

Abstract
We have shown previously that bacterial lipopolysaccharide (LPS)-mediated suppression of Phospholipase-Cβ-2 (PLCβ-2) expression is involved in M1 (inflammatory) to M2-like (wound healing) phenotypic switching of macrophages triggered by adenosine. This suppression is mediated post-transcriptionally by destabilization of PLCβ-2 mRNA. To investigate the mechanism of this LPS-mediated destabilization, we examined the roles of RNA-binding agents including microRNAs and RNA-binding proteins that are involved in regulating stability of mRNAs encoding growth factors, inflammatory mediators and proto-oncogenes. Adenylate and Uridylate (AU)-rich elements (AREs) in 3′UTRs are specific recognition sites for RNA-binding proteins including Tristetraprolin (TTP), HuR and AUF1, and for microRNAs that are involved in regulating mRNA stability. In this study, we investigated the role of TTP and AREs in regulating PLCβ-2 mRNA stability. The 3′UTR of the PLCβ-2 gene was inserted into the pLightswitch luciferase reporter plasmid and transfected into RAW264.7 cells. LPS suppressed Luciferase expression from this reporter. Luciferase expression from mutant 3′UTR constructs lacking AREs was similarly down-regulated, suggesting that these regions are not required for LPS-mediated suppression of PLCβ-2. TTP was rapidly upregulated in both primary murine macrophages and RAW264.7 cells in response to LPS. Suppression of PLCβ-2 by LPS was examined using macrophages from mice lacking TTP. LPS suppressed PLCβ-2 expression to the same extent in wild type and TTP−/− macrophages. Also, the rate of decay of PLCβ-2 mRNA in LPS-treated macrophages following transcriptional blockade was similar in wild type and TTP−/− macrophages, clearly indicating that TTP is not involved in LPS-mediated destabilization of PLCβ-2 mRNA in macrophages.
Keywords: Macrophages, Phospholipase-Cβ-2, 3′UTR, AU-rich elements, Tristetraprolin, Lipopolysaccharide (LPS)
BACKGROUND
Macrophages are tissue resident cells of myeloid origin. Macrophages take up residence in various organs and tissues during embryogenesis, and are maintained throughout life in the absence of injury or inflammation by a slow rate of cell division. In response to injury, there is a rapid influx of monocytes from the blood, which then differentiate locally into macrophages. Tissue resident macrophages participate in immune responses by killing pathogens, by producing cytokines that mediate inflammation, by producing cytokines and growth factors that regulate the tissue response to injury, and by producing angiogenic factors that regulate blood vessel growth and vascular permeability. Macrophages may exert pro-inflammatory, anti-inflammatory and wound healing phenotypes, and this phenotypic flexibility is regulated by the response of macrophages to micro-environmental stimuli that sculpt macrophage gene expression towards different polarization states.
Macrophages may be ‘classically activated’ (M1) or ‘alternatively activated’ (M2), depending upon the stimuli that they encounter. 1–8 M1 activation is induced by stimulation with IFNγ and/or LPS, is pro-inflammatory, and characterized by the expression of IL-1, IL-6, IL-12, TNFα and iNOS. M2 activation is anti-inflammatory. While the original definition of M2 activation involved induction by IL-4 or IL-13 through the IL4Rα, 3, 9–14 other pathways that induce M2-like phenotypes that are independent of IL-4Rα have been described. M2 activation is characterized by elevated expression of VEGF and IL-10, and low expression of TNFα and IL-12. Studies from our lab have described an IL-4 and IL-3 independent mode of converting M1 to M2-like macrophages that is induced by the metabolite adenosine and involves the adenosine A2A and A2B receptors. 15–21 We have shown previously that endotoxin (LPS) rapidly and specifically suppresses PLCβ-2 expression at the post-transcriptional level by destabilizing its mRNA, and that this suppression plays a role in the adenosine A2A receptor-mediated switch of macrophages from an inflammatory to an angiogenic phenotype. 1 The mechanism of destabilization of PLCβ-2 mRNA, however, is not yet known.
Tristetraprolin (TTP), also known as ZFP36, Nup475, G0S24, and TIS11, is the founding member of a small family of proteins containing tandem CCCH zinc fingers. The ZFP36 gene encodes a proline-rich, zinc finger protein of ~36 kDa with three repeats of the PPPP-motif thus giving it its common name tristetraprolin or TTP. 22 TTP has the ability to bind to AREs in the 3′UTRs of mRNAs, and target the bound mRNA for rapid degradation.23–25 TTP can negatively regulate the expression of a number of critical genes frequently overexpressed in inflammation and cancer.26 The role of TTP as a key player in promoting decay of ARE-containing mRNAs was previously established.23, 24 TTP was shown to be an immediate-early response gene induced in various cell types in response to phorbol ester, insulin, serum, and other mitogenic stimuli. 22, 27–29 TTP knockout mice exhibit a severe syndrome of growth retardation, cachexia, arthritis, inflammation and autoimmunity. 30 Most aspects of this syndrome are, however, abrogated when these mice are treated with anti-TNF antibodies right after birth. Known to target TNF-α, IL-10, GM-CSF, IL-6 mRNAs, TTP expression has also been shown to be strongly induced upon LPS treatment of RAW264.7 cells, thus making it a likely candidate for a role in the LPS-mediated PLCβ-2 mRNA destabilization in macrophages.31, 32 In this study, we investigated the role of both TTP and conserved 3′UTR AREs in the LPS-mediated destabilization of PLCβ-2 mRNA. We show here that neither TTP nor the two conserved AREs in the 3′UTR of the PLCβ2 mRNA are required for the LPS-mediated destabilization in macrophages.
MATERIALS AND METHODS
Chemicals and Reagents
Phenol extracted Escherichia coli LPS (a TLR 4 agonist free of TLR 2 agonists) was kindly provided by Dr. Stephanie Vogel (University of Maryland). For Western blotting of TTP, an antiserum provided by Dr. P. Blackshear was used that was developed against a recombinant mouse TTP-maltose binding protein fusion protein, as described previously. 33 Plasmid transfections were performed using the transfection reagent LipoD 293 from Signagen laboratories. All antibodies and plasmids were stored at −20°C.
Animals
C57BL/6J mice were purchased from Jackson laboratories and housed in the New Jersey Medical School Animal Facility. RNA from mice genetically engineered to lack the TTP gene (TTP−/− mice) was provided by Dr. Perry Blackshear (National Institute of Environmental Health Sciences, Research Triangle Park, NC). All animal procedures were reviewed and approved by the New Jersey Medical School IACUC.
Preparation of Macrophages
Mice were injected intra-peritoneally with 2.5 ml sterile Brewer’s thioglycolate broth (4% w/v, DIFCO, Detroit, MI). After four days, the mice were sacrificed by cervical dislocation, injected intraperitoneally with 3 ml ice-cold sterile phosphate buffered saline (PBS), and their peritoneal area was gently massaged. Mice were dissected to expose their peritoneal cavity, and their thioglycolate-induced terminally-differentiated peritoneal macrophages were harvested using a sterile cotton plugged Pasteur pipette. Cells were collected in a sterile polypropylene conical tube and kept on ice. Cells were then centrifuged at 300 g at 4°C for 5 minutes to form a cell pellet. They were washed twice with PBS and resuspended in RPMI 1640 (SIGMA) supplemented with 10% heat inactivated fetal bovine serum (FBS, Serum Source International), 2mM L-Glutamine (Sigma), 100μg/ml streptomycin and 100 IU/ml penicillin (Sigma)(RPMI-15%FBS) at a concentration of 1 × 106 cells/ml. Macrophages were plated at a density of 0.125 × 106 cells/cm2 in 6-well, 60 or 100 mm plates (Cell Treat Scientific Products, Shirley, MA) and incubated in 5% CO2/95% atmosphere humidified chamber at 37°C for 3 hour to promote adherence. Non-adherent cells were removed by washing with RPMI-10%FCS, followed by overnight incubation in the same medium. The medium was then changed to RPMI-1%FCS for treatment with LPS.
Cell Culture
RAW264.7 cells, a macrophage-like cell line, were obtained from American Type Culture Collection (ATCC TIB71, Manassas, VA) and maintained in RPMI 1640 medium supplemented with 10% heat inactivated FBS, 100μg/ml Streptomycin and 100 IU/ml penicillin. For treatment with LPS, cells were plated at a concentration of 0.125 × 106 cells/cm2 in 6-well, 60 mm, or 100mm plates, and incubated in 5% CO2/95% air in a humidified chamber overnight at 37°C. Cells were then treated in 1% FBS-RPMI medium alone (control) or in 1% medium containing the various test reagents.
Construction of a PLCβ-2 3′UTR Luciferase Reporter Plasmid
The 3′UTR of PLCβ-2 was cloned into the MCS of the pLightswitch_3′UTR empty vector (Switchgear Genomics) utilizing their custom cloning services. 5′ and 3′ flanking sequences (less than 100bp on either end) were added by Switchgear Genomics making the total length of the 3′UTR 1647 bp and the size of the total plasmid 5557 bp. Insertion of the PLCβ-2 3′UTR into the MCS of the empty plasmid gave rise to a hybrid transcript that contained the luciferase coding sequence driven by the RPL10 promoter fused to the PLCβ-2 3′UTR. This plasmid was termed pRPL10-luc-PLCβ-2-3′UTR. To obtain endotoxin free DNA for transfections, plasmids were prepared using an Endo free plasmid maxiprep kit (Sigma).
Construction of PLCβ-2 3′UTR mutant luciferase reporter plasmids
Three mutant plasmids were created from the pRPL10-luc-PLCβ-2-3′UTR plasmid. Two were created by deleting either of the two AREs individually, and the third had both AREs deleted. The mutant plasmids were named depending upon the position or the number of AREs deleted; pRPL10-luc-PLCβ-2-3′UTR-ΔP-ARE (proximal-ARE deleted), pRPL10-luc-PLCβ-2-3′UTR-ΔD-ARE (distal-ARE deleted), pRPL10-luc-PLCβ-2-3′UTR-Δ2-ARE (both AREs deleted). The deletions were engineered using GENEART Site-Directed Mutagenesis System (Life Technologies). Deletion primers were designed according to the manufacturer’s protocol, and comprised complementary upper and lower primers with appropriate number of base-pairs before and after the deletions. Touchdown PCR was performed, followed by isolation of the PCR product and consequent recombination and transformation into DH5α T1 bacterial cells.
Transient Transfections of RAW264.7 Cells with pRPL10-luc-PLCβ-2-3′UTR, pRPL10-luc-GAPDH-3′UTR, and with pRPL10-luc-mutant-3′UTR plasmids
RAW264.7 cells were transiently transfected with pRPL10-luc-PLCβ-2-3′UTR and pRPL10-luc-GAPDH-3′UTR (control) using the transfection reagent LipoD 293 (Signagen Labs) as follows. RAW264.7 cells were sub-cultured 24 hours before the transfection and incubated in RPMI1640-10%FBS in 100 mm dishes. RAW264.7 cells were then bulk transfected with these constructs overnight, followed by re-plating in 6-well plates with equal numbers of cells in each well the following morning. The cells were given 6–7 hours to attach in the 6-well plates. They were then treated with LPS (100ng/ml) at various time points (3h, 6h, 12h, 24h) in triplicates. One set of triplicates was left untreated to provide the constitutive luciferase expression. The samples from each well were harvested at 24 hours in 1.5ml Eppendorf tubes in 500μl of 1X Passive Lysis Buffer (Promega), vortexed for 30 seconds and centrifuged at 16,000g for 1 minute. The supernatant was transferred to fresh Eppendorf tubes and luciferase assays were performed using equal volumes of lysates and 1X Renilla Glo substrate (Promega). 1X Passive Lysis Buffer that was used to harvest the samples was used as a blank for luciferase measurements. For the luciferase assay, 10 minutes of dark adaptation and 10 seconds integration time per sample were applied.
RAW264.7 cells were then transiently transfected with pRPL10-luc-PLCβ-2-3′UTR (control), pRPL10-luc-PLCβ-2-3′UTR-ΔP-ARE, pRPL10-luc-PLCβ-2-3′UTR-ΔD-ARE, and pRPL10-luc-PLCβ-2-3′UTR-Δ2-ARE using the transfection reagent LipoD 293 (Signagen Labs) as follows: RAW264.7 cells were sub-cultured for 24 hours before transfection and incubated in RPMI1640-10% FBS in 100 mm dishes. RAW264.7 cells were then bulk transfected with these constructs overnight, followed by re-plating in 6-well plates with equal numbers of cells in each well. After 24 hours, cells were treated with LPS for 6 hours. The samples from each well were harvested in 1.5ml Eppendorf tubes in 500μl of 1X Passive Lysis Buffer (Promega), vortexed for 30 seconds and centrifuged at 16,000g for 1 minute. The supernatants were transferred to fresh Eppendorf tubes and luciferase assays were performed using equal volumes of lysates and 1X Renilla Glo substrate (Promega). 1X Passive Lysis Buffer that was used to harvest the samples was used as a blank for luciferase measurements. For the luciferase assay, 10 minutes of dark adaptation and 10 seconds integration time per sample were applied.
Western Blot Analyses
After LPS treatment, 10 × 106 murine peritoneal macrophages or RAW264.7 cells in 100mm dishes were washed twice with ice cold PBS, scraped into 1ml RIPA buffer containing Protease Inhibitor Cocktail and collected in a 1.5ml Eppendorf tube followed by passage through a 21 gauge needle (x10) to fragment the DNA. Samples were then incubated on ice for 30 minutes and centrifuged at 10,000g for 10 minutes. An aliquot of each sample was used for a Bradford Protein Assay (Bio-Rad Labs. Inc). Samples were then stored at −80°C until required for Western Blotting. For Western Blotting, cell lysates were boiled for 5 minutes with SDS-Laemmli buffer and aliquots containing 50 μg of protein were loaded onto 10% SDS-polyacrylamide gels for electrophoresis, along with an aliquot of the Precision Plus Kaleidoscope Molecular Weight marker (Bio-Rad Labs) Gels were run at 100V (constant voltage), followed by electrophoretic transfer of cell proteins onto nitrocellulose membrane using wet transfer. Transfer buffer was composed of 48mM Tris, 39mM glycine, and 20% methanol in 1 liter of distilled water and was conducted at 100mAmp (constant current) overnight at 4°C. The nitrocellulose membrane was then blocked with 5% milk in TTBS (tris buffered saline with 0.1 % tween 20 for 1 hour followed by incubation with TTP antibody diluted 1:1000 in blocking solution overnight at 4°C. The blot was washed with TTBS(x3) for 10 minutes each and then incubated with alkaline phosphatase-conjugated secondary IgG antibody (goat anti-rabbit IgG-HRP, Santa Cruz Biotechnology, Inc. sc 2004) for one hour (1:5000). Immunoreactive bands were developed using a chemiluminescent substrate ECL Plus (Amersham Biosciences), and an image was obtained using a Typhoon Imager 9400 (Amersham Biosciences). Band densities were then analyzed using ImageJ software (NIH).
Determination of the Rate of PLCβ-2 mRNA Decay in Macrophages
Macrophages were isolated from WT and TTP−/− C57Bl/6J mice and plated in 60 mm dishes. Following overnight incubation, cells were treated with LPS for various times. RNA was isolated using TRIzol (Invitrogen Corporation) according to the manufacturer’s protocol. RNA concentrations and quality were measured using the Nanodrop Spectrophotometer. RNA samples were reverse transcribed to synthesize cDNA in 50μl volume containing final concentrations of the following components:
1μg total RNA, 1X Taqman RT buffer, 5.5mM MgCl2, 2mM dNTP mix, 0.4U/μl RNAse inhibitor, random hexamers, 50U/μl Moloney Murine Leukemia Virus (MuLV) Multiscribe™ Reverse Transcriptase in RNAse-free water. Q-RT-PCR was performed using Taqman Technology using an ABI 7500 Real-time PCR system (Applied Biosystems). For each sample, mRNA levels were normalized to that of the endogenous housekeeping gene cyclophilin D since it was determined earlier that the expression of this gene does not change significantly as a result of the treatments used in our experiments. Both test and housekeeping Q-RT-PCR reactions contained final concentrations of the following components: 5μl of the cDNA synthesized in the RT step, 15μl of 2X Taqman Universal Master Mix, and 1X target assay master-mix (primers and probes for the gene to be assayed) dissolved in RNAse free water to make up the 30 μl reaction volume. Reaction tubes were placed in the ABI 7500 Real-time PCR cycler under the following PCR thermal cycling conditions: 95°C for 10 minutes; 40 cycles at [95°C (denaturation) for 15 seconds and 60°C for 60 seconds]. PLCβ-2 primers and probes were designed using Applied Biosystems Primer Express 2.0 software and synthesized by the RBHS Molecular Research Facility. 5′ ends of the probes were labeled with the fluorescent dye 6-carboxyfluorescence (FAM), and 3′ ends were coupled to the quencher molecule, Blackhole Quencher TM dye-1 (BHQ-1). The sequences of the primers and probes used for Q-RT-PCR of PLCβ-2 are:
Forward Primer 5′-CTGCCCTCGGTGCTTGTC-3′
Reverse Primer 5′-TTGTACACGGATTCTGGGAAGTC-3′
Probe 5′-FAM-TTGCAAAAGGCAAAAATGATGCTATCAACC-BHQ-3′
The PCR step was performed using an ABI-PRISM Sequence Detector System 7500. Results were normalized against the cyclophilin-D transcript as an internal control, and were used to calculate expression levels using the ΔΔCt method.
Determination of PLCβ-2 mRNA stability in Wild Type vs TTP−/− Macrophages Following Transcriptional Blockade
Macrophages were isolated from WT and TTP−/− C57Bl/6J mice as described above. Macrophages were treated with actinomycin-D (5μg/ml) for 2 hours, and then with LPS or control medium in the continued presence of actinomycin_D for various periods of time (30, 60, 90 and 120 mins). RNA was isolated, reverse transcribed and analyzed by Q-RT-PCR as described above.
Statistical Analyses
Data are presented as means ± SEM. The number of experiments performed and analyzed is indicated in the corresponding figure legends. Statistical differences between mean values were determined by one-way analysis of variance, followed by Student’s t test.
RESULTS
Bio-informatic analysis of the PLCβ-2 3′UTR reveals that putative AREs, the most common determinants of mRNA stability, are present
Since the 3′UTRs of mRNAs are implicated in the regulation of their stability, it was important to investigate the role of the PLCβ-2 3′UTR in the LPS-mediated mRNA destabilization. Bio-informatic analysis of the PLCβ-2 3′UTR revealed that it is 1481 bp long, and harbors two potential AU–rich elements (AREs). AREs are adenylate-uridylate-rich elements that serve as binding sites for various RNA-binding proteins such as TTP, HuR, and AUF1, which are involved in regulating RNA stability. One ARE present in the PLCβ-2 3′UTR is the canonical pentamer AUUUA. The other has two overlapping pentamers (Figure 1A). Investigation of conservation of these AREs in the PLCβ-2 3′UTR using Target Scan 5.2 showed that they are strongly conserved in several mammalian species (Figure 1B), suggesting that they might play an important role in regulating PLCβ-2 mRNA stability.
Figure 1. Bioinformatic Analysis of PLCβ-2 3′UTR.

A. The PLCβ-2 3′UTR (GenBank NM_177568.3) is 1481 bp long, and harbors two AU–rich elements (AREs) shown in bold. One is the canonical pentamer AUUUA, and the other has two overlapping pentamers. AREs are putative binding sites for RNA-binding proteins such as, TTP, HuR and AUF1 which are involved in regulation of mRNA stability. B. Analysis using TargetScan 7.1 shows that the second ARE is conserved across several mammalian species. Dashes indicate where sequences do not match.
3′UTR reporter assays reveal the involvement of PLCβ-2 3′UTR in its destabilization by LPS treatment
To test the role of PLCβ-2 3′UTR in destabilization of the PLCβ-2 mRNA by LPS, a luciferase reporter assay was performed using two different plasmid constructs; one, in which the PLCβ-2 3′UTR was downstream of the luciferase open reading frame, and the other which had the GAPDH 3′UTR downstream of luciferase open reading frame to serve as a control. In both these constructs, the luciferase open reading frame was under the control of the constitutive RPL10 promoter. The RPL10 gene encodes a ribosomal protein that is a component of the 60S subunit and this promoter showed only minimal regulation by LPS (data not shown). RAW264.7 cells were bulk transfected with these constructs overnight followed by re-plating in 6 well plates. The next day, cells were then treated in triplicate with LPS for various time-points (3, 6, 12 and 24 hours). One set of triplicates was left untreated to provide the constitutive luciferase expression. Samples were then harvested, and luciferase assays were performed using equal volumes of the lysates and Renilla Glo. As shown in Figure 2, Luciferase expression was significantly suppressed by 3h post-LPS treatment and reached its lowest point at 6h, followed by an increase at 12h and a return to constitutive expression levels of the untreated samples by 24 hours post-LPS treatment. This LPS-induced suppression of luciferase expression strongly suggests that the PLCβ-2 3′UTR is involved in the LPS-mediated down-regulation of PLCβ-2 mRNA expression.
Figure 2. PLCβ-2-3′UTR Regulation of Luciferase Reporter Gene Expression by LPS.

To test the role of PLCβ-2 3′UTR in the destabilization of PLCβ-2 mRNA by LPS, a luciferase reporter assay was performed using two different plasmid constructs. The first contained the PLCβ-2 3′UTR downstream of the luciferase gene, and the other the GAPDH 3′UTR down-stream of the luciferase gene to serve as control. RAW264.7 cells were transfected with these constructs and treated with LPS for various time-periods. One set of wells was left untreated to provide constitutive luciferase expression. All samples were harvested at 24 hours and luciferase assays were performed using equal volumes of lysates and Renilla Glo. The results show the relative expression of luciferase from the PLCβ-2 3′UTR construct versus that of the GAPDH 3′UTR construct, and are the means ± S.D. of two separate experiments, each performed in triplicate. (* P_ <0.01 in comparison to Untreated Control)
LPS-mediated PLCβ-2 mRNA destabilization takes place by an ARE-independent mechanism
Once the involvement of PLCβ-2 3′UTR in its destabilization was shown, the role of the 3′UTR AREs was tested. Luciferase reporter assays were performed using pRPL10-luc-PLCβ-2-3′UTR (control), and three mutant 3′UTR plasmids: pRPL10-luc-PLCβ-2-3′UTR-ΔP-ARE, pRPL10-luc-PLCβ-2-3′UTR-ΔD-ARE, and pRPL10-luc-PLCβ-2-3′UTR-Δ2-ARE. The first two mutant vectors were missing the proximal and the distal AREs respectively, and the third mutant vector was missing both the AREs. As shown in Figure 3, the degree of suppression of luciferase expression upon LPS treatment from cells transfected with mutant plasmids was not significantly lower than that of cells transfected with the control plasmid pRPL10-luc-PLCβ-2-3′UTR. LPS suppression of luciferase expression observed in cells transfected with the double mutant plasmid pRPL10-luc-PLCβ-2-3′UTR-Δ2-ARE, was in fact, slightly stronger than that of controls. This experiment shows that LPS-mediated PLCβ-2 mRNA destabilization does not require the AREs present in its 3′UTR, and must be mediated by an ARE-independent mechanism.
Figure 3. Effect of Deletions of Proximal and Distal AREs in the PLCβ-2-3′UTR on the LPS-mediated suppression of luciferase Expression.
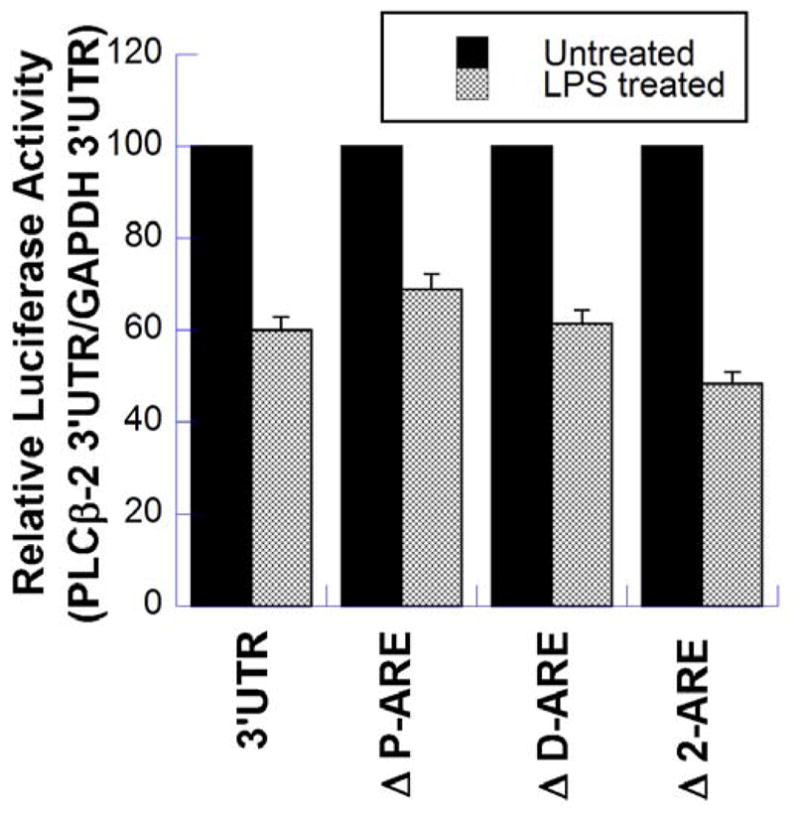
Luciferase reporter assays were performed using pRPL10-luc-PLCβ-2-3′UTR (control), and three mutant 3′UTR plasmids; pRPL10-luc-PLCβ-2-3′UTR (ΔP-ARE) which lacks the proximal ARE, pRPL10-luc-PLCβ-2-3′UTR (ΔD-ARE) which lacks the distal ARE, and pRPL10-luc-PLCβ-2-3′UTR (Δ2-ARE) which lacks both AREs. RAW264.7 cells were transfected with these constructs overnight, followed by re-plating in 6-well plates. After 24 hours, the cells were treated with LPS for 6 hours. One set of wells was left untreated to provide the constitutive luciferase expression. All samples were harvested at 24 hours and luciferase assays were performed using equal volumes of lysates and Renilla Glo. The results are the means ± S.D. of at least two separate experiments, each performed in triplicate.
LPS induces expression of TTP in RAW264.7 cells and murine peritoneal macrophages
Prior studies have shown that LPS strongly induces the expression of TTP in RAW264.7 cells and bone marrow derived macrophages. 33 We confirmed this LPS induction of TTP in RAW264.7 cells (Figures 4A). We then studied TTP expression in murine peritoneal macrophages. Macrophages from C57Bl/6J mice were treated with LPS (100 ng/ml), or with RPMI-1% FBS alone as control (C) for various times. Following treatment, the medium was removed and cells were solubilized in SDS-polyacrylamide gel (SDS-PAGE) electrophoresis sample buffer. The lysates (50 μg protein/sample) were subjected to Western blot analysis as described above, and probed with TTP antibody. An antibody to GAPDH was used as a loading control. LPS strongly induced TTP expression in murine peritoneal macrophages (Figure 4B). Elevated TTP levels were observed within 1h of treatment and peaked by 8h. TTP levels were still elevated after 24h. As expected, TTP was not expressed by macrophages from TTP−/− mice (data not shown). These data indicate that TTP is strongly induced in macrophages by LPS at both the mRNA and protein levels, and that TTP−/− mice provide a useful tool to study the effects of TTP depletion in the destabilization of PLCβ-2 mRNA by LPS.
Figure 4. LPS induces Expression of TTP in RAW264.7 Cells and Murine Peritoneal Macrophages (MPMs).

RAW264.7 cells and MPMs from C57Bl/6J mice were treated with LPS (100ng/ml), or with RPMI-1% FBS alone as control (C) for various time periods. Following treatment, media were removed, cells were solubilized in SDS-polyacrylamide gel electrophoresis sample buffer and lysates (50μg protein/sample) were Western-blotted with TTP antibody. GAPDH was used as a loading control. Each experiment was performed twice with similar results. A typical Western blot for each cell type is shown. A: RAW264.7 cells. B: Mouse peritoneal macrophages. Band intensities were quantified by using ImageQuant analysis program (GE Health care).
LPS suppresses PLCβ-2 mRNA expression in macrophages from TTP−/− mice
If TTP were responsible for LPS-mediated PLCβ-2 mRNA destabilization, macrophages from TTP knockout mice (TTP−/− mice) would be expected not to exhibit LPS-mediated suppression of PLCβ-2 mRNA expression. To investigate the role of TTP in LPS-mediated PLCβ-2 mRNA destabilization, PLCβ-2 mRNA levels from macrophages prepared from TTP−/− mice were determined using Q-RT-PCR. Following treatment of macrophages with LPS for 1.5, 4, and 8 hours, RNA was extracted from each sample, and analyzed by Q-RT-PCR. mRNA levels were normalized to those of endogenous cyclophilin-D as a housekeeping gene. As shown in Figure 5A, LPS suppressed PLCβ-2 mRNA in TTP−/− macrophages to the same extent as in wild type macrophages. As the steady state levels of PLCβ-2 mRNA are a balance between mRNA synthesis by transcription and mRNA degradation, we also determined the rate of mRNA degradation by inhibiting transcription with actinomycin-D. Macrophages from wild-type and TTP−/− mice were treated with actinomycin-D for 2 hours, followed by treatment with LPS in the presence of actinomycin-D. The half-life (t1//2) of PLCβ-2 mRNA in untreated wild type and TTP−/− macrophages was >12 hours. In both wild type and TTP−/− macrophages, LPS reduced PLCβ-2 mRNA t1//2 to 2–3 hrs (Figure 5B). These data clearly indicate that TTP is not required for the LPS-mediated down-regulation of PLCβ-2 mRNA, and suggest that other TTP-independent mechanisms must mediate the LPS-dependent destabilization of the PLCβ-2 mRNA.
Figure 5. Stability of PLCβ-2 mRNA in Macrophages from WT and TTP−/− Mice.

A. PLCβ-2 mRNA levels in mouse peritoneal macrophages from wild type and TTP−/− mice were determined by Q-RT-PCR following treatment of macrophages with LPS (100ng/ml) for 0, 1.5, 4 and 8 hours. RNA was extracted from each sample and analyzed by Q-RT-PCR. mRNA levels were normalized to those of endogenous cyclophilin-D as a house-keeping gene. The results are the means ± S.D. of at two separate experiments, each performed in triplicate.
B. Macrophages from wild-type and TTP−/− mice were treated with actinomycin-D for 2 hours, followed by treatment with LPS in the presence of actinomycin-D. PLCβ-2 mRNA levels were determined as described in A. above.
DISCUSSION
Previous studies in our lab have shown that the LPS mediated suppression of PLCβ-2 expression is an important step in the adenosine A2A-receptor dependent switch of M1 macrophages to an M2d (M2-like) phenotype in response to adenosine stimulation.1 This suppression is post-transcriptional and takes place by destabilization of PLCβ-2 mRNA. This study led us to investigate further the mechanisms involved in this LPS-mediated PLCβ-2 mRNA destabilization. The interplay between the rate of gene transcription, RNA transport, and mRNA decay determines the cytoplasmic level of an mRNA. mRNA degradation allows for the elimination of anomalous messages in addition to modulating mRNA pools in response to an ever-changing cellular environment. 34, 35 The regulation of mRNA stability in mammalian cells is one of the most powerful and rapid means of altering protein expression and maintaining cellular homeostasis. 36
Several potential mechanisms that might mediate mRNA decay. These include 3′UTR sequence elements, as well as non-3′UTR mediated processes. 37–42,43, 44,45, 46,47,48,49,50 The most common and best studied 3′UTR sequence elements involved in the regulation of mRNA stability in eukaryotic cells in response to inflammatory stimuli are the AU-rich elements or AREs found in mRNA 3′UTRs. Transcripts encoding numerous cytokines, proto-oncogenes, and transcription factors have AREs in their 3′UTRs. 51 A number of trans-acting factors in the form of ARE- Binding Proteins have been identified, including AU-rich Binding Factor-1, Tristetraprolin (TTP), KH splicing regulatory protein (KSRP), embryonic lethal abnormal vision (ELAV) proteins, and TIA-1 and TIA-1-related protein (TIAR). The commonality between their modus-operandi is the ultimate recruitment of the mRNA-decay machinery. TTP can negatively regulate the expression of a number of critical genes frequently overexpressed in inflammation and cancer. 52–61 TTP is known to target TNF-α, IL-10, GM-CSF, IL-6 mRNAs, and its expression has also been shown to be strongly induced by LPS treatment of RAW264.7 cells.31 This suggests that TTP might be an important candidate to be investigated for its role in LPS-mediated PLCβ-2 mRNA destabilization in macrophages.
Bio-informatic analysis of the PLCβ-2 3′UTR shows two AREs that are strongly conserved across mammalian species. In this study, we used luciferase reporter assays with the PLCβ-2 3′UTR to study the role of this 3′UTR in the stability of the mRNA. RAW264.7 cells were transfected with the complete 3′UTR luciferase construct and treated with LPS. The 3′UTR of the housekeeping gene GAPDH, which lacks AREs and is not regulated by LPS, was used as control. Suppression of luciferase expression by >50% was observed with the PLCβ-2 3′UTR construct in comparison to the GAPDH 3′UTR construct. This clearly demonstrates that the 3′UTR of PLCβ-2 is involved in the LPS-mediated destabilization of PLCβ-2 mRNA. To study the role of the 3′UTR AREs, site directed mutagenesis was used to delete the AREs. LPS was found to suppress luciferase expression from the mutated constructs to the same extent as in the non-mutated 3′UTR construct. These results strongly suggest that the destabilization of the mRNA is not mediated by an ARE-dependent mechanism.
Western Blot analysis showed that LPS strongly and rapidly induced the expression of TTP in murine peritoneal macrophages, in addition to confirming the previously reported LPS-mediated up-regulation of TTP in RAW264.7 cells. TTP levels were low in unstimulated macrophages, and were rapidly and strongly induced in response to LPS. Peak TTP levels were observed 4–8 hours following LPS treatment and were still markedly elevated after 24 hours. To determine the possible role of TTP in the regulation of PLCβ-2 mRNA stability, macrophages from mice genetically engineered to lack the TTP gene (TTP−/− mice) were prepared. PLCβ-2 mRNA expression in response to LPS was determined in wild type versus TTP−/− macrophages. LPS suppressed PLCβ-2 mRNA expression in TTP−/− macrophages to the same degree and with similar kinetics to that observed in wild type macrophages. In addition, direct measurement of PLCβ-2 mRNA stability in LPS treated macrophages from TTP−/− vs wild type mice where transcription was blocked clearly indicated that the stability of the PLCβ-2 mRNA was not affected by the lack of TTP (Figure). These results clearly indicate that TTP does not play an essential role in the destabilization of PLCβ-2 mRNA by LPS.
In summary, this study shows first: that the destabilization of PLCβ-2 mRNA in macrophages in response to LPS is not mediated through the conserved AREs in the 3′UTR of the PLCβ-2 mRNA; and second, that TTP, an important regulator of mRNA stability of many genes, is not required for the LPS-mediated destabilization of the PLCβ-2 mRNA. We are continuing our studies into the mechanism of this LPS-mediated destabilization of PLCβ-2 mRNA.
Acknowledgments
This study was supported in part by a grant from the US Public Health Services to SJL (R21-AI097731), a Grant-In-Aid from the American Heart Association to CSL (15GRNT23240019), and in part by the intramural research program of the NIH, NIEHS (PJB).
Abbreviations
- ARE
AU-rich element
- FBS
Fetal bovine serum
- GAPDH
Glyceraldehyde dehydrogenase
- HRP
Horseradish peroxidase
- Ifnγ
Interferon-gamma
- IL
Interleukin
- LPS
Lipopolysaccharide
- mRNA
messenger ribonucleic acid
- PBS
Phosphate buffered saline
- PLC
Phospholipase
- Q-RT-PCR
Quantitative Real Time Polymerase Chain Reaction
- RIPA
Radio immunoprecipitation assay
- RT
Reverse transcriptase
- SDS
Sodium dodecyl sulfate
- TNF
Tumor necrosis factor
- TTP
Tristetraprolin
- UTR
Untranslated region
References
- 1.Grinberg S, Hasko G, Wu D, Leibovich SJ. Suppression of PLCβ2 by endotoxin plays a role in the adenosine A(2A) receptor-mediated switch of macrophages from an inflammatory to an angiogenic phenotype. Am J Pathol. 2009;175:2439–53. doi: 10.2353/ajpath.2009.090290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goerdt S, Politz O, Schledzewski K, Birk R, Gratchev A, Guillot P, Hakiy N, Klemke CD, Dippel E, Kodelja V, Orfanos CE. Alternative versus classical activation of macrophages. Pathobiology. 1999;67:222–6. doi: 10.1159/000028096. [DOI] [PubMed] [Google Scholar]
- 3.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 4.Mantovani A, Bottazzi B, Sozzani S, Peri G, Allavena P, Dong QG, Vecchi A, Colotta F. Cytokine regulation of tumour-associated macrophages. Res Immunol. 1993;144:280–3. doi: 10.1016/0923-2494(93)80108-b. [DOI] [PubMed] [Google Scholar]
- 5.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 6.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature Revs Immunol. 2008;8:958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tietzel I, Mosser DM. The modulation of macrophage activation by tyrosine phosphorylation. Fronts Biosci. 2002;7:d1494–502. doi: 10.2741/tietzel. [DOI] [PubMed] [Google Scholar]
- 8.Zhang X, Mosser DM. Macrophage activation by endogenous danger signals. J Pathol. 2008;214:161–78. doi: 10.1002/path.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, Puel A, Biswas SK, Moshous D, Picard C, Jais JP, D’Cruz D, Casanova JL, Trouillet C, Geissmann F. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33:375–86. doi: 10.1016/j.immuni.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 11.Leibovich SJ, Chen JF, Pinhal-Enfield G, Belem PC, Elson G, Rosania A, Ramanathan M, Montesinos C, Jacobson M, Schwarzschild MA, Fink JS, Cronstein B. Synergistic up-regulation of vascular endothelial growth factor expression in murine macrophages by adenosine A(2A) receptor agonists and endotoxin. Am J Pathol. 2002;160:2231–44. doi: 10.1016/S0002-9440(10)61170-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biswas SK, Sica A, Lewis CE. Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J Immunol. 2008;180:2011–7. doi: 10.4049/jimmunol.180.4.2011. [DOI] [PubMed] [Google Scholar]
- 13.Varin A, Mukhopadhyay S, Herbein G, Gordon S. Alternative activation of macrophages by IL-4 impairs phagocytosis of pathogens but potentiates microbial-induced signalling and cytokine secretion. Blood. 2010;115:353–62. doi: 10.1182/blood-2009-08-236711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 15.Pinhal-Enfield G, Ramanathan M, Hasko G, Vogel SN, Salzman AL, Boons GJ, Leibovich SJ. An angiogenic switch in macrophages involving synergy between Toll-like receptors 2, 4, 7 and 9 and adenosine A2A receptors. Am J Pathol. 2003;163:711–21. doi: 10.1016/S0002-9440(10)63698-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Csoka B, Nemeth ZH, Virag L, Gergely P, Leibovich SJ, Pacher P, Sun CX, Blackburn MR, Vizi ES, Deitch EA, Hasko G. A2A adenosine receptors and C/EBPbeta are crucially required for IL-10 production by macrophages exposed to Escherichia coli. Blood. 2007;110:2685–95. doi: 10.1182/blood-2007-01-065870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Csoka B, Selmeczy Z, Koscso B, Nemeth ZH, Pacher P, Murray PJ, Kepka-Lenhart D, Morris SM, Jr, Gause WC, Leibovich SJ, Hasko G. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 26:376–86. doi: 10.1096/fj.11-190934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elson G, Eisenberg M, Garg C, Outram S, Ferrante CJ, Hasko G, Leibovich SJ. Induction of murine adenosine A(2A) receptor expression by LPS. analysis of the 5′ upstream promoter. Genes Immunol. 2013 doi: 10.1038/gene.2012.60. [DOI] [PubMed] [Google Scholar]
- 19.Ferrante CJ, Leibovich SJ. Regulation of macrophage polarization and wound healing. Adv in Wound Care. 2012;1:10–6. doi: 10.1089/wound.2011.0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrante CJ, Pinhal-Enfield G, Elson G, Cronstein BN, Hasko G, Outram S, Leibovich SJ. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Ralpha) signaling. Inflammation. 2013;36:921–31. doi: 10.1007/s10753-013-9621-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leibovich SJ, Chen JF, Pinhal-Enfield G, Belem PC, Elson G, Rosania A, Ramanathan M, Montesinos C, Jacobson M, Schwarzschild MA, Fink JS, Cronstein B. Synergistic up-regulation of vascular endothelial growth factor expression in murine macrophages by adenosine A2A receptor agonists and endotoxin. Am J Pathol. 2002;160:2231–44. doi: 10.1016/S0002-9440(10)61170-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lai WS, Stumpo DJ, Blackshear PJ. Rapid insulin-stimulated accumulation of an mRNA encoding a proline-rich protein. J Biol Chem. 1990;265:16556–63. [PubMed] [Google Scholar]
- 23.Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol Cell Biol. 1999;19:4311–23. doi: 10.1128/mcb.19.6.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai WS, Carballo E, Thorn JM, Kennington EA, Blackshear PJ. Interactions of CCCH zinc finger proteins with mRNA. Binding of tristetraprolin-related zinc finger proteins to Au-rich elements and destabilization of mRNA. J Biol Chem. 2000;275:17827–37. doi: 10.1074/jbc.M001696200. [DOI] [PubMed] [Google Scholar]
- 25.Brooks SA, Blackshear PJ. Tristetraprolin (TTP): interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochim Biophys Acta. 2013;1829:666–79. doi: 10.1016/j.bbagrm.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanduja S, Blanco FF, Young LE, Kaza V, Dixon DA. The role of tristetraprolin in cancer and inflammation. Front Biosci. 2012;17:174–88. doi: 10.2741/3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DuBois RN, McLane MW, Ryder K, Lau LF, Nathans D. A growth factor-inducible nuclear protein with a novel cysteine/histidine repetitive sequence. J Biol Chem. 1990;265:19185–91. [PubMed] [Google Scholar]
- 28.Ma Q, Herschman HR. A corrected sequence for the predicted protein from the mitogen-inducible TIS11 primary response gene. Oncogene. 1991;6:1277–8. [PubMed] [Google Scholar]
- 29.Varnum BC, Lim RW, Sukhatme VP, Herschman HR. Nucleotide sequence of a cDNA encoding TIS11, a message induced in Swiss 3T3 cells by the tumor promoter tetradecanoyl phorbol acetate. Oncogene. 1989;4:119–20. [PubMed] [Google Scholar]
- 30.Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. 1996;4:445–54. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 31.Tchen CR, Brook M, Saklatvala J, Clark AR. The stability of tristetraprolin mRNA is regulated by mitogen-activated protein kinase p38 and by tristetraprolin itself. J Biol Chem. 2004;279:32393–400. doi: 10.1074/jbc.M402059200. [DOI] [PubMed] [Google Scholar]
- 32.Cao H, Tuttle JS, Blackshear PJ. Immunological characterization of tristetraprolin as a low abundance, inducible, stable cytosolic protein. J Biol Chem. 2004;279:21489–99. doi: 10.1074/jbc.M400900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perez-Ortin JE, Alepuz P, Chavez S, Choder M. Eukaryotic mRNA decay: methodologies, pathways, and links to other stages of gene expression. J Mol Biol. 2013;425:3750–75. doi: 10.1016/j.jmb.2013.02.029. [DOI] [PubMed] [Google Scholar]
- 34.Schoenberg DR, Maquat LE. Regulation of cytoplasmic mRNA decay. Nature Revs Genetics. 2012;13:246–59. doi: 10.1038/nrg3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen ZJ, Malter JS. Regulation of AU-Rich Element RNA Binding Proteins by Phosphorylation and the Prolyl Isomerase Pin1. Biomolecules. 2015;5:412–34. doi: 10.3390/biom5020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Houseley J, LaCava J, Tollervey D. RNA-quality control by the exosome. Nature Revs Mol Cell biol. 2006;7:529–39. doi: 10.1038/nrm1964. [DOI] [PubMed] [Google Scholar]
- 37.Liu H, Rodgers ND, Jiao X, Kiledjian M. The scavenger mRNA decapping enzyme DcpS is a member of the HIT family of pyrophosphatases. EMBO J. 2002;21:4699–708. doi: 10.1093/emboj/cdf448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moser MJ, Holley WR, Chatterjee A, Mian IS. The proofreading domain of Escherichia coli DNA polymerase I and other DNA and/or RNA exonuclease domains. Nucleic acids Res. 1997;25:5110–8. doi: 10.1093/nar/25.24.5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fenger-Gron M, Fillman C, Norrild B, Lykke-Andersen J. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell. 2005;20:905–15. doi: 10.1016/j.molcel.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 40.Steiger M, Carr-Schmid A, Schwartz DC, Kiledjian M, Parker R. Analysis of recombinant yeast decapping enzyme. RNA. 2003;9:231–8. doi: 10.1261/rna.2151403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu JH, Yang WH, Gulick T, Bloch KD, Bloch DB. Ge-1 is a central component of the mammalian cytoplasmic mRNA processing body. RNA. 2005;11:1795–802. doi: 10.1261/rna.2142405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Badis G, Saveanu C, Fromont-Racine M, Jacquier A. Targeted mRNA degradation by deadenylation-independent decapping. Mol Cell. 2004;15:5–15. doi: 10.1016/j.molcel.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 43.Muhlrad D, Parker R. The yeast EDC1 mRNA undergoes deadenylation-independent decapping stimulated by Not2p, Not4p, and Not5p. EMBO J. 2005;24:1033–45. doi: 10.1038/sj.emboj.7600560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gatfield D, Izaurralde E. Nonsense-mediated messenger RNA decay is initiated by endonucleolytic cleavage in Drosophila. Nature. 2004;429:575–8. doi: 10.1038/nature02559. [DOI] [PubMed] [Google Scholar]
- 45.Stevens A, Wang Y, Bremer K, Zhang J, Hoepfner R, Antoniou M, Schoenberg DR, Maquat LE. Beta -Globin mRNA decay in erythroid cells: UG site-preferred endonucleolytic cleavage that is augmented by a premature termination codon. Proc Nat Acad Sci USA. 2002;99:12741–6. doi: 10.1073/pnas.192442399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stoecklin G, Lu M, Rattenbacher B, Moroni C. A constitutive decay element promotes tumor necrosis factor alpha mRNA degradation via an AU-rich element-independent pathway. Mol Cell Biol. 2003;23:3506–15. doi: 10.1128/MCB.23.10.3506-3515.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moraes KC, Wilusz CJ, Wilusz J. CUG-BP binds to RNA substrates and recruits PARN deadenylase. RNA. 2006;12:1084–91. doi: 10.1261/rna.59606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pierrat B, Lacroute F, Losson R. The 5′ untranslated region of the PPR1 regulatory gene dictates rapid mRNA decay in yeast. Gene. 1993;131:43–51. doi: 10.1016/0378-1119(93)90667-r. [DOI] [PubMed] [Google Scholar]
- 49.Herrick D, Jacobson A. A coding region segment is necessary, but not sufficient for rapid decay of the HIS3 mRNA in yeast. Gene. 1992;114:35–41. doi: 10.1016/0378-1119(92)90704-s. [DOI] [PubMed] [Google Scholar]
- 50.Khabar KS. The AU-rich transcriptome: more than interferons and cytokines, and its role in disease. J Interferon Cytokine research: the official journal of the International Society for Interferon and Cytokine Res. 2005;25:1–10. doi: 10.1089/jir.2005.25.1. [DOI] [PubMed] [Google Scholar]
- 51.Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science. 1998;281:1001–5. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 52.Carballo E, Lai WS, Blackshear PJ. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood. 2000;95:1891–9. [PubMed] [Google Scholar]
- 53.Douni E, Akassoglou K, Alexopoulou L, Georgopoulos S, Haralambous S, Hill S, Kassiotis G, Kontoyiannis D, Pasparakis M, Plows D, Probert L, Kollias G. Transgenic and knockout analyses of the role of TNF in immune regulation and disease pathogenesis. J Inflammation. 1995;47:27–38. [PubMed] [Google Scholar]
- 54.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10:4025–31. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Phillips K, Kedersha N, Shen L, Blackshear PJ, Anderson P. Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor alpha, cyclooxygenase 2, and inflammatory arthritis. Proc Nat Acad Sci USA. 2004;101:2011–6. doi: 10.1073/pnas.0400148101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suswam E, Li Y, Zhang X, Gillespie GY, Li X, Shacka JJ, Lu L, Zheng L, King PH. Tristetraprolin down-regulates interleukin-8 and vascular endothelial growth factor in malignant glioma cells. Cancer Res. 2008;68:674–82. doi: 10.1158/0008-5472.CAN-07-2751. [DOI] [PubMed] [Google Scholar]
- 57.Brennan SE, Kuwano Y, Alkharouf N, Blackshear PJ, Gorospe M, Wilson GM. The mRNA-destabilizing protein tristetraprolin is suppressed in many cancers, altering tumorigenic phenotypes and patient prognosis. Cancer Res. 2009;69:5168–76. doi: 10.1158/0008-5472.CAN-08-4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gebeshuber CA, Zatloukal K, Martinez J. miR-29a suppresses tristetraprolin, which is a regulator of epithelial polarity and metastasis. EMBO Reps. 2009;10:400–5. doi: 10.1038/embor.2009.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carrick DM, Blackshear PJ. Comparative expression of tristetraprolin (TTP) family member transcripts in normal human tissues and cancer cell lines. Arch Biochem Biophys. 2007;462:278–85. doi: 10.1016/j.abb.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 60.Sanduja S, Kaza V, Dixon DA. The mRNA decay factor tristetraprolin (TTP) induces senescence in human papillomavirus-transformed cervical cancer cells by targeting E6-AP ubiquitin ligase. Aging. 2009;1:803–17. doi: 10.18632/aging.100086. [DOI] [PMC free article] [PubMed] [Google Scholar]