

Abstract
An important event in organogenesis is the formation of signaling centers, which are clusters of growth factor-secreting cells. In the case of tooth development, sequentially formed signaling centers known as the initiation knot (IK) and the enamel knot (EK) regulate morphogenesis. However, despite the importance of signaling centers, their origin, as well as the fate of the cells composing them, remain open questions. Here, using lineage tracing of distinct epithelial populations, we found that the EK of the mouse incisor is derived de novo from a group of SRY-box 2 (Sox2)-expressing cells in the posterior half of the tooth germ. Specifically, EK progenitors are located in the posterior ventral basal layer, as demonstrated by DiI labeling of cells. Lineage tracing the formed EK with ShhCreER, which encodes an inducible Cre recombinase under the control of the Sonic hedgehog promoter, at subsequent developmental stages showed that, once formed, some EK cells in the incisor give rise to differentiated cells, whereas in the molar, EK cells give rise to the buccal secondary EK. This work thus establishes the developmental origin as well as the fate of the EK and reveals two strategies for the emergence of serially formed signaling centers: one through de novo establishment and the other by incorporation of progeny from previously formed signaling centers.
Keywords: epithelium, morphogenesis, mouse genetics, sonic hedgehog (SHH), tooth development, SOX2, enamel knot, lineage tracing, signaling center
Introduction
Organogenesis is controlled by members of multiple signaling pathways, including the Hedgehog (HH)3 family, fibroblast growth factors (FGF), bone morphogenetic proteins (BMP), and WNTs. These molecules are often produced from a single source of specialized cells known as a signaling center; examples of such structures include the apical ectodermal ridge and the zone of polarizing activity (ZPA) in the developing limb, the floor plate of the neural tube, the isthmus at the mid-hindbrain boundary, and the enamel knot (EK) in the developing tooth (1–4). Identifying the developmental origin of signaling centers, as well as their subsequent cell fate, is an important step in our quest to understand the morphogenetic processes underlying organ formation. Lineage tracing provides a powerful approach to track cells of interest (5) and previously has been used to follow cells from several signaling centers, such as the apical ectodermal ridge, the ZPA, the floor plate, Hensen's node, and the isthmus (6–11). Despite these efforts, there is still much to learn about how signaling centers arise, and the dentition provides an excellent system to address this.
A central feature of the developing tooth are interactions between the dental epithelium and the underlying mesenchyme as the tooth germ undergoes a series of morphological changes and transitions through placode, bud, cap, and bell stages (Fig. 1, A–C). This epithelial–mesenchymal interaction depends on the formation of a number of signaling centers. For instance, in the placode stage beginning at E11, several signaling molecules, such as Shh, Wnt10b, and Bmp2, are expressed in the dental epithelium (12–14), and these early expression domains correlate to rudimentary primordia of both the incisor (front teeth) (15, 16) and the molar (back teeth) (17); adult mice do not have canine or premolar teeth, although rudimentary tooth buds corresponding to premolars have been reported (17–20). As development progresses through later stages, additional distinct signaling centers are then formed. At E12.5, a signaling center is observed in the anterior portion of the developing tooth bud and expresses genes encoding several signaling molecules, including Shh (15, 16, 21). In the incisor, this signaling center is known as the initiation knot (IK) and is located at the junction between the tooth germ and the vestibular lamina that becomes the future cleft between lips and teeth (22). The IK is composed of non-proliferative cells and has been shown to regulate the size of the early tooth germ (Fig. 1, A and D) (23). At the cap stage, a second signaling center, called the EK, is formed. Similar to the IK, the EK is characterized by an array of signaling molecules, such as Shh, Bmp4, Bmp7, Wnt10a/b, Fgf4, and Fgf20, as well as the cell cycle inhibitor p21 (Fig. 1, B, C, and E) (4, 24–26). However, several differences exist between the molar and incisor EKs related to the distinct geometries of the developing structures. For example, the molar primary EK disappears as a result of apoptosis and is subsequently succeeded by the secondary EKs, which produce a similar complement of signaling molecules as the primary EK and plays a role in defining future cusp positions (4, 27, 28). In contrast, only one EK is formed in the incisor (29). These differences reflect the eventual morphological distinctions between multicuspid molars and unicuspid incisors and point to the functional significance of the EKs during tooth morphogenesis.
Figure 1.
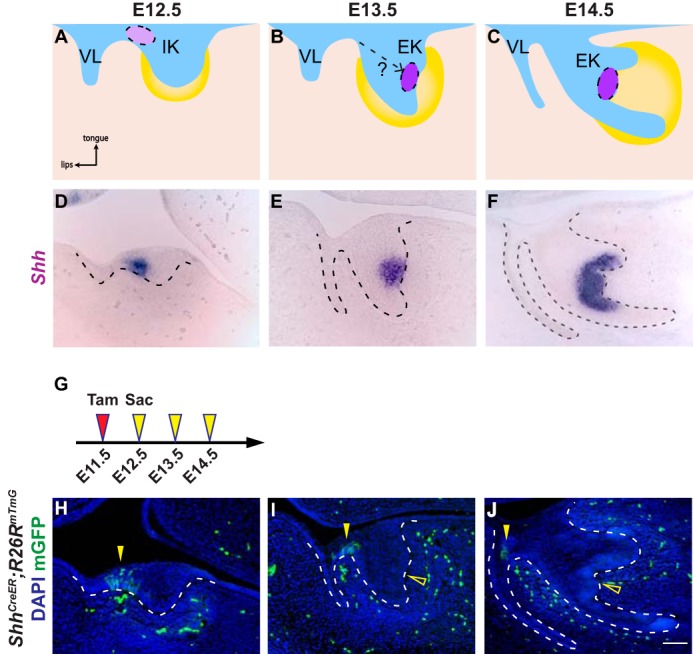
Incisor EK cells are not derived from the anterior IK population. A–C, schematic depiction of incisor development on sagittal sections. At E12.5, the dental epithelium (blue) invaginates into the underlying dental mesenchyme (yellow), forming a tooth bud. The initiation knot (light purple), which is the early signaling center of the incisor, is located at the anterior part of the incisor adjacent to the vestibular lamina (VL) (A). By E13.5, the tooth bud folds into a cap shape and the enamel knot (purple) forms (B). At E14.5, the incisor epithelium rotates posteriorly (C). D–F, in situ hybridization of Shh in the developing incisor tooth germ on sagittal sections from E12.5 to E14.5. G, timeline depicting the onset of Cre induction (Tamoxifen (Tam) injection, red arrowhead) and sample collection (yellow arrowheads). Tamoxifen concentration is 0.8 mg/30 g body weight. H–J, lineage tracing analysis of Shh-expressing cells in ShhCreER; R26RmTmG using 0.8 mg/30 g body weight of tamoxifen from E12.5 to E14.5. GFP-positive cells are only observed in the IK region (yellow arrowheads), but absent from the EK (open yellow arrowheads). n = 8 embryos for each time point. Scale bar in J represents 50 μm in D–F and H–J.
One potential mechanism for EK formation is through recruitment of cells from the IK, as the two signaling centers share many common features, and IK cells that are genetically labeled using ShhCreER appear to give rise to the EK (16). However, it is possible that the labeling of the EK observed previously could result from tamoxifen that persisted in the animal as a result of the high dosage administered. A second potential mechanism for generating the EK is by means of de novo formation. This idea was supported by a recent study that combined live imaging and the use of Fucci (fluorescent cell cycle indicator)-red reporter mice to track G0/G1 cells, which found that the EK is formed in situ as opposed to being derived from the IK (23). However, as IK cells are not permanently labeled in this experiment and only G0/G1 cells are tracked, it does not exclude the possibility that IK cells re-enter the cell cycle prior to contributing to the EK, thus eluding tracking by live imaging. As a result, the lineage connection between the IK and the EK remains an open question.
To definitively understand the lineage relationships between serially formed signaling centers, we utilized tamoxifen-inducible genetic lineage tracing and DiI labeling to track the cell fate of different populations in developing teeth. We found that there are at least two ways that new signaling centers are established. The first is exemplified by the incisor EK, which is formed de novo from E12.5 posterior ventral basal cells that are distinct from progeny of the IK. The second mechanism is by incorporating progeny from previously formed signaling centers, a process employed during formation of the molar buccal secondary EK.
Results
Shh expression marks distinct signaling centers in developing incisors
Shh is expressed during the early stages of tooth development and is essential for proper tooth morphogenesis (30). Although Shh expression is well studied in the developing molar (31, 32), its expression pattern in the incisor tooth germ across different developmental stages at a histological level has not been documented in as much detail. Therefore, we first set out to examine the expression of Shh by means of section in situ hybridization at several stages of early incisor development. At E12.5, a Shh-expressing domain corresponding to the IK was present in the anterior incisor adjacent to the vestibular lamina (Fig. 1D). This expression pattern remained unchanged at E13, despite the increased tooth germ size (supplemental Fig. S1A). However, at E13.5, a new Shh-expressing region, marking the newly formed EK, was observed in the posterior dental epithelium, which was distinct from the IK; no Shh-positive cells could be detected between the IK and the EK (Fig. 1E and supplemental Fig. S1B). At this stage, Shh expression in the IK began to quickly down-regulate and could no longer be observed at later time points (supplemental Fig. S1B). Finally, at E14.5, instead of being restricted to the EK region, the posterior Shh expression expanded laterally and was present in the cells of the inner enamel epithelium (Fig. 1F). These cells eventually become ameloblasts and the stellate reticulum, a population of cells that lies underneath the enamel epithelium.
Incisor EK cells are not derived from the IK
To investigate whether EK cells are progeny of the Shh-expressing IK cells, we generated ShhCreER; R26RmTmG mice, where membrane EGFP expression is regulated by a tamoxifen-inducible Cre recombinase that is under the control of the Shh promoter, thus allowing us to genetically label and lineage trace Shh-expressing cells at desired time points. To unambiguously label only the IK cells, but not the EK cells that form 24 h later, we set out to identify the appropriate tamoxifen concentration for CreER induction. This is a critical issue, because the time span for effective CreER-mediated recombination depends on the amount and the latency of tamoxifen in the body. The mouse limb bud presents an ideal system to test this, as the initiation of Shh in the ZPA at E9.75 is well established, and the ZPA represents the only Shh-expressing domain in the limb (33). When 2.5 mg/30 g body weight of tamoxifen was gavaged to induce CreER activation at E7.75, 48 h prior to the onset of Shh expression in the ZPA, GFP-positive cells could still be detected in the E10.5 limb bud (supplemental Fig. S2, A and B), suggesting that this concentration of tamoxifen was sufficiently high that it perdured and could label Shh-expressing cells. On the contrary, when 0.8 mg/30 g body weight of tamoxifen was administered at E7.75, no GFP-positive cells were present (supplemental Fig. S2C).
Based on these results, we labeled IK cells by providing 0.8 mg/30 g body weight of tamoxifen to female mice carrying E11.5 ShhCreER; R26RmTmG embryos. We observed GFP-positive cells in the IK at the junction of the vestibular lamina and the incisor tooth germ at E12.5 (Fig. 1, G and H). Interestingly, at both E13.5 and E14.5, the EK was devoid of GFP-positive cells, although GFP-expressing cells were still present in the anterior tooth germ where the IK was previously located (Fig. 1, I and J). Similarly, three-dimensional (3D) reconstruction of lineage traced samples using 2-photon microscopy also showed negative labeling within the EK region (supplemental Movies S1–S3), indicating that EK cells are not descendants of IK cells.
Incisor EK cells are derived from the lower posterior basal cells of the tooth bud
The results above point to the possibility that the EK is formed de novo from non-signaling cells and that EK cells may descend from the posterior portion of the incisor tooth germ. We tested this idea by first utilizing Sox2CreER; R26RmTmG embryos for lineage tracing, as Sox2 is expressed exclusively in the posterior tooth germ at E12.5 (Fig. 2A). To that end, we induced the Cre recombinase activity at E11.5 and were able to label the posterior region of the tooth germ at E12.5 (Fig. 2, D–E′). At E13.5, 48 h after Cre induction, GFP expression could be observed in both the basal and suprabasal cells of the posterior tooth germ (Fig. 2, F and F′). Importantly, several cells in the EK, as marked by the EK marker p21 (4), also expressed membrane EGFP (Fig. 2, F and F′), indicating that at least some of the EK cells are derived from the E12.5 posterior incisor epithelium. Of note, whereas some of the p21-positive cells were GFP-negative and could arise from a non-Sox2-expressing population, this mosaicism is likely due to varied CreER recombination in different cells. Similar lineage tracing results were obtained at E14.5, where GFP-positive cells became more widely distributed in the EK (Fig. 2, G and G′). This finding was unlikely to be confounded by delayed recombination taking place within the EK at E13.5 or E14.5 as a result of residual tamoxifen, as Sox2 is not normally expressed in the EK at these stages (Fig. 2, B and C) (34, 35).
Figure 2.
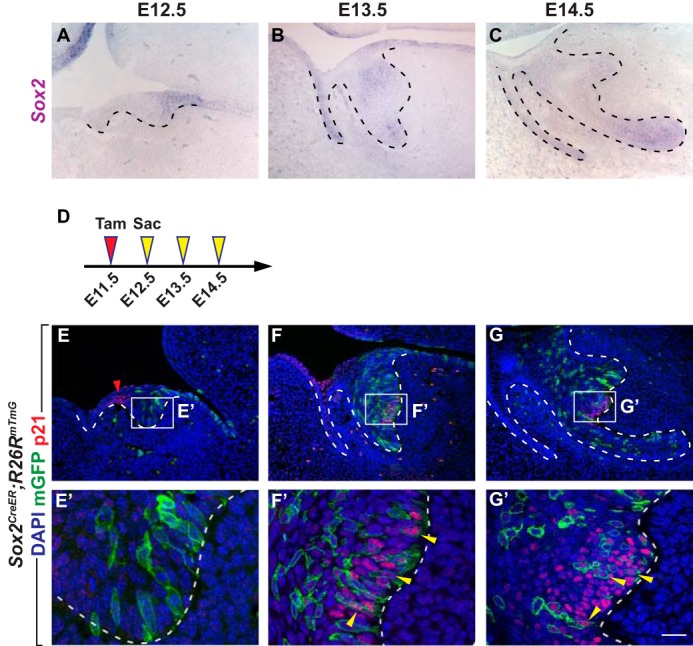
EK cells are derived from the Sox2-expressing cells at the posterior tooth bud at E12.5. A–C, in situ hybridization of Sox2 in the developing incisor tooth germ on sagittal sections from E12.5 to E14.5. D, timeline depicting the onset of Cre induction (Tamoxifen (Tam) injection, red arrowhead) and sample collections (yellow arrowheads). Tamoxifen concentration is 0.8 mg/30 g body weight. E–G′, lineage tracing analysis of cells expressing Sox2 in Sox2CreER; R26RmTmG. Induction of Cre by tamoxifen gavage at E11.5 shows labeling of the posterior tooth bud at E12.5 and subsequent labeling of EK cells at E13.5 and E14.5, which are also marked by the EK marker p21 (yellow arrowheads). p21 is also present in the IK at E12.5 (red arrowhead). n = 8 embryos for each time point. Scale bar in G′ represents 28 μm in E′–G′, and 50 μm in A–C and E–G.
The Sox2CreER lineage studies demonstrated that the posterior incisor epithelium is the source of the EK cells, but they could not resolve the exact origin of these cells because of the relatively broad expression of Sox2. Injection of the lipophilic vital dye DiI allows accurate and specific marking of a subgroup of cells that could not otherwise be distinguished from neighboring populations using genetic labeling due to the lack of cell-specific markers. The combination of DiI labeling with tissue slice culture thus provides a powerful approach to lineage trace any region of interest over a period of time (36–39). To that end, we injected DiI to E12.5 K14–actin–GFP mandible slices, which express GFP in the oral epithelium, enabling easy visualization of the tooth germ and accurate injection at the target site (Fig. 3A). Injection was carried out at four distinct locations: 1) the vestibular lamina/tooth germ junction that corresponds to the IK region (Fig. 3B); 2) the basal cells of the posterior dorsal half of tooth germ (Fig. 3C); 3) the basal cells of the posterior ventral half of tooth germ (Fig. 3D); 4) the ventral most basal cells (Fig. 3E). We then followed the movement of these cells in the GFP-positive epithelium for 2 days in culture (Fig. 3, F–I). Consistent with ShhCreER lineage tracing, DiI-labeled IK cells remained locally restricted and did not give rise to the EK, which can be visualized by Shh in situ hybridization (Fig. 3J). In comparison, many labeled tissues in the ventral tip or posterior dorsal portion of the tooth germ could be found to contribute to the stellate reticulum (suprabasal) region of the developing tooth bud after 48 h of culturing, although none moved to the EK (Fig. 3, K and M). In contrast, basal epithelium located in the posterior ventral portion of the tooth germ consistently give rise to parts of the EK, as demonstrated by overlapping DiI signal with Shh expression (Fig. 3L).
Figure 3.
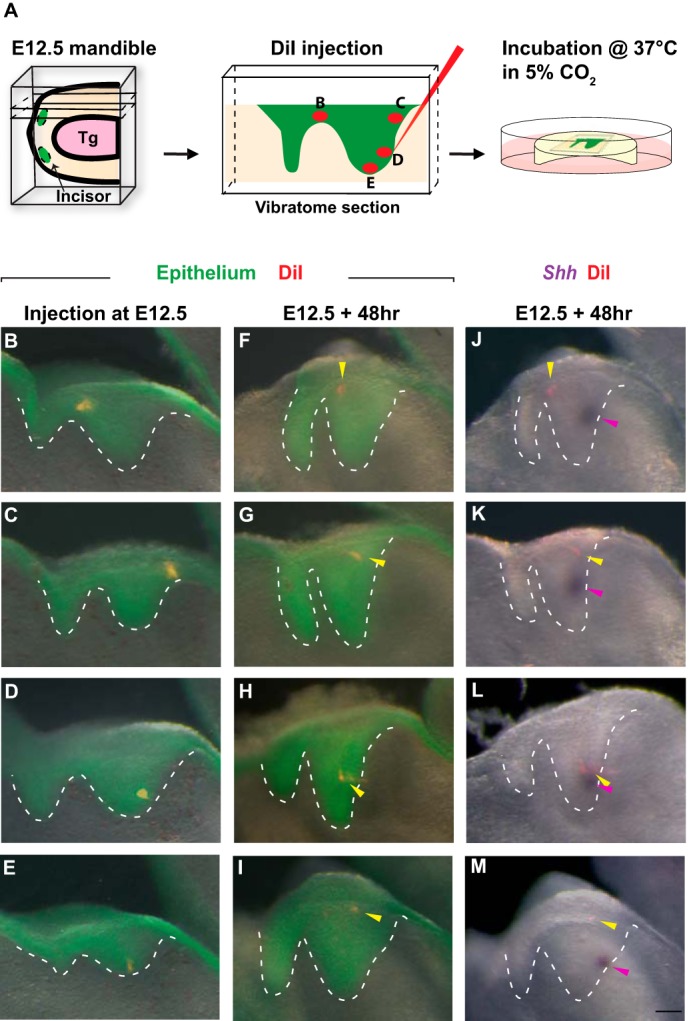
Cells from the lower portion of the posterior tooth bud contribute to the EK. A, schematic of the slice culture system with DiI injection. Tg, tongue. B–E, DiI injection at different locations of E12.5 tooth germs (epithelium is genetically labeled by actin-GFP and is green. DiI-labeled cells are red). n = 15 samples for each location. F–I, 48 h post-injection, DiI-labeled cells are observed at different locations (yellow arrowheads). J–M, a merged view showing localizations of DiI-labeled cells (yellow arrowheads) and EK cells marked by Shh in situ hybridization (purple arrowheads) after 48 h of culture. Scale bar in M represents 50 μm in B–M.
EK cells give rise to some ameloblasts but not to stem cells
Although the production of ligands from signaling centers is critical for organ development, timely termination of the signaling events is also an integral part of the regulatory process. One avenue for achieving this is by eliminating signaling cells through apoptosis, such as in the anterior neural ridge of the brain and the primary EK in the molars (4, 40, 41). To investigate whether incisor EKs undergo similar self-destruction, we performed terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling (TUNEL) staining and found that, although no apoptosis could be detected in the EK at E13.5 and E14.5, there was a slight increase in the number of apoptotic cells at E15.5 (6 ± 2.16 apoptotic cells per section, n = 4) (supplemental Fig. S3, A–C). This result is similar to what has been reported (42), although in some studies apoptosis has been observed at E13.5–E14 (29, 42), potentially due to differences in genetic backgrounds. The amount of apoptosis we observed then became diminished at E16.5, with the EK morphological bulge still retained (supplemental Fig. S3D). As a result, at least a small portion of the incisor EK was removed through cell death. However, as not all EK cells undergo apoptosis, we hypothesized that they could be incorporated into the developing epithelium and performed lineage tracing to track the fate of EK cells.
Because Shh is a key marker of the EK, we again used the ShhCreER; R26RmTmG line to address this question (Fig. 4C). We first performed lineage tracing by gavaging 0.8 mg/30 g body weight of tamoxifen to pregnant females carrying E12.5 embryos to label the EK at E13.5 without marking non-EK cells that begin to express Shh at E14.5. However, this labeling protocol yielded few GFP-positive cells in the EK at E13.5 and E14.5 (data not shown), suggesting that the denser and non-proliferating EK cells may only undergo efficient recombination when a higher concentration of tamoxifen is used. To circumvent this, 2.5 mg/30 g body weight of tamoxifen was administered at E11.5 (Fig. 4A), 72 h prior to the onset of Shh expression in non-EK cells; as at this concentration, recombination can be induced within a 48-h window after tamoxifen treatment (supplemental Fig. S2F) but is undetectable at later time points (supplemental Fig. S4, A–C), allowing us to label and track the fate of EK cells (Fig. 4, B and C). Using this regimen, we found that the movement of the EK cell progeny was relatively constrained (Fig. 4, D and F), although a small portion of the cells expanded beyond the EK bulge as ameloblasts in the labial epithelium at E16.5 (Fig. 4, D and D′). By E18.5, these EK-derived ameloblasts were largely lost, with only a few cells remaining in the epithelium (Fig. 4, F and F′, and supplemental Fig. S4, D–F). Importantly, no GFP-positive cells could be discerned in the structure at the growing end of the labial epithelium, known as the cervical loop (Fig. 4, E and G), which houses dental epithelial stem cells and newly emerged Shh-expressing transit-amplifying cells (43).
Figure 4.
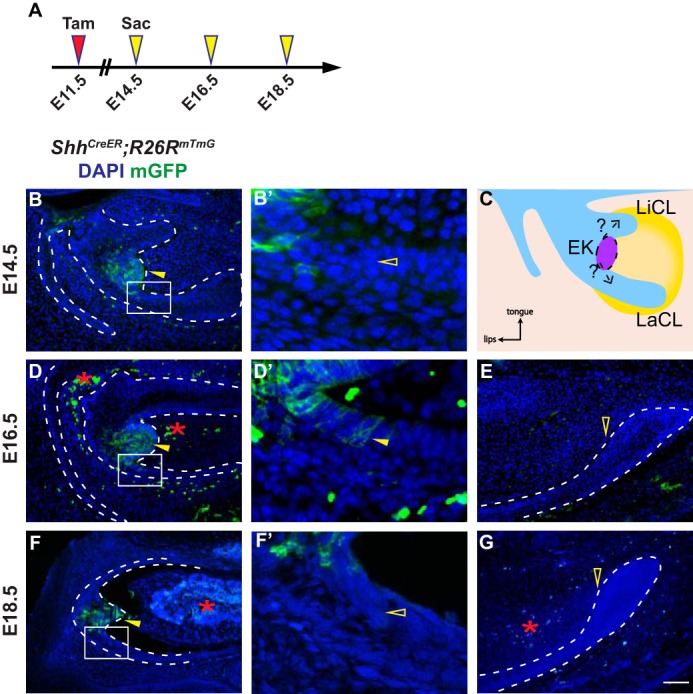
EK cells give rise to some ameloblasts, but not to stem cells in the cervical loop. A, timeline depicting the onset of Cre induction (Tamoxifen (Tam) injection, red arrowhead) and sample collections (yellow arrowheads). Tamoxifen concentration is 2.5 mg/30 g body weight. B and B′, EK cells are labeled (yellow arrowhead) in ShhCreER; R26RmTmG embryos at E14.5, but not in the labial epithelium (open yellow arrowhead). n = 8 embryos. LaCL, labial cervical loop; LiCL, lingual cervical loop. C, schematic model of the fate of EK cells on a sagittal section during incisor development. D and E, at E16.5, GFP-positive cells are detected in the EK region and in the labial epithelium (yellow arrowheads), but not in the cervical loop (open yellow arrowhead). Autofluorescent blood cells are marked by red asterisks. n = 8 embryos. F and G, by E18.5, GFP-positive cells have been replaced proximally and moved out of the ameloblast region (open yellow arrowhead in F′), whereas many cells remain in the EK region (yellow arrowhead in F). No cells were observed in the cervical loop (open yellow arrowhead in G). Autofluorescent blood cells are marked by red asterisks. n = 9 embryos. Scale bar in G represents 50 μm in B and D–G, and 12.5 μm in B′, D′, and F′.
Molar anterior buccal secondary EKs are derived from primary EKs
Our finding that the incisor EK progeny persist throughout the prenatal period and can give rise to some ameloblasts then raise the question of whether molar EKs can similarly do so, despite previous reports showing that molar primary EKs undergo apoptosis and are eliminated afterward (4, 25). We tested this by giving 1.25 mg/30 g body weight tamoxifen to ShhCreER; R26RmTmG embryos at E13.5 to genetically label the primary EK that can be observed at E14.5 (Fig. 5, A, B, D, and E). We chose this tamoxifen concentration because this is the lowest dosage that is able to efficiently induce recombination in the molar primary EK. In addition, as CreER recombination could not be detected in the dental epithelium 48 h after tamoxifen treatment (supplemental Fig. S5, A and B), we were able to lineage trace primary EKs without marking non-EK cells in the inner enamel epithelium that begin to express Shh at E15.5. Using this method, we found that at E16.5, when secondary EKs are formed (Fig. 5C), progeny of the primary EK, as marked by GFP signals, is no longer restricted to the primary EK region but populate toward the buccal side of the epithelium. This is in contrast to the lingual inner enamel epithelium and stellate reticulum, where few GFP-positive cells were observed, despite strong Shh expression (Fig. 5, F and G). The distribution of GFP-positive cells at this stage also suggests that progeny from the primary EK may contribute to the buccal secondary EK. To confirm this, we performed dual GFP immunostaining and in situ hybridization for Fgf4, a secondary EK marker, which showed that portions of the buccal secondary EK were composed of progeny from the primary EK (Fig. 5, H and I). This asymmetry of GFP-positive cells in the buccal and lingual epithelium persisted at postnatal day 0 (P0), as a number of GFP-positive ameloblasts and stellate reticulum cells were observed in and near the forming anterior buccal cusps, but not in the lingual or posterior cusps (Fig. 5, J and K, and supplemental Fig. S5, C–D′2). Together, these results show that in the molar, the primary EK cells give rise to parts of the buccal secondary EK, and these eventually contribute to molar ameloblasts and stellate reticulum cells.
Figure 5.
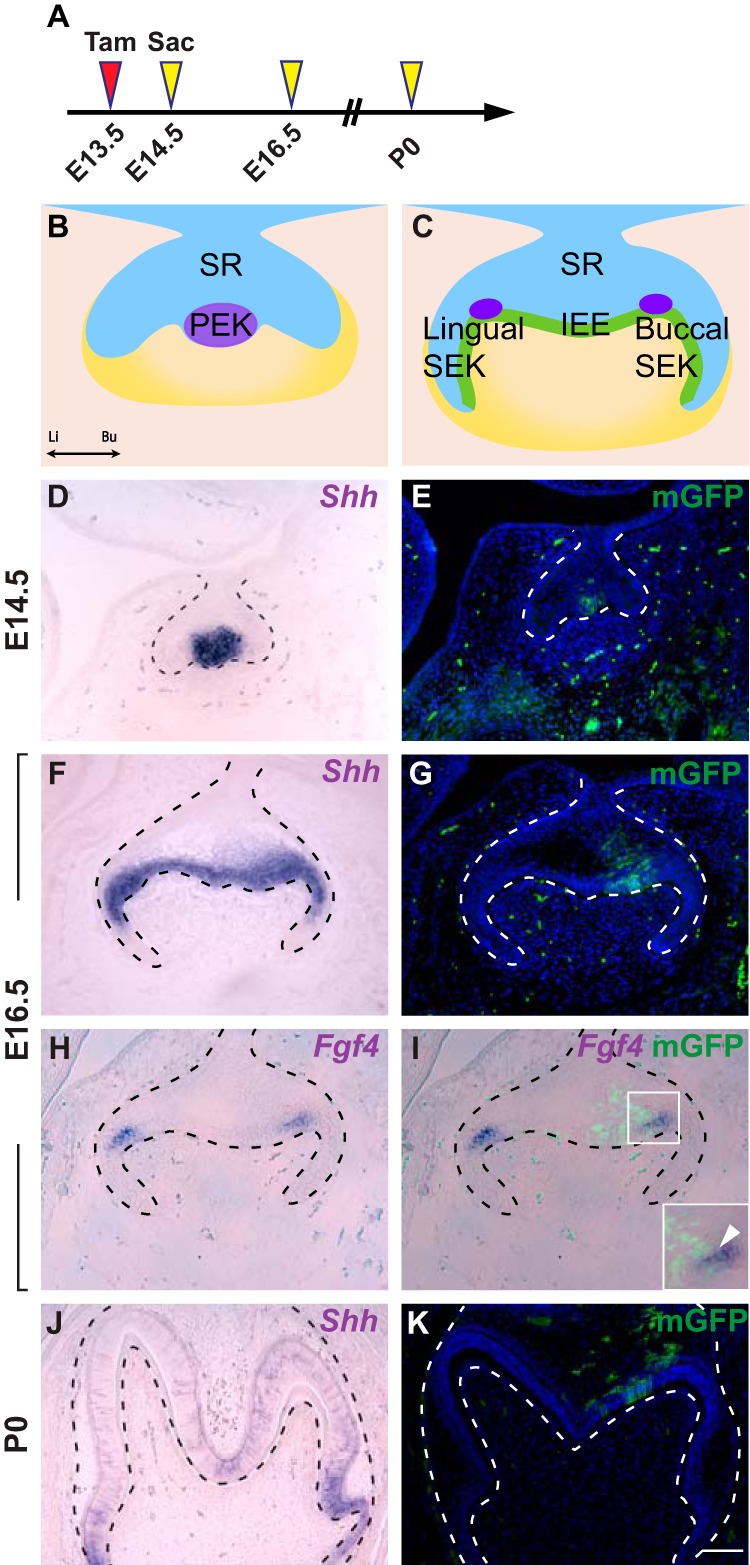
Expression of Shh and the fate of primary EK in the molar tooth germ. A, timeline depicting the onset of Cre induction (Tamoxifen (Tam) injection, red arrowhead) and sample collections (yellow arrowheads). Tamoxifen concentration is 1.25 mg/30 g body weight. B and C, schematic of primary EK and secondary EKs on a frontal section during molar development. IEE, inner enamel epithelium; PEK, primary enamel knot; SEK, secondary enamel knot; SR, stellate reticulum; Li, lingual; Bu, buccal. D–G, in situ hybridization of Shh and Fgf4 in the developing molar tooth germ on frontal sections from E14.5 to P0. H–K, lineage tracing analysis of cells expressing Shh in ShhCreER; R26RmTmG in molar tooth germ from E14.5 onwards. n = 6 embryos for each time point. Scale bar in K represents 50 μm in D–K.
Discussion
As specialized structures for ligand secretion, signaling centers must form in specific locations within developing organs to accurately pattern or regulate morphogenetic events. It is therefore important to understand the origin of signaling center cells. Here, using the mouse tooth as a model system, we found that there are at least two ways that new signaling centers are established. The first is through a de novo mechanism, as in the incisor EK, where a group of progenitor cells are induced locally to become specialized in signal secretion. The second mechanism is exemplified by the molar anterior buccal secondary EK and is by incorporating progeny from previously formed signaling centers.
We clarified the lineage relationship between the IK and EK, first by genetic lineage tracing of ShhCreER; R26RmTmG embryos and then by DiI labeling. Using these methods, we demonstrated that EK cells are not descendants of the IK. This result differs from an earlier study, where EK cells were clearly labeled after ShhCreER induction in the IK (16). However, these differences can be attributed to the more concentrated tamoxifen (9 mg/40 g body weight) used in the previous report, as well as its method of delivery using intraperitoneal (i.p.) injection (16). Indeed, when we gavaged mice with a higher concentration of tamoxifen (2.5 mg/30 g body weight) to induce ShhCreER recombination at E11.5, GFP-positive cells were apparent in the EK both at E13.5 and E14.5 (supplemental Fig. S2, D, F, and G), likely as a result of prolonged tamoxifen activity that could now activate ShhCreER in the forming EK. Importantly, both high and low tamoxifen dosage used in this study labeled the same number of cells in the IK at E12.5 (supplemental Fig. S2, E and H), suggesting that the lack of EK labeling in embryos treated with a low dosage of tamoxifen was not due to insufficient marking of early progenitor cells. It should be also noted that when a high concentration of tamoxifen (2.5 mg/30 g body weight) was given to E10.5 embryos, we could no longer detect Cre recombination in the EK at E13.5 and E14.5 (supplemental Fig. S4, A–C), despite labeling of IK cells, further indicating the de novo formation of the EK. Our results here also provided the kinetics of tamoxifen efficacy at 2.5 mg/30 g body weight, where tamoxifen is most effective in the dental epithelium within a 48-h window and its ability to induce CreER recombination is markedly reduced afterward, a result that is consistent with a previous study using 3 mg/40 g body weight of tamoxifen to study the origin of the oral vestibule (44). Last, the kinetics of tamoxifen is also determined by its delivery method, as tamoxifen administered through i.p. injection persists longer in the system than when gavage is used, and continues to activate CreER recombination in the dental epithelium even after 72 h post-injection (data not shown). Together, these results argue that EK cells are not derived from IK cells, and they demonstrate the importance of appropriate tamoxifen dosage for temporal control of lineage tracing.
Using Sox2CreER lineage tracing and DiI labeling, we subsequently found that incisor EK cells were derived from a group of basal cells present in the posterior lower tooth bud. This result is concordant with work done by Ahtiainen et al. (23), and together our data support a model where incisor EK cells are derived through a de novo mechanism at E12.5. Interestingly, the primary EK of the molar may be formed in a similar fashion, as it has been postulated that progenitors of the primary EK are located at the ventral tip of the bud prior to its formation (25), and DiI labeling of basal cells in this confined space resulted in labeling of the presumptive EK region at later stages (38). Future experiments to confirm these findings will rely on identification of markers of this pre-EK region to perform genetic labeling of these cells. Furthermore, as current EK markers, such as Shh and Bmp4 (25, 45), are expressed after the EK has formed, genetic tools based on pre-EK-specific markers will also allow us to manipulate the EK prior to its formation without affecting the rest of the dental epithelium.
Given that Shh has been reported as a key signaling molecule of the EK, it provides a genetic driver to also assess the fate of the EK cells by means of lineage tracing. In the incisor, the labeled EK cells were detected at the anterior epithelium, which is similar to previous reports using BrdU labeling for lineage tracing the non-proliferative EK cells (28). Importantly, no labeled cells were observed in the cervical loop (Fig. 4). Our observation supports a recent model that postulates a dependence on embryonic ameloblast progenitor cells located in the anterior epithelium for the initial production of differentiated ameloblasts (43). This model proposes that the subsequent generation of ameloblasts is effected by a distinct group of progenitor cells in the cervical loop that remain as stem cells for the continuous renewal of the incisor. Consequently, cells from the EK are contributors to the early ameloblasts, which are later replaced by stem cell-fueled tooth growth.
In the molar, although the primary EK ceases to exist around E15 (4, 46), we found its descendants in the buccal stellate reticulum (suprabasal cells) and secondary EK during the late cap stage to early bell stage (Fig. 5). The observation of EK progeny in the suprabasal layer is consistent with a recent report showing similar cell movements, demonstrating that cells from the basal dental epithelium can contribute to the stellate reticulum compartment (47). The notion that the secondary EK is derived from the primary EK is also supported by a previous study that set out to lineage trace BrdU-negative primary EK cells (28). However, in these studies, both lingual and buccal secondary EKs appeared to contain cells from the primary EK, and this difference can be explained by distinct labeling methods. Indeed, primary EK cells labeled by DiI were shown to move mainly to the buccal EK (37), thus approximating what we have observed here.
In summary, the data reported here demonstrate that EK progenitor cells are distinct from those present in the early IK signaling center, as the former were found to originate from the ventral-posterior basal layer of the incisor epithelium. The de novo derivation of the IK and the EK thus bears similarities to the formation of other signaling centers, such as the ZPA and the isthmus, which are formed locally in a developing organ. However, this mode of development differs from the formation of the molar buccal secondary EK, which includes descendents of an earlier signaling center, the primary EK. This lineage relationship between two signaling centers has also been shown in the lineage propagation from Hensen's node to the floor plate of the neural tube, where cells exiting from Hensen's node give rise to the floor plate (48). Consequently, signaling centers are formed through two distinct mechanisms, one by specifying a group of progenitor cells to form the entire signaling center and the other by incorporating progeny from an earlier signaling center. In the case of the molar, it is tempting to speculate that during evolution, primary EK cells were repurposed to form some of the secondary EKs that are critical for determining cusp positions and hence the shape of the tooth.
Experimental procedures
Animals
ShhCreER (6), Sox2CreER (49), R26RmTmG (50), and K14EGFP/Actb (51) mice were maintained on a mixed background of C57BL/6 and CD-1 and genotyped as reported. Mice were mated overnight, and the presence of a vaginal plug on the next day indicated E0.5. Tamoxifen was administered by oral gavage to pregnant females at a dose of 0.8, 1.25, or 2.5 mg/30 g body weight at the indicated time points. Pregnant mice were euthanized by CO2 followed by cervical dislocation. After removing the embryos from the uterus, wet body weight was determined immediately to optimize comparison between litters. All mouse work was approved by UCSF Institutional Animal Care and Use Committee.
Slice culture
Mandibles were dissected from E12.5 embryos and directly embedded in 6% low-melting agarose (Invitrogen, UltraPuret Agarose). The samples were cut in a series of 200-μm sagittal sections with a speed of 4.5 and a vibration frequency of 5.5 using the Leica VT1000 S vibrating blade microtome (Leica Microsystems GmbH). The slices were then transferred on top of a cell culture insert (Millicell), and cultured at the interface of air and media (Dulbecco's modified Eagle's medium, F-12 (DMEM/F-12) with 1% penicillin–streptomycin (Sigma)) at 37 °C and 5% CO2. After the culture, slices were fixed in 4% paraformaldehyde (PFA) and dehydrated in 100% MeOH for whole mount in situ hybridization and image analysis.
DiI labeling
DiI was kept as a stock solution (1 mg/ml of DiI in 100% DMSO) at −20 °C. Before use, the stock solution was dissolved 1:5 in 0.3 m filter-sterilized sucrose. Microinjection was performed using a capillary tubing (Borosil 1.0-mm OD × 0.75-mm ID/Fiber, FHC) that is connected to a micromanipulator (WPI, KITE-R) and a microinjector (Eppendorf, 5242). The sliced tooth germs were cultured in vitro, and the displacement of the DiI-labeled cells was observed after 24 and 48 h using a Leica MZ16F stereomicroscope equipped with a Leica DFC310 FX digital color camera.
Immunofluorescence staining
Tissue was prepared for paraffin sections by fixing embryonic heads or postnatal jaws in 4% PFA and then decalcifying in RNase-free EDTA for 3–5 days. Samples were embedded in paraffin and then sectioned at 7 μm. Paraffin sections were rehydrated, and antigen retrieval was performed by incubating in pH 6.2 citric buffer containing 10 mm citric acid, 2 mm EDTA, 0.05% Tween 20 just below boiling temperature for 15 min followed by a 30-min cool-down to room temperature. Primary antibody against GFP (1:500, Abcam, ab13970) and p21 (1:100, BD Transduction Laboratories, 556431) was used. Washes in PBST (3 × 5 min) were followed by incubation with secondary antibodies conjugated with Alexa Fluor 488 (1:250, Invitrogen) or Biotin (1:1000, Vector). TSA kit (PerkinElmer Life Sciences) was used for signal amplification for p21 detection. TUNEL staining was performed according to manufacturer's instructions (Sigma). Nuclear counterstaining was performed with DAPI (Invitrogen) and mounted with Prolong Gold antifade reagent (Invitrogen). Images were taken using a Leica DFC500 camera with a Leica DM 5000B microscope or Leica-SP5 confocal microscope.
In situ hybridization
RNA in situ hybridizations were performed either on paraffin sections or tissue slices according to standard protocols using digoxigenin-labeled probes (43, 52).
Mandible epithelium-mesenchymal dissociations
Mandibles were dissected in Hanks' medium and treated with 10 mg/ml of Dispase II (Roche Applied Science) at 37 °C for 45 min to 1 h, depending on embryonic stages. After Dispase treatment, epithelium was carefully pulled apart from the mesenchyme in Hanks' medium and fixed for 30 min in 4% PFA, then stored in 1% PFA at 4 °C until imaging.
3D reconstruction
The dissociated epithelia were embedded in 1% low-melting agarose. Z-stacked images were taken using a Nikon A1R 2-photon microscope and 3D reconstructions were made using Imaris.
Statistics and reproducibility
All experiments were repeated at least three times with similar results and representative images were shown here. Three to four litters were collected for each time point and for different tamoxifen dosage administration. More litters were collected when only 1–2 CreER-positive embryos were present in one litter. Data were shown as mean ± S.D. p values were derived from using unpaired two tailed Student's t tests.
Author contributions
W. D., J. H., and W. D. performed the experiments. W. D., J. H., and O. D. K. conceived the project and supervised the experiments. W. D., J. H., and O. D. K. wrote the manuscript. All authors contributed to the planning of the experiments and commented on the manuscript.
Supplementary Material
Acknowledgments
We thank Sarah Alto, Rebecca d'Urso, and Nicholas Wang for assistance with the mouse colony, Dr. Jeffrey Bush, Dr. Jan Prochazka, and Dr. Chunying Li for helpful discussions, members of the Klein laboratory, and Dr. Renata Peterkova and Dr. Maria Hovorakova for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R35-DE026602 (to O. D. K.) and F32-DE023705 and K99-DE025874 (to J. K. H.) from the NIDCR. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S5 and Movies S1–S3.
- HH
- Hedgehog
- FGF
- fibroblast growth factor
- BMP
- bone morphogenetic protein
- ZPA
- zone of polarizing activity
- EK
- enamel knot
- IK
- initiation knot
- PFA
- paraformaldehyde
- Shh
- Sonic hedgehog
- EGFP
- enhanced green fluorescent protein
- DiI
- 1,1′-dioctadecyl 3,3,3′,3′-tetramethylindocarbocyanine perchlorate.
References
- 1. Tabin C. (1995) The initiation of the limb bud: growth factors, Hox genes, and retinoids. Cell 80, 671–674 [DOI] [PubMed] [Google Scholar]
- 2. Jessell T. M. (2000) Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat. Rev. Genet. 1, 20–29 [DOI] [PubMed] [Google Scholar]
- 3. Wassef M., and Joyner A. L. (1997) Early mesencephalon/metencephalon patterning and development of the cerebellum. Perspect. Dev. Neurobiol. 5, 3–16 [PubMed] [Google Scholar]
- 4. Jernvall J., Aberg T., Kettunen P., Keränen S., and Thesleff I. (1998) The life history of an embryonic signaling center: BMP-4 induces p21 and is associated with apoptosis in the mouse tooth enamel knot. Development 125, 161–169 [DOI] [PubMed] [Google Scholar]
- 5. Kretzschmar K., and Watt F. M. (2012) Lineage tracing. Cell 148, 33–45 [DOI] [PubMed] [Google Scholar]
- 6. Harfe B. D., Scherz P. J., Nissim S., Tian H., McMahon A. P., and Tabin C. J. (2004) Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 118, 517–528 [DOI] [PubMed] [Google Scholar]
- 7. Patten I., Kulesa P., Shen M. M., Fraser S., and Placzek M. (2003) Distinct modes of floor plate induction in the chick embryo. Development 130, 4809–4821 [DOI] [PubMed] [Google Scholar]
- 8. Louvi A., Alexandre P., Métin C., Wurst W., and Wassef M. (2003) The isthmic neuroepithelium is essential for cerebellar midline fusion. Development 130, 5319–5330 [DOI] [PubMed] [Google Scholar]
- 9. Alexandre P., and Wassef M. (2003) The isthmic organizer links anteroposterior and dorsoventral patterning in the mid/hindbrain by generating roof plate structures. Development 130, 5331–5338 [DOI] [PubMed] [Google Scholar]
- 10. Guo Q., Loomis C., and Joyner A. L. (2003) Fate map of mouse ventral limb ectoderm and the apical ectodermal ridge. Dev. Biol. 264, 166–178 [DOI] [PubMed] [Google Scholar]
- 11. Kimmel R. A., Turnbull D. H., Blanquet V., Wurst W., Loomis C. A., and Joyner A. L. (2000) Two lineage boundaries coordinate vertebrate apical ectodermal ridge formation. Genes Dev. 14, 1377–1389 [PMC free article] [PubMed] [Google Scholar]
- 12. Dassule H. R., and McMahon A. P. (1998) Analysis of epithelial-mesenchymal interactions in the initial morphogenesis of the mammalian tooth. Dev. Biol. 202, 215–227 [DOI] [PubMed] [Google Scholar]
- 13. Shirokova V., Jussila M., Hytönen M. K., Perälä N., Drögemüller C., Leeb T., Lohi H., Sainio K., Thesleff I., and Mikkola M. L. (2013) Expression of Foxi3 is regulated by ectodysplasin in skin appendage placodes. Dev. Dyn. 242, 593–603 [DOI] [PubMed] [Google Scholar]
- 14. Mucchielli M. L., Mitsiadis T. A., Raffo S., Brunet J. F., Proust J. P., and Goridis C. (1997) Mouse Otlx2/RIEG expression in the odontogenic epithelium precedes tooth initiation and requires mesenchyme-derived signals for its maintenance. Dev. Biol. 189, 275–284 [DOI] [PubMed] [Google Scholar]
- 15. Hovorakova M., Prochazka J., Lesot H., Smrckova L., Churava S., Boran T., Kozmik Z., Klein O., Peterkova R., and Peterka M. (2011) Shh expression in a rudimentary tooth offers new insights into development of the mouse incisor. J. Exp. Zool. B Mol. Dev. Evol. 316, 347–358 [DOI] [PubMed] [Google Scholar]
- 16. Hovorakova M., Smrckova L., Lesot H., Lochovska K., Peterka M., and Peterkova R. (2013) Sequential Shh expression in the development of the mouse upper functional incisor. J. Exp. Zool. B Mol. Dev. Evol. 320, 455–464 [DOI] [PubMed] [Google Scholar]
- 17. Prochazka J., Pantalacci S., Churava S., Rothova M., Lambert A., Lesot H., Klein O., Peterka M., Laudet V., and Peterkova R. (2010) Patterning by heritage in mouse molar row development. Proc. Natl. Acad. Sci. U.S.A. 107, 15497–15502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peterková R., Lesot H., Vonesch J. L., Peterka M., and Ruch J. V. (1996) Mouse molar morphogenesis revisited by three dimensional reconstruction: I. analysis of initial stages of the first upper molar development revealed two transient buds. Int. J. Dev. Biol. 40, 1009–1016 [PubMed] [Google Scholar]
- 19. Peterková R., Peterka M., Viriot L., and Lesot H. (2002) Development of the vestigial tooth primordia as part of mouse odontogenesis. Connect. Tissue Res. 43, 120–128 [DOI] [PubMed] [Google Scholar]
- 20. Viriot L., Lesot H., Vonesch J. L., Ruch J. V., Peterka M., and Peterková R. (2000) The presence of rudimentary odontogenic structures in the mouse embryonic mandible requires reinterpretation of developmental control of first lower molar histomorphogenesis. Int. J. Dev. Biol. 44, 233–240 [PubMed] [Google Scholar]
- 21. Jussila M., and Thesleff I. (2012) Signaling networks regulating tooth organogenesis and regeneration, and the specification of dental mesenchymal and epithelial cell lineages. Cold Spring Harb. Perspect. Biol. 4, a008425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hovorakova M., Lochovska K., Zahradnicek O., Domonkosova Tibenska K., Dornhoferova M., Horakova-Smrckova L., and Bodorikova S. (2016) One odontogenic cell-population contributes to the development of the mouse incisors and of the oral vestibule. PLoS ONE 11, e0162523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ahtiainen L., Uski I., Thesleff I., and Mikkola M. L. (2016) Early epithelial signaling center governs tooth budding morphogenesis. J. Cell Biol. 214, 753–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Häärä O., Harjunmaa E., Lindfors P. H., Huh S. H., Fliniaux I., Åberg T., Jernvall J., Ornitz D. M., Mikkola M. L., and Thesleff I. (2012) Ectodysplasin regulates activator-inhibitor balance in murine tooth development through Fgf20 signaling. Development 139, 3189–3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vaahtokari A., Aberg T., Jernvall J., Keränen S., and Thesleff I. (1996) The enamel knot as a signaling center in the developing mouse tooth. Mech. Dev. 54, 39–43 [DOI] [PubMed] [Google Scholar]
- 26. Li C. Y., Hu J., Lu H., Lan J., Du W., Galicia N., and Klein O. D. (2016) αE-catenin inhibits YAP/TAZ activity to regulate signalling centre formation during tooth development. Nat. Commun. 7, 12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jernvall J., Keränen S. V., and Thesleff I. (2000) Evolutionary modification of development in mammalian teeth: quantifying gene expression patterns and topography. Proc. Natl. Acad. Sci. U.S.A. 97, 14444–14448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Coin R., Lesot H., Vonesch J. L., Haikel Y., and Ruch J. V. (1999) Aspects of cell proliferation kinetics of the inner dental epithelium during mouse molar and incisor morphogenesis: a reappraisal of the role of the enamel knot area. Int. J. Dev. Biol. 43, 261–267 [PubMed] [Google Scholar]
- 29. Kieffer S., Peterkova R., Vonesch J. L., Ruch J. V., Peterka M., and Lesot H. (1999) Morphogenesis of the lower incisor in the mouse from the bud to early bell stage. Int. J. Dev. Biol. 43, 531–539 [PubMed] [Google Scholar]
- 30. Cobourne M. T., Hardcastle Z., and Sharpe P. T. (2001) Sonic hedgehog regulates epithelial proliferation and cell survival in the developing tooth germ. J. Dent. Res. 80, 1974–1979 [DOI] [PubMed] [Google Scholar]
- 31. Bitgood M. J., and McMahon A. P. (1995) Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev. Biol. 172, 126–138 [DOI] [PubMed] [Google Scholar]
- 32. Dassule H. R., Lewis P., Bei M., Maas R., and McMahon A. P. (2000) Sonic hedgehog regulates growth and morphogenesis of the tooth. Development 127, 4775–4785 [DOI] [PubMed] [Google Scholar]
- 33. Echelard Y., Epstein D. J., St-Jacques B., Shen L., Mohler J., McMahon J. A., and McMahon A. P. (1993) Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 75, 1417–1430 [DOI] [PubMed] [Google Scholar]
- 34. Juuri E., Saito K., Ahtiainen L., Seidel K., Tummers M., Hochedlinger K., Klein O. D., Thesleff I., and Michon F. (2012) Sox2+ stem cells contribute to all epithelial lineages of the tooth via Sfrp5+ progenitors. Dev. Cell 23, 317–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang L., Yuan G., Liu H., Lin H., Wan C., and Chen Z. (2012) Expression pattern of Sox2 during mouse tooth development. Gene Expr. Patterns 12, 273–281 [DOI] [PubMed] [Google Scholar]
- 36. Matalova E., Antonarakis G. S., Sharpe P. T., and Tucker A. S. (2005) Cell lineage of primary and secondary enamel knots. Dev. Dyn. 233, 754–759 [DOI] [PubMed] [Google Scholar]
- 37. Cho S. W., Lee H. A., Cai J., Lee M. J., Kim J. Y., Ohshima H., and Jung H. S. (2007) The primary enamel knot determines the position of the first buccal cusp in developing mice molars. Differentiation 75, 441–451 [DOI] [PubMed] [Google Scholar]
- 38. Mitsiadis T. A., Tucker A. S., De Bari C., Cobourne M. T., and Rice D. P. (2008) A regulatory relationship between Tbx1 and FGF signaling during tooth morphogenesis and ameloblast lineage determination. Dev. Biol. 320, 39–48 [DOI] [PubMed] [Google Scholar]
- 39. Bossing T., and Technau G. M. (1994) The fate of the CNS midline progenitors in Drosophila as revealed by a new method for single cell labelling. Development 120, 1895–1906 [DOI] [PubMed] [Google Scholar]
- 40. Nonomura K., Yamaguchi Y., Hamachi M., Koike M., Uchiyama Y., Nakazato K., Mochizuki A., Sakaue-Sawano A., Miyawaki A., Yoshida H., Kuida K., and Miura M. (2013) Local apoptosis modulates early mammalian brain development through the elimination of morphogen-producing cells. Dev. Cell 27, 621–634 [DOI] [PubMed] [Google Scholar]
- 41. Vaahtokari A., Aberg T., and Thesleff I. (1996) Apoptosis in the developing tooth: association with an embryonic signaling center and suppression by EGF and FGF-4. Development 122, 121–129 [DOI] [PubMed] [Google Scholar]
- 42. Miard S., Peterková R., Vonesch J. L., Peterka M., Ruch J. V., and Lesot H. (1999) Alterations in the incisor development in the Tabby mouse. Int. J. Dev. Biol. 43, 517–529 [PubMed] [Google Scholar]
- 43. Klein O. D., Lyons D. B., Balooch G., Marshall G. W., Basson M. A., Peterka M., Boran T., Peterkova R., and Martin G. R. (2008) An FGF signaling loop sustains the generation of differentiated progeny from stem cells in mouse incisors. Development 135, 377–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hayashi S., and McMahon A. P. (2002) Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 244, 305–318 [DOI] [PubMed] [Google Scholar]
- 45. Hardcastle Z., Mo R., Hui C. C., and Sharpe P. T. (1998) The Shh signalling pathway in tooth development: defects in Gli2 and Gli3 mutants. Development 125, 2803–2811 [DOI] [PubMed] [Google Scholar]
- 46. Lesot H., Vonesch J. L., Peterka M., Turecková J., Peterková R., and Ruch J. V. (1996) Mouse molar morphogenesis revisited by three-dimensional reconstruction: II. spatial distribution of mitoses and apoptosis in cap to bell staged first and second upper molar teeth. Int. J. Dev. Biol. 40, 1017–1031 [PubMed] [Google Scholar]
- 47. Morita R., Kihira M., Nakatsu Y., Nomoto Y., Ogawa M., Ohashi K., Mizuno K., Tachikawa T., Ishimoto Y., Morishita Y., and Tsuji T. (2016) Coordination of cellular dynamics contributes to tooth epithelium deformations. PLoS ONE 11, e0161336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Le Douarin N. M., and Halpern M. E. (2000) Discussion point. Origin and specification of the neural tube floor plate: insights from the chick and zebrafish. Curr. Opin. Neurobiol. 10, 23–30 [DOI] [PubMed] [Google Scholar]
- 49. Arnold K., Sarkar A., Yram M. A., Polo J. M., Bronson R., Sengupta S., Seandel M., Geijsen N., and Hochedlinger K. (2011) Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 9, 317–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Muzumdar M. D., Tasic B., Miyamichi K., Li L., and Luo L. (2007) A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 [DOI] [PubMed] [Google Scholar]
- 51. Vaezi A., Bauer C., Vasioukhin V., and Fuchs E. (2002) Actin cable dynamics and Rho/Rock orchestrate a polarized cytoskeletal architecture in the early steps of assembling a stratified epithelium. Dev. Cell 3, 367–381 [DOI] [PubMed] [Google Scholar]
- 52. Riddle R. D., Johnson R. L., Laufer E., and Tabin C. (1993) Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 75, 1401–1416 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.