

Abstract
Cardiovascular complications are a leading cause of morbidity and mortality in type 2 diabetes mellitus (T2DM) and are associated with alterations of blood vessel structure and function. Although endothelial dysfunction and aortic stiffness have been documented, little is known about the effects of T2DM on coronary microvascular structural remodeling. The renin–angiotensin–aldosterone system plays an important role in large artery stiffness and mesenteric vessel remodeling in hypertension and T2DM. The goal of this study was to determine whether the blockade of AT1R signaling dictates vascular smooth muscle growth that partially underlies coronary arteriole remodeling in T2DM. Control and db/db mice were given AT1R blocker losartan via drinking water for 4 weeks. Using pressure myography, we found that coronary arterioles from 16-week db/db mice undergo inward hypertrophic remodeling due to increased wall thickness and wall-to-lumen ratio with a decreased lumen diameter. This remodeling was accompanied by decreased elastic modulus (decreased stiffness). Losartan treatment decreased wall thickness, wall-to-lumen ratio, and coronary arteriole cell number in db/db mice. Losartan treatment did not affect incremental elastic modulus. However, losartan improved coronary flow reserve. Our data suggest that Ang II–AT1R signaling mediates, at least in part, coronary arteriole inward hypertrophic remodeling in T2DM without affecting vascular mechanics, further suggesting that targeting the coronary microvasculature in T2DM may help reduce cardiac ischemic events.
Keywords: Vascular remodeling, Type 2 diabetes mellitus, Angiotensin receptor blocker, Coronary arterioles
1. Introduction
In patients with type 2 diabetes mellitus (T2DM), cardiovascular complications are the leading cause of death and account for the greatest amount in health care expenditures [16,23]. Diabetes-related vasculopathies include both micro- and macrovascular complications resulting from structural and functional deficits [13,22,29]. Macro-vascular diseases in diabetes include structurally related alterations in lumen diameter (atherosclerosis, thrombosis) and passive mechanical properties (aortic stiffness) [22,29]. Likewise, microvascular complications, which include retinopathy, nephropathy and neuropathy, involve both structural (increased wall thickness and decreased lumen) and functional (endothelial dysfunction and increased myogenic tone) alterations [13,22]. T2DM dependent functional deficits in endothelial and vascular smooth muscle cells (VSMCs) lead to impaired endothelium-dependent relaxation, enhanced myogenic tone and/or changes in responses to vasoactive factors. Collectively, these impair the ability of the vessel to maintain proper tissue perfusion [39] leading to reduced blood flow and increased peripheral resistance, followed by tissue damage, and necrosis [5].
Most functional studies have been performed in the aorta and renal and mesenteric resistance beds, which are likely to respond in a vascular bed-specific manner due to distinct patterns of receptor expression, redox-sensitive cascades, and mechanical properties [6,7,32]. However, the impact of microvascular dysfunction and cardiovascular complications in patients with T2DM is poorly understood, especially with regard to microvascular structural remodeling. A reduced ability to respond to increased metabolic demand in coronary resistance arteries in diabetes is due to both altered function and structure [33,35]. These alterations may lead to myocardial injury, decreased cardiac perfusion, and reduced coronary flow reserve. Evidence from clinical trails suggests that, when faced with increased myocardial demand during exercise or exertion, coronary flow reserve is reduced in diabetic patients [33,35,49]. Alterations in structure and function also include altered vessel stiffness and reduced coronary flow reserve in diabetic coronary resistance arteries [19,43,45,52].
The renin–angiotensin–aldosterone system (RAAS) is an important neurohormonal system that oversees cardiovascular and renal functions not only by regulating systemic blood pressure and fluid volume, but also via direct actions on tissues. More recently, local paracrine or tissue activation of the RAAS has direct effects on vascular smooth muscle and endothelial cells that are blood pressure independent. Augmented activity of the RAAS leads to cardiovascular diseases (hypertension, left ventricular hypertrophy, and atherosclerosis) and cardiovascular events (myocardial infarction, congestive heart failure, and stroke). The systemic and paracrine biological actions of angiotensin II (Ang II), the major active component in the RAAS, are transduced via two known receptor subtypes, angiotensin II type 1 receptor (AT1R) and angiotensin II type 2 receptor (AT2R). Both receptors regulate VSMC function, though they vary in their actions. The AT1R is highly expressed in the vasculature, specifically in VSMCs and is responsible for many of the normal physiological and pathophysiological effects associated with Ang II, including blood pressure regulation, vasoconstriction, cell growth, and extracellular matrix deposition [41,44]. On the other hand, the AT2R is associated with apoptosis and vasodilation [40]. To date the mechanisms that dictate cardiovascular microvessel (coronary arteriole) remodeling are not fully understood.
A role for the RAAS and Ang II in diabetes-induced vasculopathies has been reported in both animal models and humans [2,4,12,14]. Many clinical trials with ACE inhibitors and AT1R blockers (ARBs) have shown that RAAS blockade not only has a protective effect on cardiovascular complications of T2DM, but can also reduce the incidence of novel T2DM onset in hypertensive patients [17,25,46]. For example, ARB treatment in diabetic patients resulted in reduced arterial medial thickening of microvessels obtained from gluteal biopsies [38]. Therefore, in the current study we hypothesized that Ang II-dependent signaling through the AT1R mediates T2DM-induced coronary arteriole remodeling. In order to test this hypothesis, we treated db/db mice with losartan, an ARB, at a sub-pressor dose for 4 weeks and investigated coronary arteriole remodeling via pressure myography.
2. Materials and methods
2.1. Animals
In the pilot study presented in Fig. 1, db/db mice were crossed with mice harboring a global knockout (kindly provided by Dr. LM Harrison-Barnard) to generate heterozygous db/dbAT1Ra+/− mice and double homozygous (db/dbAT1Ra−/−) mice [36]. Please note that this colony was lost during Hurricane Katrina, preventing further experimentation. All other experiments were conducted on 12 or 16 week-old male control, non-diabetic heterozygous (Db/db; BKS.Cg-m+/+Leprdb/J) and male diabetic, homozygous and obese (db/db; BKS.Cg-m+/+Leprdb/J) mice from The Jackson Laboratory (Bar Harbor, ME). These db/db mice exhibit a diabetic phenotype: obese by 4 weeks of age, hyperglycemic by 8 weeks of age, overt DM by 12 weeks of age, as well as hyperlipidemia, obesity, and insulin resistance. Heterozygous mice were used as controls since previous data indicated no morphological or physiological difference from wild type mice (data not shown). All mice were housed under a 12-hour light/dark cycle. Mice were allowed ad libitum access to standard, low-fat chow and water. This study was conducted in accordance with the National Institutes of Health Guidelines, and it was approved by the Institutional Animal Care and Use Committee (IACUC) at Nationwide Children’s Hospital.
Fig. 1.

AT1R blockade and AT1R KO reduce diabetes-induced coronary arteriole remodeling. H&E-stained sections of septal coronary arterioles from 16 week control (A), db/db (B), db/db + candesartan (C), db/dbAT1R+/− (D), and db/dbAT1R−/− (E) mice. Representative of n = 4 mice per group.
2.2. Quantitative real time PCR (qPCR) analysis
Coronary vessel pools were lysed by repeated sonication at 4 °C in Qiazol extraction buffer (Qiagen). Following chloroform extraction and centrifugation, RNA from the aqueous phase was purified using commercially available kits and protocols (Qiagen RNeasy Microarray Tissue mini kit), followed by quantitation using a NanoDrop 2000 (Thermo Scientific). RNA was reverse transcribed using the Maxima™ First Strand cDNA Synthesis Kit and protocol (Thermo Scientific), and equivalent amounts of first strand cDNA were amplified in duplicate for each pool using appropriate Roche Universal Probe/primer pairs for each target gene (Rpl13a [NM_009438]: forward TCCCTGCTGCTC TCAAGG, reverse GCCCCAGGTAAGCAAACTT, average control Ct = 19.3; AT1Ra [NM_177322]: forward GGCTTGAGTCCTGGTCCAC, reverse CAGCCATTTTATACCAATCTTTCA, average control Ct = 28.7; elastin and collagen 1a1 as reported previously [19]) and Maxima Probe qPCR master mix (Thermo Scientific). Amplifications were carried out for 40–45 cycles using an Eppendorf MasterCycler-ep Realplex thermocycler. Parallel amplifications using non-reverse transcribed samples were performed to rule out genomic DNA contamination. Data were analyzed for relative expression using the 2−ΔΔct method, with the ribosomal protein transcript Rpl13a serving as the internal control [37] and the average Het value for the aorta serving as the second normalizer.
2.3. Drug treatment
Control or db/db mice were administered vehicle water or losartan (3 mg/kg/day) (Sigma, 61188), treated water for 4 weeks, beginning at 12 weeks of age. Water bottles were changed 2 times a week. This dose of losartan was chosen based upon a previous report that doses ≤ 10 mg/kg/day in diabetic mice do not affect blood pressure [48].
2.4. Blood glucose, insulin and plasma Ang II measurements
Prior to end point experiments, fasting blood glucose (8–10 hour food withdrawal during the light cycle) was measured from tail vein blood using the Accu-Chek Advantage meter (Roche, Indianapolis, Indiana). Insulin levels were measured using an ELISA kit from Mercodia (Winston-Salem, NC). The provided protocol was followed exactly, with the following exception: All db/db samples were diluted 1:3 with Calibrator 0 solution prior to assay (i.e. 20 μL plasma + 40 μL Calibrator 0). This was done in anticipation of db/db mouse insulin levels being very high. Samples were read on the SpectraMax M5 spectrophotometer. Plasma Ang II concentrations were measured by radioimmunoassay at Hypertension Core lab at Wake Forest University.
2.5. Coronary arteriole isolation and measurement of structural and passive mechanical properties
At the end of treatment (16 weeks) mice were anesthetized using 3% isoflurane, vaporized with 100% oxygen. The heart was excised and dissected in cold physiologic salt solution (PSS). The right ventricle was removed and septal coronary arterioles (<120 μm internal diameter) at the level of the superior papillary muscle were isolated, excised and mounted onto two glass microcannulas within a pressure myograph chamber (Living Systems, Burlington, VT) as previously described by our lab [19]. One vessel was isolated per animal. Prior to any measurements, vessels were equilibrated for 30 min under constant intraluminal pressure (50 mm Hg) at 37 °C in PSS. Internal diameter and left and right wall thickness (WT) were continuously monitored by a video image analyzer (Living Systems) and recorded using LabCart 6 data acquisition software connected to a PowerLab 16/30 (ADInstruments, Inc., Colorado Springs, CO).
All experiments were performed in Ca2+-free PSS in the presence of 2 mM EGTA and 100 μM sodium nitroprusside. A passive pressure–diameter curve was generated by increasing intraluminal pressure from 0 to 125 mm Hg. Coronary wall thickness (WT) and internal diameters (Di) were recorded at each pressure. The following structural and mechanical parameters were calculated as previously described [19]:
External diameter (De) = Di + 2WT
Wall/lumen ratio = (WT/Di) × 100
Cross sectional area , where De is the external diameter
† Growth index = (CSAC − CSAd)/CSAC, where C is control and d is diabetic
Circumferential stress (σ) = (P × Di)/(2WT), where P is pressure in dynes per square centimeter.
Circumferential strain (ε) = (Di − D0)/D0, where Di is the internal diameter for a given intraluminal pressure and D0 is the original diameter measured at 10 mm Hg of intraluminal pressure.
Elastic modulus (E) = stress (σ)/strain (ε) is used to determine arterial stiffness. However, since the stress–strain relationship was non-linear we obtained the tangential or incremental elastic modulus (Einc), or simply the slope of the stress–strain relationship (Δσ/Δε).
2.6. Nuclear staining of coronary arteriole sections
LV septal sections (5 μm) from formalin-fixed hearts (n = 6) were stained with DAPI and an α-smooth muscle actin antibody (Sigma A2547, St. Louis, MO). Cells positive for both DAPI and α-smooth muscle actin were designated VSMCs, counted by two blinded individuals and averaged. Data are expressed as the number of VSMC/medial CSA.
2.7. Coronary blood flow and flow reserve
Coronary blood flow (CBF) was measured non-invasively by high-frequency, high-resolution ultrasound (Vevo2100, Visual Sonics, Toronto, Canada) equipped with a 30 MHz probe, at baseline and under conditions of maximum flow (hyperemia) as described previously [19]. Mice were anesthetized with 2% isoflurane vaporized with 100% oxygen. Following induction, isoflurane was reduced to 1% to determine baseline coronary flow, and then increased to 3% to measure maximal coronary flow. Data were analyzed offline by one observer to eliminate inter-observer variability. CBF was calculated using the equation:
CBF (mL/min) = ((π/4) × D2 × VTI × HR)/1000, where D is the internal coronary diameter (in mm) measured in B-mode ultrasound images, VTI is the velocity–time-integral (in mm), or area under the curve of the Doppler blood flow velocity tracing, and HR is the heart rate.
Coronary flow reserve (CFR) = CBFhyperemia/CBFbaseline where CBFhyperemia is coronary flow measured during 3% isoflurane administration.
2.8. Blood pressure telemetry
Radiotelemetry transmitters (PA-C10, Data Sciences, St. Paul, MN) were implanted into mice as described by our lab [19]. Briefly, mice were anesthetized with 2% isoflurane, and the right carotid artery was isolated and cannulated with a telemeter catheter connected to a radio-telemetry transmitter. Since db/db mice are more sensitive to surgical stressors, data recording commenced after the return of normal diurnal blood pressure rhythms (7–10 days). Data were collected for 10 s every 15 min for a total of 4 weeks using DataQuest A.R.T. 4.2 software.
2.9. Elastin immunofluorescence
Paraffin-embedded heart sections (5 μm thick) from 16-week animals were incubated with an elastin antibody (1:75, ab21610, Abcam, Cambridge, MA) overnight at 4 °C. Slides were then incubated for 1 h at room temperature with an Alexa Fluor 568 conjugated secondary antibody (1:150, A-11036, Life Technologies, Carlsbad, CA). Images were taken at 60× magnification using the same exposure time.
2.10. Capillary density
Paraffin-embedded heart sections (5 μm thick) from 16-week animals were incubated overnight at 4 °C with a CD31 antibody (1:100, sc-1506, Santa Cruz, CA). Secondary antibody, ABC reagents and 3,3′-Diaminobenzidine (DAB) were used to detect CD31 antibody and Hematoxylin (blue) was used to counterstain. Images were taken at 40× magnification and capillaries <15 μm, labeled in brown, were counted and normalized to area by two blinded individuals.
2.11. Statistical analysis
All data are represented as mean ± SEM with a probability of p < 0.05 used for significance. Coronary structural measurements and calculations were analyzed using two way ANOVA followed by a Bonferroni post-test. All other measurements were analyzed either using two way ANOVA followed by a Sidak post-test or by a Student t test. All statistical analyses were conducted using Prism 6.0 (GraphPad, La Jolla, CA).
3. Results
3.1. Angiotensin receptor blocker and AT1R transgenic deletion reduce coronary arteriole remodeling in db/db mice
We performed pilot studies comparing the effectiveness of angiotensin receptor blockade (candesartan) vs. genetic deletion of the AT1R in the db/db background. Control and db/db mice were treated with the AT1R blocker, candesartan cilexetil (1 mg/kg/day) in the drinking water from 10 to 16 weeks. As previously shown [19], morphological analysis of septal coronary arteriole indicated that db/db coronary arterioles (Fig. 1B) displayed an increase in remodeling over controls (Fig. 1A). AT1R blockade (Fig. 1C) and homozygous AT1R deletion (Fig. 1E) attenuated inward hypertrophic remodeling (increased wall thickness, decreased lumen diameter) in db/db mice to a similar extent, while heterozygous AT1R deletion (Fig. 1D) resulted in an intermediate morphological phenotype. Given these data we chose to focus on more clinically relevant pharmacological treatment.
3.2. Differential expression of AT1R between vascular beds
As we have previously shown that aortic vessel beds remodel differently than coronary arterioles in response to diabetes or MetS [19,43], and since AT1R deletion had an impact on morphological analysis of coronary arterioles, we used q-PCR to investigate AT1R expression levels in these beds in both control and db/db mice. Expression levels did not differ significantly between control or db/db mice in either vascular bed; however coronary expression levels of the AT1R were 10- to 20-fold higher than aortic expression levels when normalized to total RNA content (Fig. 2).
Fig. 2.
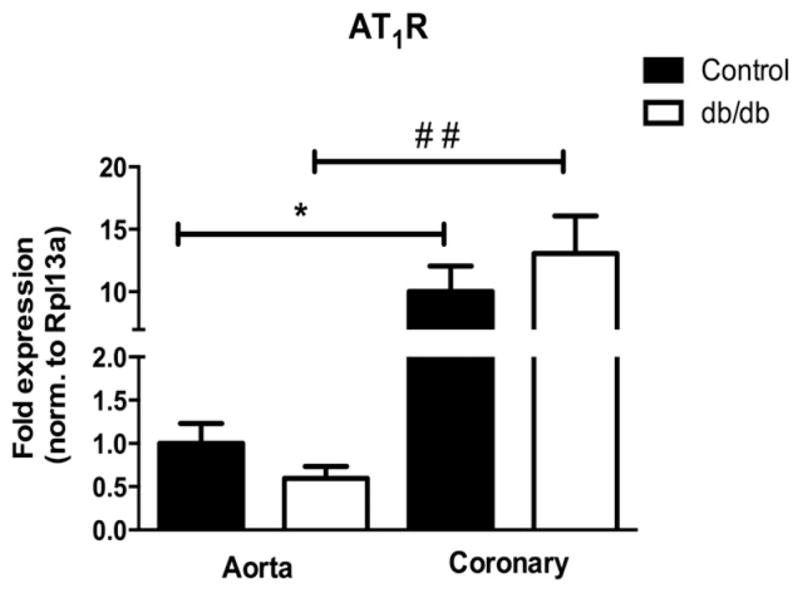
Differential expression of AT1R between vascular beds. mRNA expression was quantified by real-time PCR and normalized to the internal reference Rpl13a. Values are expressed as fold change (control aorta set to 1.0). Values are mean ± SEM, *p < 0.05 to control aorta; ##p < 0.01 to db/db aorta. N = 3 or 4 per group.
3.3. Phenotypic characteristics of losartan-treated control and db/db mice
Body weight, fasting blood glucose, and plasma insulin levels were all significantly increased in 16-week db/db mice with or without losartan treatment when compared to controls (Table 1). To confirm that the dose of losartan used (3 mg/kg/day) did not affect blood pressure, we used telemetry probes to measure blood pressure and heart rate in all treatment groups. No significant differences in blood pressure or heart rate were detected between db/db and control mice, whether or not mice were treated with losartan (Table 1). In addition, plasma Ang II concentrations were not different between control and db/db mice, and Ang II levels were unaffected by losartan treatment.
Table 1.
Body weight, fasting blood glucose levels, plasma insulin, systolic and diastolic blood pressure, heart rate and plasma Ang II.
| Control | db/db | Los Control | Los db/db | |
|---|---|---|---|---|
| Body weight (g) | 31 ± 1 | 47 ± 2 * | 30 ± 1 | 47 ± 2 * |
| Fasting BG (mg/dL) | 129 ± 3 | 499 ± 28 * | 130 ± 4 | 546 ± 22 * |
| Plasma insulin (μg/L) | 0.16 ± 0.03 | 1.89 ± 0.34 * | 0.19 ± 0.03 | 2.19 ± 0.55 *, # |
| SBP (mm Hg) | 128 ± 5 | 128 ± 3 | 123 ± 8 | 127 ± 3 |
| DBP (mm Hg) | 98 ± 5 | 99 ± 4 | 97 ± 4 | 99 ± 4 |
| Heart rate (beats per min) | 435 ± 22 | 469 ± 17 | 454 ± 11 | 464 ± 23 |
| Plasma Ang II (pg/mL) | 359.9 ± 94.5 | 374 ± 110.1 | 253.9 ± 13.8 | 307.4 ± 83.1 |
Body weight, fasting blood glucose levels, systolic and diastolic blood pressure, heart rate and plasma Ang II levels in 16 week control and db/db mice ± 4 week losartan treatment. Values are mean ± SEM;
p < 0.0001 vs. control;
p < 0.05 Los Control vs. Los db/db; n = 24 mice per group for blood glucose and body weight, n = 5 mice per group for blood pressure, n = 7–12 for insulin levels, n = 7 for plasma Ang II levels. BG; blood glucose, SBP; systolic blood pressure, DBP; diastolic blood pressure.
3.4. Angiotensin receptor blockade reduces coronary arteriole inward hypertrophic remodeling
We next used pressure myography to measure passive structural properties of coronary arterioles. Since no differences in structural remodeling (wall thickness, lumen diameter, or medial cross sectional area) were found between control mice and losartan-treated control mice (data not shown) we excluded the losartan control data in order to highlight the differences between db/db mice with and without losartan treatment.
As previously shown [19], db/db mice (n = 10) exhibited inward hypertrophic remodeling as evidenced by reduced lumen diameter, increased wall thickness (control: 6.6 ± 0.4 vs. db/db: 9.2 ± 0.5) and increased wall/lumen ratio (control: 5.4 ± 0.3 vs. db/db: 8.8 ± 0.5) (Fig. 3). Losartan-treated db/db mice (n = 10) reduced the inward hypertrophic remodeling by normalizing wall thickness (Los db/db: 7.1 ± 0.3) and wall/lumen ratio (Los db/db: 6.5 ± 0.3) (Fig. 3). Due to the change in wall thickness and wall/lumen ratio, the medial CSA in losartan-treated db/db mice was not significantly reduced compared to untreated db/db mice and no different than control mice (Fig. 3E).
Fig. 3.

T2DM coronary arteriole passive structural measurements: lumen diameter (A), external diameter (B), wall thickness (C), wall:lumen ratio (D), and medial cross sectional area (E). *p < 0.05; ***p < 0.001 vs. control; ^p < 0.05; ^^p < 0.01 vs. db/db untreated; n = 9–10 per group.
3.5. Losartan attenuates coronary arteriole growth
Growth index, a measure of vessel growth, was increased in db/db mice across a range of pressures, which was reduced by 78% with losartan treatment at 100 mm Hg (Fig. 4A). DAPI and αSMA double-stained coronary arteriole VSMCs normalized to medial CSA were increased in db/db mice as previously published (Fig. 4B) [19]. Losartan-treated db/db mice exhibited a decreased number of VSMCs within the vascular wall when compared to untreated db/db mice (Fig. 4B).
Fig. 4.

Losartan blocked the growth of coronary arterioles in db/db mice. Decreased growth index of isolated coronary arterioles under a range of pressures (A). These indices are calculated as a percent over the averaged control; thus only db/db and Los db/db values are plotted and statistics are not performed. Losartan treatment reduced coronary arteriole cell numbers (cells positive for both alpha smooth muscle actin and DAPI) (B) Values are mean ± SEM, *p < 0.05 vs. control; ^p < 0.05 vs. untreated db/db; n = 3–5.
3.6. Effect of losartan on passive mechanical properties
Next we determined whether the structural remodeling of losartan-treated db/db coronary arterioles was associated with changes in the passive mechanical properties of the vessel wall. In keeping with our previous findings [19], db/db mice displayed a decrease in circumferential wall stress, stress/strain slope, and incremental elastic modulus (Fig. 5A, C–D). Strain was unchanged among all groups (Fig. 5B). Losartan treatment was able to correct the decrease in wall stress seen in the db/db mice (Fig. 5A). Conversely, losartan treatment in db/db mice had no effect on the decreased stiffness in untreated db/db coronary arterioles as measured by the stress–strain curve and the incremental elastic modulus (Fig. 5C & D). The stress–strain curve was shifted further right in losartan-treated db/db mice than the untreated db/db or control mice (Fig. 5C) indicating a less stiff vessel. However, the incremental elastic modulus of elasticity, representing a slope of the stress–strain curve, was significantly lower in db/db coronary arterioles, which was reduced with losartan treatment, suggesting that losartan does not affect coronary arteriole stiffness (Fig. 5D).
Fig. 5.

T2DM coronary arteriole passive mechanical properties: circumferential stress (A), and strain (B) over a range of pressures. Coronary arteriole stress–strain curves (C) and incremental modulus of elasticity (D). *p < 0.05, ***p < 0.001 vs. control; ^p < 0.05 vs. db/db untreated; n = 9–10 per group.
3.7. Expression of extracellular matrix components in losartan-treated coronary arterioles
Since losartan did not have a significant effect on coronary stiffness, we undertook additional experiments to determine a possible mechanism. q-PCR analysis of pooled septal coronary arterioles (n = 6 of 4–5 pooled coronaries) was used to examine the expression of collagen 1 (col1a1) and elastin. Collagen 1 expression was not significantly different between control and db/db mice as previously published [19]; losartan treatment also did not affect collagen 1 expression (Fig. 6). Elastin mRNA expression was increased in db/db mice relative to control mice [19]. Losartan treatment did not have any significant effect on elastin expression (Fig. 6).
Fig. 6.

Differential expression of extracellular matrix components. mRNA expression was quantified by q-PCR and normalized to Rpl13a. Values are relative to control for each target. Values are mean ± SEM, *p < 0.05 relative to control. N = 6 per group.
3.8. Losartan does not affect elastin content
In addition to evaluating the elastin mRNA content in losartan-treated coronaries, we determined the elastin content and organization in coronary vessels. Immunofluorescence staining revealed increased elastin in db/db mice compared to controls (Fig. 7A & B); however, consistent with elastin mRNA expression, losartan treatment did appear to alter the elastin content relative to db/db vessels alone (Fig. 7C). Furthermore, there were no obvious differences in elastin organization in any of the groups.
Fig. 7.

Elastin staining of septal coronary arterioles shows increase in db/db (B) over control (A). Losartan treatment (C) shows no difference in elastin staining compared to db/db mice. Representative of n = 3 per group.
3.9. Losartan improves coronary flow reserve
In order to determine the functional significance of coronary remodeling, we directly measured baseline and maximal (hyperemia) coronary blood flow velocity by non-invasive transthoracic high-frequency pulse wave Doppler imaging. Although no change in baseline coronary flow velocity was observed among the groups, there was a significant decrease in maximal coronary flow velocity in db/db and losartan-treated db/db mice (control: 1.7 ± 0.1 vs. db/db: 0.9 ± 0.05 vs. Los db/db: 1.1 ± 0.1) (Fig. 8A), suggesting vascular impairment at maximum dilation. In addition, untreated db/db mice showed a significantly lower coronary flow reserve (CFR) than untreated control mice (control: 7.3 ± 0.4 vs. db/db: 4.4 ± 0.3) as previously published [19]. In contrast, losartan-treated db/db mice showed an increase in CFR over untreated db/db mice (Los db/db: 6.0 ± 0.7) (Fig. 8B).
Fig. 8.

Losartan increases coronary flow reserve in db/db mice. Coronary blood flow was reduced in db/db mice during hyperemic conditions using 3% isoflurane (A). Coronary flow reserve was reduced in db/db mice. Losartan increased coronary flow reserve (B). ***p < 0.001 from control; ^p < 0.05 vs. db/db untreated; n = 7–8 per group.
3.10. Losartan does not affect capillary density
Chronic diabetes is associated with coronary rarefaction, which would contribute to the decrease in coronary flow reserve in db/db mice. To address this issue, we assessed capillary density in left ventricular tissue sections by immunostaining with an antibody against CD31. Capillaries were defined as those vessels <15 μm in diameter immunostaining brown (Fig. 9A). Capillary counts revealed no significant differences in any groups at this time point (Fig. 9B).
Fig. 9.

LV tissue sections were immunostained with an antibody to CD31 (A). Capillaries (<15 μm), labeled in brown, were counted in 2 fields per slide. Representative of n = 3 per group.
4. Discussion
In this study, we showed for the first time that losartan treatment from 12 to 16 weeks reduces coronary arteriole inward hypertrophic remodeling in db/db mice. This reduced remodeling is associated with an improved CFR but had no effect on coronary arteriole stiffness. These data suggest that losartan treatment causes a distinct pattern of remodeling in the diabetic coronary microvasculature. We chose 12 weeks of age as our time point to start treatment because previous studies in our lab [19] characterized an initial trend towards vessel remodeling at this time point, which then became statistically significant by 16 weeks of age. In addition, at 12 weeks of age, endothelial dysfunction is observed in db/db mice [51], potentially signaling the start of microvascular complications. Also, at 16 weeks of age db/db mice do not exhibit characteristics usually associated with advanced diabetic complications such as overt hypertension, macrovascular remodeling (i.e. atherosclerosis) and capillary rarefaction [26]. Therefore, the 16-week time point in db/db mice likely represents an intermediate stage of disease progression.
The reduced inward remodeling with losartan treatment is consistent with the well-known effects of AT1R blockers in reducing VSMC growth. Two types of VSMC growth have been documented in resistance artery remodeling — hypertrophy and hyperplasia [8,39] Hypertrophic growth involves increased cell size without cell division and is an important component of medial thickening. Hyperplasia results from cell proliferation and is involved in medial thickening, neointima formation and inward remodeling. Several studies have implicated Ang II and oxidative stress as mediators of VSMC growth in T2DM [47]. However, most studies examining VSMC growth have utilized VSMCs isolated from large conduit arteries of non-diabetic animals, rather than VSMCs isolated from coronary resistance arteries of diabetic animals. This is a concern, as there is clearly evidence that VSMCs isolated from resistance arteries are functionally distinct compared to VSMC from conduit arteries [3,9]. In addition, published data from our laboratory indicate that vascular-bed specific remodeling occurs in T2DM mice [19] and Ossabaw swine [43]. The reduction in growth index, and the decrease in VSMC cell number (DAPI-positive nuclei in αSMA positive cells) suggest that reduced Ang II-dependent VSMC hypertrophy/hyperplasia is the most likely mechanism that normalized medial wall thickness in losartan-treated db/db mice. We attempted to measure VSMC proliferation by a variety of methods, including immunostaining of tissue sections, but we were unable to detect any changes (data not shown). In retrospect, immunostaining of cross-sections from a single “snapshot” in time may not be the best way to determine vascular smooth muscle cell proliferation given that cell cycle rates and turnover are extremely slow [34]. For example, a study by McEwan et al. reported that a two-week infusion of a pressor dose of Ang II and BrdU with miniosmotic pumps was necessary to label the proliferating VSMCs [30]. In their study, losartan treatment decreased the number of BrdU-positive cells in intramyocardial coronary arteries. Future studies in our mice will use long-term BrdU infusion in order to determine the contribution of vascular smooth muscle cell proliferation to the observed medial thickening in coronary arterioles.
VSMC growth is a hallmark of increased Ang II signaling in a variety of diseases including atherosclerosis, hypertension and neointimal formation following balloon injury [31]. In diabetes, AT1R dependent VSMC growth has been linked to the JAK/STAT and MAPK signaling pathways [27] since blockade of these pathways prevented Ang II-induced VSMC hypertrophy and proliferation [1,28]. Therefore, our findings of reduced medial thickness associated with a decreased growth index in response to AT1R blockade suggest that these Ang II-dependent signaling pathways are activated under normal conditions in diabetic coronary arterioles. In this study the capacity of losartan to reduce wall thickness likely integrates with vascular mechanics, where increasing coronary arteriole wall thickness reduces wall stress, perhaps to compensate for increased extravascular forces during contraction of a stiffer myocardium.
Since aberrant Ang II is normally associated with pro-fibrotic actions in the vessel wall, we expected losartan treatment to alter coronary stiffness. However, losartan treatment did not alter coronary arteriole wall stiffness in db/db mice — a finding that was unanticipated. One possible explanation could be that we observed no change in elastin mRNA expression or elastin protein content/organization in db/db coronary arterioles treated with losartan. Another explanation for this finding is that ARB treatment has been shown to increase plasma Ang II. It is possible that some of the observed effects of losartan could reflect activation of the AT2R signaling, which has been shown to inhibit VSMC growth and proliferation [24]. However we observed no differences in plasma Ang II levels, although limited tissue availability precluded measurements of tissue Ang II levels. Another possibility is that growth responses via Ang II–AT1R are mediated in part by ERK1/2 MAP kinases, JNK and Akt, while its pro-fibrotic actions are mediated through TGF-β dependent signaling cascades [11]. Therefore, one possibility for the unchanged mechanics is that the Ang II–TGF-β axis is disrupted in db/db coronary arterioles. In preliminary studies, we saw no change in mRNA expression of TGF-β receptor 1 and receptor 2 in coronary arterioles between control and db/db mice with and without losartan treatment (data not shown) although TGF-β expression was undetermined. It is possible that other mediators increased in T2DM (i.e. hyperglycemia and TNF-α) may antagonize the pro-fibrotic actions of Ang II at the level of TGF-β or other signaling pathways. Future studies in coronary arteriole VSMCs are necessary to identify the effects of Ang II on VSMC growth and extracellular matrix production.
Although the beneficial effects of ARB therapy on coronary flow reserve in hypertensive patients are well known, the effects of ARBs in diabetic patients are more equivocal. A study by Kawata et al. reported that ACE inhibition with temocapril, but not the ARB candesartan, improved coronary flow velocity reserve (CFVR) in a small cohort of Japanese diabetic patients [20]. However, it is interesting to note that 6 of 12 patients in the ARB group did show an increase in CFVR. In a study of 14 patients, Kjaer et al. reported no effects of short-term losartan treatment on coronary vasomotor function, although no healthy subjects were included as controls [21]. Conversely, Hinoi et al. reported that the ARB telmisartan improved CFVR in patients with essential hypertension and insulin resistance [15] while Dr. Tune’s laboratory demonstrated that telmisartan increased coronary blood flow, in part by decreasing Ang II-induced vasoconstriction [50]. Collectively, these dissimilar results point to the need for larger prospective trials comparing both traditional (i.e. ACE inhibitors and ARBs) and more novel (e.g. aldosterone) RAAS inhibitors on coronary microvascular structure and function in T2DM.
One limitation of this study is that it is focused solely on one member of one class of RAAS inhibitors, ARBs. It is possible that other classes may affect microvascular remodeling, such as ACE inhibitors and aldosterone antagonists, as they also have beneficial effects on CFVR [18,20]. Moreover, newer generations of ARBs, such as telmisartan, may be more effective than losartan in T2DM patients given its dual action as an ARB and a PPAR-γ agonist. Finally, we chose an intermediate time point in T2DM in order to avoid more chronic complications such as hypertension and severe nephropathy. It is possible that different drug efficacy may be dependent upon the point of administration in the disease progression.
5. Conclusions
The results from this study identify a novel role for AT1R blockade in reducing diabetes-induced coronary arteriole remodeling in type 2 diabetic mice independent of blood pressure lowering. While coronary wall thickness, wall-to-lumen ratio and medial CSA were reduced; stiffness was unchanged with losartan treatment of db/db mice. The reduced inward remodeling was associated with a decrease in growth index. Therefore AT1R mediates in part, coronary arteriole inward hypertrophic remodeling in db/db mice without affecting vascular mechanics. We reported a nearly identical remodeling pattern in db/db mice subjected to aerobic exercise [42], suggesting that angiotensin receptor blockade is equally effective as exercise in reducing coronary microvascular remodeling. Thus, although exercise is deemed the “real polypill” [10], pharmacological therapy may offer several advantages (increased patient compliance, patients unable to exercise) in the clinical diabetic population. Future studies are necessary to determine the molecular mechanisms that account for changes in microvascular remodeling as well as the relative contribution of neurohormonal agents, inflammation, hyperglycemia and insulin resistance.
Acknowledgments
We thank T. Aaron West and Ian L. Sunyecz for their technical expertise. This work was supported by the National Institutes of Health (K99HL116769 to AJT and R01HL056046 to PAL), American Heart Association (13SDG16840035 to AJT), and internal funding from the Research Institute at Nationwide Children’s Hospital (to PAL and AJT).
Footnotes
Calculated from group data, therefore statistical analysis cannot be performed.
References
- 1.Amiri F, Shaw S, Wang X, Tang J, Waller JL, Eaton DC, Marrero MB. Angiotensin II activation of the JAK/STAT pathway in mesangial cells is altered by high glucose. Kidney Int. 2002;61:1605–1616. doi: 10.1046/j.1523-1755.2002.00311.x. [DOI] [PubMed] [Google Scholar]
- 2.Anderson S. Role of local and systemic angiotensin in diabetic renal disease. Kidney Int Suppl. 1997;63:S107–S110. [PubMed] [Google Scholar]
- 3.Archer SL. Diversity of phenotype and function of vascular smooth muscle cells. J Lab Clin Med. 1996;127:524–529. doi: 10.1016/s0022-2143(96)90142-0. [DOI] [PubMed] [Google Scholar]
- 4.Cohn JN, Tognoni G. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med. 2001;345:1667–1675. doi: 10.1056/NEJMoa010713. [DOI] [PubMed] [Google Scholar]
- 5.Deedwania P, Srikanth S. Diabetes and vascular disease. Expert Rev Cardiovasc Ther. 2008;6:127–138. doi: 10.1586/14779072.6.1.127. [DOI] [PubMed] [Google Scholar]
- 6.Deng DX, Tsalenko A, Vailaya A, Ben-Dor A, Kundu R, Estay I, Tabibiazar R, Kincaid R, Yakhini Z, Bruhn L, Quertermous T. Differences in vascular bed disease susceptibility reflect differences in gene expression response to atherogenic stimuli. Circ Res. 2006;98:200–208. doi: 10.1161/01.RES.0000200738.50997.f2. [DOI] [PubMed] [Google Scholar]
- 7.Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev. 2008;88:1009–1086. doi: 10.1152/physrev.00045.2006. [DOI] [PubMed] [Google Scholar]
- 8.Endemann DH, Pu Q, De Ciuceis C, Savoia C, Virdis A, Neves MF, Touyz RM, Schiffrin EL. Persistent remodeling of resistance arteries in type 2 diabetic patients on antihypertensive treatment. Hypertension. 2004;43:399–404. doi: 10.1161/01.HYP.0000112029.03691.e7. [DOI] [PubMed] [Google Scholar]
- 9.Fisher SA. Vascular smooth muscle phenotypic diversity and function. Physiol Genomics. 2010;42A:169–187. doi: 10.1152/physiolgenomics.00111.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiuza-Luces C, Garatachea N, Berger NA, Lucia A. Exercise is the real polypill. Physiology (Bethesda) 2013;28:330–358. doi: 10.1152/physiol.00019.2013. [DOI] [PubMed] [Google Scholar]
- 11.Gao X, He X, Luo B, Peng L, Lin J, Zuo Z. Angiotensin II increases collagen I expression via transforming growth factor-beta1 and extracellular signal-regulated kinase in cardiac flbroblasts. Eur J Pharmacol. 2009;606:115–120. doi: 10.1016/j.ejphar.2008.12.049. [DOI] [PubMed] [Google Scholar]
- 12.Hayashi T, Sohmiya K, Ukimura A, Endoh S, Mori T, Shimomura H, Okabe M, Terasaki F, Kitaura Y. Angiotensin II receptor blockade prevents microangiopathy and preserves diastolic function in the diabetic rat heart. Heart. 2003;89:1236–1242. doi: 10.1136/heart.89.10.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He Z, King GL. Microvascular complications of diabetes. Endocrinol Metab Clin N Am. 2004;33(215–238):xi–xii. doi: 10.1016/j.ecl.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 14.Heeneman S, Sluimer JC, Daemen MJ. Angiotensin-converting enzyme and vascular remodeling. Circ Res. 2007;101:441–454. doi: 10.1161/CIRCRESAHA.107.148338. [DOI] [PubMed] [Google Scholar]
- 15.Hinoi T, Tomohiro Y, Kajiwara S, Matsuo S, Fujimoto Y, Yamamoto S, Shichijo T, Ono T. Telmisartan, an angiotensin II type 1 receptor blocker, improves coronary microcirculation and insulin resistance among essential hypertensive patients without left ventricular hypertrophy. Hypertens Res. 2008;31:615–622. doi: 10.1291/hypres.31.615. [DOI] [PubMed] [Google Scholar]
- 16.Hogan P, Dall T, Nikolov P. Economic costs of diabetes in the US in 2002. Diabetes Care. 2003;26:917–932. doi: 10.2337/diacare.26.3.917. [DOI] [PubMed] [Google Scholar]
- 17.Investigators, H.O.P.E.S. Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Lancet. 2000;355:253–259. [PubMed] [Google Scholar]
- 18.Joffe HV, Kwong RY, Gerhard-Herman MD, Rice C, Feldman K, Adler GK. Beneficial effects of eplerenone versus hydrochlorothiazide on coronary circulatory function in patients with diabetes mellitus. J Clin Endocrinol Metab. 2007;92:2552–2558. doi: 10.1210/jc.2007-0393. [DOI] [PubMed] [Google Scholar]
- 19.Katz PS, Trask AJ, Souza-Smith FM, Hutchinson KR, Galantowicz ML, Lord KC, Stewart JA, Jr, Cismowski MJ, Varner KJ, Lucchesi PA. Coronary arterioles in type 2 diabetic (db/db) mice undergo a distinct pattern of remodeling associated with decreased vessel stiffness. Basic Res Cardiol. 2011;106:1123–1134. doi: 10.1007/s00395-011-0201-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawata T, Daimon M, Hasegawa R, Teramoto K, Toyoda T, Sekine T, Yamamoto K, Uchida D, Himi T, Yoshida K, Komuro I. Effect on coronary flow velocity reserve in patients with type 2 diabetes mellitus: comparison between angiotensin-converting enzyme inhibitor and angiotensin II type 1 receptor antagonist. Am Heart J. 2006;151:798 e799–715. doi: 10.1016/j.ahj.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 21.Kjaer A, Kristoffersen US, Tarnow L, Parving HH, Hesse B. Short-term oral treatment with the angiotensin II receptor antagonist losartan does not improve coronary vasomotor function in asymptomatic type 2 diabetes patients. Diabetes Res Clin Pract. 2009;84:34–38. doi: 10.1016/j.diabres.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 22.Krentz AJ, Clough G, Byrne CD. Interactions between microvascular and macrovascular disease in diabetes: pathophysiology and therapeutic implications. Diabetes Obes Metab. 2007;9:781–791. doi: 10.1111/j.1463-1326.2007.00670.x. [DOI] [PubMed] [Google Scholar]
- 23.Laing SP, Swerdlow AJ, Slater SD, Burden AC, Morris A, Waugh NR, Gatling W, Bingley PJ, Patterson CC. Mortality from heart disease in a cohort of 23,000 patients with insulin-treated diabetes. Diabetologia. 2003;46:760–765. doi: 10.1007/s00125-003-1116-6. [DOI] [PubMed] [Google Scholar]
- 24.Lemarie CA, Schiffrin EL. The angiotensin II type 2 receptor in cardiovascular disease. J Renin-Angiotensin-Aldosterone Syst. 2010;11:19–31. doi: 10.1177/1470320309347785. [DOI] [PubMed] [Google Scholar]
- 25.Lindholm LH, Ibsen H, Dahlof B, Devereux RB, Beevers G, de Faire U, Fyhrquist F, Julius S, Kjeldsen SE, Kristiansson K, Lederballe-Pedersen O, Nieminen MS, Omvik P, Oparil S, Wedel H, Aurup P, Edelman J, Snapinn S. Cardiovascular morbidity and mortality in patients with diabetes in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:1004–1010. doi: 10.1016/S0140-6736(02)08090-X. [DOI] [PubMed] [Google Scholar]
- 26.Marin P, Andersson B, Krotkiewski M, Bjorntorp P. Muscle fiber composition and capillary density in women and men with NIDDM. Diabetes Care. 1994;17:382–386. doi: 10.2337/diacare.17.5.382. [DOI] [PubMed] [Google Scholar]
- 27.Marrero MB, Fulton D, Stepp D, Stern DM. Angiotensin II-induced signaling pathways in diabetes. Curr Diabetes Rev. 2005;1:197–202. doi: 10.2174/1573399054022802. [DOI] [PubMed] [Google Scholar]
- 28.Marrero MB, Schieffer B, Li B, Sun J, Harp JB, Ling BN. Role of Janus kinase/signal transducer and activator of transcription and mitogen-activated protein kinase cascades in angiotensin II- and platelet-derived growth factor-induced vascular smooth muscle cell proliferation. J Biol Chem. 1997;272:24684–24690. doi: 10.1074/jbc.272.39.24684. [DOI] [PubMed] [Google Scholar]
- 29.Marso SP, Stern DM. Diabetes and Cardiovascular Disease: Integrating Science and Clinical Medicine. Lippincott Williams & Wilkins; Philadelphia: 2004. [Google Scholar]
- 30.McEwan PE, Gray GA, Sherry L, Webb DJ, Kenyon CJ. Differential effects of angiotensin II on cardiac cell proliferation and intramyocardial perivascular fibrosis in vivo. Circulation. 1998;98:2765–2773. doi: 10.1161/01.cir.98.24.2765. [DOI] [PubMed] [Google Scholar]
- 31.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 32.Methe H, Balcells M, del Alegret MC, Santacana M, Molins B, Hamik A, Jain MK, Edelman ER. Vascular bed origin dictates flow pattern regulation of endothelial adhesion molecule expression. Am J Physiol Heart Circ Physiol. 2007;292:H2167–H2175. doi: 10.1152/ajpheart.00403.2006. [DOI] [PubMed] [Google Scholar]
- 33.Nahser PJ, Jr, Brown RE, Oskarsson H, Winniford MD, Rossen JD. Maximal coronary flow reserve and metabolic coronary vasodilation in patients with diabetes mellitus. Circulation. 1995;91:635–640. doi: 10.1161/01.cir.91.3.635. [DOI] [PubMed] [Google Scholar]
- 34.Neese RA, Misell LM, Turner S, Chu A, Kim J, Cesar D, Hoh R, Antelo F, Strawford A, McCune JM, Christiansen M, Hellerstein MK. Measurement in vivo of proliferation rates of slow turnover cells by 2H2O labeling of the deoxyribose moiety of DNA. Proc Natl Acad Sci U S A. 2002;99:15345–15350. doi: 10.1073/pnas.232551499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nitenberg A, Valensi P, Sachs R, Dali M, Aptecar E, Attali JR. Impairment of coronary vascular reserve and ACh-induced coronary vasodilation in diabetic patients with angiographically normal coronary arteries and normal left ventricular systolic function. Diabetes. 1993;42:1017–1025. doi: 10.2337/diab.42.7.1017. [DOI] [PubMed] [Google Scholar]
- 36.Park S, Bivona BJ, Harrison-Bernard LM. Compromised renal microvascular reactivity of angiotensin type 1 double null mice. Am J Physiol Ren Physiol. 2007;293:F60–F67. doi: 10.1152/ajprenal.00049.2007. [DOI] [PubMed] [Google Scholar]
- 37.Ragni E, Vigano M, Rebulla P, Giordano R, Lazzari L. What is beyond a qRT-PCR study on mesenchymal stem cell differentiation properties: how to choose the most reliable housekeeping genes. J Cell Mol Med. 2013;17:168–180. doi: 10.1111/j.1582-4934.2012.01660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schiffrin EL, Park JB, Intengan HD, Touyz RM. Correction of arterial structure and endothelial dysfunction in human essential hypertension by the angiotensin receptor antagonist losartan. Circulation. 2000;101:1653–1659. doi: 10.1161/01.cir.101.14.1653. [DOI] [PubMed] [Google Scholar]
- 39.Schofield I, Malik R, Izzard A, Austin C, Heagerty A. Vascular structural and functional changes in type 2 diabetes mellitus: evidence for the roles of abnormal myogenic responsiveness and dyslipidemia. Circulation. 2002;106:3037–3043. doi: 10.1161/01.cir.0000041432.80615.a5. [DOI] [PubMed] [Google Scholar]
- 40.Siragy HM. The role of the AT2 receptor in hypertension. Am J Hypertens. 2000;13:62S–67S. doi: 10.1016/s0895-7061(00)00274-0. [DOI] [PubMed] [Google Scholar]
- 41.Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52:639–672. [PubMed] [Google Scholar]
- 42.Trask AJ, Delbin MA, Katz PS, Zanesco A, Lucchesi PA. Differential coronary resistance microvessel remodeling between type 1 and type 2 diabetic mice: impact of exercise training. Vasc Pharmacol. 2012;57:187–193. doi: 10.1016/j.vph.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trask AJ, Katz PS, Kelly AP, Galantowicz ML, Cismowski MJ, West TA, Neeb ZP, Berwick ZC, Goodwill AG, Alloosh M, Tune JD, Sturek M, Lucchesi PA. Dynamic micro- and macrovascular remodeling in coronary circulation of obese Ossabaw pigs with metabolic syndrome. J Appl Physiol (1985) 2012;113:1128–1140. doi: 10.1152/japplphysiol.00604.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tunon J, Ruiz-Ortega M, Egido J. Regulation of matrix proteins and impact on vascular structure. Curr Hypertens Rep. 2000;2:106–113. doi: 10.1007/s11906-000-0067-2. [DOI] [PubMed] [Google Scholar]
- 45.Vavuranakis M, Stefanadis C, Triandaphyllidi E, Toutouzas K, Toutouzas P. Coronary artery distensibility in diabetic patients with simultaneous measurements of luminal area and intracoronary pressure: evidence of impaired reactivity to nitro-glycerin. J Am Coll Cardiol. 1999;34:1075–1081. doi: 10.1016/s0735-1097(99)00331-9. [DOI] [PubMed] [Google Scholar]
- 46.Weir MR. Effects of renin–angiotensin system inhibition on end-organ protection: can we do better? Clin Ther. 2007;29:1803–1824. doi: 10.1016/j.clinthera.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 47.Wolf G. Growth factors and the development of diabetic nephropathy. Curr Diabetes Rep. 2003;3:485–490. doi: 10.1007/s11892-003-0012-2. [DOI] [PubMed] [Google Scholar]
- 48.Yang J, Tan Y, Zhao F, Ma Z, Wang Y, Zheng S, Epstein PN, Yu J, Yin X, Zheng Y, Li X, Miao L, Cai L. Angiotensin II plays a critical role in diabetic pulmonary fibrosis most likely via activation of NADPH oxidase-mediated nitrosative damage. Am J Physiol Endocrinol Metab. 2011;301:E132–E144. doi: 10.1152/ajpendo.00629.2010. [DOI] [PubMed] [Google Scholar]
- 49.Yokoyama I, Momomura S, Ohtake T, Yonekura K, Nishikawa J, Sasaki Y, Omata M. Reduced myocardial flow reserve in non-insulin-dependent diabetes mellitus. J Am Coll Cardiol. 1997;30:1472–1477. doi: 10.1016/s0735-1097(97)00327-6. [DOI] [PubMed] [Google Scholar]
- 50.Zhang C, Knudson JD, Setty S, Araiza A, Dincer UD, Kuo L, Tune JD. Coronary arteriolar vasoconstriction to angiotensin II is augmented in prediabetic metabolic syndrome via activation of AT1 receptors. Am J Physiol Heart Circ Physiol. 2005;288:H2154–H2162. doi: 10.1152/ajpheart.00987.2004. [DOI] [PubMed] [Google Scholar]
- 51.Zhang H, Park Y, Zhang C. Coronary and aortic endothelial function affected by feedback between adiponectin and tumor necrosis factor alpha in type 2 diabetic mice. Arterioscler Thromb Vasc Biol. 2010;30:2156–2163. doi: 10.1161/ATVBAHA.110.214700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zoneraich S. Small-vessel disease, coronary artery vasodilator reserve, and diabetic cardiomyopathy. Chest. 1988;94:5–7. doi: 10.1378/chest.94.1.5. [DOI] [PubMed] [Google Scholar]