

Abstract
Dioscorea villosa (wild yam) is native to North America and has been widely used as a natural alternative for estrogen replacement therapy to improve women’s health as well as to treat inflammation, muscle spasm, and asthma. Diosgenin and dioscin (glycoside form of diosgenin) are reported to be the pharmacologically active compounds. Despite the reports of significant pharmacological properties of dioscin and diosgenin in conditions related to inflammation, cancer, diabetes, and gastrointestinal ailments, no reports are available on ADME properties of these compounds. This study was carried out to determine ADME properties of diosgenin and dioscin and their effects on major drug metabolizing enzymes (CYP 3A4, 2D6, 2C9, and 1A2). The stability was determined in simulated gastric and intestinal fluids (SGF, pH 1.2 and SIF, pH 6.8), and intestinal transport was evaluated in Caco-2 model. Phase I and phase II metabolic stability was determined in human liver microsomes and S9 fractions, respectively. Quantitative analysis of dioscin and diosgenin was performed by UPLC-MS system. Dioscin degraded up to 28.3% in SGF and 12.4% in SIF, which could be accounted for by its conversion to diosgenin (24.2%. in SGF and 2.4% in SIF). The depletion of diosgenin in SGF and SIF was < 10%. Diosgenin was stable in HLM but disappeared in S9 fraction with a half-life of 11.3 min. In contrast, dioscin was stable in both HLM and S9 fractions. Dioscin showed higher permeability across Caco-2 monolayer with no significant efflux, while diosgenin was subjected to efflux mediated by P-glycoprotein. Diosgenin and dioscin inhibited CYP3A4 with IC50 values of 17 and 33 μM, respectively, while other CYP enzymes were not affected. In conclusion, dioscin showed better intestinal permeability. Conversion of dioscin to diosgenin was observed in both gastric and intestinal fluids. No phase I metabolism was detected for both compounds. The disappearance of diosgenin in S9 fraction indicated phase II metabolism.
Keywords: Dioscorea villosa, Dioscoreaceae, diosgenin, dioscin, Caco-2, metabolic stability, CYP inhibition, intestinal transport
Introduction
Dioscorea villosa L. (wild yam) (Dioscoreaceae) is native to North America. Traditionally, wild yam root and rhizome extract has been used to relieve symptoms associated with menopause and rheumatoid arthritis. Steroidal saponins are the main class of compounds found in D. villosa, and the pharmacologically active compounds are reported to be diosgenin and dioscin (Fig. 1).
Fig. 1.

Chemical structures of diosgenin and dioscin.
Since diosgenin has structural similarities with cholesterol and other endogenous steroids, it has been used as a precursor for synthetic steroids in pharmaceutical industry. Additionally, it is also used in synthesis of steroid like estrogens and DHEA [1]. Several preclinical studies have implicated the potential use of diosgenin in several ailments like cancer, diabetes, hypercholesterolemia, gastrointestinal disorders, and inflammatory conditions [2–5]. In addition, diosgenin and dioscin have been reported to show antifungal properties [6]. Dioscin is the glycoside form of diosgenin and has been shown to possess anti-oxidant and anti-cancer properties [7]. Given the wide spectrum of the uses of these two compounds in several therapeutic areas, studying ADME properties is of paramount importance [8].
The oral bioavailability of any drug is mainly governed by its stability in biological fluids (gastric and intestinal fluids), intestinal absorption, and metabolic stability. The stability of a drug in biological fluids is determined by incubating the drug with simulated gastric and intestinal fluids and quantifying the amount of drug loss [9]. Although several in vitro methods are available to predict the intestinal absorption, studying the bi-directional transport of a drug across Caco-2 cell (human colorectal adenocarcinoma) monolayers to predict its absorption is the most widely used method [10]. When cultured (21 days), Caco-2 cells differentiate into polarized monolayers which resemble small intestinal mucosa in expressing tight junctional proteins and several influx and efflux transporters. The permeability coefficient values obtained from the Caco-2 transport studies correlate well with the intestinal absorption in vivo [10–12]. The next important parameter in characterizing ADME properties of a drug is determining the Phase I and Phase II metabolic stability. This is achieved by incubating drug in presence of either human liver microsomes or S9 fraction and determining the half-life and intrinsic clearance [13].
This study was aimed to determine ADME properties of dioscin and its aglycone diosgenin which included stability in biological fluids, intestinal absorption, and metabolic stability. Since cytochrome P450 enzymes (CYPs, mainly 1A2, 2C19, 3A4, and 2D6) are involved in the metabolism of the majority of drugs and most of the drug-drug interactions are caused by the effects of concomitantly used drugs on these enzymes, the inhibitory potential of diosgenin and dioscin towards these enzymes was also determined employing C-DNA baculovirus-expressed enzymes and fluorescence substrates.
Results and Discussion
For orally administered drugs, stability in SGF and SIF contributes significantly to the intestinal permeability and overall bioavailability. The extent of gastric stability determines the amount of drug available for absorption through intestinal mucosa. This can be determined by incubating the drug with the gastric fluid for specified time period and quantifying the amount of remaining drug [14]. The stability of dioscin and its aglycone diosgenin was evaluated in SGF (pH 1.2) in presence of pepsin for 2 hours and SIF (pH 6.8) in presence of pancreatin for 3 hours. Dioscin degraded up to 28.3% in SGF during 3-hour incubation. The glycosides have been shown to undergo hydrolysis in gastric conditions and convert to the aglycone form [15]. Similar to the previous observations with glycosides, disappearance of dioscin in SGF can be accounted for by its conversion to 24.2% of diosgenin (Table 1). It is also important to note that 71.7% of dioscin was still detected after 3-hour incubation in SGF indicating that a significant amount of dioscin will be available to permeate through the intestine. In SIF, 12.4% of dioscin disappeared, with only 2.4% being converted to aglycone (Table 2). In accordance with the biopharmaceutical classification system that compounds which degrade more than 5% may have stability issues [14], dioscin can be placed in a category of drugs that show poor stability in SGF and SIF. Previous pharmacokinetic study of dioscin in rats has shown that dioscin has low oral bioavailability [16]. The instability of dioscin in gastric and intestinal fluids may contribute to its low systemic availability. On the other hand, the aglycone diosgenin was degraded by 8.5% in SGF (Table 1), which is slightly higher than the recommended value of 5%, while in SIF, it was found to be stable with only 4.8% degradation after 3 hours of incubation (Table 2).
Table 1.
Stability of dioscin and diosgenin in SGF (pH 1.2).
| Incubation time (min) | Dioscin (glycoside) | Diosgenin (aglycone) | |||
|---|---|---|---|---|---|
| Amount of dioscin in SGF (μg/mL) | Formation of diosgenin with time (%) | % RD of dioscin depletion | Amount of diosgenin in SGF (μg/mL) | % RD of diosgenin depletion | |
| 0 | 45.0 ± 4.0 | – | – | 52.4 ± 0.9 | – |
| 30 | 37.5 ± 1.2 | 6.3 | 16.7 | 50.5 ± 0.9 | 3.62 |
| 60 | 34.4 ± 1.1 | 19.8 | 23.6 | 48.0 ± 1.9 | 8.31 |
| 120 | 32.3 ± 1.0 | 24.2 | 28.3 | 47.9 ± 1.1 | 8.59 |
Data are expressed as mean ± SD of triplicate samples from one experiment. RD = Relative difference.
Table 2.
Stability of dioscin and diosgenin in SIF (pH 6.8).
| Incubation time (min) | Dioscin (glycoside) | Diosgenin (aglycone) | |||
|---|---|---|---|---|---|
| Amount of dioscin in SIF (μg/mL) | Formation of diosgenin with time (%) | % RD of dioscin depletion | Amount of diosgenin in SIF (μg/mL) | % RD of diosgenin depletion | |
| 0 | 41.1 ± 0.8 | 0.0 | – | 57.0 ± 1.0 | – |
| 60 | 39.3 ± 1.0 | 0.0 | 4.3 | 56.2 ± 1.9 | 1.28 |
| 120 | 37.0 ± 1.3 | 2.4 | 9.9 | 54.6 ± 2.0 | 4.21 |
| 180 | 36.3 ± 1.1 | 2.9 | 11.6 | 54.2 ± 3.2 | 4.80 |
Data are expressed as mean ± SD of triplicate samples from one experiment. RD = Relative difference.
Next, we evaluated the in vitro intestinal permeability of diosgenin and dioscin using 21-day Caco-2 model. Intestinal absorption of a drug can be predicted by Papp values quantified across Caco-2 monolayers. Compounds with Papp value greater than 1 × 10−6 cm/s are considered to have good intestinal absorption; incomplete absorption occurs if the values are close to 1 × 10−7 cm/s [10, 17]·In our initial transport experiments performed in transport buffer (HBSS), no diosgenin levels were detected in either the apical or basolateral side after 2-hour incubation. This is in accordance to the reports that diosgenin exhibits poor aqueous solubility [1], which may affect the detection of low levels of drug. To overcome this, we have used 1% HPβCD as a solubilizing agent. Cyclodextrins form hydrophilic complexes with the lipophilic compounds and thereby increase the solubility in aqueous fluids [18,19]. Based on the previous reports, the use of 1% HPβCD in Caco-2 transport studies brought no significant alteration in either the integrity or structural properties of the monolayers [20]. In agreement with these observations, no alterations in TEER values of monolayers or Papp values of positive control atenolol and marker Ly were observed in our experiments. Accordingly, 1% HPβCD was used in dissolving our test drugs and controls for transport studies. These observations also confirm that the use of cyclodextrins as solubilizing agents is a useful strategy to study intestinal transport of poorly soluble compounds.
In the present study, diosgenin showed a Papp value of 9.6 × 10−6 cm/s in the absorptive direction (A–B) and 48 × 10−6 cm/s in the secretory direction (B–A) as shown in Table 3. The transport was higher in the secretory direction (21%) compared to the absorptive direction (3.8%). The low permeability marker compound atenolol showed apparent permeability values of 2.3 × 10−6 cm/s (2.8% transport) in A–B direction and 1.31 × 10−6 cm/s (1.4% transport) in B–A direction (Table 3), which are similar to previous reports [21]. In addition, at the end of the transport experiments, the monolayer integrity was confirmed by measuring the TEER across the monolayer, and by measuring Papp value of Ly, which was in the range of 2.0 ± 0.5 × 10−6 cm/s. The permeability values of diosgenin in the absorptive direction are significantly higher than the permeability of atenolol (Table 3), but the values are low in comparison to the reported values of propranolol (32 × 10−6 cm/s) [22] which is considered as a marker for high permeability in Caco-2 transport studies. The Papp values of dioscin were 15 × 10−6 cm/s (7.5% transport) in A–B and 20 × 10−6 cm/s (11% transport) in B–A, which is twofold higher than those for diosgenin. Similar to diosgenin, permeability values of dioscin were higher than atenolol and lower than propranolol. These results suggest that both diosgenin and dioscin exhibit a moderate intestinal permeability across Caco-2 monolayers. The two compounds showed a linear transport across Caco-2 monolayers for 2 hours as shown in Fig. 2. The diosgenin levels were not detected until 60 min suggesting slower absorption across the intestinal layers compared to dioscin (Fig. 2).
Table 3.
Transport of diosgenin and dioscin in absorptive (A–B) and secretory (B–A) directions across Caco-2 cell monolayers.
| Compound | Papp ×10−6 cm/sec | % Transport | Efflux ratio | ||
|---|---|---|---|---|---|
| Absorptive direction (A–B) | Secretory direction (B–A) | Absorptive direction (A–B) | Secretory direction (A–B) | ||
| Diosgenin | 9.6 ± 0.7 | 48 ± 4*** | 3.8 ± 0.8 | 21 ± 1 | 5.0 |
| Dioscin | 15 ± 1 | 20 ± 3 | 7.5 ± 0.8 | 11 ± 1 | 1.3 |
| Atenolol | 2.3 ± 0.5 | 2.8 ± 0.8 | 1.3 ± 0.1 | 1.4 ± 0.4 | 1.2 |
Papp values were determined for diosgenin and dioscin as described in Materials and Methods. Atenolol was used as a positive control in each experiment. Data are expressed as mean ± SD of 3 experiments. Papp values in the secretory direction were compared with Papp values in the absorptive direction by Student’s t-test;
p > 0.05; Efflux ratio = Papp B–A/Papp A–B.
Fig. 2.
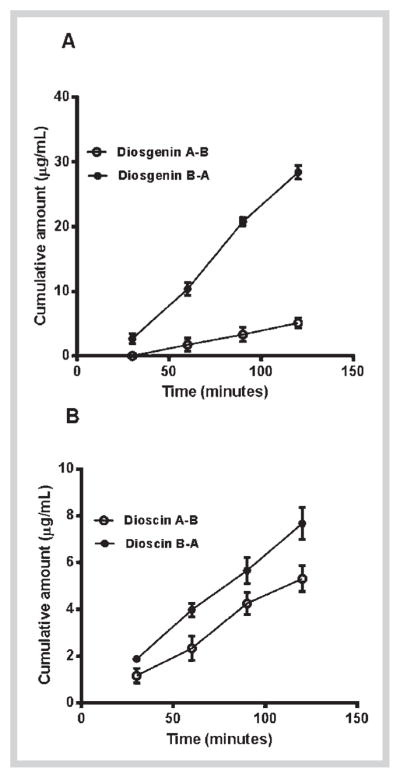
Cumulative amount of diosgenin (A) and dioscin (B) transported across Caco-2 cell monolayers for 2 h. The data are represented as mean ± SD of 3 experiments (n = 1 in each experiment).
The next objective was to see if there are any efflux mechanisms involved in the transport of diosgenin and dioscin. Efflux ratios (Papp B–A/Papp A–B) quantified in transport studies are an indirect measurement of involvement of any efflux mechanisms [23, 24]. Drugs with efflux ratios close to unity suggest that there is an equal directional flux and passive diffusion is the predominant mechanism involved in the absorption [25]. For drugs with efflux ratio greater than 2, there is a definite contribution of efflux transporters in transport mechanisms. In our study, a high efflux ratio of 5.0 for diosgenin indicates that efflux transporters are involved in the transport of diosgenin which could result in its limited intestinal absorption. An efflux ratio of 1.3 for dioscin is suggestive of passive diffusion as a primary mechanism involved in the intestinal absorption of dioscin. Since the efflux ratio of diosgenin was greater than 2, we investigated the role of major efflux transporters in its transport. The three major efflux transporters belonging to ABC transporter family are P-gp, multidrug resistant protein1 and 2 (MRP1, MRP2), and BCRP. These transporters are expressed on the apical side of the intestinal mucosa and play a pivotal role in altering the bioavailability of substrate drugs [26]. To study the role of these transporters, the transport of diosgenin was monitored in presence of inhibitors of P-gp (verapamil), MRP1 and 2 (MK-571), and BCRP (KO143). The addition of verapamil significantly increased (3 fold) the permeability of diosgenin in the absorptive direction (A–B) with no effect on the permeability in secretory direction (B–A) as demonstrated in Fig. 2. Compounds such as grepafloxacin show a similar trend where flux changes only in absorptive direction in the presence of inhibitors, hence, diosgenin can be placed in the same class [27]. The addition of MK571 and KO143 increased the Papp value of diosgenin in the absorptive direction by 1.84 and 1.33 fold, respectively (Fig. 3A), without affecting the Papp value in secretory direction, similar to verapamil. In addition, the efflux ratio of diosgenin (5.0) was significantly lower in presence of verapamil (2.1), while the presence of MK571 (3.8) and KO143 (4.0) had no significant effect on the efflux ratio, as shown in Fig. 3B. These results indicate that P-gp is the primary transporter involved in the efflux of diosgenin and the use of P-gp inhibitors may be a useful strategy to enhance its in vivo bioavailability. The possibility of involvement of additional isoforms of MRP proteins (MRP 3–6) and solute carrier transporters that are also expressed in Caco-2 cells cannot be ruled out, which may also contribute to the high efflux of diosgenin. Previous studies have indicated that glycosides like hesperidin and genistin show poor intestinal permeation compared to their corresponding aglycones (hesperetin and genistein) [28,29]. In contrast to these reports, our results show that the glycoside dioscin has better intestinal permeability than its aglycone form. Dioscin has been previously shown to inhibit P-gp [12] which could be responsible for its better absorption. In addition, the predicted Log P values of diosgenin and dioscin are 5.12 and 1.12, respectively. Compounds with Log P values between 1 and 4 have been shown to have ideal liphophilicity and aqueous solubility to penetrate biological membranes. Further, compounds with Log P greater than 4 have been predicted to have poor aqueous solubility and intestinal absorption [30]. Based on this theory, diosgenin is expected to have lower permeability than dioscin, which is in accordance to our observations.
Fig. 3.
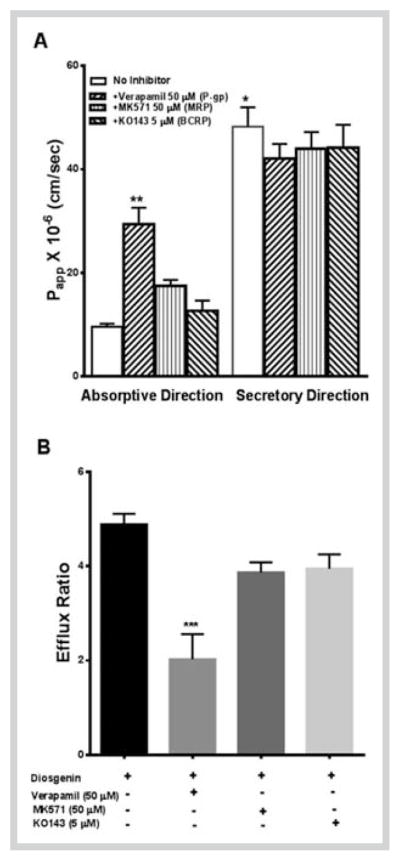
Permeability (A) and efflux ratios (B) of diosgenin across Caco-2 monolayer in presence and absence of P-gp (Verapamil), MRP (MK 571), and BCRP (KO143) inhibitors. *** P < 0.001, ** p < 0.05, determined by Kruskal-Wallis test; * p < 0.01, determined by Mann-Whitney test. The data are expressed as mean ± SD of 3 experiments (n = 1 in each experiment).
Next, we determined the metabolic stability of diosgenin and dioscin in HLM and S9 fractions. Diosgenin was stable in HLM (Fig. 4) but degraded in S9 fraction with a half-life of 11.3 min and intrinsic clearance of 7 mL·min−1·kg−1 (Fig. 4). This indicates that diosgenin could be mainly metabolized by phase II enzymes. In contrast, the aglycone dioscin was found to be relatively stable in both HLM and S9 fractions with more than 50% of the compound detected after incubation (Figs. 4 and 5). This observation suggests that dioscin is metabolically stable. However, extra-hepatic mechanisms may be involved in the elimination of dioscin as shown in a previous in vivo study [16]. The high metabolic stability may explain the long half-life of dioscin (25.6 h) after oral administration as observed in a previously reported pharmacokinetic study of dioscin in rat [31].
Fig. 4.

Phase I metabolic depletion of diosgenin and dioscin in pooled human liver microsomes with respect to time. Testosterone was used as positive control. The data are expressed as mean ± SD of 3 experiments (n = 1 in each experiment).
Fig. 5.

Phase II metabolic depletion of diosgenin and dioscin in S9 fractions with respect to time. 7-Hydroxycoumarin was used as a positive control. The data are expressed as mean ± SD of triplicate samples in one experiment.
Finally, we investigated if diosgenin and dioscin interact with major drug metabolizing enzymes such as CYP 3A4, 2D6, 2C9, and 1A2. The alteration in the activity of these enzymes by concomitantly administered drugs is responsible for the majority of drug-drug interactions [32]. Diosgenin and dioscin inhibited catalytic activity of CYP3A4 with IC50 values of 9 μg/mL (17 μM) and 29 μg/mL (33 μM), respectively (Table 4, Fig. 6), while other CYP enzymes were not affected. At these high concentrations, the possibility of diosgenin and dioscin to cause drug-drug interactions seems to be remote.
Table 4.
IC50 values of inhibition of cytochrome P450 enzymes by diosgenin and dioscin. The assay conditions are described in Materials and Methods.
| Compound | IC50 (μM) 3A4 |
2D6 | 2C19 | 1A2 |
|---|---|---|---|---|
| Diosgenin | 17 ± 4 | NE | NE | NE |
| Dioscin | 33 ± 4 | NE | NE | NE |
| Quinidine* | – | 0.04 ± 0.01 | – | – |
| Ketoconazole* | 0.05 ± 0.01 | – | – | – |
| Tranylcypromine* | – | – | 0.9 ± 0.0 | – |
| Furafylline* | – | – | – | 1.12 ± 0.01 |
Data are expressed as mean ± SD of triplicate samples from one experiment. NE = Not effective up to 50 μg/mL.
Positive controls.
Fig. 6.
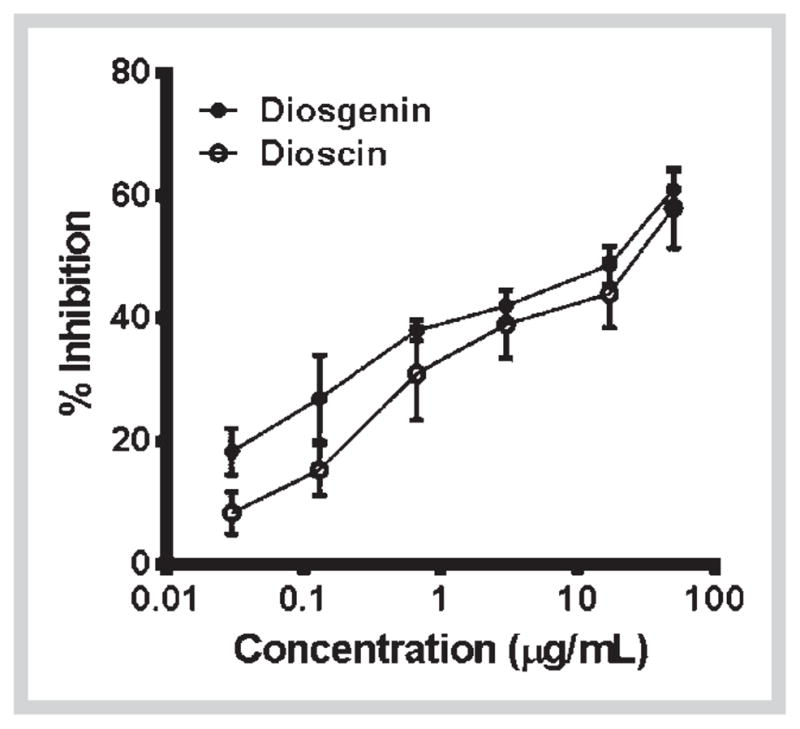
Dose-response curves of CYP3A4 enzyme inhibition by diosgenin and dioscin. The data shown are mean ± SD of triplicate treatments in one experiment.
In conclusion, dioscin showed better intestinal permeation than diosgenin, however, its bioavailability may be impeded due to its instability in SGF and SIF. Diosgenin was found to be a substrate for P-gp, which seems to limit its intestinal permeability and bioavailability. A weak interaction with CYP 3A4 and no effect on other CYPs are indicative of drug safety if used in combination with other drugs.
Materials and Methods
Caco-2 cells were obtained from American Type Culture Collection (ATCC). DMEM, MEM, HBSS, HEPES, trypsin EDTA, penicillin-streptomycin, and sodium pyruvate were obtained from GIB-CO BRL Invitrogen Corporation. FBS was from Hyclone Lab Inc. CYPremes™ human liver microsomes and S9 fractions (pooled mixed sex) were from In Vitro Technologies Inc. CYP1A2/CEC, CYP2C9/MFC, CYP3A4/BQ, and CYP2D6/AMMC high throughput inhibitor screening kits were from BD Gentest. Transwell® plates (12 well, 1.12 mm diameter, 0.4 μM pore size) were from Costar Corp. G-6-PDH, glucose-6-phosphate, NADP+, UDPGA, atenolol, and all other chemicals were from Sigma Chem. Co. KO143 was purchased from Tocris Biosciences. Strata-X filter plates for extraction of diosgenin and dioscin in transport experiments were purchased from Phenomenex. The purity of the positive controls was ≥ 98%.
Diosgenin and dioscin were isolated from the extract of Dioscorea villosa rhizomes as described earlier, and the purity of both the compounds was confirmed by chromatographic analysis to be ≥ 95% [33]. A 10 mM stock solution of diosgenin and dioscin was prepared in DMSO for transport experiments (final DMSO 1%) or methanol for stability studies and CYP inhibition assays (final methanol 0.1%).
Stability of diosgenin and dioscin in simulated gastric and intestinal fluids
Previously described USP specifications were used to prepare simulated gastric and intestinal fluids as described below [34].
Preparation of SGF
Sodium chloride (1.0 g) and purified pepsin (1.6 g) (derived from porcine stomach mucosa) were dissolved in 3.5 mL of 0.1 N hydrochloric acid (HCl), and the pH was adjusted to 1.2 with 0.1 N HCl. The final volume was made up to 500 mL with nanopure water.
Preparation of SIF
A solution of monobasic potassium phosphate (6.8 g) was made in 250 mL of distilled water. To this solution, 77 mL of 0.2 N sodium hydroxide and 500 mL of water were added and mixed well followed by the addition of 10 g of pancreatin (from porcine pancreas). The pH of simulated SIF was adjusted to 6.8 ± 0.1 with 0.2 N sodium hydroxide or 0.2 N hydrochloric acid. The final volume was made up to 1000 mL with nanopure water.
Determination of diosgenin and dioscin stability in SGF and SIF
A solution of diosgenin and dioscin was prepared in SGF or SIF (0.5 mg/mL). Aliquots (1 mL) of these solutions were pipetted into glass tubes and placed in a 37°C shaking water bath (120 RPM). The incubation times were 0, 30, 60, and 120 min for SGF and 0, 60, 120, and 180 min for SIF. The experiment was done in triplicates. At the corresponding time points, 100 μL of the sample was taken out and added to 200 μL of ice cold methanol and mixed well. Samples were centrifuged at 12000 RPM for 5 min, and the supernatant was used for UPLC-MS analysis. The relative difference (RD), which determines the % of drug remaining at the end of incubation period compared to the initial amount, was calculated using the following equation
where Ci is the initial concentration of drug determined at zero time and Cf is the concentration determined at the end of incubation period.
Cell culture and bi-directional transport assay across Caco-2 monolayers
Cell culture conditions for Caco-2 cells were similar as described earlier [21]. Cells between passage numbers 30–42 were used for transport studies. Transport experiments were performed in 12-well Transwell® plates. Cells were seeded at a density of 63000 cells/cm2 and grown for 21 days, with change of media every 3 days. Physiologically and morphologically confluent monolayers with TEER values greater than 400 ohm/cm2 were used for the experiment. HBSS with 10 mM HEPES was used as transport buffer. For bidirectional transport, compounds (100–200 μM) were added to apical side for determining apical to basolateral transport (A–B; absorptive direction) and basal side for determining basolateral to apical transport (B-A; secretory direction). Volume of apical and basolateral chambers were 0.6 and 1.5 mL, respectively. Aliquots of 200 μL were taken out from basolateral (for AB transport) or from apical (for B-A transport) chamber at 30, 60, 90, and 120 min. An equal volume was replaced with transport buffer at every time point. At the end of the experiment, an aliquot was also taken out from apical or basolateral chamber for analysis of drug. To determine the integrity of monolayers, TEER values were measured before and after the experiment. In addition, permeability of Ly, a fluorescent paracellular diffusion marker, was also determined across the monolayers after the transport experiment. Furthermore, cytotoxicity of dioscin and diosgenin was determined to Caco-2 cells using MTS proliferation assay (Supporting Information, Fig. 1S).
Papp (cm/sec) was calculated from the following equation:
where dq/dt is the rate of transport, Co is the initial concentration in the donor compartment, and A is the surface area of the filter. To quantify the rate of transport, cumulative amount of test compounds was plotted against time (min).
The solubility of diosgenin was very poor in the transport buffer. To overcome this, 1% cyclodextrin was used as a solubilizing agent. To maintain the homogeneity, all the experimental compounds were dissolved in transport buffer containing 1% HPβCD including the controls. The samples from bi-directional transport assays were extracted with 100% methanol containing 0.05% formic acid by solid phase extraction method using Strata-X filter plates.
Assay for metabolic stability in human liver microsomes and S9 fractions
To determine Phase I and Phase II metabolic stability of diosgenin and dioscin, human liver microsomes and S9 fractions were used, respectively. The assay conditions and reaction mixtures were similar as described previously [35]. Testosterone and 7-hydroxy coumarin were used as positive controls for Phase I and Phase II metabolism, respectively. The reaction mixture was primed for 5 min at 37°C and then diosgenin (100 μM), dioscin (50 μM), or controls (10 μM) were added. Aliquots of 100 μL were collected from reaction volumes at predetermined time points of 0, 15, 30, 60, 90, 120, and 180 min and extracted with 200 μL of ice cold acetonitrile/methanol (50:50). After centrifuging for 15 min at 12000 RPM (4°C), the supernatants were analyzed by UPLC-MS. Elimination half life (T1/2) and CL’int (mL/min/kg) were calculated as reported previously [13].
where k is the slope of the line obtained by plotting natural logarithmic percentage (Ln %) of diosgenin and dioscin remaining in the reaction mixture versus incubation time (min).
Assay for inhibition of cytochrome P450 (1A2, 2C19, 3A4, and 2D6)
The assay conditions were followed as described previously [36]. The assay was performed in a total volume of 200 μL using 96-well microtiter plates. Test compounds (50–0.2 μM) or positive control (5–0.02 μM), cofactors mix, and G-6-PDH were added to the wells and pre incubated for 10 min at 37°C, and the plates were read to account for any fluorescence caused by the test compounds (0-min readouts, both diosgenin and dioscin had no measurable auto fluorescence). Reaction was initiated by adding the enzyme substrate mixture and incubated for 30 min. Reaction was stopped by adding ice cold acetonitrile/0.5 M Trisbase (80:20). Fluorescence was measured on a Spectramax M5 plate reader (Molecular Devices) at specified excitation and emission wavelengths. IC50 values were obtained from dose curves generated by plotting percent inhibition against tested concentrations.
Analytical method
All the samples and controls were analyzed using Waters Acquity ultra performance-liquid chromatography-UV/mass spectrometry (UPLC-UV/MS) (Waters Corp.). The system binary solvent manager, sampler manager, column compartment, and PDA detector were connected to Waters Empower 2 data station. For the analysis of diosgenin and dioscin, an Acquity UPLC BEH Shield RP18 column (100 mm × 2.1 mm I.D., 1.7 μm) was used. A column temperature of 40°C and sample temperature of 15°C were maintained during sample analysis. The mobile phase consisted of water (A) and acetonitrile (B), both containing 0.05% formic acid at a flow rate of 0.27 mL/min, which were applied in the following gradient elution (Waters curve type 6): 80% A: 20% B at 0 min, 50% A: 50% B in next 5 min, 20% A: 80% B in next 2 min, and 100% B in next 3 min. Separation was followed by a 3-min washing procedure with 100% B and re-equilibration period of 3.5 min. The total run time for analysis was 13 min. The injection volume was 10 μL. The effluent from the LC column was directed into the ESI probe. Mass spectrometer conditions were optimized to obtain maximal sensitivity. The source temperature and the desolvation temperature were maintained at 150°C and 350°C, respectively. The probe voltage (capillary voltage), cone voltage, and extractor voltage were fixed at 3.0 kV, 30 V, and 3 V, respectively. Nitrogen was used as the source of desolvation gas (650 L/hr) and drying gas (25 L/hr). Compounds were confirmed in selected ion recording (SIR) mode. [M + H]+ = 415.3 ion for diosgenin and 913.4 [M − H]− ion in negative mode for dioscin were selected as detecting ions. The lower limit of quantification and detection of diosgenin and dioscin were 0.2 μg/mL and 0.1 μg/mL, respectively. Mass spectra were obtained at a dwell time of 0.1 sec in SIR and 500 Da/sec of scan rate. The analysis of atenolol, testosterone, and 7-hydroxycoumarin was done according to method described earlier [37].
Statistical methods
All values are represented as mean ± SEM (n = 3). Papp values in B–A direction were compared with Papp values in A–B direction by Student’s t-test. Non-parametric data were analysed by the Mann-Whitney test and Kruskall-Wall test using Graph Pad Prism Version 5. p < 0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgments
We acknowledge the United States Department of Agriculture, Agricultural Research Service, Specific Cooperative Agreement No. 58–6408–2–0009, the FDA “Science Based Authentication of Dietary Supplements” award number 1 U01 FD004246–02, and the University of Illinois Botanical Research Center (BRC) entitled “Botanical Identification, Characterization, Quality Assurance and Quality Control”, funded by NIH, prime award number 1P50 AT006268–02.
Abbreviations
- ADME
absorption, distribution, metabolism, and excretion
- Caco-2
human colonic adenocarcinoma
- CYP
cytochrome P450
- Papp
apparent permeability
- Ly
lucifer yellow
- P-gp
P-glycoprotein
- BCRP
breast cancer resistant protein
- MRP
multidrug resistant protein
- HLM
human liver microsomes
- CL’int
intrinsic clearance
- DHEA
dehydroepiandrosterone
- HPβCD
hydroxypropoyl-β-cyclodextrin
- ABC
ATP binding cassette
- G-6-PDH
glucose-6-phosphate dehydrogenase
- TEER
trans-epithelial electrical resistance
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting information available online at http://www.thieme-connect.de/ejournals/toc/plantamedica
Data regarding cytotoxicity of diosgenin and dioscin in Caco-2 cells are available as Supporting Information.
References
- 1.Komesaroff PA, Black CV, Cable V, Sudhir K. Effects of wild yam extract on menopausal symptoms, lipids and sex hormones in healthy menopausal women. Climacteric. 2001;4:144–150. [PubMed] [Google Scholar]
- 2.Al-Matubsi HY, Nasrat NA, Oriquat GA, Abu-Samak M, Al-Mzain KA, Salim M. The hypocholesterolemic and antioxidative effect of dietary diosgenin and chromium chloride supplementation on high-cholesterol fed Japanese quails. Pak J Biol Sci. 2011;14:425–432. doi: 10.3923/pjbs.2011.425.432. [DOI] [PubMed] [Google Scholar]
- 3.Das S, Dey KK, Dey G, Pal I, Majumder A, Maitichoudhury S, Kundu SC, Mandal M. Antineoplastic and apoptotic potential of traditional medicines thymoquinone and diosgenin in squamous cell carcinoma. PLoS One. 2012;7:e46641. doi: 10.1371/journal.pone.0046641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim DS, Jeon BK, Lee YE, Woo WH, Mun YJ. Diosgenin induces apoptosis in HepG2 cells through generation of reactive oxygen species and mitochondrial pathway. Evid Based Complement Alternat Med. 2012;2012:981675. doi: 10.1155/2012/981675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jan TR, Wey SP, Kuan CC, Liao MH, Wu HY. Diosgenin, a steroidal sapogenin, enhances antigen-specific IgG2a and interferon-gamma expression in ovalbumin-sensitized BALB/c mice. Planta Med. 2007;73:421–426. doi: 10.1055/s-2007-967169. [DOI] [PubMed] [Google Scholar]
- 6.Sautour M, Mitaine-Offer AC, Miyamoto T, Dongmo A, Lacaille-Dubois MA. Antifungal steroid saponins from Dioscorea cayenensis. Planta Med. 2004;70:90–92. doi: 10.1055/s-2004-815467. [DOI] [PubMed] [Google Scholar]
- 7.Kim YS, Kim EA, Park KG, Lee SJ, Kim MS, Sohn HY, Lee TJ. Dioscin sensitizes cells to TRAIL-induced apoptosis through downregulation of c-FLIP and Bcl-2. Oncol Rep. 2012;28:1910–1916. doi: 10.3892/or.2012.1962. [DOI] [PubMed] [Google Scholar]
- 8.Manda VK, Avula B, Ali Z, Smillie TJ, Khan IA, Khan SI. Characterization of ADME properties of diosgenin and dioscin: bioactive constituents of Dioscorea villosa (Wild yam) International Conference on the Science of Botanicals, Oxford Planta Med. 2013;79:79. doi: 10.1055/s-0033-1350699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jimenez P, Tejero J, Cabrero P, Cordoba-Diaz D, Girbes T. Differential sensitivity of d-galactose-binding lectins from fruits of dwarf elder (Sambucus ebulus L) to a simulated gastric fluid. Food Chem. 2013;136:794–802. doi: 10.1016/j.foodchem.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 10.Artursson P, Palm K, Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv Drug Deliv Rev. 2001;46:27–43. doi: 10.1016/s0169-409x(00)00128-9. [DOI] [PubMed] [Google Scholar]
- 11.Smetanova L, Stetinova V, Svoboda Z, Kvetina J. Caco-2 cells, biopharmaceutics classification system (BCS) and biowaiver. Acta Med (Hradec Kralove) 2011;54:3–8. [PubMed] [Google Scholar]
- 12.Sun BT, Zheng LH, Bao YL, Yu CL, Wu Y, Meng XY, Li YX. Reversal effect of Dioscin on multidrug resistance in human hepatoma HepG2/adriamycin cells. Eur J Pharmacol. 2011;654:129–134. doi: 10.1016/j.ejphar.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 13.Chao P, Uss AS, Cheng KC. Use of intrinsic clearance for prediction of human hepatic clearance. Expert Opin Drug Metab Toxicol. 2010;6:189–198. doi: 10.1517/17425250903405622. [DOI] [PubMed] [Google Scholar]
- 14.FDA. Guidance for industry, waiver of in vivo bioavailability and bioequivalence studies for immediate release solid oral dosage forms containing certain active moieties/active ingriedients based on biopharmaceutical classification system. Rockville, Maryland, USA: FDA; 2000. Food and Drug Administration Center for Drug Evaluation and Research. [Google Scholar]
- 15.Kuhlmann J, Abshagen U, Rietbrock N. Cleavage of glycosidic bonds of digoxin and derivatives as function of pH and time. Naunyn Schmiedebergs Arch Pharmacol. 1973;276:149–156. doi: 10.1007/BF00501187. [DOI] [PubMed] [Google Scholar]
- 16.Li K, Tang Y, Fawcett JP, Gu J, Zhong D. Characterization of the pharmacokinetics of dioscin in rat. Steroids. 2005;70:525–530. doi: 10.1016/j.steroids.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 17.Artursson P, Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun. 1991;175:880–885. doi: 10.1016/0006-291x(91)91647-u. [DOI] [PubMed] [Google Scholar]
- 18.Dahan A, Miller JM, Hoffman A, Amidon GE, Amidon GL. The solubility-permeability interplay in using cyclodextrins as pharmaceutical solubilizers: mechanistic modeling and application to progesterone. J Pharm Sci. 2010;99:2739–2749. doi: 10.1002/jps.22033. [DOI] [PubMed] [Google Scholar]
- 19.Loftsson T, Vogensen SB, Brewster ME, Konradsdottir F. Effects of cyclodextrins on drug delivery through biological membranes. J Pharm Sci. 2007;96:2532–2546. doi: 10.1002/jps.20992. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi Y, Kondo H, Yasuda T, Watanabe T, Kobayashi S, Yokohama S. Common solubilizers to estimate the Caco-2 transport of poorly water-soluble drugs. Int J Pharm. 2002;246:85–94. doi: 10.1016/s0378-5173(02)00347-2. [DOI] [PubMed] [Google Scholar]
- 21.Madgula VL, Avula B, Pawar RS, Shukla YJ, Khan IA, Walker LA, Khan SI. In vitro metabolic stability and intestinal transport of P57AS3 (P57) from Hoodia gordonii and its interaction with drug metabolizing enzymes. Planta Med. 2008;74:1269–1275. doi: 10.1055/s-2008-1074580. [DOI] [PubMed] [Google Scholar]
- 22.Madgula VL, Avula B, Choi YW, Pullela SV, Khan IA, Walker LA, Khan SI. Transport of Schisandra chinensis extract and its biologically-active constituents across Caco-2 cell monolayers – an in-vitro model of intestinal transport. J Pharm Pharmacol. 2008;60:363–370. doi: 10.1211/jpp.60.3.0012. [DOI] [PubMed] [Google Scholar]
- 23.Makhey VD, Guo A, Norris DA, Hu P, Yan J, Sinko PJ. Characterization of the regional intestinal kinetics of drug efflux in rat and human intestine and in Caco-2 cells. Pharm Res. 1998;15:1160–1167. doi: 10.1023/a:1011971303880. [DOI] [PubMed] [Google Scholar]
- 24.Sun H, Pang KS. Permeability, transport, and metabolism of solutes in Caco-2 cell monolayers: a theoretical study. Drug Metab Dispos. 2008;36:102–123. doi: 10.1124/dmd.107.015321. [DOI] [PubMed] [Google Scholar]
- 25.Pang KS, Maeng HJ, Fan J. Interplay of transporters and enzymes in drug and metabolite processing. Mol Pharm. 2009;6:1734–1755. doi: 10.1021/mp900258z. [DOI] [PubMed] [Google Scholar]
- 26.Taipalensuu J, Tornblom H, Lindberg G, Einarsson C, Sjoqvist F, Melhus H, Garberg P, Sjostrom B, Lundgren B, Artursson P. Correlation of gene expression of ten drug efflux proteins of the ATP-binding cassette transporter family in normal human jejunum and in human intestinal epithelial Caco-2 cell monolayers. J Pharmacol Exp Ther. 2001;299:164–170. [PubMed] [Google Scholar]
- 27.Yamaguchi H, Yano I, Hashimoto Y, Inui KI. Secretory mechanisms of grepafloxacin and levofloxacin in the human intestinal cell line caco-2. J Pharmacol Exp Ther. 2000;295:360–366. [PubMed] [Google Scholar]
- 28.Kobayashi S, Tanabe S, Sugiyama M, Konishi Y. Transepithelial transport of hesperetin and hesperidin in intestinal Caco-2 cell monolayers. Biochim Biophys Acta. 2008;1778:33–41. doi: 10.1016/j.bbamem.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 29.Walle UK, French KL, Walgren RA, Walle T. Transport of genistein-7-glucoside by human intestinal CACO-2 cells: potential role for MRP2. Res Commun Mol Pathol Pharmacol. 1999;103:45–56. [PubMed] [Google Scholar]
- 30.Alvarez-Figueroa MJ, Pessoa-Mahana CD, Palavecino-Gonzalez ME, Mella-Raipan J, Espinosa-Bustos C, Lagos-Munoz ME. Evaluation of the membrane permeability (PAMPA and skin) of benzimidazoles with potential cannabinoid activity and their relation with the Biopharmaceutics Classification System (BCS) AAPS PharmSciTech. 2011;12:573–578. doi: 10.1208/s12249-011-9622-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li K, Wang Y, Gu J, Chen X, Zhong D. Determination of dioscin in rat plasma by liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;817:271–275. doi: 10.1016/j.jchromb.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka E. Clinically important pharmacokinetic drug-drug interactions: role of cytochrome P450 enzymes. J Clin Pharm Ther. 1998;23:403–416. doi: 10.1046/j.1365-2710.1998.00086.x. [DOI] [PubMed] [Google Scholar]
- 33.Ali Z, Smillie TJ, Khan IA. Cholestane steroid glycosides from the rhizomes of Dioscorea villosa (wild yam) Carbohydr Res. 2013;370:86–91. doi: 10.1016/j.carres.2012.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.USP. Test solutions. Rockville: US Pharmacopeial Convention; 2008. United States Pharmacopoeia XXX1/National Formulary 21, USP/NF 2008; p. 817. [Google Scholar]
- 35.Madgula VL, Avula B, Pawar RS, Shukla YJ, Khan IA, Walker LA, Khan SI. Characterization of in vitro pharmacokinetic properties of hoodigogenin A from Hoodia gordonii. Planta Med. 2010;76:62–69. doi: 10.1055/s-0029-1185949. [DOI] [PubMed] [Google Scholar]
- 36.Crespi CL, Miller VP, Penman BW. Microtiter plate assays for inhibition of human, drug-metabolizing cytochromes P450. Anal Biochem. 1997;248:188–190. doi: 10.1006/abio.1997.2145. [DOI] [PubMed] [Google Scholar]
- 37.Madgula VL, Avula B, Reddy VLN, Khan IA, Khan SI. Transport of decursin and decursinol angelate across Caco-2 and MDR-MDCK cell monolayers: in vitro models for intestinal and blood-brain barrier permeability. Planta Med. 2007;73:330–335. doi: 10.1055/s-2007-967137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.