. the corpus callosum (A and C), anterior commissure (I and K), and cerebellum (Q and S)). Prominent and profuse PLP staining was observed in normal corpus callosum (B and C), anterior commissure (J and K), and cerebellum (R and S). Insets show perikaryonic Cx47 staining observed in parallel to the PLP-stained myelinated axon fibers (D, L, and T (thin arrow)). MHV-A59–infected mice showed disrupted Cx47 staining, specifically in the corpus callosum (E and G). Anterior commissure (M and O) and cerebellum (U and W) showed Cx47 staining, which appeared to be normal, but the number of Cx47-positive puncta was marginally reduced. The PLP staining was reduced significantly in specific areas of the corpus callosum (F and G), but only marginal loss was evident in anterior commissure (N and O) and cerebellum (V and W). The insets show the altered expression pattern of Cx47 (H, P, and X (thick arrow)). Depletion of Cx47 staining was noticeably associated with the loss of PLP staining in the corpus callosum (H).
Discussion
The current studies demonstrate two main, novel findings. First, MHV-A59 infection altered Cx43 trafficking to the cell surface in a MT-dependent manner. We report that MHV-A59 association with the MT network restricted Cx43 trafficking to the cell membrane. Second, the oligodendrocytic GJ coupling partner of Cx43, Cx47, was down-regulated in MHV-A59 infection, and this depletion was associated with loss of the myelin marker PLP at the chronic stage of viral infection.
The MT network is known to perform important functions in bidirectional transport within the cell. ERGIC structures move from ER exit sites to the Golgi by tracking along MTs (37), and vesicles emerging from the Golgi reach the cell surface with the help of the same cytoskeletal network. As a result, trafficking of Cx43 molecules residing in these organelles was inhibited, preventing Cx43 from reaching the cell surface due to MT depolymerization induced by colchicine treatment. In contrast, in normal primary astrocytes, Cx43 did localize at the cell periphery and was organized in a beads-on-a-string array on MT threads. Results suggest that Cx43 molecules directly bind to β-tubulin at the cell surface, where it anchors MTs at their distal ends (15). Inside the cytoplasm, Cx43 molecules are reported to be associated with MTs plus the tracking protein EB1, which, in turn, interacts with p150 (Glued). This Glued protein is a component of the dynein–dynactin complex, and this complex can tether microtubules to adherens junctions, resulting in the delivery of Cx43 at the cell surface (14). Similarly, in control primary astrocytes in the current studies, it was observed that Cx43 was aligned along the MTs. Upon infection, this alignment was perturbed, and colocalization of Cx43-β-tubulin was significantly reduced. Interestingly, at the same time, viral particles were observed to be colocalized with the MT network, specifically at the cell periphery. In early time of infection (6 and 12 h p.i.), colocalization was mostly observed at the distal parts of the cells. Interestingly, at 18 and 24 h p.i., the colocalization points were more spread out, with a drastic increase in colocalization at 24 h p.i. The distribution of colocalization points between viral N and β-tubulin near the cell periphery suggests that the organelle carriers containing Cx43 might be in dynamic, competitive interaction with trafficking viral particles for MTs. These findings were further supported by TIRF microscopy showing the presence of the viral particles residing in the vicinity of the cell surface. Herpes simplex virus 1 also uses MTs for the trafficking of virions, based on TIRF microscopy showing that these virions were clustered at specific sites along the adherent cell surface (22). Similarly, in the current study, MHV-A59 particles were confirmed to be aligned along the MTs, specifically at the periphery (near 100 nm). At the surface of uninfected astrocytes, Cx43 was observed to be associated along the same conduit. In contrast, in MHV-A59–infected cells, the presence of Cx43 was not detected in the vicinity of the cell surface, by TIRF microscopy, thus providing further evidence to suggest that viral particles used the MT network as a conduit to reach the cell surface, replacing the Cx43 molecules.
Previous studies showed that another demyelinating strain of MHV, JHM, specifically uses the MT network for transneuronal spread and viral protein trafficking (20). Previous studies have also demonstrated that cytoskeletal molecules like actin filaments or MTs are exploited by various viruses to promote entry or spread from cell to cell. A rotavirus membrane glycoprotein, NSP-4, binds to MTs and arrests normal ER-to-Golgi trafficking (38). Ebola virus matrix protein VP40 is reported to interact directly with MTs (39). Although multiple studies show direct or indirect interaction of virus with MTs, there are limited data available on the specific cellular proteins that might be disrupted due to MT-mediated viral trafficking. The co-IP experiment performed here confirmed that the molecular interaction between Cx43 and β-tubulin was significantly diminished upon viral infection. Co-IP with β-tubulin and N-protein demonstrated biochemical evidence of interaction of virus with the MT network. Taken together, the phenomenon of viral interaction with MTs and transport along MTs might be an important mechanism of the normal cell-to-cell propagation pathway of MHV-A59. To the best of our knowledge, this is the first report showing MT network–mediated trafficking of MHV-A59 directly affecting trafficking of Cx43 molecules to the cell surface.
Whereas this study showed that MHV-A59 interacted with MTs and down-regulated interaction between Cx43 and β-tubulin, it is feasible that one or more accessory proteins are also involved in this interaction. Prior studies suggested that the dynein–dynactin complex has an important role in Cx43 trafficking. It has also been shown that negative-stranded RNA virus, Hantaan virus, N protein uses MTs for intracellular trafficking and that the movement occurs via molecular motors, such as dynein (40). In addition, during adenovirus infection, cytoplasmic dynein is reported to mediate interaction between viral capsid and MTs (25). Thus, there may be a crucial role of molecular motors and other MT-associated proteins involved in altered trafficking of Cx43. This is supported by the current studies, where the inhibition of cytoplasmic dynein by ciliobrevin D resulted in impaired delivery of Cx43 to the cell surface. Hence, further investigation is warranted to determine the specific molecular motors involved in transport of virus that may mediate retention of Cx43 in the ER and/or ERGIC.
In contrast to other Cxs, Cx43 is specifically known to interact with tubulins (15). There are other studies available that show that Cx26 is highly dependent on the MT network for GJ formation, compared with partial dependence of cell surface delivery of other Cxs, like Cx32 and Cx43 (41). In addition, nocodazole-induced destabilization of the MT network also affected Cx30-mediated GJ plaque formation, proving its partial dependence on MTs for delivery to the cell surface (42). Astrocytes are reported to express Cx30 and Cx26, along with a major expression of Cx43 (43). Hence, whether MHV-A59 infection also disrupts MT-dependent transport of other astrocytic connexins can be an important focus of future work.
This study provides important information about the interaction between the MT network and MHV-A59, and results support its direct involvement in diminishing Cx43-specific GJIC formation. MHV-A59–induced neuroinflammation in the acute stage of infection leads to demyelination and axonal loss in the chronic stage (28). MHV infection produces persistent, productive infection in primary astroglial cell cultures, but the pathological importance of glial cell infection remains unclear (44). Meningeal fibroblasts also express a large amount of Cx43 (45). Virus infection might therefore also disrupt glia-meningeal fibroblast GJ communication and have a crucial role in altered blood–brain barrier permeability observed during neuroinflammation.5
Recent findings show that astrocytic Cx43 not only forms GJICs between astrocytes by Cx43–Cx43 channels, but also with oligodendrocytes by Cx43–Cx47 channels (46). Cx43–Cx47–mediated GJICs have a crucial role in maintaining oligodendrocytic K+ buffering and nutrient homeostasis (3). Moreover, Cx43 has been shown to control Cx47 phosphorylation and stability in GJs, and the loss of Cx43 may result in the secondary loss of Cx47 (32). According to a recent study on multiple sclerosis, disruption of Cx43–Cx47–mediated GJIC is hypothesized to be a mechanism by which demyelinating plaques expand (47). Our study was extended to observe the fate of Cx47 during MHV-A59–induced neuroinflammation. It was found that Cx47 was down-regulated both at acute and chronic phases of MHV-A59 infection. In contrast to Cx43 expression, which was replenished at day 30 p.i., a significant 35.83 ± 6.5% depletion was observed for Cx47 expression even at day 30 p.i. (peak of demyelination) (Fig. 10). Interestingly, it was reported that Cx47 expression is very dynamic during both development and the de- or remyelination process in the adult brain (48). In situ immunofluorescence data here demonstrated that Cx47 was depleted around infected areas at day 5 p.i. At day 30 p.i., the Cx47 signal was lost in and around white-matter regions, albeit a moderate overexpression of Cx47 was observed in the thalamus.
In recent clinical investigations of multiple sclerosis, it was observed that oligodendrocyte GJs were lost in myelinated fibers, and astrocyte-oligodendrocyte GJ connectivity was disrupted in multiple sclerosis lesions. From acute to chronic phases of demyelinating disease, oligodendrocytic GJs only recover partially. In contrast, as a part of the host remyelination process, oligodendrocyte precursor cells are recruited and express Cx47. These Cx47-expressing cells appear to re-establish limited metabolic connectivity with astrocytes (49). Our study showed that the loss of Cx47 staining in white matter was associated with loss of the myelin marker PLP at the chronic phase of MHV-A59 infection. The Cx47 characteristic perikaryonic expression in oligodendrocyte somata and proximal processes was noticeably disrupted in the white-matter regions (Fig. 12). Loss of functional Cx47 is also reported to be associated with microglial activation (34). Microglial activation and microglia-induced myelin stripping are also hallmarks of MHV-A59–induced demyelination (50). MHV-A59–induced demyelination is driven by multimechanistic pathways, including oligodendroglial and neuronal infection, death and damage, and microglial activation (50, 51). Along with these factors, MHV-A59 infection in astrocytes may affect glial GJ communication. Whether the loss of panglial GJ communication plays a pivotal role in the process of myelin loss warrants further investigation.
Experimental procedures
Preparation of mixed glial cultures
Mixed glial cultures were established from newborn mice (day 0 to 1) using a protocol described previously (52). Briefly, following removal of meninges, brain tissues were homogenized and incubated in a rocking water bath set at 37 °C for 30 min in Hanks' balanced salt solution (Gibco), containing 300 μg/ml DNase I (Sigma) and 0.25% trypsin (Sigma). Enzyme-dissociated cells were triturated in the presence of 0.25% FBS, followed by a wash and centrifugation (300 × g for 10 min). The pellet was again resuspended in Hanks' balanced salt solution and passed through a 70-μm nylon mesh. A second wash and centrifugation (300 × g for 10 min) was performed, and finally the cell pellet was diluted with astrocyte-specific medium (DMEM containing 1% penicillin–streptomycin, 1% l-glutamine, and 10% FBS). Cells were plated and allowed to adhere for 1 day in a humidified CO2 incubator at 37 °C. After 24 h, all non-adherent cells were removed, and fresh astrocyte-specific medium was added. Adherent cells were maintained as mixed glial culture in astrocyte-specific medium until confluence, with a medium change every 3–4 days.
Isolation of primary astrocytes from mixed glia
When the mixed glia culture was observed to be confluent, the addition of new medium was stopped for 10 days to allow differential adhesion of astrocytes and microglia. To dislodge the microglia, which grow over a strongly adherent astrocyte layer, the culture flask was thoroughly agitated in an orbital incubator shaker (180 rpm for 45 min at 37 °C). Weakly adherent microglia cells came off into suspension, and these cells suspended in the culture medium were removed. The remaining adherent monolayers of cells were used as enriched astrocyte cultures for further experimentation (19).
Infection of primary astrocytes with MHV-A59
A neurotropic demyelinating strain of coronavirus, MHV-A59, was used to understand the mechanism of retention of Cx43 in the intracellular compartment, similar to previous studies (19). Primary astrocytes were infected with inoculation medium (DMEM containing 1% penicillin–streptomycin and 1% glutamine with 2% FBS) containing MHV-A59 at an MOI of 2. Virus particles were allowed to adhere for 1 h at 37 °C in a humidified CO2 incubator. After 1 h of incubation, infected cells were maintained in astrocyte-specific medium containing 10% normal serum. Infected cultures were subjected to different studies at specified time points p.i.
Immunofluorescence microscopy
Immunofluorescence studies were done according to a protocol described previously (19). For standard immunofluorescence, primary astrocytes were plated on etched glass coverslips. The cells were fixed with 4% paraformaldehyde (PFA), followed by permeabilization with PBS containing 0.5% Triton X-100. Then cells were blocked with PBS containing 0.5% Triton X-100 and 2.5% heat-inactivated goat serum and then incubated with primary antisera diluted in blocking solution for 1 h. To remove nonspecifically bound antibody, cells were washed and then labeled with secondary antisera diluted in blocking solution. Finally, the cells were washed with PBS, mounted with mounting medium containing DAPI (VectaShield, Vector Laboratories), and visualized using an Axio Observer microscope with the Apotome module (Carl Zeiss). Images were acquired and processed with Zen 2012 software (Carl Zeiss) or using a Zeiss confocal microscope (LSM710). The sources and dilutions of primary antibodies are listed in Table 1.
Table 1.
Primary antibodies used for immunofluorescence, co-IP, and Western blotting
Secondary antibodies were as described in the text.
| Antibodies | Dilution/Amount used | Source |
|---|---|---|
| Polyclonal anti Cx43 antibody (rabbit) | Immunofluorescence: 1:200 | C6219, Sigma |
| Western blotting: 1:1000 | ||
| Polyclonal anti β-tubulin antibody (rabbit) | Immunofluorescence: 1:100 | BB-AB0119, Biobharati LifeSciences Pvt. Ltd. (Kolkata, India) |
| Co-IP: 3 μg | ||
| Monoclonal anti β-tubulin antibody (mouse) | Immunofluorescence: 1:200 | Clone TUB 2.1, T4026, Sigma |
| Co-IP: 2 μg | ||
| Monoclonal anti Nucleocapsid (N) antibody (mouse) | Immunofluorescence: 1:50 | Kindly provided by Dr. Julian Leibowitz |
| Western blotting: 1:100 | ||
| Polyclonal anti-Cx47 antibody (rabbit) | Immunofluorescence: 1:50 | Invitrogen (catalog number: 36-4700) |
| Western blotting: 1:1000 | ||
| Anti-PLP antibody (rat) | Immunofluorescence: 1:1 | Kindly provided by Judith B. Grinspan (Children's Hospital of Philadelphia, Philadelphia, PA) |
TIRF microscopy
Primary astrocytes were plated on uncoated glass-bottom chambers and allowed to grow until confluence. The cells were double-immunolabeled for either Cx43 and β-tubulin or β-tubulin and N protein. Cells were counterstained with DAPI. One ml of PBS was added onto the chamber, and cells were imaged with an Olympus IX83 inverted microscope with a CellTIRF system. The images were taken with a ×100, 1.51 numeric aperture TIRF objective, using a 488-nm laser. Epifluorescence images were captured in parallel for blue, green, and red channels using the DAPI, FITC, and TRITC filter cubes. All TIRF image processing was performed with cellSens (Olympus Life Science) and ImageJ software (National Institutes of Health).
Co-immunoprecipitation
co-IPs were performed according to a protocol described previously (14) with minor modifications. Primary astrocytes were washed with PBS and harvested with PBS containing protease inhibitors. Following centrifugation, cells were resuspended with PBS containing 1% Triton X-100, 1 mm EDTA, 1× cOmplete protease inhibitor (Roche Applied Science), and phosphatase inhibitors (1 mm NaVO4 and 10 mm NaF) at 4 °C. Cells were homogenized at regular intervals, and after successful lysis in non-denaturing conditions, samples were centrifuged at 1000 × g for 5 min at 4 °C. Then the supernatant was incubated with MagnaBind IgG beads (Thermo Fisher Scientific, Rockford, IL) for 1 h at 4 °C, to clear any protein that binds nonspecifically to the beads. After preclearing, samples were incubated with 2–3 μg of primary antibody overnight at 4 °C. The primary antibody-specific MagnaBind particles were then added to the samples and incubated for 2 h at 4 °C. The primary antibody-bound MagnaBind IgG particles were isolated using a magnetic separator (Miltenyi Biotec, Tubingen, Germany). The beads were washed three times, and finally the bound material was eluted in SDS-PAGE sample buffer. Samples were boiled and separated by SDS-PAGE, transferred to PVDF, and probed for target protein by Western blotting, as described previously (19). The sources and dilutions of primary antibodies are listed in Table 1.
Inoculation of mice
Use of animals and all experimental procedures were reviewed and approved by the Indian Institute of Science Education and Research Kolkata. Animal protocols were followed according to the guidelines of the Committee for the Purpose of Control And Supervision of Experiments on Animals, India. MHV-free, 4-week-old, C57BL/6 mice were intracranially inoculated with one-half of the 50% lethal dose (one-half LD50) of MHV-A59 (2000 pfu). The mice were monitored daily for signs and symptoms of disease. Mice were mock-infected with PBS-BSA and were maintained in parallel. Mice were sacrificed at the peak of inflammation (day 5 p.i.) and peak of demyelination (day 30 p.i.). Brain tissues were harvested for experimentation.
Isolation of total protein from brain
To analyze Cx47 expression in vivo, total protein was extracted from brain as described previously, with minor modifications (53). Brains were flash-frozen in liquid N2 and immediately dissolved in PBS containing 2% SDS, 1× EDTA-free Complete protease inhibitor (Roche Applied Science), and phosphatase inhibitors (1 mm NaVO4 and 10 mm NaF). Lysates were sonicated on ice at 30% amplitude of 30 kHz for 0.5-s intervals using a Sartorius Labsonic M sonicator. Samples were centrifuged for 20 min at maximum speed at 4 °C in an Eppendorf 5415 R centrifuge. Supernatants were taken, and total protein content was estimated with a Pierce BCA protein assay kit (Thermo Scientific). Twenty micrograms of total protein were loaded for each sample, and they were probed for Cx47 using rabbit polyclonal anti-Cx47 antibody (Table 1) as well as for an internal control, γ-actin, by Western blotting.
Tissue processing and double-label immunofluorescence of frozen sections
Mock– and MHV-A59–infected mice (at day 5 and day 30 p.i.) were perfused transcardially with PBS, followed by cold PBS containing 4% PFA. Brains were harvested in 4% PFA for 6 h and then placed at 4 °C for 4 h in 10% sucrose, followed by 30% sucrose overnight. Tissues were embedded with OCT medium (Tissue Tek, Hatfield, PA), sectioned sagitally with the help of a cryotome (Thermo Scientific) to 10-μm thickness, and mounted on charged glass slides. Immunostaining was done as described previously (54). Frozen tissue sections were washed with PBS at room temperature to remove cryomatrix. Tissues then were incubated for 1 h at room temperature with 1 m glycine in PBS to reduce nonspecific cross-linking, followed by a 10-min incubation at room temperature with 1 mg/ml NaBH4 in PBS to reduce autofluorescence. Slides were washed with PBS and incubated with blocking serum containing PBS with 0.5% Triton X-100 and 2.5% goat serum. The sections were incubated overnight at 4 °C with a primary antisera diluted in blocking serum, washed, and subsequently incubated with secondary antiserum diluted in PBS with goat serum for 2 h at room temperature. All incubations were carried out in a humidified chamber. After PBS washing, sections were mounted with DAPI containing mounting medium and were imaged using a Zeiss confocal microscope (LSM710), as specified. The images were processed with ImageJ or Zen 2010 (Carl Zeiss) software.
Statistical analysis
All values shown are mean values ± S.D., and data are represented as points in the scatter plots. Student's unpaired t test was used to validate significance between two groups, and the tests were two-tailed. Kruskal–Wallis ANOVA was performed for multiple-group comparison in colocalization studies, after which pairwise comparisons were made using the post hoc Mann–Whitney U test for multiple testing. For all experiments, statistical significance was set at p < 0.05.
Author contributions
R. B. designed and performed the experiments, analyzed the data, and wrote the manuscript. A. B. performed the experiments and analyzed the data. D. T. performed the analysis of the data and assisted in rewriting of the manuscript. J. D. S. led all aspects of this work, including experimental design; participated in or supervised all experimental procedures; analyzed and interpreted data; and critically revised the manuscript.
Supplementary Material
Acknowledgments
We thank the Indian Institute of Science Education and Research Kolkata apoptome and confocal facility and Ritabrata Ghosh for apoptome and confocal microscopy assistance. We also thank Dr. Bidisha Sinha and Arikta Biswas for the TIRF facility and for assistance in TIRF imaging. We also thank Kenneth S. Shindler for critically reading and editing the manuscript. The IISER-K TIRF facility is supported by Wellcome Trust DBT-India Alliance Grant IA/I/13/1/500885 T.
This work was supported by Research Grants BT/PR14260/MED/30/437/2010 and BT/PR4530/MED/30/715/2012 from the Department of Biotechnology, India; Research Grant RG3774A2/1 from the National Multiple Sclerosis Society (United States); and a grant from the Indian Institute of Science Education and Research Kolkata (IISER-K) India start-up fund (to J. D. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1 and S2.
R. Basu, A. Bose, D. Thomas, and J. Das Sarma, manuscript in preparation.
- MT
- microtubule
- GJ
- gap junction
- Cx
- connexin
- ER
- endoplasmic reticulum
- GJIC
- gap junction intercellular communication
- MHV
- mouse hepatitis virus
- ERGIC
- endoplasmic reticulum Golgi intermediate complex
- p.i.
- postinfection
- N
- nucleocapsid
- ANOVA
- analysis of variance
- TIRF
- total internal reflection fluorescence
- IP
- immunoprecipitation
- MOI
- multiplicity of infection
- PFA
- paraformaldehyde.
References
- 1. Das Sarma J., Wang F., and Koval M. (2002) Targeted gap junction protein constructs reveal connexin-specific differences in oligomerization. J. Biol. Chem. 277, 20911–20918 [DOI] [PubMed] [Google Scholar]
- 2. Iacobas D. A., Urban-Maldonado M., Iacobas S., Scemes E., and Spray D. C. (2003) Array analysis of gene expression in connexin-43 null astrocytes. Physiol. Genomics 15, 177–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lutz S. E., Zhao Y., Gulinello M., Lee S. C., Raine C. S., and Brosnan C. F. (2009) Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J. Neurosci. 29, 7743–7752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sofroniew M. V., and Vinters H. V. (2010) Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Charles A. C., Merrill J. E., Dirksen E. R., and Sanderson M. J. (1991) Intercellular signaling in glial cells: calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron 6, 983–992 [DOI] [PubMed] [Google Scholar]
- 6. Charles A. C., Naus C. C., Zhu D., Kidder G. M., Dirksen E. R., and Sanderson M. J. (1992) Intercellular calcium signaling via gap junctions in glioma cells. J. Cell Biol. 118, 195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Finkbeiner S. (1992) Calcium waves in astrocytes-filling in the gaps. Neuron 8, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 8. Sáez J. C., Connor J. A., Spray D. C., and Bennett M. V. (1989) Hepatocyte gap junctions are permeable to the second messenger, inositol 1,4,5-trisphosphate, and to calcium ions. Proc. Natl. Acad. Sci. U.S.A. 86, 2708–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tabernero A., Giaume C., and Medina J. M. (1996) Endothelin-1 regulates glucose utilization in cultured astrocytes by controlling intercellular communication through gap junctions. Glia 16, 187–195 [DOI] [PubMed] [Google Scholar]
- 10. Naus C. C., Bechberger J. F., Zhang Y., Venance L., Yamasaki H., Juneja S. C., Kidder G. M., and Giaume C. (1997) Altered gap junctional communication, intercellular signaling, and growth in cultured astrocytes deficient in connexin43. J. Neurosci. Res. 49, 528–540 [DOI] [PubMed] [Google Scholar]
- 11. Giaume C., and McCarthy K. D. (1996) Control of gap-junctional communication in astrocytic networks. Trends Neurosci. 19, 319–325 [DOI] [PubMed] [Google Scholar]
- 12. Dermietzel R., Gao Y., Scemes E., Vieira D., Urban M., Kremer M., Bennett M. V., and Spray D. C. (2000) Connexin43 null mice reveal that astrocytes express multiple connexins. Brain Res. Brain Res. Rev. 32, 45–56 [DOI] [PubMed] [Google Scholar]
- 13. Lauf U., Giepmans B. N., Lopez P., Braconnot S., Chen S. C., and Falk M. M. (2002) Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc. Natl. Acad. Sci. U.S.A. 99, 10446–10451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shaw R. M., Fay A. J., Puthenveedu M. A., von Zastrow M., Jan Y. N., and Jan L. Y. (2007) Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 128, 547–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Giepmans B. N., Verlaan I., Hengeveld T., Janssen H., Calafat J., Falk M. M., and Moolenaar W. H. (2001) Gap junction protein connexin-43 interacts directly with microtubules. Curr. Biol. 11, 1364–1368 [DOI] [PubMed] [Google Scholar]
- 16. Crow D. S., Beyer E. C., Paul D. L., Kobe S. S., and Lau A. F. (1990) Phosphorylation of connexin43 gap junction protein in uninfected and Rous sarcoma virus-transformed mammalian fibroblasts. Mol. Cell. Biol. 10, 1754–1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Köster-Patzlaff C., Hosseini S. M., and Reuss B. (2007) Persistent Borna disease virus infection changes expression and function of astroglial gap junctions in vivo and in vitro. Brain Res. 1184, 316–332 [DOI] [PubMed] [Google Scholar]
- 18. Fatemi S. H., Folsom T. D., Reutiman T. J., and Sidwell R. W. (2008) Viral regulation of aquaporin 4, connexin 43, microcephalin and nucleolin. Schizophr. Res. 98, 163–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Basu R., Banerjee K., Bose A., and Das Sarma J. (2015) Mouse hepatitis virus infection remodels connexin43-mediated gap junction intercellular communication in vitro and in vivo. J. Virol. 90, 2586–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pasick J. M., Kalicharran K., and Dales S. (1994) Distribution and trafficking of JHM coronavirus structural proteins and virions in primary neurons and the OBL-21 neuronal cell line. J. Virol. 68, 2915–2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Biswas K., and Das Sarma J. (2014) Effect of microtubule disruption on neuronal spread and replication of demyelinating and nondemyelinating strains of mouse hepatitis virus in vitro. J. Virol. 88, 3043–3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mingo R. M., Han J., Newcomb W. W., and Brown J. C. (2012) Replication of herpes simplex virus: egress of progeny virus at specialized cell membrane sites. J. Virol. 86, 7084–7097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hollinshead M., Rodger G., Van Eijl H., Law M., Hollinshead R., Vaux D. J., and Smith G. L. (2001) Vaccinia virus utilizes microtubules for movement to the cell surface. J. Cell Biol. 154, 389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yea C., Dembowy J., Pacione L., and Brown M. (2007) Microtubule-mediated and microtubule-independent transport of adenovirus type 5 in HEK293 cells. J. Virol. 81, 6899–6908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kelkar S. A., Pfister K. K., Crystal R. G., and Leopold P. L. (2004) Cytoplasmic dynein mediates adenovirus binding to microtubules. J. Virol. 78, 10122–10132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiao P. J., and Samulski R. J. (2012) Cytoplasmic trafficking, endosomal escape, and perinuclear accumulation of adeno-associated virus type 2 particles are facilitated by microtubule network. J. Virol. 86, 10462–10473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Das Sarma J. (2010) A mechanism of virus-induced demyelination. Interdiscip. Perspect. Infect. Dis. 2010, 109239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lavi E., Gilden D. H., Wroblewska Z., Rorke L. B., and Weiss S. R. (1984) Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology 34, 597–603 [DOI] [PubMed] [Google Scholar]
- 29. Bugiani M., Al Shahwan S., Lamantea E., Bizzi A., Bakhsh E., Moroni I., Balestrini M. R., Uziel G., and Zeviani M. (2006) GJA12 mutations in children with recessive hypomyelinating leukoencephalopathy. Neurology 67, 273–279 [DOI] [PubMed] [Google Scholar]
- 30. Uhlenberg B., Schuelke M., Rüschendorf F., Ruf N., Kaindl A. M., Henneke M., Thiele H., Stoltenburg-Didinger G., Aksu F., Topaloğlu H., Nürnberg P., Hübner C., Weschke B., and Gärtner J. (2004) Mutations in the gene encoding gap junction protein α12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am. J. Hum. Genet. 75, 251–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Orthmann-Murphy J. L., Abrams C. K., and Scherer S. S. (2008) Gap junctions couple astrocytes and oligodendrocytes. J. Mol. Neurosci. 35, 101–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. May D., Tress O., Seifert G., and Willecke K. (2013) Connexin47 protein phosphorylation and stability in oligodendrocytes depend on expression of Connexin43 protein in astrocytes. J. Neurosci. 33, 7985–7996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Firestone A. J., Weinger J. S., Maldonado M., Barlan K., Langston L. D., O'Donnell M., Gelfand V. I., Kapoor T. M., and Chen J. K. (2012) Small-molecule inhibitors of the AAA+ ATPase motor cytoplasmic dynein. Nature 484, 125–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tress O., Maglione M., Zlomuzica A., May D., Dicke N., Degen J., Dere E., Kettenmann H., Hartmann D., and Willecke K. (2011) Pathologic and phenotypic alterations in a mouse expressing a connexin47 missense mutation that causes Pelizaeus-Merzbacher-like disease in humans. PLoS Genet. 7, e1002146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li X., Penes M., Odermatt B., Willecke K., and Nagy J. I. (2008) Ablation of Cx47 in transgenic mice leads to the loss of MUPP1, ZONAB and multiple connexins at oligodendrocyte-astrocyte gap junctions. Eur. J. Neurosci. 28, 1503–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Markoullis K., Sargiannidou I., Gardner C., Hadjisavvas A., Reynolds R., and Kleopa K. A. (2012) Disruption of oligodendrocyte gap junctions in experimental autoimmune encephalomyelitis. Glia 60, 1053–1066 [DOI] [PubMed] [Google Scholar]
- 37. Brandizzi F., and Barlowe C. (2013) Organization of the ER-Golgi interface for membrane traffic control. Nat. Rev. Mol. Cell Biol. 14, 382–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu A., Bellamy A. R., and Taylor J. A. (2000) Immobilization of the early secretory pathway by a virus glycoprotein that binds to microtubules. EMBO J. 19, 6465–6474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ruthel G., Demmin G. L., Kallstrom G., Javid M. P., Badie S. S., Will A. B., Nelle T., Schokman R., Nguyen T. L., Carra J. H., Bavari S., and Aman M. J. (2005) Association of ebola virus matrix protein VP40 with microtubules. J. Virol. 79, 4709–4719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ramanathan H. N., Chung D. H., Plane S. J., Sztul E., Chu Y. K., Guttieri M. C., McDowell M., Ali G., and Jonsson C. B. (2007) Dynein-dependent transport of the hantaan virus nucleocapsid protein to the endoplasmic reticulum-Golgi intermediate compartment. J. Virol. 81, 8634–8647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martin P. E., Blundell G., Ahmad S., Errington R. J., and Evans W. H. (2001) Multiple pathways in the trafficking and assembly of connexin 26, 32 and 43 into gap junction intercellular communication channels. J. Cell Sci. 114, 3845–3855 [DOI] [PubMed] [Google Scholar]
- 42. Qu C., Gardner P., and Schrijver I. (2009) The role of the cytoskeleton in the formation of gap junctions by Connexin 30. Exp. Cell Res. 315, 1683–1692 [DOI] [PubMed] [Google Scholar]
- 43. Altevogt B. M., and Paul D. L. (2004) Four classes of intercellular channels between glial cells in the CNS. J. Neurosci. 24, 4313–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lavi E., Suzumura A., Hirayama M., Highkin M. K., Dambach D. M., Silberberg D. H., and Weiss S. R. (1987) Coronavirus mouse hepatitis virus (MHV)-A59 causes a persistent, productive infection in primary glial cell cultures. Microb. Pathog. 3, 79–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Arishima H., Sato K., and Kubota T. (2002) Immunohistochemical and ultrastructural study of gap junction proteins connexin26 and 43 in human arachnoid villi and meningeal tumors. J. Neuropathol. Exp. Neurol. 61, 1048–1055 [DOI] [PubMed] [Google Scholar]
- 46. Orthmann-Murphy J. L., Freidin M., Fischer E., Scherer S. S., and Abrams C. K. (2007) Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J. Neurosci. 27, 13949–13957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Markoullis K., Sargiannidou I., Schiza N., Hadjisavvas A., Roncaroli F., Reynolds R., and Kleopa K. A. (2012) Gap junction pathology in multiple sclerosis lesions and normal-appearing white matter. Acta Neuropathol. 123, 873–886 [DOI] [PubMed] [Google Scholar]
- 48. Parenti R., Cicirata F., Zappalà A., Catania A., La Delia F., Cicirata V., Tress O., and Willecke K. (2010) Dynamic expression of Cx47 in mouse brain development and in the cuprizone model of myelin plasticity. Glia 58, 1594–1609 [DOI] [PubMed] [Google Scholar]
- 49. Kleopa K. A., Sargiannidou I. and Markoullis K. (2013) Connexin pathology in chronic multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Neuroimmunol. 4, 45–58 [Google Scholar]
- 50. Das Sarma J., Kenyon L. C., Hingley S. T., and Shindler K. S. (2009) Mechanisms of primary axonal damage in a viral model of multiple sclerosis. J. Neurosci. 29, 10272–10280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kenyon L. C., Biswas K., Shindler K. S., Nabar M., Stout M., Hingley S. T., Grinspan J. B., and Das Sarma J. (2015) Gliopathy of demyelinating and non-demyelinating strains of mouse hepatitis virus. Front. Cell. Neurosci. 9, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Marek R., Caruso M., Rostami A., Grinspan J. B., and Das Sarma J. (2008) Magnetic cell sorting: a fast and effective method of concurrent isolation of high purity viable astrocytes and microglia from neonatal mouse brain tissue. J. Neurosci. Methods 175, 108–118 [DOI] [PubMed] [Google Scholar]
- 53. Ezan P., André P., Cisternino S., Saubaméa B., Boulay A. C., Doutremer S., Thomas M. A., Quenech'du N., Giaume C., and Cohen-Salmon M. (2012) Deletion of astroglial connexins weakens the blood-brain barrier. J. Cereb. Blood Flow Metab. 32, 1457–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Das Sarma J., Iacono K., Gard L., Marek R., Kenyon L. C., Koval M., and Weiss S. R. (2008) Demyelinating and nondemyelinating strains of mouse hepatitis virus differ in their neural cell tropism. J. Virol. 82, 5519–5526 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.