

Abstract
Skin cancer is the most common cancer, and exposure to ultraviolet (UV) radiation, namely UVA and UVB, is the major risk factor for skin cancer development. UVA is significantly less effective in causing direct DNA damage than UVB, but UVA has been shown to increase skin cancer risk. The mechanism by which UVA contributes to skin cancer remains unclear. Here, using RNA-Seq, we show that UVA induces autophagy and lysosomal gene expression, including the autophagy receptor and substrate p62. We found that UVA activates transcription factor EB (TFEB), a known regulator of autophagy and lysosomal gene expression, which, in turn, induces p62 transcription. Next, we identified a novel relationship between p62 and cyclooxygenase-2 (COX-2), a prostaglandin synthase critical for skin cancer development. COX-2 expression was up-regulated by UVA-induced p62, suggesting that p62 plays a role in UVA-induced skin cancer. Moreover, we found that p62 stabilizes COX-2 protein through the p62 ubiquitin-associated domain and that p62 regulates prostaglandin E2 production in vitro. In a syngeneic squamous cell carcinoma mouse model, p62 knockdown inhibited tumor growth and metastasis. Furthermore, p62-deficient tumors exhibited reduced immune cell infiltration and increased cell differentiation. Because prostaglandin E2 is known to promote pro-tumorigenic immune cell infiltration, increase proliferation, and inhibit keratinocyte differentiation in vivo, this work suggests that UVA-induced p62 acts through COX-2 to promote skin tumor growth and progression. These findings expand our understanding of UVA-induced skin tumorigenesis and tumor progression and suggest that targeting p62 can help prevent or treat UVA-associated skin cancer.
Keywords: autophagy, cancer, metastasis, oncogene, p62 (sequestosome 1(SQSTM1)), COX-2, skin cancer, skin tumor, tumor growth, ultraviolet radiation
Introduction
Skin cancer is the most common cancer, with about 3.5 million cases diagnosed each year. Unlike many cancers, the incidence of skin cancer is rising worldwide. Exposure to ultraviolet (UV) radiation, namely UVA (315–400 nm) and UVB (285–315 nm), is the major risk factor for the development of skin cancer. Of these, UVA accounts for ∼95% of UV in sunlight, and tanning beds emit UVA in doses 12-fold higher than the sun (1). However, UVA is significantly less effective in causing direct DNA damage than UVB, which led many to believe that it was non-tumorigenic. It has since been shown that UVA can induce skin carcinogenesis in vivo (2–4), and indoor tanning, even intermittently, significantly increases skin cancer risk (5, 6). However, the mechanism of UVA's contribution to skin cancer remains unclear.
Macroautophagy (hereafter referred to as autophagy) is a cellular self-eating process that targets unwanted or damaged organelles and proteins to lysosomes for degradation through autophagosomes (7, 8). The protein p62, a multidomain protein also known as sequestosome 1 (SQSTM1), acts as an autophagy adaptor and substrate for the selective inclusion of cargo. During autophagy, p62 binds to LC3 in the autophagosomal membrane through the LC3-interacting region, as well as to polyubiquitinated proteins and protein aggregates bound for degradation through the ubiquitin-associated (UBA)2 domain (9, 10). In addition to its role as an autophagy adaptor, p62 forms interactions with a number of proteins to activate pro-tumorigenic signaling pathways. p62 is found to be up-regulated in several human cancers, including lung cancer, breast cancer, melanoma, and skin squamous cell carcinoma (11–15). Recent studies have demonstrated that p62 promotes tumor formation and progression (16, 17) through regulating NF-κB (18, 19) and NRF2 (20–22). Furthermore, p62 expression was induced by Ras activation during tumorigenesis (19). Recently, we have found that p62 binds to the oncogenic transcription factor Twist1 and promotes Twist1 stabilization (15). This interaction promotes the epithelial-mesenchymal transition and, hence, skin tumor growth and metastasis (15). Identifying the underlying molecular and cellular mechanisms of p62 regulation and function may elucidate mechanisms key to skin tumorigenesis and tumor progression.
One p62 regulator is a member of the microphthalmia-associated transcription factor family, transcription factor EB (TFEB), a master regulator of autophagy and lysosomal gene expression (23–25). TFEB binds to the coordinated lysosomal expression and regulation (CLEAR) binding site found in the promoter of many autophagy and lysosomal genes to activate gene transcription and ultimately the degradation of autophagy substrates (23, 25, 26). TFEB activation is regulated primarily through phosphorylation. Under nutrient-rich conditions, TFEB is primarily cytosolic, phosphorylated, and inactive (27). Upon nutrient deprivation, TFEB rapidly translocates to the nucleus to induce transcription of autophagy and lysosomal genes (27). However, the role of TFEB in UVA response is unknown.
Another crucial oncogene in skin cancer is cyclooxygenase 2 (COX-2). COX-2 is an inducible prostaglandin synthase that catalyzes the rate-limiting step of prostaglandin E2 (PGE2) synthesis. COX-2 expression is induced by a number of stimuli, including UVA (28), and is negatively regulated by the ubiquitin-proteasome system (29). COX-2 acts through PGE2 signaling to promote proliferation (30), invasion (31), and inflammation (32, 33). Overexpression of COX-2 occurs in many cancers, including skin cancer (34, 35), and is correlated with poor prognosis (34). Transgenic mice with overexpression of COX-2 are highly susceptible to spontaneous skin tumor formation (36), and knockdown of COX-2 reduces susceptibility to experimentally induced tumorigenesis (36). Furthermore, inhibition of COX-2 prevents UV-induced skin tumorigenesis in humans (37, 38), even in patients at high risk of non-melanoma skin cancers (39).
Here, we show that TFEB is a UVA-responsive factor responsible for the activation of autophagy–lysosomal genes, including p62, in keratinocytes following UVA exposure. p62 bound to COX-2 and stabilized COX-2 through the UBA domain of p62. p62-mediated COX-2 stabilization promotes increased PGE2 production and may be responsible for increased tumor growth and metastasis in vivo. Elucidating this link between p62 and COX-2 has therefore uncovered a novel oncogenic pathway that may be a key to the development and metastasis of skin cancer.
Results
RNA-Seq analysis of UVA radiation response identifies autophagy–lysosome pathway
To identify genes regulated by UVA radiation, RNA-Seq was performed on sham- or UVA-irradiated normal human epidermal keratinocytes (NHEKs). A comparison of sham- with UVA-irradiated NHEKs identified >4,000 differentially expressed genes (Fig. 1, A and B). Pathway analysis shows that one of the up-regulated pathways following UVA exposure is the lysosome/autophagy pathway (Fig. 1C). UVA was found to induce the expression of a number of autophagy-related genes, including TFEB, p62, LC3, ULK1, ULK2, LAMP1, and LAMP2 (Fig. 1D). These data indicate that UVA activates transcription of the autophagy–lysosomal pathway, including the expression of p62.
Figure 1.

UVA induces expression of autophagy and lysosomal genes. A, schematic of RNA-Seq analysis of normal human epidermal keratinocytes (NHEK) at 6 h post-sham or -UVA (20 J/cm2) radiation. Gene expression and pathway analysis were performed using the Tuxedo pipeline and DAVID. B, volcano plot representing genes significantly differentially expressed between sham- and UVA-irradiated NHEKs as determined by RNA-Seq. p < 0.05. C, pathway analysis of the significantly up-regulated genes by RNA-Seq identified highly enriched pathways following UVA irradiation in NHEKs. D, real-time PCR validation of RNA-Seq results in NHEKs treated with UVA. *, p < 0.05; **, p < 0.01 (Student's t test). Error bars, S.D.
p62 regulation is independent of autophagy in UVA response
Because p62 is up-regulated in skin cancer and regulates skin tumor growth and metastasis (15), we elected to focus on p62 induction by UVA. UVA irradiation increased the protein levels of p62 in a time-dependent (Fig. 2A) and dose-dependent (Fig. 2B) manner. In addition, UVA also increased p62 protein levels in mouse epidermis in vivo (Fig. 2C) and skin cancer cells (Fig. 2D). To determine the mechanism by which UVA regulates p62 expression, we assessed the role of autophagy, because p62 is a selective autophagy substrate, and thus inhibition of autophagy leads to an increase in p62 protein levels (9, 10). We next assessed whether p62 up-regulation depended on the inhibition of autophagic flux. Treatment with the lysosome inhibitor bafilomycin A1, which inhibits autophagic flux, increased basal and UVA-induced p62 protein levels (Fig. 2E). In addition, knockdown of the essential autophagy gene Atg7 (Fig. 2F) or Atg5 (Fig. 2G) increased both basal and UVA-induced p62 up-regulation. These data indicate that suppression of autophagy did not impair UVA-induced p62 up-regulation.
Figure 2.

p62 is up-regulated by UVA independent of autophagy. A, p62 protein levels in NHEKs 0, 1.5, and 6 h after UVA radiation (10 J/cm2). B, p62 protein levels 6 h after irradiation with 0, 5, 10, or 15 J/cm2 UVA. C, immunohistochemical staining of p62 in mouse skin after UVA irradiation (30 J/cm2) every other day for a total of three treatments and collected at 72 h after the final UVA irradiation. D, p62 protein levels in the SCC cell line PAM212 at 0, 1.5, and 6 h after UVA irradiation. E, p62 protein levels in irradiated PAM212 cells treated with vehicle or the autophagic flux inhibitor bafilomycin A1. F, p62 protein levels in PAM212 cells transfected with shCon or shAtg7 after sham or UVA irradiation. G, p62 levels in iBMK cells transfected with shCon or shAtg5 after sham or UVA irradiation.
TFEB is a UVA-responsive transcription factor regulating p62 expression
To determine the mechanism by which UVA regulates p62 expression, we assessed the role of the transcription factor TFEB (Fig. 1C), because TFEB expression was induced by UVA in our RNA-Seq analysis (Fig. 1C). TFEB controls expression of a number of autophagy–lysosomal genes, including p62, through the CLEAR element within the promoter (25). TFEB activation is negatively regulated by phosphorylation, with dephosphorylation triggering the nuclear translocation of TFEB and activation of CLEAR network/TFEB target genes. Following UVA exposure, TFEB protein exhibited a shift in molecular weight suggestive of dephosphorylation (Fig. 3A). Furthermore, there was also an increase in TFEB nuclear localization after UVA irradiation (Fig. 3B), supporting an increase in UVA-induced TFEB activation. TFEB knockdown prevented UVA-induced p62 up-regulation at the protein level (Fig. 4A). Knockdown of TFEB also prevented UVA-induced expression of p62 as well as LC3 (Fig. 4, B–D). Inhibition of RNA synthesis by actinomycin D (Fig. 4E) blocked UVA-induced up-regulation of p62 mRNA, indicating that p62 is transcriptionally up-regulated by UVA. Furthermore, UVA induced p62 up-regulation in PAM212 skin cancer cells (Fig. 4F). ChIP analysis of the p62 promoter indicated that UVA increased the binding of TFEB to the p62 promoter (Fig. 4G). These findings demonstrate that TFEB activation is required for UVA-induced p62 expression.
Figure 3.
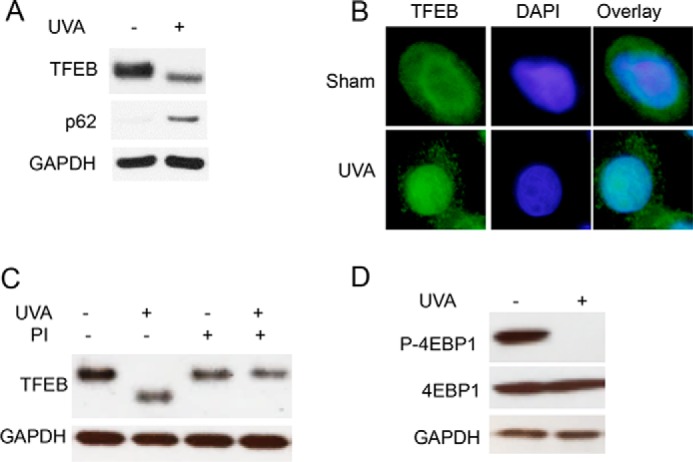
TFEB is activated by UVA. A, TFEB and p62 protein expression in NHEKs 6 h after treatment with sham or UVA (20 J/cm2) irradiation. B, immunofluorescence analysis of TFEB localization in NHEKs 6 h after sham or UVA irradiation. C, TFEB protein expression in NHEKs treated with phosphatase inhibitor (PI) calyculin A (50 nm) 0.5 h after sham or UVA radiation (20 J/cm2). D, phosphorylation of mechanistic target of rapamycin (mTOR) target gene 4EBP1 0.5 h after UVA exposure (20 J/cm2) in NHEKs.
Figure 4.

TFEB is activated by UVA to regulate expression of p62 and LC3. A, TFEB and p62 protein levels in NHEKs transfected with siCon or siTFEB treated with sham or UVA irradiation. Shown are TFEB (B), p62 (C), and LC3 mRNA (D) in NHEKs transfected with siCon or siTFEB and treated with sham or UVA irradiation. n.s., not significant; *, p < 0.05; **, p < 0.01. E, qPCR analysis of p62 mRNA in NHEKs treated with sham or UVA radiation with or without actinomycin D (1 μg/ml) for 6 h. F, p62 mRNA levels in PAM212 squamous cell carcinoma cells 6 h after sham or UVA irradiation. G, ChIP analysis of TFEB binding to the p62 promoter in NHEKs treated with sham or UVA irradiation. **, p < 0.01. Error bars, S.D.
p62 regulates COX-2 expression
Next, we determined the function of p62 induction in UVA response. Using a candidate gene approach, we found that COX-2 was up-regulated in response to UVA, in parallel with p62 induction in NHEK cells (Fig. 5A) and PAM212 skin cancer cells (Fig. 5B). This was accompanied by a moderate increase in COX-2 transcription following UVA (Fig. 5, C and D). Because previous reports have shown that COX-2 induction is required for the development of skin cancer (36), we next asked whether p62 regulates COX-2 in skin cancer cells. Knockdown of p62 led to a decrease in COX-2 protein levels in PAM212 cells (Fig. 5E). Basal COX-2 transcription was also reduced in p62-deficient cells (Fig. 5F). These data indicate that p62 regulates COX-2 protein and transcription.
Figure 5.

COX-2 is induced by UVA concomitant with p62 up-regulation. A, COX-2 and p62 protein levels in NHEKs treated with sham or UVA irradiation (20 J/cm2). B, p62 and COX-2 protein in PAM212 cells after UVA irradiation (10 J/cm2). Shown is qPCR analysis of p62 and COX-2 protein in NHEKs (C) and PAM212 cells (D). *, p < 0.05; **, p < 0.01. E, COX-2 protein levels in PAM212 cells transfected with shCon and shp62. F, qPCR analysis of COX-2 RNA levels in PAM212 cells transfected with shCon and shp62. ***, p < 0.001. Error bars, S.D.
p62 binds to COX-2 and regulates COX-2 stability
Because increased COX-2 protein levels and activity are required for skin cancer development (36–38), we further examined the regulation of COX-2 protein levels by p62. We have shown that p62 stabilizes Twist1 via the direct interaction of the UBA domain of p62 with polyubiquitinated Twist1 (15). Therefore, we asked whether p62 similarly regulated COX-2 protein stability through the p62 UBA domain. Expression of p62 in HeLa cells, which lack endogenous p62, COX-2, and Twist1 expression, was sufficient to increase the protein stability of exogenously expressed COX-2 (Fig. 6, A and B). This increase in COX-2 protein stability was lost in cells transfected with a mutant p62 construct that lacks the UBA domain (Fig. 6, C and D), suggesting that the regulation of COX-2 by p62 is dependent on the p62 UBA domain. It appears that the UBA domain loss also decreased basal COX-2 protein level (Fig. 6D). Co-immunoprecipitation (co-IP) of endogenous p62 and COX-2 in skin cancer cells shows that p62 interacts with COX-2 (Fig. 6E). Western blotting of p62 co-IP results showed the binding of p62 with COX-2 (Fig. 6E). We were not able to detect the binding of p62-dUBA with COX-2 (data not shown). This could be due to the loss of binding due to UBA deletion or due to the low basal COX-2 protein levels (Fig. 6D). In addition, p62 also regulates COX-2 transcription through the UBA domain (Fig. 6F). These results suggest that p62 regulates COX-2 transcription and protein stability.
Figure 6.

p62 regulates COX-2 protein stability through the UBA domain. A, HeLa cells were transiently transfected with COX-2 and either empty vector or HA–p62. COX-2 stability was measured over time by treating with CHX for the time indicated. B, quantification of the stability of COX-2 protein in A. C, HA-tagged WT or mutant p62 lacking the UBA domain (dUBA) were transfected into HeLa cells. D, COX-2 stability was measured over time after treating with CHX for 0, 4, or 8 h and quantified. E, co-immunoprecipitation of p62 and COX-2 in PAM212 cells treated with the proteasome inhibitor MG132 (10 μm) for 6 h. F, transcription of COX-2 in HeLa cells transiently transfected with COX-2 and HA–p62 or HA–p62–dUBA 48 h after transcription. IB, immunoblotting. Error bars, S.D. *, p < 0.05; ***, p < 0.001.
p62 regulates PGE2 production
COX-2 activity leads to production of PGE2, which promotes tumorigenesis through autocrine and paracrine signaling. To determine whether p62 regulates PGE2 production as a result of COX-2 regulation, we examined PGE2 levels in skin cancer cells following p62 knockdown. We found that p62 knockdown decreased PGE2 production (Fig. 7A), and this deficit was reversed by re-expression of COX-2 (Fig. 7A). Similarly, expression of both HA–p62 and COX-2 in HeLa cells increased PGE2 levels beyond the PGE2 levels in cells expressing COX-2 alone (Fig. 7B). Co-expression of COX-2 with mutant p62 lacking the UBA domain reduced PGE2 levels to that of cells lacking COX-2 (Fig. 7B). Therefore, p62 regulates PGE2 production by stabilizing COX-2 through the UBA domain of p62.
Figure 7.

p62 regulates PGE2 production but not proliferation or migration in vitro. A, PGE2 production in PAM212 cells stably transfected with shCon or shp62. shp62 cells were transiently transfected with COX-2 (shp62+COX-2). n.s., not significant; *, p < 0.05; **, p < 0.01. B, PGE2 production in HeLa cells transiently transfected with EV, HA–p62, COX-2, and HA–p62–dUBA, as indicated. n.s., not significant; *, p < 0.05; **, p < 0.01. C, proliferation of PAM212 cells stably transfected with shCon or shp62 measured over 3 days using an MTS proliferation assay. D, migration of WT, shCon, and shp62 PAM212 cells measured using a Transwell migration assay in serum-free medium. Error bars, S.D.
p62 does not regulate proliferation or migration in vitro
Considering the known role of PGE2 production in proliferation and migration, we assessed whether p62 knockdown impacted these functions in skin cancer cells. In both the Cell Counting Kit-8 assay (Fig. 7C) and the MTS assay (data not shown), p62 knockdown in skin cancer cells had no effect on proliferation. Similarly, p62 knockdown had no effect on migration in Transwell migration assays (Fig. 7D), even when PGE2 was used as a chemoattractant (data not shown). Whereas this data suggests that p62-mediated effects on PGE2 production do not affect the cancer cell proliferation and migration, PGE2 is a known paracrine signaling mediator and may affect neighboring cells in the tumor microenvironment.
p62 is required for tumor growth and metastasis in vivo
To test the requirement for p62 in skin tumor growth and progression, we utilized a syngeneic mouse model of skin cancer. In this model, we injected control and p62-knockdown PAM212 skin cancer cells into BALB/c mice. Measurement of tumor growth over 10 weeks showed that p62 knockdown inhibited tumor growth (Fig. 8A) and metastasis to the lung (Fig. 8B). These data indicate that p62 is required for tumor growth and metastasis. Histological analysis of tumor samples suggested that p62-knockdown tumors exhibit decreased immune cell infiltration and increased cell differentiation (Fig. 8C, arrows). Therefore, we next performed immunohistochemical (IHC) analysis using immune and differentiation markers. Staining for myeloperoxidase (MPO), a marker of myeloid cells, showed that p62-knockdown tumors exhibited lower levels of MPO-positive cells (Fig. 8D). Staining for differentiation marker keratin 10 (K10) also showed that p62-knockdown tumors had an increase in K10-positive differentiated cells as compared with controls (Fig. 8E). These findings support an oncogenic role of p62 in tumor growth and progression.
Figure 8.

p62 regulates tumor growth, metastasis, and immune infiltration in vivo. A, shCon and shp62 PAM212 cells were injected into syngeneic BALB/c mice, and tumor growth was measured over 10 weeks. n = 6 mice/group. *, p < 0.05, Student's t test, significant difference as compared with the shCon group. B, metastasis to the lungs of mice injected with shCon and shp62 PAM212 cells was measured using H&E staining. n = 6 mice/group. *, p < 0.05, Student's t test, significant difference. C, H&E staining of tumors from mice injected with shCon and shp62 PAM212 cells. Black arrow, immune cell infiltration. Gray arrow, differentiated keratinocytes. Magnification was ×20; scale bar, 100 μm. Inset, ×40 magnification; scale bar, 50 μm. D and E, IHC staining for MPO (D) and K10 (E) in tumors from mice injected with shCon and shp62 PAM212 cells. Magnification was ×20; scale bar, 100 μm. Inset, ×40 magnification; scale bar, 50 μm; error bars, S.D.
Discussion
UVA radiation is known to cause skin cancer. However, the signaling pathways underlying UVA response are unknown. Here we show that UVA radiation activates the transcription of an autophagy and lysosomal gene program, including p62, through the transcription factor TFEB. p62 regulates COX-2 by two mechanisms: 1) promoting COX-2 expression and 2) stabilizing COX-2 protein by binding to COX-2. Knockdown of p62 inhibits skin tumor growth and metastasis, in association with a decrease in immune cell infiltration in the tumors. Our findings suggest that targeting p62 may be an effective method to prevent tumor growth and metastasis after UVA.
We found that UVA irradiation up-regulates p62 at the transcription levels, independent of autophagy, because 1) UVA increased the level of p62 mRNA, 2) inhibiting RNA synthesis abolished the UVA-induced p62 mRNA increase, and 3) UVA induced the up-regulation of p62 in lysosome-inhibited cells and cells with a genetic autophagy deficiency. Previous studies have shown that in keratinocytes, UVA induces autophagy (40), and this work suggests that p62 functions independently of autophagy in UVA response.
In examining the potential functions of p62 in UVA response, we have identified a novel relationship between p62 and COX-2. As we previously reported with Twist1 (15), p62 binds and stabilizes polyubiquitinated COX-2 through the p62 UBA domain. COX-2 is primarily degraded at the proteasome via ER-associated degradation (29, 41). The interaction between COX-2 and the UBA domain of p62 may prevent this degradation of COX-2 at the proteasome, because we have demonstrated that p62 can prevent Twist1 degradation at the proteasome (15). This emerging pattern of positive regulation of protein stability by p62 suggests a mechanism by which p62 differentiates between polyubiquitinated proteins bound for degradation in autophagy and those to be stabilized. Further work will determine how p62 differentiates between these two sets of proteins and whether other oncogenic proteins are similarly stabilized by p62.
COX-2 stabilization by p62 also increases the production of PGE2. PGE2 is the key effector of COX-2 activity and acts to promote proliferation (30), invasion (31), and pro-tumorigenic immune infiltration (32, 33) while inhibiting cancer cell differentiation. Similar to COX-2 knockdown (36), our studies show that knockdown of p62 led to decreased tumor growth, metastasis, and immune infiltration as well as increased cancer cell differentiation. It is possible that p62 is acting through COX-2-mediated PGE2 production to promote skin cancer growth and metastasis. Further study of the p62-COX-2 signaling axis could determine whether this pathway is critical for skin tumor progression and could provide a novel target for the prevention and treatment of skin cancers.
In addition to p62, our RNA-Seq analysis in primary human keratinocytes identified multiple genes in the autophagy–lysosome pathways. Whereas we have shown that p62 is required for the transcription and protein stability of COX-2 and for skin tumor growth and metastasis in vivo, other UVA-regulated genes may also have important roles in UVA-associated skin cancers. In particular, the function of the autophagy–lysosomal genes is unknown and needs to be investigated in vitro and in vivo. In addition to p62, UVA-induced TFEB nuclear translocation also mediates LC3 up-regulation at the mRNA levels. The roles of TFEB and LC3 in skin cancer are unknown and deserve to be investigated in the future. Recent studies have shown that TFEB is up-regulated in pancreatic cancers and is required for pancreatic cancer growth. The MiT/TFE family of transcription factors, including TFEB, mediates cancer cell metabolic reprograming to maintain amino acid pools (42, 43). LC3 has been shown to be up-regulated in several cancers, including esophageal, gastric, and colorectal cancers (44). However, future studies are required to investigate the mechanism and function of LC3 up-regulation in these cancers and skin cancers.
Experimental procedures
Cell lines and drug treatments
NHEKs were grown in KGM-Gold BulletKit medium (Lonza) according to the manufacturer's protocol. NHEKs were cultured for less than four passages. PAM212 (squamous cell carcinoma), HeLa, and iBMK (immortalized mouse baby kidney epithelium, kindly provided by Dr. Eileen White, Cancer Institute of New Jersey, New Brunswick, NJ) cells were maintained in DMEM supplemented with 10% fetal bovine serum (Hyclone), 1% non-essential amino acids (Invitrogen), 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). PAM212 and HeLa cells were maintained in monolayer culture at 37 °C and in 95% air, 5% CO2 (v/v). iBMK cells were maintained at 38.5 °C in 8.5% CO2.
To inhibit transcription, NHEKs were treated with 1 μg/ml actinomycin D (Fisher) for 1 h prior to sham or UVA radiation. To inhibit autophagic flux, cells were treated with 25 nm bafilomycin A1 (Sigma) for 1 h prior to UVA radiation. Protein stability was assessed by treating cells with 100 ng/μl cyclohexamide (CHX) for the indicated times. Cells were treated with 10 μm MG132 (Sigma) for 6 h before lysing cells for co-IP.
siRNA and plasmid transfection
NHEKs were transiently transfected with siRNA targeting TFEB (Santa Cruz Biotechnology) or p62 (Dharmacon) using an Amaxa Nucleofector kit according to the manufacturer's protocol. Mouse shCon, shp62, shAtg5, and shAtg7 constructs in pLKO.1 vector were purchased from Sigma. Lentivirus was produced by co-transfecting HEK-293T cells with the lentiviral construct, pCMV8.2 packaging plasmid, and pVSV-G envelope plasmid. Supernatant was collected 24–48 h after transfection and used to infect cells in the presence of 8 μg/ml Polybrene (Sigma). Stable clones were selected using 2 μg/ml puromycin (Santa Cruz Biotechnology) treatment for 2 weeks as described previously (15).
HA–p62 in pcDNA4 was obtained from Qing Zhong (University of California, Berkeley, CA) (Addgene plasmid 28027). Mutant HA–p62–dUBA was generated from HA–p62–pCDNA4 as described previously (15). HeLa cells were transfected with HA–p62 and HA–p62–dUBA constructs using X-tremeGENE 9 (Roche Applied Science) as described previously (45). COX-2 (human) in pCDNA5 vector was generously provided by William Smith (University of Michigan) (46). pCMV6-AC-GFP COX-2 (mouse) was purchased from Origene. COX-2 constructs were transiently transfected into HeLa cells (human COX-2) and PAM212 cells (mouse COX-2) using X-tremeGENE 9 HP transfection reagent (Roche Applied Science) as described previously (45).
UVA treatments
For UVA irradiation, four parallel PUVA lamps were used, and doses were measured using a Goldilux UV meter equipped with UVA and UVB detector (Oriel). Contamination from UVB irradiation was eliminated using a 0.13-mm Mylar filter material from Cope Plastics. This filter limits UVB exposure to 0.003% of the total emitted UV radiation (47, 48). We have found that these lamps emit no UVC radiation (100–280 nm). The UVA dosages used here are relevant to human exposure. The dose of UVA that will cause erythema is 30 J/cm2 for human skin (1), and the UVA dose (20 J/cm2) used in the in vitro studies equates to about 1 h in the midday sun during the summer at latitude 48 ° north in Paris, France (49). In our laboratory, obtaining 20 J/cm2 of UVA irradiation requires ∼1 h. Therefore, the UVA dose used in our application is relevant to human exposure.
RNA-Seq analysis
NHEKs were exposed to sham or UVA irradiation (20 J/cm2). 6 h after irradiation, RNA was prepared from these cells using an RNeasy Plus kit (Qiagen), according to the manufacturer's protocol. Two biological repeats were included for each treatment group. RNA quality assessment, library preparation, and sequencing were performed by the University of Chicago Functional Genomics Facility. RNA quality assessment was performed using an Agilent Bioanalyzer. An oligo(dT)-selected library was prepared, and sequencing was performed on an Illumina HiSeq2000 platform with 50-bp single-end reads.
The established Tuxedo protocol was used to analyze the RNA-Seq data (50). In this pipeline, quality control analysis of raw RNA-Seq data were performed in FastQ Groomer. Reads were then aligned to the human reference genome (hg38) using Tophat2. Transcript assembly was performed using Cufflinks. Cufflinks assemblies were combined using CuffMerge for all treatments. CuffDiff was used to calculate the difference in gene expression between sham- and UVA-irradiated samples, with a false discovery rate of 10% and p < 0.05 as a standard of significance. CummeRbund was used to perform analyses of differentially expressed genes. Gene ontology and KEGG pathway analysis were performed using DAVID.
Real-time PCR
Quantitative real-time PCR assays were performed using a CFX Connect real-time system (Bio-Rad) using Bio-Rad iQ SYBR Green Supermix (Bio-Rad, 1708880) (1). The threshold cycle number (CT) for each sample was determined in triplicate. The CT values were normalized against Gapdh as described previously (15, 45). Amplification primers were as follows: GAPDH, 5′-ACC ACA GTC CAT GCC ATC AC-3′ (forward) and 5′-TCC ACC ACC CTG TTG CTG TA-3′ (reverse); TFEB, 5′-GCT GAT CCC CAA GGC CAA T-3′ (forward) and 5′-TCT CCA GCT CCC TGG ACT TT-3′ (reverse); p62, 5′-CAG AGA AGC CCA TGG ACA G-3′ (forward) and 5′-AGC TGC CTT GTA CCC ACA TC-3′ (reverse); LC3, 5′-AGA CCT TCA AGC AGC GCC G-3′ (forward) and 5′-ACA CTG ACA ATT TCA TCC CG-3′ (reverse); ULK1, 5′-TCG AGT TCT CCC GCA AGG-3′ (forward) and 5′-CGT CTG AGA CTT GGC GAG GT-3′ (reverse); ULK2, 5′-TGG GTC CTC CCA ACT ATC TAC AAG T-3′ (forward) and 5′-CGA GAT GTT GTG TGG CAC CAA-3′ (reverse); LAMP1, 5′-TCT CAG TGA ACT ACG ACA CCA-3′ (forward) and 5′-AGT GTA TGT CCT CTT CCA AAA GC-3′ (reverse); LAMP2, 5′-GAA AAT GCC ACT TGC CTT TAT GC-3′ (forward) and 5′-AGG AAA AGC CAG GTC CGA AC-3′ (reverse).
Western blotting
Prior to Western blot analysis, cells were treated as indicated and then lysed using radioimmune precipitation lysis buffer containing protease and phosphatase inhibitors (Thermo). Supernatant was removed for analysis or frozen for later use. Normalization of total protein levels was performed using the BCA assay. Lysate was separated on 4–12% gradient SDS-PAGE gels (Novex) and blotted onto nitrocellulose membranes (Novex). For expression analysis, the following antibodies were used: p62 (GP62-C, Progen), GAPDH (FL-335, Santa Cruz Biotechnology), COX-2 (ab15191, Abcam), LC3A/B (4108, Cell Signaling), and TFEB (A303-673A-M). Anti-HRP secondary antibodies (Cell Signaling) were used for visualization of proteins. Film was used for visualization.
ChIP
IP was performed on 5 × 106 cells following treatment with sham or UVA (20 J/cm2) irradiation. ChIP was performed using an EZ-Magna ChIP A Kit (Millipore), according to the manufacturer's protocol. The lysate was sonicated 12 times for 10 s each, with a 30-s rest between each sonication. IP was performed with TFEB (ab2636, Abcam) antibody as well as with positive and negative control antibodies included in the EZ-Magna ChIP kit. qPCR analysis of IP samples was performed using the following primers: 5′-CAC AGG CCT TCC TTG TGT C-3′ (forward) and 5′-GCA GAG GCT GTG GCC TA-3′ (reverse).
Immunofluorescence microscopy
For immunofluorescence analysis, cells were seeded onto glass coverslips overnight prior to treatment. Following treatment, cells were fixed using 10% neutral buffered formalin, permeabilized using Triton X-100, and incubated with primary antibody overnight. Alexa Fluor fluorescence-conjugated secondary antibodies were added for 2 h before imaging samples. Samples were examined using an inverted microscope with fluorescence function and dedicated analysis software (Olympus, model IX71).
PGE2 production assay
PGE2 production was measured using a PGE2 parameter assay kit (KGE004B, R&D Systems). HeLa cells were transiently transfected with empty vector (EV), EV and HA-tagged p62, EV and COX-2, HA–p62 and COX-2, or HA–p62–dUBA and COX-2 using X-tremeGENE 9 transfection reagent. 48 h after transfection, cells were serum-starved for 24 h before taking samples for this assay. PAM212 cells with stable knockdown of p62 (shp62) were transiently transfected with mouse COX-2 (shp62–COX-2). 48 h after transfection, cells were serum-starved for 24 h prior to taking medium for this assay.
Migration and proliferation assays
Cell proliferation was measured using the CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (MTS) (Promega) according to the manufacturer's protocol as described previously (15). Cell viability was measured using the Cell Counting Kit-8 assay (Sigma) according to the manufacturer's protocol. Migration was measured using Transwell inserts (Corning) with serum-free medium above and below the insert, with medium supplemented with 10% FBS below the insert, and with serum-free medium containing PGE2 below the insert. Migrated cells were measured 16 h after seeding.
Mouse studies
All animal procedures were approved by the University of Chicago institutional animal care and use committee. BALB/c mice were purchased from Charles River Laboratories. 5 million PAM212 cells, which are syngeneic with BALB/c mice, were injected subcutaneously into the flanks of BALB/c mice. Tumor growth was measured over 10–12 weeks using a caliper, and at 1 cm3 volume, tumors were harvested along with the lungs for histological analysis. Tumors and lungs were fixed in 10% formalin for IHC analysis. For UVA treatment studies in mice, SKH1 hairless mice received sham or 15-J/cm2 UVA irradiation every other day for a total of three treatments, and then skin samples were harvested for immunohistochemical analysis 72 h after the final treatment.
IHC analysis
For H&E, MPO, and K10 staining, tumors and lungs (or skin in UVA experiments) were harvested and fixed in 10% formalin. Paraffin-embedding, sectioning, and H&E staining were performed by the Human Tissue Resource Center at the University of Chicago. MPO (Abcam ab45977, 1:500) and K10 (Covance MMS-159S, 1:1000) staining was performed as described previously (51). Stained samples were scanned by the Integrated Light Microscopy Core Facility at the University of Chicago.
Statistical analysis
Statistical analyses for qPCR and migration, proliferation, viability, and PGE2 production assays were performed using GraphPad Prism version 5. Data were expressed as mean of at least three independent experiments and analyzed by Student's t test. Error bars indicate S.D. p < 0.05 was considered statistically significant.
Author contributions
A. S., B. Z., and Y. Y. H. conceived and coordinated the study, analyzed the data, and wrote the paper. A. S. and B. Z. designed, performed, and analyzed the experiments shown in all figures. L. Q. provided technical assistance and critical reagents. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Norbert Fusenig for providing the HaCaT cells (human keratinocytes and epithelial cells), Dr. Seungmin Hwang for providing the pLKO.1 shAtg5 (mouse) and pLKO.1 shAtg7 (mouse) vectors, Dr. Eileen White for providing the iBMK cells, Dr. William Smith for the COX-2 plasmid (human), Terri Li for immunohistochemistry, and Dr. Ann Motten for critical reading of the manuscript.
This work was supported by NIEHS, National Institutes of Health (NIH), Grants ES024373 and ES016936 (to Y. Y. H.), American Cancer Society Grant RSG-13-078-01 (to Y. Y. H.), NCI, NIH, Grant P30 CA014599 (University of Chicago Cancer Research Center), NIH Clinical and Translational Science Award (CTSA) UL1 TR000430, and the University of Chicago Friends of Dermatology Endowment Fund. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- UBA
- ubiquitin-associated
- MPO
- myeloperoxidase
- TFEB
- transcription factor EB
- CLEAR
- coordinated lysosomal expression and regulation
- COX-2
- cyclooxygenase-2
- PGE2
- prostaglandin E2
- NHEK
- normal human epidermal keratinocyte
- IP
- immunoprecipitation
- IHC
- immunohistochemical
- MPO
- myeloperoxidase
- CHX
- cyclohexamide
- qPCR
- quantitative PCR
- EV
- empty vector
- mTOR
- mechanistic target of rapamycin
- K10
- keratin 10
- MTS
- [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt].
References
- 1. van Weelden H., de Gruijl F. R., van der Putte S. C., Toonstra J., and van der Leun J. C. (1988) The carcinogenic risks of modern tanning equipment: is UV-A safer than UV-B? Arch. Dermatol. Res. 280, 300–307 [DOI] [PubMed] [Google Scholar]
- 2. de Laat A., van der Leun J. C., and de Gruijl F. R. (1997) Carcinogenesis induced by UVA (365-nm) radiation: the dose-time dependence of tumor formation in hairless mice. Carcinogenesis 18, 1013–1020 [DOI] [PubMed] [Google Scholar]
- 3. Sterenborg H. J. C. M., and van der Leun J. C. (1990) Tumorigenesis by a long wavelength UV-A source. Photochem. Photobiol. 51, 325–330 [DOI] [PubMed] [Google Scholar]
- 4. Kelfkens G., de Gruijl F. R., and van der Leun J. C. (1991) Tumorigenesis by short-wave ultraviolet A: papillomas versus squamous cell carcinomas. Carcinogenesis 12, 1377–1382 [DOI] [PubMed] [Google Scholar]
- 5. Lazovich D., Vogel R. I., Berwick M., Weinstock M. A., Anderson K. E., and Warshaw E. M. (2010) Indoor tanning and risk of melanoma: a case-control study in a highly exposed population. Cancer Epidemiol. Biomarkers Prev. 19, 1557–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang M., Qureshi A. A., Geller A. C., Frazier L., Hunter D. J., and Han J. (2012) Use of tanning beds and incidence of skin cancer. J. Clin. Oncol. 30, 1588–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Klionsky D. J. (2007) Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 8, 931–937 [DOI] [PubMed] [Google Scholar]
- 8. Mizushima N., Levine B., Cuervo A. M., and Klionsky D. J. (2008) Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., and Johansen T. (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J. A., Outzen H., Øvervatn A., Bjørkøy G., and Johansen T. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates. J. Biol. Chem. 282, 24131–24145 [DOI] [PubMed] [Google Scholar]
- 11. Inoue D., Suzuki T., Mitsuishi Y., Miki Y., Suzuki S., Sugawara S., Watanabe M., Sakurada A., Endo C., Uruno A., Sasano H., Nakagawa T., Satoh K., Tanaka N., Kubo H., et al. (2012) Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 103, 760–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thompson H. G. R., Harris J. W., Wold B. J., Lin F., and Brody J. P. (2003) p62 overexpression in breast tumors and regulation by prostate-derived Ets factor in breast cancer cells. Oncogene 22, 2322–2333 [DOI] [PubMed] [Google Scholar]
- 13. Rolland P., Madjd Z., Durrant L., Ellis I. O., Layfield R., and Spendlove I. (2007) The ubiquitin-binding protein p62 is expressed in breast cancers showing features of aggressive disease. Endocr. Relat. Cancer 14, 73–80 [DOI] [PubMed] [Google Scholar]
- 14. Ellis R. A., Horswell S., Ness T., Lumsdon J., Tooze S. A., Kirkham N., Armstrong J. L., and Lovat P. E. (2014) Prognostic impact of p62 expression in cutaneous malignant melanoma. J. Invest. Dermatol. 134, 1476–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qiang L., Zhao B., Ming M., Wang N., He T. C., Hwang S., Thorburn A., He Y. Y. (2014) Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc. Natl. Acad. Sci. U.S.A. 111, 9241–9246 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16. Moscat J., and Diaz-Meco M. T. (2009) p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 137, 1001–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moscat J., and Diaz-Meco M. T. (2012) P62: a versatile multitasker takes on cancer. Trends Biochem. Sci. 37, 230–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mathew R., Kongara S., Beaudoin B., Karp C. M., Bray K., Degenhardt K., Chen G., Jin S., and White E. (2007) Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 21, 1367–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Duran A., Linares J. F., Galvez A. S., Wikenheiser K., Flores J. M., Diaz-Meco M. T., and Moscat J. (2008) The Signaling adaptor p62 is an important NF-κB mediator in tumorigenesis. Cancer Cell 13, 343–354 [DOI] [PubMed] [Google Scholar]
- 20. Inami Y., Waguri S., Sakamoto A., Kouno T., Nakada K., Hino O., Watanabe S., Ando J., Iwadate M., Yamamoto M., Lee M. S., Tanaka K., and Komatsu M. (2011) Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 193, 275–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Komatsu M., Kurokawa H., Waguri S., Taguchi K., Kobayashi A., Ichimura Y., Sou Y. S., Ueno I., Sakamoto A., Tong K. I., Kim M., Nishito Y., Iemura S., Natsume T., Ueno T., et al. (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213–223 [DOI] [PubMed] [Google Scholar]
- 22. Lau A., Wang X. J., Zhao F., Villeneuve N. F., Wu T., Jiang T., Sun Z., White E., and Zhang D. D. (2010) A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol. Cell. Biol. 30, 3275–3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Settembre C., Zoncu R., Medina D. L., Vetrini F., Erdin S., Erdin S., Huynh T., Ferron M., Karsenty G., Vellard M. C., Facchinetti V., Sabatini D. M., and Ballabio A. (2012) A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sardiello M., Palmieri M., di Ronza A., Medina D. L., Valenza M., Gennarino V. A., Di Malta C., Donaudy F., Embrione V., Polishchuk R. S., Banfi S., Parenti G., Cattaneo E., and Ballabio A. (2009) A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 [DOI] [PubMed] [Google Scholar]
- 25. Palmieri M., Impey S., Kang H., di Ronza A., Pelz C., Sardiello M., and Ballabio A. (2011) Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 20, 3852–3866 [DOI] [PubMed] [Google Scholar]
- 26. Settembre C., De Cegli R., Mansueto G., Saha P. K., Vetrini F., Visvikis O., Huynh T., Carissimo A., Palmer D., Klisch T. J., Wollenberg A. C., Di Bernardo D., Chan L., Irazoqui J. E., and Ballabio A. (2013) TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 15, 647–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martina J. A., Chen Y., Gucek M., and Puertollano R. (2012) MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bachelor M. A., Silvers A. L., and Bowden G. T. (2002) The role of p38 in UVA-induced cyclooxygenase-2 expression in the human keratinocyte cell line, HaCaT. Oncogene 21, 7092–7099 [DOI] [PubMed] [Google Scholar]
- 29. Rockwell P., Yuan H., Magnusson R., and Figueiredo-Pereira M. E. (2000) Proteasome inhibition in neuronal cells induces a proinflammatory response manifested by upregulation of cyclooxygenase-2, its accumulation as ubiquitin conjugates, and production of the prostaglandin PGE2. Arch. Biochem. Biophys. 374, 325–333 [DOI] [PubMed] [Google Scholar]
- 30. Cao Y., and Prescott S. M. (2002) Many actions of cyclooxygenase-2 in cellular dynamics and in cancer. J. Cell. Physiol. 190, 279–286 [DOI] [PubMed] [Google Scholar]
- 31. Singh B., Berry J. A., Shoher A., Ramakrishnan V., and Lucci A. (2005) COX-2 overexpression increases motility and invasion of breast cancer cells. Int. J. Oncol. 26, 1393–1399 [PubMed] [Google Scholar]
- 32. Tjiu J.-W., Chen J. S., Shun C. T., Lin S. J., Liao Y. H., Chu C. Y., Tsai T. F., Chiu H. C., Dai Y. S., Inoue H., Yang P. C., Kuo M. L., Jee S. H. (2009) Tumor-associated macrophage-induced invasion and angiogenesis of human basal cell carcinoma cells by cyclooxygenase-2 induction. J. Invest. Dermatol. 129, 1016–1025 [DOI] [PubMed] [Google Scholar]
- 33. Zelenay S., van der Veen A. G., Böttcher J. P., Snelgrove K. J., Rogers N., Acton S. E., Chakravarty P., Girotti M. R., Marais R., Quezada S. A., Sahai E., and Reis e Sousa C. (2015) Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162, 1257–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. An K. P., Athar M., Tang X., Katiyar S. K., Russo J., Beech J., Aszterbaum M., Kopelovich L., Epstein E. H. Jr., Mukhtar H., and Bickers D. R. (2002) Cyclooxygenase-2 expression in murine and human nonmelanoma skin cancers: implications for therapeutic approaches. Photochem. Photobiol. 76, 73–80 [DOI] [PubMed] [Google Scholar]
- 35. Higashi Y., Kanekura T., and Kanzaki T. (2000) Enhanced expression of cyclooxygenase (COX)-2 in human skin epidermal cancer cells: evidence for growth suppression by inhibiting COX-2 expression. Int. J. Cancer 86, 667–671 [DOI] [PubMed] [Google Scholar]
- 36. Tiano H. F., Loftin C. D., Akunda J., Lee C. A., Spalding J., Sessoms A., Dunson D. B., Rogan E. G., Morham S. G., Smart R. C., and Langenbach R. (2002) Deficiency of either cyclooxygenase (COX)-1 or COX-2 alters epidermal differentiation and reduces mouse skin tumorigenesis. Cancer Res. 62, 3395–3401 [PubMed] [Google Scholar]
- 37. Wright T. I., Spencer J. M., and Flowers F. P. (2006) Chemoprevention of nonmelanoma skin cancer. J. Am. Acad. Dermatol. 54, 933–946 [DOI] [PubMed] [Google Scholar]
- 38. Pentland A. P., Schoggins J. W., Scott G. A., Khan K. N. M., and Han R. (1999) Reduction of UV-induced skin tumors in hairless mice by selective COX-2 inhibition. Carcinogenesis 20, 1939–1944 [DOI] [PubMed] [Google Scholar]
- 39. Elmets C. A., Viner J. L., Pentland A. P., Cantrell W., Lin H. Y., Bailey H., Kang S., Linden K. G., Heffernan M., Duvic M., Richmond E., Elewski B. E., Umar A., Bell W., and Gordon G. B. (2010) Chemoprevention of nonmelanoma skin cancer with celecoxib: a randomized, double-blind, placebo-controlled trial. J. Natl. Cancer Inst. 102, 1835–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao Y., Zhang C. F., Rossiter H., Eckhart L., König U., Karner S., Mildner M., Bochkov V. N., Tschachler E., and Gruber F. (2013) Autophagy is induced by UVA and promotes removal of oxidized phospholipids and protein aggregates in epidermal keratinocytes. J. Invest. Dermatol. 133, 1629–1637 [DOI] [PubMed] [Google Scholar]
- 41. Yuan C., and Smith W. L. (2015) A cyclooxygenase-2-dependent prostaglandin E2 biosynthetic system in the golgi apparatus. J. Biol. Chem. 290, 5606–5620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perera R. M., Stoykova S., Nicolay B. N., Ross K. N., Fitamant J., Boukhali M., Lengrand J., Deshpande V., Selig M. K., Ferrone C. R., Settleman J., Stephanopoulos G., Dyson N. J., Zoncu R., Ramaswamy S., et al. (2015) Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Klein K., Werner K., Teske C., Schenk M., Giese T., Weitz J., and Welsch T. (2016) Role of TFEB-driven autophagy regulation in pancreatic cancer treatment. Int. J. Oncol. 49, 164–172 [DOI] [PubMed] [Google Scholar]
- 44. Yoshioka A., Miyata H., Doki Y., Yamasaki M., Sohma I., Gotoh K., Takiguchi S., Fujiwara Y., Uchiyama Y., and Monden M. (2008) LC3, an autophagosome marker, is highly expressed in gastrointestinal cancers. Int. J. Oncol. 33, 461–468 [PubMed] [Google Scholar]
- 45. Qiang L., Zhao B., Shah P., Sample A., Yang S., and He Y. Y. (2016) Autophagy positively regulates DNA damage recognition by nucleotide excision repair. Autophagy 12, 357–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mbonye U. R., Wada M., Rieke C. J., Tang H. Y., Dewitt D. L., and Smith W. L. (2006) The 19-amino acid cassette of cyclooxygenase-2 mediates entry of the protein into the endoplasmic reticulum-associated degradation system. J. Biol. Chem. 281, 35770–35778 [DOI] [PubMed] [Google Scholar]
- 47. Wischermann K., Popp S., Moshir S., Scharfetter-Kochanek K., Wlaschek M., de Gruijl F., Hartschuh W., Greinert R., Volkmer B., Faust A., Rapp A., Schmezer P., and Boukamp P. (2008) UVA radiation causes DNA strand breaks, chromosomal aberrations and tumorigenic transformation in HaCaT skin keratinocytes. Oncogene 27, 4269–4280 [DOI] [PubMed] [Google Scholar]
- 48. White A. L., and Jahnke L. S. (2004) Removing UV-A and UV-C radiation from UV-B fluorescent lamp emissions: differences in the inhibition of photosynthesis in the marine alga Dunaliella tertiolecta using chromate versus cellulose acetate-polyester filters. Photochem. Photobiol. 80, 340–345 [DOI] [PubMed] [Google Scholar]
- 49. Jeanmougin M., and Civatte J. (1987) [Dosimetry of solar ultraviolet radiation. Daily and monthly changes in Paris]. Ann. Dermatol. Venereol. 114, 671–676 [PubMed] [Google Scholar]
- 50. Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D. R., Pimentel H., Salzberg S. L., Rinn J. L., and Pachter L. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Qiang L., Wu C., Ming M., Viollet B., and He Y. Y. (2013) Autophagy controls p38 activation to promote cell survival under genotoxic stress. J. Biol. Chem. 288, 1603–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]