

Abstract
Tissue factor (TF) is expressed in vascular and nonvascular tissues and functions in several pathways, including embryonic development, inflammation, and cell migration. Many risk factors for atherosclerosis, including hypertension, diabetes, obesity, and smoking, increase TF expression. To better understand the TF-related mechanisms in atherosclerosis, here we investigated the role of 12/15-lipoxygenase (12/15-LOX) in TF expression. 15(S)-hydroxyeicosatetraenoic acid (15(S)-HETE), the major product of human 15-LOXs 1 and 2, induced TF expression and activity in a time-dependent manner in the human monocytic cell line THP1. Moreover, TF suppression with neutralizing antibodies blocked 15(S)-HETE–induced monocyte migration. We also found that NADPH- and xanthine oxidase–dependent reactive oxygen species (ROS) production, calcium/calmodulin-dependent protein kinase IV (CaMKIV) activation, and interactions between nuclear factor of activated T cells 3 (NFATc3) and FosB proto-oncogene, AP-1 transcription factor subunit (FosB) are involved in 15(S)-HETE–induced TF expression. Interestingly, NFATc3 first induced the expression of its interaction partner FosB before forming the heterodimeric NFATc3–FosB transcription factor complex, which bound the proximal AP-1 site in the TF gene promoter and activated TF expression. We also observed that macrophages from 12/15-LOX−/− mice exhibit diminished migratory response to monocyte chemotactic protein 1 (MCP-1) and lipopolysaccharide compared with WT mouse macrophages. Similarly, compared with WT macrophages, monocytes from 12/15-LOX−/− mice displayed diminished trafficking, which was rescued by prior treatment with 12(S)-HETE, in a peritonitis model. These observations indicate that 15(S)-HETE–induced monocyte/macrophage migration and trafficking require ROS-mediated CaMKIV activation leading to formation of NFATc3 and FosB heterodimer, which binds and activates the TF promoter.
Keywords: gene expression, migration, monocyte, signal transduction, trafficking
Introduction
Tissue factor (TF)2 is a transmembrane glycoprotein and expresses in several cell types of vascular and nonvascular tissues (1–3). In addition to its function in the extrinsic coagulation pathway, TF plays a role in embryonic development, wound repair, angiogenesis, inflammation, cell migration, and tumor metastasis (4–7). Furthermore, many risk factors for atherosclerosis, including hyperlipidemia, hypertension, diabetes, obesity, and smoking, all increase the expression of TF, and atherothrombosis, which occurs following plaque rupture, is the leading cause of death in the western world (8–10). In addition, many reports have shown that TF in vascular cells and macrophages within the atherosclerotic lesions is responsible for the thrombogenicity associated with the plaque rupture (11, 12). Recent studies have also shown that TF, particularly of monocytic origin, plays a role in various inflammatory diseases, including glomerulonephritis, inflammatory bowel disease, systemic lupus erythematosus, and sepsis (13–16). The findings that although lipid molecules such as oxidized low density lipoprotein (oxLDL) and oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (oxPAPC) induce TF expression in endothelial cells, monocytes, and macrophages, and high density lipoprotein (HDL) blocks its expression, provide additional lines of evidence for the possible role of TF in atherosclerosis (17–19). The link between TF and atherogenesis is further supported by studies demonstrating that HMG-CoA reductase inhibitors, namely statins, reduce the expression of TF in vascular smooth muscle cells, monocytes, and endothelial cells (17, 20).
Since the discovery that human atherosclerotic plaques contain abundant levels of both mRNA and protein levels of 15-lipoxygenase (15-LOX), many studies using pharmacological and genetic approaches have shown a role for this enzyme in the pathogenesis of atherosclerosis (21–23). Because atherosclerotic plaques produce 15(S)-hydroxyeicosatetraenoic acid (15(S)-HETE) as a predominant eicosanoid, a major arachidonic acid metabolite of 15-LOX1/2 (24, 25), we asked the question whether there is any link between 15(S)-HETE and TF in the modulation of inflammation, an underlying factor in both atherogenesis and atherothrombosis. In this study, we report that 15(S)-HETE induces TF expression and its activity in human monocytes and inhibition or depletion of TF levels blocks 15(S)-HETE–induced monocyte migration. In addition, we show that 15(S)-HETE–induced TF expression and its activity require reactive oxygen species (ROS)–dependent calcium/calmodulin-dependent protein kinase IV (CaMKIV)–mediated nuclear factor of activated T cells 3 (NFATc3) and FosB interactions and their occupancy of AP-1 site in the TF promoter. Our observations also suggest that the migration of monocytes to inflamed peritoneum is dependent on 12/15 lipoxygenase (12/15-LOX)–mediated TF expression. Thus, these findings provide evidence for the role of TF in 12/15-LOX–induced monocyte migration in vitro and monocyte trafficking in vivo.
Results
TF expression increases with the progression of atherosclerotic lesions (10, 11). Because many studies including our own have shown a role for 12/15-LOX in atherogenesis (21, 23, 26), we asked the question whether there is any link between 12/15-LOX and TF in the pathogenesis of atherosclerosis, particularly monocyte/macrophage buildup in the vessel wall. To address this notion, we tested the effect of 15(S)-HETE, a major arachidonic acid metabolite of human 15-LOX1/2, on TF expression in THP1 cells, a human monocytic cell line. 15(S)-HETE not only induced the expression of TF at both mRNA and protein levels, but also increased its activity in a time-dependent manner (Fig. 1A). The specificity experiments have revealed that 15(S)-HETE is the most potent HETE in inducing TF expression as compared with other HETEs (Fig. 1B). To understand the functional significance of TF expression by 15(S)-HETE, we tested its involvement in 15(S)-HETE–induced monocyte migration, as it is a key player in vascular inflammation, atherogenesis, and atherothrombosis. Neutralizing TF antibodies (anti-human TF Ab-D3) (27) besides abolishing 15(S)-HETE–induced Factor Xa generation also suppressed its effects on monocyte migration (Fig. 1C). In addition, depletion of TF levels by its antisense oligonucleotides (ASOs) attenuated 15(S)-HETE–induced monocyte migration (Fig. 1C).
Figure 1.

TF mediates 15(S)-HETE–induced THP1 cell migration. A, quiescent THP1 cells were treated with vehicle or 15(S)-HETE (0.1 μm) for the indicated time periods and either RNA was isolated or cell extracts were prepared. The RNA and cell extracts were analyzed by RT-PCR and Western blotting for TF mRNA and protein levels using its specific primers and antibodies, respectively. TF activity in cell extracts was measured using a kit as described in “Materials and methods.” B, quiescent THP1 cells were treated with vehicle or the indicated HETE (0.1 μm) for 2 h, and TF mRNA, protein, and activity were measured as described in A. The mRNA and protein levels in A and B were normalized to β-actin mRNA and β-tubulin protein levels, respectively. C, quiescent THP1 cells were incubated with IgG or TF neutralizing antibodies (anti-human TF Ab-D3, 5 μg/ml) for 30 min, treated with vehicle or 15(S)-HETE (0.1 μm) for either 2 h and TF activity was measured or 8 h and cell migration was measured. Cells were also transfected with control or TF ASOs (100 nm), quiesced, and treated with vehicle or 15(S)-HETE, and migration was measured. The bar graphs represent mean ± S.D. values of three experiments. *, p < 0.01 versus vehicle control or control ASO; **, p < 0.01 versus 15(S)-HETE, TF, or control ASO + 15(S)-HETE.
We have previously demonstrated a role for NADPH oxidase and xanthine oxidase–mediated ROS production in 15(S)-HETE–induced THP1 cell migration (23). Therefore, to explore the mechanisms underlying 15(S)-HETE–induced TF expression, we have studied the role of NADPH oxidase and xanthine oxidase. Apocynin, diphenyleneiodonium (DPI), and allopurinol, the NADPH oxidase and xanthine oxidase inhibitors, respectively (23), suppressed 15(S)-HETE–induced TF expression and its activity (Fig. 2A). To confirm these findings, we used an ASO approach. Depletion of either p47Phox, a component of NADPH oxidase (28), or xanthine oxidase substantially attenuated 15(S)-HETE–induced TF expression and its activity (Fig. 2B). Inhibition or depletion of NADPH oxidase or xanthine oxidase by pharmacological and ASO approaches, respectively, also negated the effect of 15(S)-HETE on monocyte migration (Fig. 2, C and D).
Figure 2.

NADPH oxidase and xanthine oxidase-dependent ROS production is required for 15(S)-HETE–induced TF expression and its activity in mediating THP1 cell migration. A, quiescent THP1 cells are treated with vehicle or 15(S)-HETE (0.1 μm) in the presence and absence of apocynin (100 μm), DPI (10 μm), or allopurinol (100 μm) for 2 h, and TF expression (mRNA and protein levels) and its activity were measured. B, cells were transfected with control, p47Phox, or xanthine oxidase ASOs, quiesced, and treated with vehicle or 15(S)-HETE (0.1 μm) for 2 h, and TF expression (mRNA and protein levels) and its activity were measured. C and D, all the conditions were the same as in A or B except that cells were treated with vehicle or 15(S)-HETE (0.1 μm) for 8 h, and cell migration was measured. The mRNA and protein levels in A and B were normalized to β-actin mRNA and β-tubulin protein levels, respectively. The bar graphs represent mean ± S.D. of three experiments. *, p < 0.01 versus vehicle control or control ASO; **, p < 0.01 versus 15(S)-HETE or control ASO + 15(S)-HETE.
A large number of studies have shown that ROS play a role in the activation of both serine/threonine and tyrosine kinases in modulating cellular signal transduction (29). In addition, a recent study has shown that ROS mediate activation of calcium/calmodulin-dependent protein kinases (CaMKs) (30). Because nothing is known on the role of CaMKs in TF expression and having found that 15(S)-HETE–induced TF expression and activation require ROS production, we wanted to test the effect of 15(S)-HETE on the activation of CaMKs. 15(S)-HETE, while having no major effect on the phosphorylation of CaMKI or CaMKII, induced the serine phosphorylation of CaMKIV in a time-dependent manner with maximum effect at 30 min (Fig. 3A). Based on this observation, we tested the role of CaMKIV in 15(S)-HETE–induced TF expression and monocyte migration. Our results showed that STO609, a potent inhibitor of CaMKIV (31), suppressed 15(S)-HETE–induced TF expression and its activity (Fig. 3B). In line with these observations, STO609 also blocked 15(S)-HETE–induced monocyte migration (Fig. 3B). To confirm these observations we also used an ASO approach. Knockdown of CaMKIV by its ASOs inhibited 15(S)-HETE–induced expression and enzymatic activity of TF, as well as monocyte migration (Fig. 3C). To understand the link between ROS and CaMKIV, we further examined the role of NADPH oxidase and xanthine oxidase in 15(S)-HETE–induced CaMKIV activation. Inhibition or depletion of either NADPH oxidase or xanthine oxidase attenuated the effect of 15(S)-HETE on CaMKIV phosphorylation (Fig. 3D). These results indicate that NADPH oxidase and xanthine oxidase–mediated ROS production is required for CaMKIV activation in TF expression facilitating monocyte migration in response to 15(S)-HETE.
Figure 3.

15(S)-HETE–induced TF expression and its activity require CaMKIV activation. A, quiescent THP1 cells treated with vehicle or 15(S)-HETE (0.1 μm) for the indicated time periods, and cell extracts were prepared. Equal amounts of protein from control and each treatment were analyzed by Western blotting for the phosphorylation of the indicated CaMKs using their specific antibodies and normalized to their total levels. B, quiescent THP1 cells were treated with vehicle or 15(S)-HETE (0.1 μm) in the presence and absence of STO609 (10 μm) either for 2 h and TF expression (mRNA and protein levels) and its activity were measured, or for 8 h and cell migration was measured. C, upper panel, THP1 cells were transfected with control or the indicated ASOs of CaMKIV, and 48 h later cell extracts were prepared and analyzed by Western blotting for CaMKIV levels using its specific antibodies and normalized to β-tubulin levels. Lower panels, THP1 cells that were transfected with control or the indicated CaMKIV ASO and quiesced were treated with vehicle or 15(S)-HETE (0.1 μm) either for 2 h and analyzed for TF expression (mRNA and protein levels) and its activity or for 8 h and cell migration was measured. D, quiescent THP1 cells were treated with vehicle or 15(S)-HETE (0.1 μm) in the presence and absence of apocynin (100 μm), DPI (10 μm), or allopurinol (100 μm) for 1 h or transfected with control, p47Phox, or xanthine oxidase ASO and quiesced before subjecting to treatment with vehicle or 15(S)-HETE (0.1 μm) for 1 h and cell extracts were prepared. Equal amounts of protein from control and each treatment were analyzed by Western blotting for pCaMKIV levels using its specific antibodies and normalized to its total levels. The mRNA and protein levels in B and C were normalized to β-actin mRNA and β-tubulin protein levels. The bar graphs represent mean ± S.D. values of three independent experiments. *, p < 0.01 versus vehicle control or control ASO; **, p < 0.01 versus vehicle control + 15(S)-HETE or control ASO + 15(S)-HETE.
Many studies have reported a role for various transcriptional mechanisms in the regulation of TF expression in response to a wide array of stimulants in a variety of cell types (18, 32, 33). To find out which of these transcriptional mechanisms are involved in 15(S)-HETE–induced TF expression, we cloned ∼2.1 kb human TF promoter and performed TRANSFAC analysis. TRANSFAC analysis showed the presence of four SP1-binding sites, three NFAT-binding sites, one NFκB-binding site, one CREB-binding site, and three AP-1–binding sites (Fig. 4A). Using ∼2.1 kb TF promoter-luciferase construct, we observed a 7-fold increase in the promoter activity in response to 15(S)-HETE as compared with vehicle control (Fig. 4B). This result infers a role for transcriptional mechanisms in 15(S)-HETE–induced TF expression. To identify the regulatory elements involved in 15(S)-HETE–induced TF promoter activity, we performed serial promoter deletion analysis. Promoter deletion analysis revealed the presence of 15(S)-HETE–responsive element(s) between 0.23 kb and 0.19 kb region of the promoter from the transcription start site. Because two AP-1 sites are present in this region and to find whether these sites are involved in 15(S)-HETE–induced TF promoter activity, we performed site-directed mutagenesis. We found that disruption of the proximal but not the distal AP-1 site attenuates 15(S)-HETE–induced TF promoter activity (Fig. 4B). These findings reveal the importance of the proximal AP-1 site in 15(S)-HETE–induced transcriptional regulation of TF expression. To identify the AP-1 components mediating 15(S)-HETE–induced TF promoter activity, we studied the time course effect of 15(S)-HETE on the expression of the Fos and Jun family of proto-oncogenes. 15(S)-HETE, although having little effect on cFos, Fra1, cJun, and JunD levels, induced the expression of FosB, Fra2, and JunB levels (Fig. 4C). Based on these observations we tested the role of these proto-oncogenes in 15(S)-HETE–induced TF promoter activity. Depletion of FosB but not Fra2 or JunB levels using their respective ASOs blocked 15(S)-HETE–induced 0.23 kb TF promoter-luciferase activity (Fig. 4D). In accordance with these findings, depletion of FosB but not JunB or Fra2 attenuated 15(S)-HETE–induced TF expression and its activity as well as monocyte migration (Fig. 4E). In addition, pharmacological inhibition or ASO-mediated depletion of NADPH oxidase, xanthine oxidase, or CaMKIV suppressed FosB expression and 0.23 kb TF promoter-luciferase activity (Fig. 4, F–K). These observations show that NADPH oxidase and xanthine oxidase-mediated ROS production and CaMKIV activation are required for FosB induction in enhancing AP-1–dependent TF expression at transcriptional level.
Figure 4.



FosB mediates 15(S)-HETE–induced TF promoter activity. A, TF promoter encompassing from −2174 nt to +116 nt was cloned and sequenced. B, TF promoter-luciferase constructs with serial 5′ truncations or site-directed insertion of mutations into the indicated AP-1 site in the pGL3-hTFp (0.23 kb)-Luc construct were made, transfected into THP1 cells, quiesced, treated with vehicle or 15(S)-HETE (0.1 μm) for 6 h and their activities were measured. AP-1p and AP-1d represent the proximal and distal AP-1–binding sites located at −205, and −217 nt, respectively. C, quiescent THP1 cells were treated with vehicle or 15(S)-HETE (0.1 μm) for the indicated time periods, and the cell extracts were prepared and analyzed by Western blotting for the indicated proteins using their specific antibodies. D, THP1 cells were transfected with control or the indicated JunB, FosB, or Fra2 ASOs and 48 h later cell extracts were prepared and analyzed by Western blotting for JunB, FosB, or Fra2 levels using their specific antibodies and normalized to β-tubulin. THP1 cells were co-transfected with TF promoter-luciferase construct in combination with or without control, JunB, FosB, or Fra2 ASOs, quiesced, treated with vehicle or 15(S)-HETE (0.1 μm) for 6 h, and analyzed for luciferase activity. E, THP1 cells that were transfected with control, JunB, FosB, or Fra2 ASOs and quiesced were treated with vehicle or 15(S)-HETE (0.1 μm) either for 2 h and analyzed for TF expression (mRNA and proteins levels) and its activity or for 8 h and cell migration was measured. F–I, the role of NADPH oxidase, xanthine oxidase, and CaMKIV in 15(S)-HETE–induced FosB expression was determined using pharmacological and ASO approaches as described in Fig. 2, A and B, and Fig. 3, B and C, respectively. The blots in F–I are reprobed with β-tubulin for normalization. J, THP1 cells were transfected with empty vector or pGL3-hTFp0.23-Luc construct, quiesced, treated with vehicle or 15(S)-HETE (0.1 μm) in the presence and absence of apocynin (100 μm), DPI (10 μm), or allopurinol (100 μm) for 6 h, and cell extracts were prepared and analyzed for luciferase activity. K, THP1 cells were co-transfected with empty vector or pGL3-hTFp0.23-Luc in combination with and without control or CaMKIV ASO, quiesced, treated with vehicle or 15(S)-HETE (0.1 μm) for 6 h, and cell extracts were prepared and analyzed for luciferase activity. *, p < 0.01 versus vehicle control or control ASO or empty vector; **, p < 0.01 versus vehicle control + 15(S)-HETE or control ASO + 15(S)-HETE or pGL3-hTF + 15(S)-HETE.
Although Jun proteins can form active AP-1 complex via either homodimerization or heterodimerization involving the Jun or Fos family of proteins, the Fos family members by themselves cannot form functional AP-1 and therefore require partnership with Jun proteins or another family of transcriptional factors (34, 35). To identify the binding partner(s) for FosB in 15(S)-HETE–induced transcriptional transactivation of TF promoter, we performed co-immunoprecipitation experiments. As shown in Fig. 5A, FosB was found to exist in complex with cFos, Fra1, cJun, JunB, JunD, ATF2, CREB, NFATc1, NFATc2, NFATc4, p65, PPARγ, and STAT1 constitutively, and 15(S)-HETE had no effect on these interactions. However, FosB association with NFATc3 was found to be increased in a time-dependent manner in response to 15(S)-HETE (Fig. 5A). Therefore, we next sought to study the role of NFATc3 on TF expression and activity. Blockade of NFATs by CsA, a potent inhibitor of calcineurin (35), suppressed 15(S)-HETE–induced TF promoter activity and its expression (Fig. 5B). Interestingly, blockade of NFATs also decreased 15(S)-HETE–induced FosB expression (Fig. 5B). In line with these results, depletion of NFATc3 levels using its ASOs attenuated 15(S)-HETE–induced FosB and TF expression as well as TF activity (Fig. 5B). Furthermore, depletion of NFATc3 levels reduced 15(S)-HETE–induced TF promoter activity and monocyte migration (Fig. 5, C and D). In addition, we observed that 15(S)-HETE induces the translocation of NFATc3 from the cytoplasm to the nucleus in a time-dependent manner and this translocation requires NADPH oxidase, xanthine oxidase, and CaMKIV activation (Fig. 5, E and F).
Figure 5.

NFATc3 mediates 15(S)-HETE–induced TF expression and activity. A, equal amounts of protein from control and the indicated time periods of 15(S)-HETE (0.1 μm)–treated cells were immunoprecipitated with anti-FosB antibodies, and the immunocomplexes were analyzed by Western blotting for the indicated proteins using their specific antibodies followed by normalization to FosB. B, quiescent THP1 cells were treated with vehicle or 15(S)-HETE (0.1 μm) in the presence or absence of CsA (10 μm) for 2 h and analyzed for TF expression (mRNA and protein levels) and its activity. All the conditions were the same as in the upper panel except that cells were transfected with control or NFATc3 ASOs and quiesced before subjecting to treatment with and without 15(S)-HETE. C, THP1 cells were transfected with control or NFATc3 ASOs, quiesced, and subjected to 15(S)-HETE (0.1 μm)–induced migration. D, THP1 cells were co-transfected with empty vector or pGL3-hTFp in combination with and without control or NFTAc3 ASOs, quiesced, treated with vehicle or 15(S)-HETE (0.1 μm) for 6 h; cell extracts were prepared and analyzed for luciferase activity. E, quiesced THP1 cells were treated with vehicle or 15(S)-HETE (0.1 μm) for the indicated time periods, and the cytoplasmic extracts (CE) and nuclear extracts (NE) were prepared and analyzed by Western blotting for the indicated proteins using their specific antibodies. F, THP1 cells were transfected with control, p47phox, xanthine oxidase, or CaMKIV ASOs, quiesced, treated with vehicle or 15(S)-HETE (0.1 μm) for the indicated time periods, and the CE and NE were prepared and analyzed by Western blotting for the indicated proteins using their specific antibodies. *, p < 0.01 versus control ASO; **, p < 0.01 versus control ASO + 15(S)-HETE.
To find whether both FosB and NFATc3 bind to the proximal AP-1 site, we studied the time course effect of 15(S)-HETE on AP-1 DNA–binding activity using proximal AP-1 element as a biotin-labeled probe. 15(S)-HETE induced AP-1 DNA–binding activity in a time-dependent manner with a maximum effect at 2 h (Fig. 6A). In addition, supershift analysis indicated that only FosB and NFATc3 but not Fra2 or JunB bind to this AP-1 site (Fig. 6A). To confirm these observations, we performed ChIP and chromatin re-immunoprecipitation (re-ChIP) assays. ChIP assay showed that FosB and NFATc3 bind to the same region of the TF promoter containing the proximal AP-1 site (Fig. 6B). The re-ChIP analysis of anti-FosB or anti-NFATc3 chromatin immunocomplexes for the presence of NFATc3 and FosB, respectively, further confirmed that NFATc3 and FosB bind to the same region (Fig. 6B). In addition, depletion of NADPH oxidase, xanthine oxidase, or CaMKIV using their ASOs blocked the recruitment of NFATc3 and FosB to the TF promoter in response to 15(S)-HETE (Fig. 6, C and D). Based on all these observations, it may be suggested that NFATc3, by inducing and partnering with FosB, binds to AP-1 site on TF promoter and enhances its activity in a manner that is dependent on NADPH oxidase and xanthine oxidase–mediated ROS production and CaMKIV activation.
Figure 6.

NFATc3 and FosB interact with each other and bind to AP-1 site in the TF promoter. A, nuclear extracts of control and the indicated time periods or 2 h of 15(S)-HETE (0.1 μm)–treated cells were analyzed for AP-1 and DNA activity and for the presence of FosB, JunB, Fra2, or NFATc3 in the AP-1-DNA complexes by EMSA and supershift EMSA, respectively, using AP-1–binding site at −205 nt in the TF promoter as a biotin-labeled probe. B, THP1 cells were treated with vehicle or 15(S)-HETE (0.1 μm) for the indicated time periods and subjected to ChIP and re-ChIP assay of TF promoter using anti-FosB or anti-NFATc3 antibodies. C and D, THP1 cells were transfected with control, p47phox, xanthine oxidase, or CaMKIV ASOs, quiesced, treated with vehicle or 15(S)-HETE (0.1 μm) for 2 h and subjected to ChIP and re-ChIP assays for FosB and NFATc3 binding to TF promoter as described in B. *, p < 0.01 versus control ASO; **, p < 0.01 versus control ASO + 15(S)-HETE. FP, free probe.
To explore the functional significance of 15(S)-HETE and TF link in vivo, we isolated peritoneal macrophages from WT and 12/15-LOX−/− mice and subjected them to monocyte chemotactic protein 1 (MCP-1) and LPS-induced migration. It is interesting to note that both MCP-1 and LPS stimulate the migration of only WT and not 12/15-LOX−/− macrophages (Fig. 7A). To substantiate the role of TF in macrophage migration in vivo, we also measured MCP-1 and LPS-induced efflux of macrophages from the peritoneal cavity using the method described by Cao et al. (36). In this assay, the number of macrophages remaining in the peritoneal cavity following an intraperitoneal injection of MCP-1 or LPS is determined and compared to that of macrophages remaining after PBS injection. Both MCP-1 and LPS induced the efflux of macrophages from the peritoneal cavity of WT mice but not 12/15-LOX−/− mice (Fig. 7B). We also observed that inhibiting TF activity by injecting its neutralizing antibody into the peritoneum reduces the efflux of macrophages from the peritoneal cavity in WT mice (Fig. 7B). These findings suggest that macrophage migration in vivo is also dependent on 12/15-LOX–induced TF expression and its activity. These results also suggest that MCP-1 and LPS-induced macrophage migration depends on 12/15-LOX–mediated TF expression. Consistent with this notion, both MCP-1 and LPS induced TF expression only in WT but not 12/15-LOX−/− mice (Fig. 7C).
Figure 7.

12/15-LOX/15(S)-HETE is required for monocyte/macrophage trafficking. A, peritoneal macrophages from WT and 12/15-LOX−/− mice were isolated and subjected to MCP-1 (20 ng/ml) and LPS (100 ng/ml)–induced migration. B, in vivo migration of peritoneal macrophages induced by MCP-1 or LPS in presence and absence of TF neutralizing antibodies (anti-mouse TF Ab-1H1) was performed using macrophage efflux model as described in “Materials and methods.” C, peritoneal macrophages from WT and 12/15-LOX−/− mice were isolated and treated with and without MCP-1 (20 ng/ml) or LPS (100 ng/ml) for 2 h. Cell extracts were prepared and analyzed by Western blotting for TF levels using its specific antibodies, and the blot was normalized for β-tubulin. D, PBMC from WT and 12/15-LOX−/− mice were isolated, incubated with TF nAb for 1 h at room temperature, treated with and without MCP-1, LPS, or 12(S)-HETE for 2 h, labeled with PKH67, and injected via tail vein into peritonitis recipient WT mice. After 24 h, peritoneal cells were harvested and the number of PKH67-positive as well as PKH67+Mac-1–positive cells were quantified by flow cytometry. *, p < 0.01 versus vehicle control; **, p < 0.01 versus WT + MCP-1 or WT + LPS or WT + 12(S)-HETE or 12/15-LOX−/− + 12(S)-HETE.
To determine whether 12/15-LOX–dependent TF expression and activity are required for monocyte trafficking, we performed adoptive transfer of mononuclear cells from blood to peritoneum under the influence of thioglycolate-induced peritonitis. We isolated mononuclear cells from both WT and 12/15-LOX−/− mice, treated with and without MCP-1, LPS, or 12(S)-HETE in the presence and absence of TF neutralizing antibodies and labeled with cell tracker PKH67, injected via tail vein into thioglycolate-treated WT mice; 24 h later the PKH67 and PKH67/Mac-1–positive cells in the peritoneum were counted. It was found that the number of PKH67 and PKH67/Mac-1–positive cells from WT mice were substantially higher than those from 12/15-LOX−/− mice in the peritoneum of the thioglycolate-treated WT mice (Fig. 7D). Furthermore, prior treatment with MCP-1 or LPS enhanced the migratory capacity of WT but not 12/15-LOX−/− macrophages into the peritoneum of the thioglycolate-treated WT mice by several-fold, and these effects were found to be sensitive to inhibition by TF neutralizing antibodies (Fig. 7D). However, mononuclear cells from both WT and 12/15-LOX−/− mice treated with 12(S)-HETE prior to their injection into the recipient WT mice exhibited almost the same capacity of trafficking and incubation with TF neutralizing antibodies before treatment with 12(S)-HETE negated their mobility (Fig. 7D). These findings indicate that 12/15-LOX–dependent TF expression and activity play an important role in the trafficking of monocytes from blood to the inflamed tissue.
Discussion
Atherothrombotic complications are the leading causes of death in the world (37, 38). Although the risk factors for these diseases have been identified, the disease initiation and progression mechanisms appear to be complex, intertwined with genetic and nutritional factors (39). Increased co-localization of 15-LOX with the epitopes of the oxidized low-density lipoprotein particles and the growing number of studies using both pharmacological and genetic approaches provides convincing evidence for the role of lipoxygenases, particularly 12/15-LOX, in these diseases (21, 22, 27). In addition, a large body of literature supports a role for macrophages in the pathogenesis of both atherosclerosis and atherothrombosis (40–42). It has been reported that TF expression increases with the progression of atherosclerotic lesions and plays a key role in thrombus formation following plaque rupture (41, 42).
In the present study we show that 15(S)-HETE, the major arachidonic acid metabolite of human 15-LOX1/2, induces the expression and activity of TF in THP1 cells. This result suggests that besides its role in atherosclerosis, 12/15-LOX via its effect on TF expression may also play a role in thrombosis following plaque rupture. A recent study supports this view as it has demonstrated a role for 15(S)-HETE in thrombin generation and platelet aggregation (43). Because macrophages appear to be involved in atherogenesis and atherothrombosis and 15(S)-HETE–induced migration of monocytes is dependent on TF expression, it is likely that 12/15-LOX and 12/15(S)-HETE could be involved in these vascular lesions via TF expression and monocyte migration. The finding that neutralizing TF antibodies blocks 15(S)-HETE–induced monocyte migration may imply that TF activity is required for the effect of 15(S)-HETE on monocyte migration. This is a novel finding, as many previous studies have suggested that TF-induced tumor cell migration and invasion depend on its cytoplasmic domain rather than its proteolytic activity (44). It is quite possible that both TF expression and activity might be involved in cytoskeleton remodeling in the modulation of monocyte migration by 15(S)-HETE. In fact, many studies have reported that TF in complex with Factor VII binds and mediates PAR2 signaling in the modulation of cell migration (45). Alternatively, TF and Factor VII–mediated thrombin generation could also play a role in 15(S)-HETE–induced monocyte migration (46). It was also demonstrated that TF directly binds to actin-binding protein 280, leading to activation of focal adhesion kinase (FAK) and cell migration (47).
A large body of data suggests that oxidative stress plays a notable role in atherogenesis and atherothrombosis (48). In this context, in the present study we found that 15(S)-HETE–induced TF expression and activation require ROS production. These observations also suggest that ROS production acts upstream to TF expression in 15(S)-HETE–induced monocyte migration. A large body of data shows that ROS, particularly H2O2, plays a role as signaling molecule via its capacity to enhance phosphorylation and activation of various kinases, including the serine/threonine kinases and tyrosine kinases (30, 31). In the present study we found that 15(S)-HETE stimulates serine phosphorylation of CaMKIV via NADPH oxidase and xanthine oxidase–dependent ROS production in mediating TF expression and monocyte cell migration. Actin cytoskeletal remodeling is essential for cell motility, and CaMKIV via phosphorylation of cofilin and its inhibition plays a role in actin polymerization (49). Based on these findings, it is likely that NADPH oxidase and xanthine oxidase–dependent H2O2 production via CaMKIV activation and TF expression may lead to cytoskeleton remodeling facilitating monocyte migration. It should be pointed out that we have reported previously that 15(S)-HETE induces ROS production in monocytes via an interdependent activation of both NADPH oxidase and xanthine oxidase (23). In line with this observation, recently we have also demonstrated a similar interdependency between NADPH oxidase and xanthine oxidase activation in the production of ROS by cholesterol crystals in mediating CD36 expression and foam cell formation (50). Many growth factors and cytokines produce ROS in modulating cell proliferation, migration, or apoptosis mostly via activation of NADPH oxidase (51, 52). However, it is not clear whether any cross talk exists between NADPH oxidase and xanthine oxidase in the ROS production by growth factors or cytokines. Therefore, it would be quite interesting to find whether interdependency exists between NADPH oxidase and xanthine oxidase activation in the production of ROS in mediating cell proliferation, migration, or apoptosis in response to growth factors or cytokines and whether it is a unique mechanism of ROS production.
Many studies have demonstrated a role for transcriptional mechanisms in the regulation of TF expression in response to a wide array of stimulants in several cell types (33). In addition, a role for various transcriptional factors, including AP-1, Egr1, NFκB, and STAT, have been reported in the regulation of TF expression (18, 33, 34). Toward this end, our data show that although 15(S)-HETE triggers the induction of JunB, Fra2, and FosB only FosB was involved in 15(S)-HETE–induced TF promoter activity and its expression. Various studies have shown the importance of AP-1 sites and cFos-cJun or cFos-JunD heterodimers in mediating TF promoter activity in monocytes and endothelial cells (33). Blockade of NADPH oxidase, xanthine oxidase, or CaMKIV suppresses 15(S)-HETE–induced FosB expression, which suggests that ROS-mediated CaMKIV activation is involved in 15(S)-HETE–induced FosB/AP-1 activation in TF promoter activity and its expression in mediating monocyte migration. A role for ROS involvement in TF expression has been reported previously (53). In addition, we have reported previously that NADPH oxidase and xanthine oxidase–dependent ROS production is required for CREB-mediated IL-17A expression in 15(S)-HETE–induced monocyte migration and their adhesion to endothelial cells (23). Based on our previous and present observations, it may be speculated that ROS via TF and IL-17A expression might be involved in modulating various aspects of cell motility such as generation of local cellular guidance cues and promoting cytoskeletal remodeling in 15(S)-HETE–induced monocyte migration, as cell migration is a collective process of various cellular events (54, 55). However, further studies are required to find which of these various cellular events the TF and IL-17A are targeting in the modulation of monocyte migration by 15(S)-HETE.
Although Jun proteins can form active AP-1 complex via either homodimerization or heterodimerization involving the Jun or Fos family of proteins, the Fos family members by themselves cannot form functional AP-1. When analyzed for the association of transcription factors, whose binding sites are present in TF promoter, with FosB, we found that NFATc3 forms a complex with FosB in response to 15(S)-HETE. Furthermore, our results revealed that NFATc3 plays a role in 15(S)-HETE–induced TF promoter activity and its expression in mediating monocyte migration. These findings are also consistent with the upstream signaling events in TF expression as depletion of NADPH oxidase, xanthine oxidase, or CaMKIV blocked 15(S)-HETE–induced NFATc3 nuclear translocation, indicating the role of ROS-CaMKIV signaling in NFATc3 activation by 15(S)-HETE. What is more interesting is that 15(S)-HETE–induced FosB expression also requires NFATc3 activation. This result is indeed exciting as NFATc3 induces its partner FosB levels and then forms a complex with it in enhancing TF expression in response to 15(S)-HETE. Previous studies have shown that NFATs interact with the Fos or Jun family of proteins and bind to AP-1 and NFAT composite DNA-binding elements in enhancing gene expression (35). In this context, our EMSA, supershift EMSA, ChIP, and re-ChIP analyses show that NFATc3 in concert with FosB binds to a consensus AP-1 element rather than a composite AP-1 and NFAT element in TF promoter and enhances its expression in response to 15(S)-HETE.
Monocyte/macrophage migration to, and their buildup in, the vessel wall is a critical event in vascular inflammation and its diseases (37). In this regard, our results demonstrate not only a role for 15(S)-HETE–induced TF expression and its activity in monocyte migration in vitro, but also the importance of 12/15-LOX–12/15(S)-HETE–mediated TF expression in monocyte/macrophage migration in vivo. The lack of the responsiveness of 12/15-LOX−/− monocytes/macrophages to MCP-1 and LPS-induced migration as compared with those from WT mice and the capacity of 12(S)-HETE to restore their migratory response in TF-sensitive manner suggests that 12/15-LOX-12/15(S)-HETE–mediated TF expression is also required for a chemokine and inflammatory molecules–induced migration of these cells. This conclusion can be further corroborated by the observations that both MCP-1 and LPS induce the expression of TF only in WT but not 12/15-LOX−/− macrophages. The defective trafficking of 12/15-LOX−/− monocytes/macrophages to thioglycolate-instilled peritoneum as compared with those of WT monocytes/macrophages suggests that the monocyte/macrophage trafficking to the inflamed peritoneum is dependent on 12/15-LOX. Because prior treatment with 12(S)-HETE restores the capacity of 12/15-LOX−/− monocyte/macrophage trafficking in TF-dependent manner further indicates that 12/15-LOX–12/15(S)-HETE–mediated TF expression is required for the inflammatory responsiveness of these cells. In summary, as depicted in Fig. 8, our findings suggest that 12/15(S)-HETE via NADPH oxidase and xanthine oxidase–dependent ROS production and CaMKIV activation leads to NFATc3 stimulation, which, in turn, by inducing and partnering with FosB binds to and enhances TF promoter activity and its expression in mediating monocyte/macrophage migration and trafficking both in vitro and in vivo.
Figure 8.
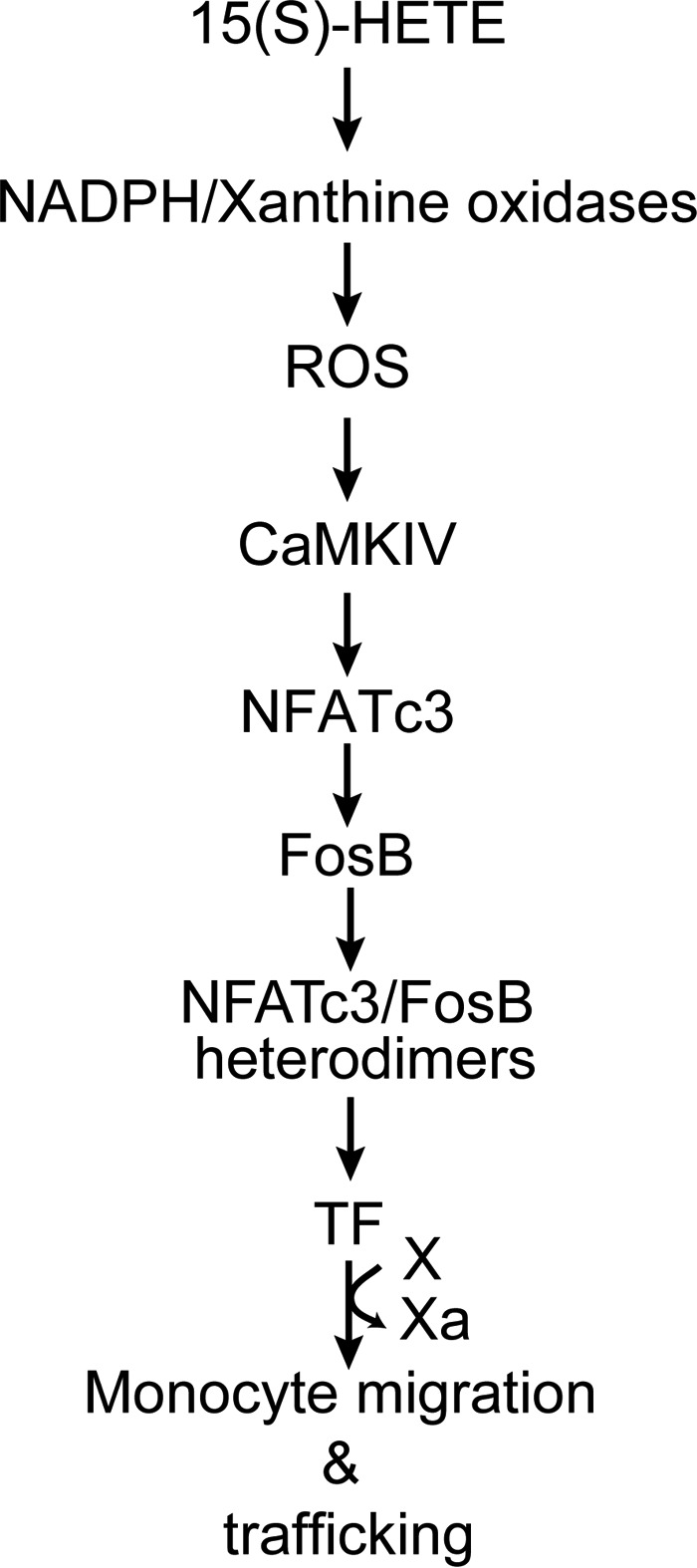
Schematic diagram depicting the potential mechanism of 15(S)-HETE–induced TF expression and monocyte migration/trafficking.
Materials and methods
Reagents
Anti-pCaMKI (ab62215), anti-pCaMKIV (ab59424), anti-PE–Cy5–Mac-1 (ab25533), and anti-TF (ab104513) antibodies were obtained from Abcam (Cambridge, MA). Anti-NFATc1 antibodies (MA3–024) were purchased from Affinity BioReagents (Golden, CO). Anti-pCaMKII (3361) antibody was bought from Cell Signaling Technology (Danvers, MA). Anti-peroxisome proliferator-activated receptor δ antibodies (NBP1–39684) were purchased from Novus Biologicals (Littleton, CO). Anti-ATF2 (SC-6223), anti-CaMKI (SC-33165), anti-CaMKII (SC-9035), anti-CaMKIV (SC-166156), anti-CREB (SC-186), anti-cFos (SC-52), anti-FosB (SC-48), anti-Fra1 (SC-605), anti-Fra2 (SC-604), anti-HIF-1α (SC-12542), anti-cJun (SC-44), anti-JunB (SC-46), anti-JunD (SC-74), anti-NFATc2 (SC-13034), anti-NFATc3 (SC-8321), anti-NFATc4 (SC-271127), anti-p47Phox (SC14015), anti-p65 (SC-56735), anti-PPARγ (SC7196), anti-STAT1 (SC-464), anti-STAT2 (SC-476), anti-STAT5 (SC-836), anti-β-tubulin (SC-9104), and anti-xanthine oxidase (H110) antibodies; normal mouse serum (SC-45051); and normal rabbit serum (SC-3888) were obtained from Santa Cruz Biotechnology (Dallas, TX). Tissue factor chromogenic activity kit (CT1002b) was purchased from Assaypro (St. Charles, MO). 5(S)-HETE (34230), 12(S)-HETE (34570), 15(R)-HETE (34710), and 15(S)-HETE (34720) were purchased from Cayman Chemicals (Ann Arbor, MI). Recombinant mouse MCP-1 (578406) was obtained from Biolegend (San Diego, CA). Apocynin (A10809), allopurinol (A8003), LPS (L2637), and PKH67 (PKH67GL) were procured from Sigma-Aldrich. Diphenyleneiodonium chloride (BML-CN240) was purchased from Enzo Life Sciences (Farmingdale, NY). STO-609 acetate (1551) was bought from Tocris Bioscience (Bristol, UK). Lipofectin transfection reagent (15596018) and SYBR Green PCR Master Mix (4309195) were obtained from Invitrogen. pGL3 basic vector and Luciferase Assay System (E4530) were purchased from Promega (Madison, WI). Biotin 3′ End DNA Labeling Kit (89818) and LightShift Chemiluminescent EMSA Kit (20148) were procured from Pierce Biotechnology (Rockford, IL). The Amersham ECL Western Blotting Detection Reagents (RPN2106) were obtained from GE Healthcare. All the phosphorothioate-modified antisense oligonucleotides (ASOs) and primers were synthesized by IDT (Coralville, IA). The phosphorothioate-modified ASOs used in this study are as follows: hControl ASO, 5′-GGGGGUTCTCTGCGTACGGTGCUAGU-3′; hCaMKIV ASO1 (NM_001744), 5′-GUGGAACCCCAGGGUACUGCGCACCTGAA-3′; ASO2, 5′-AAUUCAATGCCCGGCGUAAGCUUAA-3′; hp47Phox ASO (NM_000265), 5′-GUUGGGCTCAGGGTCTTCCGUCUC-3′; and hXO ASO (NM_000379), 5′-GCCUCCTCCCATTCTCTTCACUCG-3′.
Cell culture
THP1 cells were purchased from American Type Culture Collection (Manassas, VA) and cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 50 units/ml penicillin, 50 μg/ml streptomycin, and 50 μm β-mercaptoethanol. Cultures were maintained in a humidified 95% air and 5% CO2 atmosphere at 37 °C. Cells were quiesced in RPMI 1640 medium containing 50 units/ml penicillin, 50 μg/ml streptomycin, 50 μm β-mercaptoethanol, and 1% heat-inactivated FBS for 12 h and used to perform the experiments unless otherwise indicated.
Animals
WT and Alox15 knock-out (12/15-LOX−/−) mice (56) on C57BL/6 background were bred and maintained according to the Institutional Animal Care and Use Committee facility guidelines. The experiments involving the use of animals were approved by the Institutional Animal Care and Use Committee of the University of Tennessee Health Science Center, Memphis, Tennessee.
Transfections
Transfections were performed as described previously (23). Briefly, cells were transfected with the indicated ASO at 100 nm concentration using Lipofectin transfection reagent for 6 h following the manufacturer's instructions. After transfection, cells were maintained in complete RPMI 1640 medium for 36 h followed by growth arresting in serum-free medium overnight before being used for the experiments.
RT-PCR
Total cellular RNA was extracted from THP1 cells using TRIzol reagent according to the manufacturer's protocol. Reverse transcription was performed with a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Complementary DNA (cDNA) was then used as a template for amplification using the following primers: human TF (NM_1993), forward: 5′-ACCTCACCGACGAGATTGT-3′ and reverse: 5′-TACCTGGAGACAAACCTCGG-3′; human β-actin (NM_001101), forward: 5′-AGCCATGTACGTTGCTAT-3′ and reverse: 5′-GATGTCCACGTCACACTTCA-3′. The amplification was performed using GeneAMP PCR System 2400 (Applied Biosystems). The amplified PCR products were separated on 1.5% agarose gels and stained with ethidium bromide, and the images were captured using a Kodak In-Vivo Imaging System.
Western blotting
Cell extracts containing an equal amount of protein were resolved by electrophoresis on 0.1% (w/v) SDS and 10% (w/v) polyacrylamide gels. The proteins were transferred electrophoretically to a nitrocellulose membrane. After blocking in either 5% (w/v) nonfat dry milk or 5% (w/v) BSA, the membrane was probed with appropriate primary antibodies followed by incubation with horseradish peroxidase–conjugated secondary antibodies. The antigen-antibody complexes were detected using enhanced chemiluminescence detection reagent kit.
Tissue factor activity
Tissue factor activity was measured using a kit following the supplier's protocol.
Cell migration
A modified Boyden chamber method was used to measure cell migration (23). After growth arresting overnight in serum-free RPMI 1640 medium, THP1 cells were resuspended in serum-free RPMI 1640 medium and plated on Matrigel-coated cell culture inserts at 5 × 104 cells per insert well. 15(S)-HETE was added to a final concentration of 0.1 μm to both the upper and the lower chambers, and the cells were incubated for 8 h at 37 °C. The inserts were then lifted, nonmigrated cells on the upper surface of the membrane were removed with a cotton swab, and the membrane was fixed in methanol and stained with 4′,6-diamidino-2-phenylindole (DAPI) to visualize the nuclei. Whenever ASOs were used, cells were transfected first with the indicated ASOs for 6 h in serum-free RPMI 1640 medium, recovered in complete medium for 36 h, and subjected to synchronization in serum-free medium for overnight before being analyzed for migration. Whenever pharmacological inhibitors or neutralizing antibodies were used, cells were exposed to the indicated inhibitor or neutralizing antibody for 30 min before the treatment. The DAPI-positive cells were counted under an inverted microscope (Carl Zeiss AxioVision Observer.Z1) and the cell migration was expressed as the number of cells per field of view.
TF promoter cloning
The human TF promoter sequence encompassing −2184 to +116 nt relative to the transcription start site was amplified from human THP1 cell genomic DNA using forward primer (hTFpF1) 5′-GGTACCTTCTCAGAGCCTCCAAGATAGA-3′ incorporating a KpnI restriction enzyme site at the 5′-end and a reverse primer (hTFpR1) 5′-CTCGAGAGTCCATGTCTACCAGTTGGCG-3′ incorporating an XhoI restriction enzyme site at the 5′-end. The boldface letters indicate KpnI and XhoI sites in both the forward and the reverse primers, respectively. The PCR product was digested with KpnI and XhoI, and the purified fragment was cloned into KpnI and XhoI sites of the pGL3 basic vector (Promega) to yield pGL3-hTFp2.1 kb plasmid. To generate 5′-truncated promoter fragments starting from pGL3-hTFp1.6 kb, pGL3-hTFp0.68 kb, pGL3-hTFp0.38 kb, pGL3-hTFp0.25 kb, pGL3-hTFp0.23 kb, pGL3-hTFp0.19 kb to +116 nt, the following forward 5′-GGTACCCAGCTGGCAGAGAGCTCAAAG-3′, 5′-GGTACCGTCACACCGTGGCTGACACC-3′, 5′-GGTACCAGAATTCTTCCAAGGCG-3′, 5′-GGTACCAGAATTCTTCCAAGGCG-3′, 5′-GGTACCGCTCGGTGGCGCGGTTGAAT-3′, 5′-GGTACCGCGGTTGAATCACTGGGGT-3′, 5′-GGTACCTCCCGGAGTTTCCTA-3′, and reverse 5′-CGAGAGTCCATGTCTACCAGTTGGCG-3′ primers were used for PCR amplification using pGL3-hTFp2.1 kb plasmid as a template. The PCR products were digested with KpnI and XhoI and cloned into KpnI and XhoI sites of the pGL3 basic vector. Site-directed mutations within the AP-1–binding sites at −205 nt and −217 nt (pGL3-hTFp0.23-M1 and M2, respectively) were introduced using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) according the manufacturer's instructions using the following primers: forward, 5′-CTGGGGGCCGTCATCCCTTGC-3′ and reverse, 5′-GCAAGGGATGACGGCCCCCAG-3′, for the AP-1 site at −205 nt and forward, 5′-TGGCGCGGCTCCATGACTGGGG-3′ and reverse, 5′-CCCCAGTCATGGAGCCGCGCCA-3′ for the AP-1 site at −217 nt, respectively. The italic boldface letters indicate the mutated bases. The nucleotide sequence of each construct was verified by DNA sequencing.
Luciferase assay
THP1 cells were transfected with pGL3 empty vector or indicated TF promoter constructs using Lipofectamine transfection reagent in serum-free medium for 6 h. After 36 h recovery in complete medium, cells were growth arrested in serum-free medium for 12 h, treated with and without 0.1 μm 15(S)-HETE for 6 h, washed with cold PBS, and lysed in 200 μl of lysis buffer. The cell extracts were cleared by centrifugation at 12,000 rpm for 2 min at 4 °C. The supernatants were assayed for luciferase activity using Luciferase Assay System (Promega) and a single tube luminometer (TD20/20; Turner Designs, Sunnyvale, CA) and expressed as relative luciferase units.
EMSA
Nuclear extracts of THP1 cells with and without the indicated treatments were prepared as described previously (57). The protein content of the nuclear extracts was determined using a micro-BCA method (Pierce). Biotin-labeled double-stranded oligonucleotides encompassing AP-1–binding element at −205 nt (sense, 5′-CTGGGGTGAGTCATCCCTTGCAGGTCCCGGAGTT-3′ and antisense, 5′-AACTCCGGGACCTGCAAGGGATGACTCACCCCAG-3′) were used as a probe for the EMSA. Briefly, 5 μg of nuclear extract was incubated in a binding buffer (10 mm Tris-HCl, pH 7.9, 50 mm KCl, 1 mm DTT, 15% glycerol) with 2.5 nm biotin-labeled probe and 2 μg of poly(dI:dC) for 30 min at room temperature in a total volume of 20 μl on ice, and the DNA-protein complexes were analyzed by electrophoresis on 6% polyacrylamide gels and visualized by chemiluminescence imaging. To perform a supershift EMSA, the complete reaction mix was incubated with 2 μg of the indicated antibody for 1 h on ice before separating it by electrophoresis. Normal serum was used as a negative control.
ChIP and re-ChIP assays
ChIP assay was performed on THP1 cells with and without the indicated treatments using a kit following the supplier's protocol (Upstate Biotechnology Inc., Lake Placid, NY). FosB and NFATc3-DNA complexes were immunoprecipitated using anti-FosB and anti-NFATc3 antibodies, respectively. Pre-immune mouse serum or rabbit serum were used as negative controls. For re-ChIP assay, the chromatin complexes were eluted from the first ChIP with 10 mm DTT at 37 °C for 30 min and diluted 20 times with ChIP dilution buffer (1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, 150 mm NaCl) and re-immunoprecipitated with the indicated antibodies in a sequential manner. The immunoprecipitated DNA was uncross-linked, subjected to proteinase K digestion, and purified using QIAquick columns (28104, Qiagen, Valencia, CA). The ChIP and re-ChIP samples were analyzed by quantitative PCR using the following primers: forward, 5′-CTGGGGTGAGTCATCCCTTGC-3′ and reverse, 5′-TATGCTGGGCGGCGAGT-3′ that would amplify a 94-bp fragment encompassing the AP-1–binding site at −205 nt. Quantitative PCR was performed on 7300 Real-Time PCR systems (Applied Biosystems) using SYBR Green PCR Master Mix. The reactions were carried out with following conditions: 95 °C for 10 min followed by 40 cycles at 95 °C for 15 s with extension at 60 °C for 1 min. The test antibodies (anti-FosB and anti-NFATc3) and control IgG Ct values were normalized to input DNA Ct values to obtain ΔCt values. Then the ΔΔCt values were calculated by subtracting the ΔCt values of control from the experimental ΔCt values. The fold enrichment of FosB and NFATc3-bound TF promoter is calculated by using 2(−ΔΔCt).
Isolation of peritoneal macrophages
Mice were injected with 1 ml of 4% thioglycolate intraperitoneally, and 4 days later the animals were anesthetized with ketamine and xylazine and the peritoneal lavage was collected in RPMI 1640 medium. Cells were incubated in 100-mm Petri dishes (3 × 105 cells/cm2) in RPMI medium containing penicillin (50 units/ml) and streptomycin (50 μg/ml). After 3 h, floating cells (mostly red blood cells) were removed by washing with PBS and the adherent cells (macrophages) were collected and used as required.
Isolation of peripheral blood mononuclear cells (PBMCs)
WT or 12/15-LOX−/− mice were anesthetized with ketamine and xylazine. Blood was collected by cardiac puncture in a 1-ml syringe containing 0.05 ml of 0.5 m EDTA and diluted with equal volume of PBS. Blood was then overlaid on the Lymphoprep and centrifuged at 1500 rpm for 30 min at 4 °C. PBMC layer was collected, treated with RBC lysis buffer, washed twice with PBS, resuspended in PBS, and used as required. Wherever required, after isolation and appropriate treatments, cells were washed with PBS and labeled with PKH67 according to the manufacturer's instructions.
In vivo monocyte/macrophage migration
In vivo monocyte/macrophage migration was measured according to the method of Cao et al. (36). WT or 12/15-LOX−/− mice were injected intraperitoneally (IP) with 1 ml of 4% thioglycolate, and 4 days later mice were administered with and without MCP-1 (200 μg/kg body weight) or LPS (1.25 μg/mouse) in the presence and absence of neutralizing anti-mouse TF antibody 1H1 (17) (20 mg/kg body weight) via IP. Whenever antibodies were used, they were injected 1 h before MCP-1 or LPS was introduced. Four h later, the peritoneal macrophages were collected and counted using a Beckman Coulter particle counter. The cells were then stained with PE-cy5–conjugated anti-Mac-1 antibody and counted by flow cytometer. Migration index was determined by using the following formula: [1-(number of peritoneal macrophages from treatment group/number of peritoneal macrophages from control group)] × 100.
Adoptive transfer of mononuclear cells
Recipient mice (WT) were lightly anesthetized with ketamine and xylazine and injected with 4% thioglycolate into the peritoneal cavity to stimulate peritoneal inflammation. Four days after thioglycolate administration the mice received, via tail vein injection, peripheral blood mononuclear cells (1.4 × 105 cells/mouse) isolated from WT or 12/15-LOX−/− mice; treated with and without MCP-1 (20 ng/ml), LPS (100 ng/ml), or 12(S)-HETE (0.1 μm); and labeled with PKH67. Whenever the effect of neutralizing TF Ab was tested, cells were pretreated with the antibodies (2 μg/ml) for 1 h before the treatments. After 24 h, peritoneal cells were isolated, washed, re-suspended in FACS buffer, and stained with PE-cy5–conjugated anti-Mac-1 antibody, and the number of PKH67-positive macrophages were quantified by flow cytometer.
Statistics
All the experiments were repeated three times and the data are presented as mean ± S.D. The treatment effects were analyzed by two-way analysis of variance (ANOVA) test, and the p values < 0.05 were considered statistically significant. In the case of EMSA, supershift-EMSA, and Western blotting, one representative set of data is shown.
Author contributions
S. K. performed all the experiments. N. K. S. contributed in preparing the rough draft of the manuscript. D. K. provided critical reagents, participated in the discussions, and corrected the manuscript. G. N. R. conceived the overall goal of the project, designed the experiments, interpreted the data, and wrote the manuscript.
This work was supported in part by National Institutes of Health Grants HL064165 and HL103575 (to G. N. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- TF
- tissue factor
- 12/15-LOX
- 12/15-lipoxygenase
- 15(S)-HETE
- 15(S)-hydroxyeicosatetraenoic acid
- ROS
- reactive oxygen species
- CaMKIV
- calcium/calmodulin-dependent protein kinase IV
- NFATc3
- nuclear factor of activated T cells 3
- MCP-1
- monocyte chemotactic protein 1
- 15-LOX
- 15-lipoxygenase
- ASO
- antisense oligonucleotide
- CaMK
- calcium/calmodulin-dependent protein kinase
- nt
- nucleotide
- Ct
- threshold cycle.
References
- 1. Ruf W., and Edgington T. S. (1994) Structural biology of tissue factor, the initiator of thrombogenesis in vivo. FASEB J. 8, 385–390 [PubMed] [Google Scholar]
- 2. Pawlinski R., Fernandes A., Kehrle B., Pedersen B., Parry G., Erlich J., Pyo R., Gutstein D., Zhang J., Castellino F., Melis E., Carmeliet P., Baretton G., Luther T., Taubman M., Rosen E., and Mackman N. (2002) Tissue factor deficiency causes cardiac fibrosis and left ventricular dysfunction. Proc. Natl. Acad. Sci. U.S.A. 99, 15333–15338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nishibe T., Parry G., Ishida A., Aziz S., Murray J., Patel Y., Rahman S., Strand K., Saito K., Saito Y., Hammond W. P., Savidge G. F., Mackman N., and Wijelath E. S. (2001) Oncostatin M promotes biphasic tissue factor expression in smooth muscle cells: Evidence for Erk-1/2 activation. Blood 97, 692–699 [DOI] [PubMed] [Google Scholar]
- 4. Parry G. C., and Mackman N. (2000) Mouse embryogenesis requires the tissue factor extracellular domain but not the cytoplasmic domain. J. Clin. Invest. 105, 1547–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen J., Kasper M., Heck T., Nakagawa K., Humpert P. M., Bai L., Wu G., Zhang Y., Luther T., Andrassy M., Schiekofer S., Hamann A., Morcos M., Chen B., Stern D. M., Nawroth P. P., and Bierhaus A. (2005) Tissue factor as a link between wounding and tissue repair. Diabetes 54, 2143–2154 [DOI] [PubMed] [Google Scholar]
- 6. Versteeg H. H., Spek C. A., Peppelenbosch M. P., and Richel D. J. (2004) Tissue factor and cancer metastasis: the role of intracellular and extracellular signaling pathways. Mol. Med. 10, 6–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chu A. J. (2005) Tissue factor mediates inflammation. Arch. Biochem. Biophys. 440, 123–132 [DOI] [PubMed] [Google Scholar]
- 8. Matetzky S., Tani S., Kangavari S., Dimayuga P., Yano J., Xu H., Chyu K. Y., Fishbein M. C., Shah P. K., and Cercek B. (2000) Smoking increases tissue factor expression in atherosclerotic plaques: Implications for plaque thrombogenicity. Circulation 102, 602–604 [DOI] [PubMed] [Google Scholar]
- 9. Samad F., Pandey M., and Loskutoff D. J. (1998) Tissue factor gene expression in the adipose tissues of obese mice. Proc. Natl. Acad. Sci. U.S.A. 95, 7591–7596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Celi A., Cianchetti S., Dell'Omo G., and Pedrinelli R. (2010) Angiotensin II, tissue factor and the thrombotic paradox of hypertension. Expert Rev. Cardiovasc. Ther. 8, 1723–1729 [DOI] [PubMed] [Google Scholar]
- 11. Wilcox J. N., Smith K. M., Schwartz S. M., and Gordon D. (1989) Localization of tissue factor in the normal vessel wall and in the atherosclerotic plaque. Proc. Natl. Acad. Sci. U.S.A. 86, 2839–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Taubman M. B., Fallon J. T., Schecter A. D., Giesen P., Mendlowitz M., Fyfe B. S., Marmur J. D., and Nemerson Y. (1997) Tissue factor in the pathogenesis of atherosclerosis. Thromb. Haemost. 78, 200–204 [PubMed] [Google Scholar]
- 13. Tipping P. G., Erlich J. H., Apostolopoulos J., Mackman N., Loskutoff D., Holdsworth S. R. (1995) Glomerular tissue factor expression in crescentic glomerulonephritis. Correlations between antigen, activity, and mRNA. Am. J. Pathol. 147, 1736–1748 [PMC free article] [PubMed] [Google Scholar]
- 14. Palkovits J., Novacek G., Kollars M., Hron G., Osterode W., Quehenberger P., Kyrle P. A., Vogelsang H., Reinisch W., Papay P., and Weltermann A. (2013) Tissue factor exposing microparticles in inflammatory bowel disease. J. Crohns Colitis 7, 222–229 [DOI] [PubMed] [Google Scholar]
- 15. Segal J., Kickler T., Petri M. (2000) Tissue factor activity in patients with systemic lupus erythematosus: Association with disease activity. J. Rheumatol. 27, 2827–2832 [PubMed] [Google Scholar]
- 16. Taylor F. B. Jr., Chang A., Ruf W., Morrissey J. H., Hinshaw L., Catlett R., Blick K., and Edgington T. S. (1991) Lethal E. coli septic shock is prevented by blocking tissue factor with monoclonal antibody. Circ. Shock 33, 127–134 [PubMed] [Google Scholar]
- 17. Owens A. P. 3rd, Passam F. H., Antoniak S., Marshall S. M., McDaniel A. L., Rudel L., Williams J. C., Hubbard B. K., Dutton J. A., Wang J., Tobias P. S., Curtiss L. K., Daugherty A., Kirchhofer D., Luyendyk J. P., et al. (2012) Monocyte tissue factor-dependent activation of coagulation in hypercholesterolemic mice and monkeys is inhibited by simvastatin. J. Clin. Invest. 122, 558–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bochkov V. N., Mechtcheriakova D., Lucerna M., Huber J., Malli R., Graier W. F., Hofer E., Binder B. R., and Leitinger N. (2002) Oxidized phospholipids stimulate tissue factor expression in human endothelial cells via activation of ERK/EGR-1 and Ca++/NFAT. Blood 99, 199–206 [DOI] [PubMed] [Google Scholar]
- 19. Viswambharan H., Ming X. F., Zhu S., Hubsch A., Lerch P., Vergères G., Rusconi S., and Yang Z. (2004) Reconstituted high-density lipoprotein inhibits thrombin-induced endothelial tissue factor expression through inhibition of RhoA and stimulation of phosphatidylinositol 3-kinase but not Akt/endothelial nitric oxide synthase. Circ. Res. 94, 918–925 [DOI] [PubMed] [Google Scholar]
- 20. Mulhaupt F., Matter C. M., Kwak B. R., Pelli G., Veillard N. R., Burger F., Graber P., Lüscher T. F., and Mach F. (2003) Statins (HMG-CoA reductase inhibitors) reduce CD40 expression in human vascular cells. Cardiovasc. Res. 59, 755–766 [DOI] [PubMed] [Google Scholar]
- 21. Ylä-Herttuala S., Rosenfeld M. E., Parthasarathy S., Glass C. K., Sigal E., Witztum J. L., and Steinberg D. (1990) Colocalization of 15-lipoxygenase mRNA and protein with epitopes of oxidized low density lipoprotein in macrophage-rich areas of atherosclerotic lesions. Proc. Natl. Acad. Sci. U.S.A. 87, 6959–6963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sendobry S. M., Cornicelli J. A., Welch K., Bocan T., Tait B., Trivedi B. K., Colbry N., Dyer R. D., Feinmark S. J., and Daugherty A. (1997) Attenuation of diet-induced atherosclerosis in rabbits with a highly selective 15-lipoxygenase inhibitor lacking significant antioxidant properties. Br. J. Pharmacol. 120, 1199–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kotla S., Singh N. K., Heckle M. R., Tigyi G. J., and Rao G. N. (2013) The transcription factor CREB enhances interleukin-17A production and inflammation in a mouse model of atherosclerosis. Sci. Signal. 6, ra83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Simon T. C., Makheja A. N., and Bailey J. M. (1989) Formation of 15-hydroxyeicosatetraenoic acid (15-HETE) as the predominant eicosanoid in aortas from Watanabe Heritable Hyperlipidemic and cholesterol-fed rabbits. Atherosclerosis 75, 31–38 [DOI] [PubMed] [Google Scholar]
- 25. Henriksson P., Hamberg M., and Diczfalusy U. (1985) Formation of 15-HETE as a major hydroxyeicosatetraenoic acid in the atherosclerotic vessel wall. Biochim. Biophys. Acta 834, 272–274 [DOI] [PubMed] [Google Scholar]
- 26. Cyrus T., Witztum J. L., Rader D. J., Tangirala R., Fazio S., Linton M. F., and Funk C. D. (1999) Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. J. Clin. Invest. 103, 1597–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kirchhofer D., Moran P., Chiang N., Kim J., Riederer M. A., Eigenbrot C., and Kelley R. F. (2000) Epitope location on tissue factor determines the anticoagulant potency of monoclonal anti-tissue factor antibodies. Thromb. Haemost. 84, 1072–1081 [PubMed] [Google Scholar]
- 28. Drummond G. R., Selemidis S., Griendling K. K., and Sobey C. G. (2011) Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 10, 453–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Holmström K. M., and Finkel T. (2014) Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15, 411–421 [DOI] [PubMed] [Google Scholar]
- 30. Howe C. J., Lahair M. M., McCubrey J. A., and Franklin R. A. (2004) Redox regulation of the calcium/calmodulin-dependent protein kinases. J. Biol. Chem. 279, 44573–44581 [DOI] [PubMed] [Google Scholar]
- 31. Tokumitsu H., Inuzuka H., Ishikawa Y., Ikeda M., Saji I., and Kobayashi R. (2002) STO-609, a specific inhibitor of the Ca2+/calmodulin-dependent protein kinase. J. Biol. Chem. 277, 15813–15818 [DOI] [PubMed] [Google Scholar]
- 32. Oeth P., Parry G. C., and Mackman N. (1997) Regulation of the tissue factor gene in human monocytic cells. Role of AP-1, NF-κ B/Rel, and Sp1 proteins in uninduced and lipopolysaccharide-induced expression. Arterioscler. Thromb. Vasc. Biol. 17, 365–374 [DOI] [PubMed] [Google Scholar]
- 33. Moll T., Czyz M., Holzmüller H., Hofer-Warbinek R., Wagner E., Winkler H., Bach F. H., and Hofer E. (1995) Regulation of the tissue factor promoter in endothelial cells. Binding of NFκB-, AP-1-, and Sp1-like transcription factors. J. Biol. Chem. 270, 3849–3857 [DOI] [PubMed] [Google Scholar]
- 34. Kouzarides T., and Ziff E. (1988) The role of the leucine zipper in the fos–jun interaction. Nature 336, 646–651 [DOI] [PubMed] [Google Scholar]
- 35. Macián F., López-Rodríguez C., and Rao A. (2001) Partners in transcription: NFAT and AP-1. Oncogene 20, 2476–2489 [DOI] [PubMed] [Google Scholar]
- 36. Cao C., Lawrence D. A., Strickland D. K., and Zhang L. (2005) A specific role of integrin Mac-1 in accelerated macrophage efflux to the lymphatics. Blood 106, 3234–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Libby P., Ridker P. M., and Hansson G. K. (2011) Progress and challenges in translating the biology of atherosclerosis. Nature 473, 317–325 [DOI] [PubMed] [Google Scholar]
- 38. Furie B., and Furie B. C. (2008) Mechanisms of thrombus formation. N. Engl. J. Med. 359, 938–949 [DOI] [PubMed] [Google Scholar]
- 39. McGillicuddy F. C., and Roche H. M. (2012) Nutritional status, genetic susceptibility, and insulin resistance—important precedents to atherosclerosis. Mol. Nutr. Food Res. 56, 1173–1184 [DOI] [PubMed] [Google Scholar]
- 40. Takahashi K., Takeya M., and Sakashita N. (2002) Multifunctional roles of macrophages in the development and progression of atherosclerosis in humans and experimental animals. Med. Electron Microsc. 35, 179–203 [DOI] [PubMed] [Google Scholar]
- 41. Fuster V., Fayad Z. A., Moreno P. R., Poon M., Corti R., and Badimon J. J. (2005) Atherothrombosis and high-risk plaque. Part II: Approaches by noninvasive computed tomographic/magnetic resonance imaging. J. Am. Coll. Cardiol. 46, 1209–1218 [DOI] [PubMed] [Google Scholar]
- 42. Fuster V., Moreno P. R., Fayad Z. A., Corti R., and Badimon J. J. (2005) Atherothrombosis and high-risk plaque. Part I: Evolving concepts. J. Am. Coll. Cardiol. 46, 937–954 [DOI] [PubMed] [Google Scholar]
- 43. Vijil C., Hermansson C., Jeppsson A., Bergström G., and Hultén L. M. (2014) Arachidonate 15-lipoxygenase enzyme products increase platelet aggregation and thrombin generation. PLoS One 9, e88546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ott I., Weigand B., Michl R., Seitz I., Sabbari-Erfani N., Neumann F. J., and Schömig A. (2005) Tissue factor cytoplasmic domain stimulates migration by activation of the GTPase Rac1 and the mitogen-activated protein kinase p38. Circulation 111, 349–355 [DOI] [PubMed] [Google Scholar]
- 45. Hjortoe G. M., Petersen L. C., Albrektsen T., Sorensen B. B., Norby P. L., Mandal S. K., Pendurthi U. R., and Rao L. V. (2004) Tissue factor-factor VIIa-specific up-regulation of IL-8 expression in MDA-MB-231 cells is mediated by PAR-2 and results in increased cell migration. Blood 103, 3029–3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. López-Pedrera C., Barbarroja N., Dorado G., Siendones E., and Velasco F. (2006) Tissue factor as an effector of angiogenesis and tumor progression in hematological malignancies. Leukemia 20, 1331–1340 [DOI] [PubMed] [Google Scholar]
- 47. Ott I., Fischer E. G., Miyagi Y., Mueller B. M., Ruf W. (1998) A role for tissue factor in cell adhesion and migration mediated by interaction with actin-binding protein 280. J. Cell Biol. 140, 1241–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Madamanchi N. R., Hakim Z. S., and Runge M. S. (2005) Oxidative stress in atherogenesis and arterial thrombosis: The disconnect between cellular studies and clinical outcomes. J. Thromb. Haemost. 3, 254–267 [DOI] [PubMed] [Google Scholar]
- 49. Takemura M., Mishima T., Wang Y., Kasahara J., Fukunaga K., Ohashi K., and Mizuno K. (2009) Ca2+/calmodulin-dependent protein kinase IV-mediated LIM kinase activation is critical for calcium signal-induced neurite outgrowth. J. Biol. Chem. 284, 28554–28562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kotla S., Singh N. K., and Rao G. N. (2017) ROS via BTK-p300-STAT1-PPARγ signaling activation mediates cholesterol crystals-induced CD36 expression and foam cell formation. Redox Biol. 11, 350–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Papaharalambus C. A., and Griendling K. K. (2007) Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends Cardiovasc. Med. 17, 48–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liou G. Y., and Storz P. (2010) Reactive oxygen species in cancer. Free Radic. Res. 44, 479–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Golino P., Ragni M., Cirillo P., Avvedimento V. E., Feliciello A., Esposito N., Scognamiglio A., Trimarco B., Iaccarino G., Condorelli M., Chiariello M., and Ambrosio G. (1996) Effects of tissue factor induced by oxygen free radicals on coronary flow during reperfusion. Nat. Med. 2, 35–40 [DOI] [PubMed] [Google Scholar]
- 54. Berzat A., and Hall A. (2010) Cellular responses to extracellular guidance cues. EMBO J. 29, 2734–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Friedl P., and Gilmour D. (2009) Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 10, 445–457 [DOI] [PubMed] [Google Scholar]
- 56. Sun D., and Funk C. D. (1996) Disruption of 12/15-lipoxygenase expression in peritoneal macrophages. Enhanced utilization of the 5-lipoxygenase pathway and diminished oxidation of low density lipoprotein. J. Biol. Chem. 271, 24055–24062 [PubMed] [Google Scholar]
- 57. Kotla S., and Rao G. N. (2015) Reactive oxygen species (ROS) mediate p300-dependent STAT1 protein interaction with peroxisome proliferator-activated receptor (PPAR)-γ in CD36 protein expression and foam cell formation. J. Biol. Chem. 290, 30306–30320 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]