

Abstract
INTRODUCTION
Aneuploidy, also known as somatic copy number alterations (SCNAs), is widespread in human cancers and has been proposed to drive tumorigenesis. The relationship between SCNAs and the characteristic functional features or “hallmarks” of cancer is not well understood. Among these cancer hallmarks is immune evasion, which is accomplished by neoantigen editing, defects in antigen presentation and inhibition of tumor infiltration, and/or cytotoxic activities of immune cells. Whether and how tumor SCNA levels influence immune evasion is of particular interest as this information could potentially be used to improve the efficacy of immune checkpoint blockade, a therapy that has produced durable responses in a subset of cancer patients.
RATIONALE
Understanding how SCNAs and mutation load affect tumor evolution, and through what mechanisms, is a key objective in cancer research. To explore the relationships between SCNA levels, tumor mutations, and cancer hallmarks, we examined data from 5255 tumor/normal samples representing 12 cancer types from The Cancer Genome Atlas project. We assigned each tumor an SCNA score and looked for correlations with the number and types of tumor mutations. We also compared the gene expression profiles of tumors with high versus low SCNA levels to identify differences in cellular signaling pathways.
RESULTS
First, we found that, for most tumors, there was a positive correlation between SCNA levels and the total number of mutations. Second, tumors harboring activating oncogenic mutations in the receptor tyrosine kinase–RAS–phosphatidylinositol 3-kinase pathway showed fewer SCNAs, a finding at odds with the hypothesis of oncogene-driven genomic instability. Third, we found that tumors with high levels of SCNAs showed elevated expression of cell cycle and cell proliferation markers (cell cycle signature) and reduced expression of markers for cytotoxic immune cell infiltrates (immune signature). The increased expression level of the cell cycle signature was primarily predicted by focal SCNAs, with a lesser contribution of arm and whole-chromosome SCNAs. In contrast, the lower expression level of the immune signature was primarily predicted by high levels of arm and whole-chromosome SCNAs. SCNA levels were a stronger predictor of markers of cytotoxic immune cell infiltration than tumor mutational load. Finally, through analysis of data from two published clinical trials of immunotherapy in melanoma patients, we found that high SCNA levels in tumors correlated with poorer survival of patients. The combination of the tumor SCNA score and the tumor mutational load was a better predictor of survival after immunotherapy than either biomarker alone.
CONCLUSION
We found that two hallmarks of cancer, cell proliferation and immune evasion, are predicted by distinct types of aneuploidy that likely act through distinct mechanisms. Proliferation markers mainly correlated with focal SCNAs, implying a mechanism related to the action of specific genes targeted by these SCNAs. Immune evasion markers mainly correlated with arm- and chromosome-level SCNAs, consistent with a mechanism related to general gene dosage imbalance rather than the action of specific genes. A retrospective analysis of melanoma patients treated with immune checkpoint blockade anti–CTLA-4 (cytotoxic T lymphocyte–associated protein 4) therapy revealed that high SCNA levels were associated with a poorer response, suggesting that tumor aneuploidy might be a useful biomarker for predicting which patients are most likely to benefit from this therapy.
Graphical abstract
Genetic events associated with two cancer hallmarks: cell proliferation and immune evasion. Across several human tumor types, high SCNA levels correlate with increased expression of cell cycle markers and decreased expression of markers of cytotoxic immune cell infiltrates. A high load of tumor neoantigens (reflecting a high level of point mutations) promotes the detection of tumors by the immune system, limiting immune evasion. The relative contribution of focal, arm/chromosome, and neoantigen load to the prediction of proliferation and immune evasion is shown.
Many human solid tumors are characterized by aneuploidy, the presence of an abnormal number of chromosomes (1). Additional somatic copy number alterations (SCNAs), known as segmental aneuploidies, are also widespread in tumors; these include chromosome arm and focal SCNAs smaller than a chromosome arm (2, 3). There is increasing evidence that aneuploidy is a driving force in tumorigenesis (4–9). We recently showed that the recurrent patterns of focal and arm/chromosome SCNAs can, in part, be explained by the distribution and potency of cancer driver genes as predicted by their point mutational patterns in sporadic tumors (10, 11). An abnormally high number of chromosomes are associated with high-grade tumors and poor prognosis (12–15). Paradoxically, experimentally induced aneuploidy is known to have negative effects on cellular physiology, including higher basal proteotoxic stress (16–20). It is unknown whether the deleterious effects of aneuploidy on protein homeostasis are simply outweighed by protumorigenic gene dosage effects or whether they might even have a distinct benefit in the context of tumorigenesis.
Immune evasion is an important cancer hallmark and has long been recognized as a fundamental process in tumor formation and progression (21, 22). Cancer immune evasion occurs through the selection of tumor variants that become resistant to an immune attack primarily mediated by T cells and natural killer (NK) cells (21, 22). One approach to overcome this evasion is to block the inhibitory molecules that the immune system naturally uses to restrain excessive immune responses. These inhibitory molecules represent the basis for immune checkpoint blockade therapy (23). Antibody-mediated inactivation of inhibitory molecules, such as cytotoxic T lymphocyte–associated protein 4 (CTLA-4) and programmed death-ligand 1 (PD-L1), has produced durable responses in a subset (20 to 30%) of patients with advanced tumors (23).
The identification of biomarkers that predict patient response to CTLA-4 and PD-1/PD-L1 blockade is a key focus of current research. Some progress has been made. For example, the response to CTLA-4 and PD-L1 blockade in cancer patients has been shown to correlate with the expression of CD8+ and T helper type 1–associated molecular markers such as IFN-γ (interferon- γ), PRF1, TAP1, and GZMB (24–26). Furthermore, the clinical benefit of PD-1 and CTLA-4 blockade in melanoma and non–small cell lung melanoma is associated with tumor neoantigen burden (26–28). Cytolytic immune infiltrates have also been shown to correlate with the total number of mutations in certain human tumor types, although the mechanisms controlling immune infiltration are not well understood (29).
Here, our objective is to better understand the evolution of cancer by identifying which molecular properties of cancer cells might influence SCNAs and, conversely, which aspects of cancer biology might be influenced by SCNAs. We have uncovered unanticipated relationships of SCNA levels with mutation number, with classes of cancer drivers, and with cell proliferation and immune infiltration signatures. These relationships illuminate how cancer evolves and how SCNA levels might affect cancer therapy and patient survival.
Relationships between SCNAs and other genetic features of cancer
To uncover relationships between known molecular characteristics of tumors and SCNA levels that might inform cancer evolution, we first developed a means by which to stratify tumors based on their SCNA levels. We examined data from 5255 tumor/normal samples from 12 cancer types from The Cancer Genome Atlas (TCGA) project. We assigned each tumor an overall SCNA level score, defined as the sum of three standardized SCNA level scores: chromosome SCNA level, arm SCNA level, and focal SCNA level (focal SCNAs defined as <50% of a chromosome arm; tables S1 and S7; see Materials and methods) (3, 30). The different SCNA classes (focal, arm, and chromosome) were considered independently because they arise from biologically distinct mechanisms (31, 32). Hereafter, we use the terms “aneuploidy” and “SCNA level” interchangeably.
In contrast with a previous report showing that SCNAs are more abundant in tumors with a low mutation burden (33), we found a positive correlation between the number (n) of mutations and SCNA level (Fig. 1A) in 8 of 12 tumor types. Only colorectal carcinoma (CRC) and uterine corpus endometrial carcinoma (UCEC) exhibited a statistically significant negative correlation between the number of mutations and the SCNA level. The distribution of the number of mutations in these tumor types is bimodal, with most of the samples bearing ~100 exonic mutations on average and the remaining samples having a ~10-fold higher number of mutations (hypermutated; fig. S1A and table S1A; see Materials and methods). The negative correlation between mutations and SCNAs in CRC and UCEC tumors was dependent on the presence of these hypermutated samples (Fig. 1A, see dotted line). Thus, within individual cancer types, the number of mutations tends to positively correlate with the SCNA level.
Fig. 1. Relationship between SCNA level and point mutations.
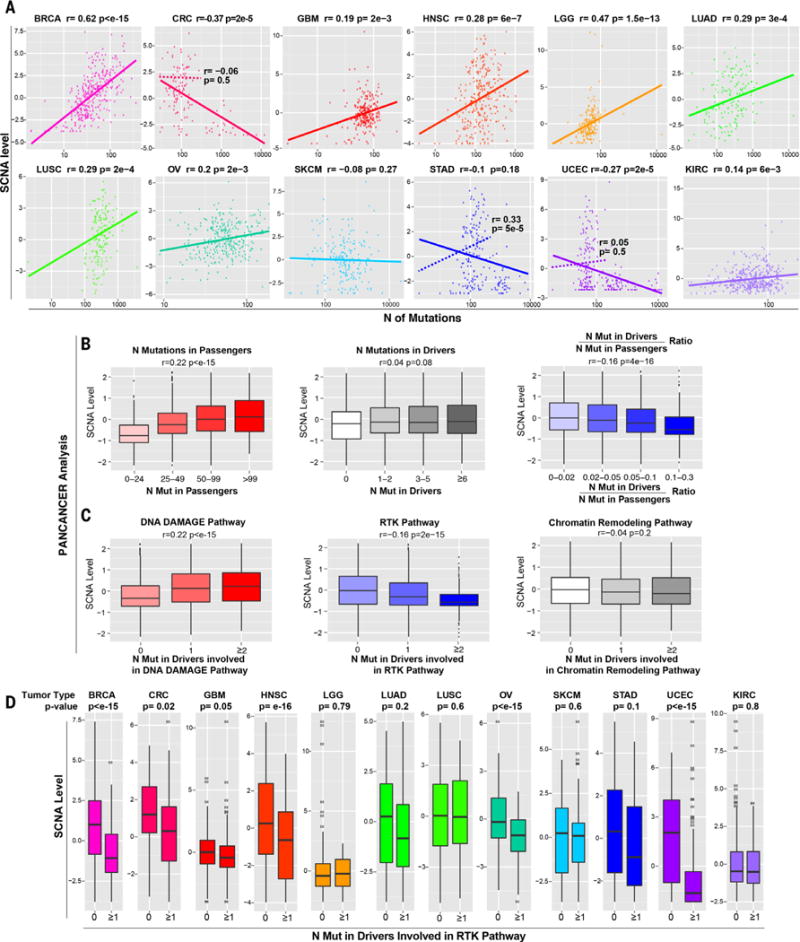
(A) For each tumor type, a plot is shown containing the calculated SCNA level (see Materials and methods) versus the total number of mutations in exons. The Spearman correlation coefficient and P value are shown. For CRC, STAD, and UCEC, two plots are shown, based either on all tumors (solid line) or after excluding hypermutated samples (dashed line; see also fig. S1A, table S1, and Materials and methods). (B and C) Pan-cancer analysis showing the relationship between the SCNA level and the number of mutations in passenger genes, functionally relevant mutations in OG and TSG drivers predicted by TUSON (11) or the ratio between the number of mutations in these drivers and passengers (B), and the number of drivers involved in the indicated pathways (C) (table S2B). Only tumor samples with a total number of exonic mutations lower than 400 were considered to exclude hypermutated samples. The Spearman correlation coefficient and P value are shown (see table S2 for the same analysis including hypermutated samples). (D) The relationship between the SCNA level and the presence of at least one functionally relevant mutation in TSGs or OGs involved in the RTK pathway [as in (C); see table S2, B and D] is shown as a box plot (P values refer to Wilcoxon test). The hypermutated samples were excluded (see Materials and methods).
Next, we separated mutations in driver genes, that is, tumor suppressor genes (TSGs) or oncogenes (OGs), as predicted by TUSON Explorer (11), and mutations in passenger genes, that is, genes not predicted to be cancer drivers. Consistent with the data in Fig. 1A, SCNA levels positively correlated with the number of mutations in passenger genes (Fig. 1B and table S2E) but not with the number of mutant cancer driver genes (Fig. 1B). Similar results were found in most tumor types when analyzed individually (table S2C).
To investigate how alterations in different classes of cancer drivers relate to aneuploidy, we analyzed the correlations between SCNA levels and mutations in sets of TSGs and OGs acting in 14 different cancer pathways (table S2B). Mutations in driver genes involved in the DNA damage response pathway were positively correlated with SCNA levels (Fig. 1C and fig. S1B), consistent with a putative role of this pathway in genomic stability (34, 35). Mutations in only one pathway showed a negative correlation with SCNAs: the receptor tyrosine kinase (RTK) pathway, which also includes phosphatidylinositol 3-kinase (PI3K) and RAS pathway genes (Fig. 1, C and D, and table S2D; P = 2 × 10−15). This inverse relationship is at odds with the notion that the RTK-RAS-PI3K pathway activation generates genomic instability, as previously proposed in the OG-induced genomic instability hypothesis (see Discussion) (36–38).
Aneuploidy correlates with tumor cell proliferation and reduced cytotoxic immune infiltration
To determine which cancer hallmarks are associated with aneuploidy, we searched for cellular signaling pathways with altered regulation in tumors with high aneuploidy. We compared the gene expression profiles of tumors with high (higher than 70th percentile) and low (lower than 30th percentile) SCNA levels (39). We used Gene Set Enrichment Analysis (GSEA) (40) to identify gene sets whose expression is enriched or depleted in high aneuploidy tumors. This analysis was performed across all tumor samples (pan-cancer analysis), as well as within each tumor type, taking into account the tumor histological types (tumor type–specific analysis; tables S3 and S4).
Among the gene sets up-regulated in high versus low aneuploidy tumors in both the pan-cancer analysis and in 11 of 12 individual tumor types were those implicated in DNA replication, cell cycle regulation, G2-M checkpoints, mitosis, and chromosome maintenance (figs. S2, A to D, and S3 and tables S3, A and B, and S4A). The level of pS345Chk1, a physical marker of S phase, was also increased in high aneuploidy tumors (fig. S2C). These results suggest that there is increased cell proliferation in high aneuploidy tumors, consistent with previous observations (41, 42).
Most of the gene expression signatures showing significant down-regulation in high aneuploidy tumors [false discovery rate (FDR) < 10−5] were signatures associated with the immune system (Fig. 2A and table S3, A and C). Because the genes comprising these signatures are predominantly expressed in immune cells and not tumor cells, this observation reflects the degree of infiltration and potential activity by the immune cells in the tumor microenvironment (Fig. 2, A and B, see below). Three related but distinct groups of gene sets affected by aneuploidy emerged from this analysis. First, the expression of genes associated with adaptive immunity was significantly decreased in high aneuploidy tumors (Fig. 2A). For example, the expression of genes encoding components of the T cell receptor (TCR) complex, such as CD3E, CD3D, CD3G, and CD247, was decreased more than twofold (FDR < 10−4; table S3, A and C). The expression of B cell–specific genes, including genes encoding the Igα and Igβ proteins of the B cell antigen receptor complex, was also significantly reduced (table S3). Second, the expression of genes specific for cytotoxic activities mediated by CD8+ T cells and NK cells was significantly reduced in high aneuploidy tumors, with an FDR of <10−4 and an enrichment score (ES) equal to or lower than −0.8 (Fig. 2A). Genes mediating cytotoxic functions, such as granzymes GZMA, GZMB, GZMH, GZMK, and GZMM, all showed mRNA reduction by twofold or greater in tumors with high versus low SCNA levels [log2 fold change (log2FC) ≤ −1; Fig. 2B and table S3A]. The expression of genes involved in the IFN-γ pathway (including JAK2, IRF1, IRF2, GBP4, and GBP5) was also significantly lower (Fig. 2B and table S3A). Third, genes in pathways related to the presence of an ongoing immune response and a cytokine-rich microenvironment [which included proinflammatory cytokines such as interleukin-1β (IL-1β), IL-16, IL-18, C-C motif chemokine ligand-4 (CCL4), CCL3, CCL19, CCL7, CCL11, CCL22, and CCL13] showed reduced expression in high aneuploidy tumors (Fig. 2B and table S3A).
Fig. 2. Depletion of cytotoxic immune infiltrates in high versus low SCNA level tumors.
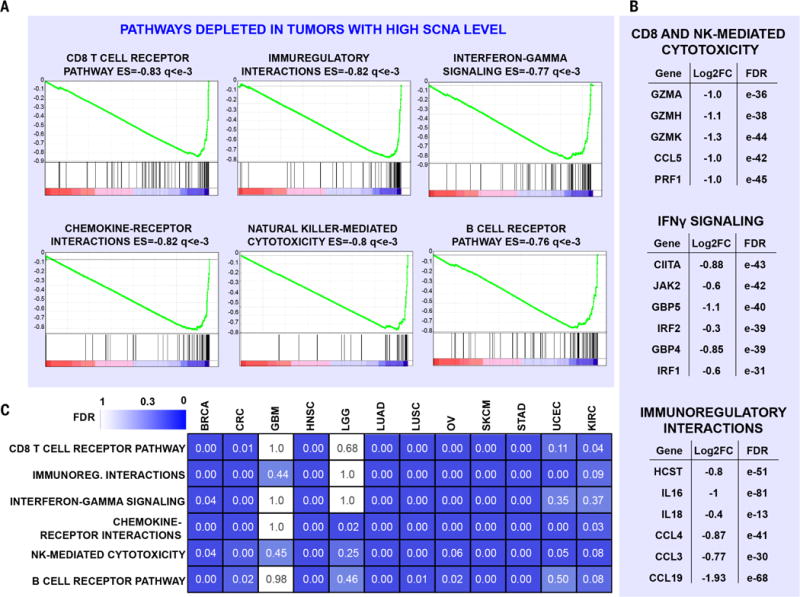
(A and B) RNA sequencing (RNA-seq) analysis was performed comparing high (higher than 70th percentile) versus low (lower than 30th percentile) SCNA level tumors (pan-cancer analysis), considering tumor type as a covariate. GSEA plots, ES, and FDR (q) are shown for representative gene sets depleted in high versus low SCNA level tumors (A). In (B), the log2FC values between high versus low SCNA level tumors and corresponding FDR are shown for representative genes. See also table S3, A and C. (C) For each tumor type, RNA-seq analysis and GSEA were performed as in (A), and the FDR values for the indicated tumor types and pathways are shown as a heat map. 0.00 indicates an FDR of <10−2. See also table S4B.
Analysis of individual tumor types yielded consistent results: Expression of genes in signatures characteristic of the adaptive immune system and immune-mediated cytolytic/cytotoxic activities was significantly decreased in epithelial tumor types and melanomas (Fig. 2C, fig. S4, and table S4B). Notable exceptions were brain cancers such as glioblastoma multiforme (GBM) and lower-grade glioma (LGG), where a reduction in the expression of immune-related gene sets was not observed. Overall, this analysis indicates that high aneuploidy tumors display a lower level of markers associated with immune-mediated cytotoxic functions than do low aneuploidy tumors.
To investigate the changes in immune cell type composition responsible for the observed changes in immune gene expression, we used the ImmGen gene expression database (43) to derive gene sets that distinguish different classes of immune cells. For each type of immune cell, we identified genes specifically expressed in that cell type and not in other immune cells or epithelial cells (table S4, C and D; see Materials and methods). Markers expressed specifically by CD8+ T cells and NK cells showed the strongest reduction in tumors with high SCNA levels relative to low SCNA level tumors (P = 10−27). Expression of genes specific for CD4+ T cells (Fig. 3A) was reduced, as were regulatory T cell (Treg)–specific genes, although to a lesser extent than genes specific for CD8+ T cells. Expression of markers specific for dendritic cells and macrophages was also significantly reduced (Fig. 3A). We infer from these cell type expression changes that the numbers of specific cell types expressing these signatures are correspondingly reduced.
Fig. 3. Markers of specific immune cells in high versus low SCNA level tumors.
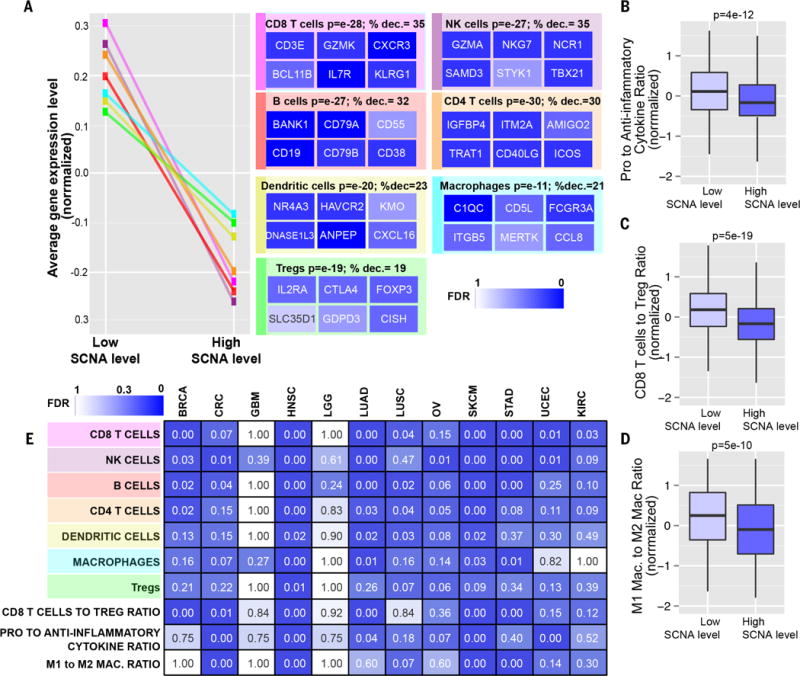
(A) Sets of genes specific for different types of immune cells (see table S4D) were analyzed for their expression in tumors with low or high SCNA levels in the pan-cancer analysis. Representative genes characterizing each immune cell type are shown with a heat map representing the corresponding FDR derived from the RNA-seq analysis (% dec., percent decrease). (B to D) Box plots of the gene expression ratios (normalized, presented as Z score) of (B) proinflammatory (IFN-γ, IL-1A, IL-1B, and IL-2) versus anti-inflammatory genes (TGFB1, IL-10, IL-4, and IL-11), (C) CD8+ Tcell–specific versus Treg-specific genes (table S4D), and (D) M1 macrophage–specific versus M2 macrophage–specific genes (see Materials and methods and table S4D). (E) RNA-seq analysis was performed comparing tumors with high versus low SCNA level for the indicated cancer types. Gene sets representing markers specific for the indicated immune cell types were used to perform a GSEA analysis as in (A). For the signatures “CD8+ T cell to Treg ratio,” “pro- to anti-inflammatory cytokines ratio,” and “M1 to M2 macrophage ratio,” a similar analysis to the one shown in (B) to (D) was performed. The FDR is shown using a heat map.
The ratio between immune cell types with a protumorigenic versus antitumorigenic function, rather than the absolute number of immune cell types, is thought to determine whether the net effect of these cells is tumor promotion versus inhibition (22). We therefore looked at the ratios between different types of immune cells and between immune molecules known to have immunosuppressive/protumorigenic or immunostimulatory/antitumorigenic functions. We found that the ratio between the mRNA levels of CD8+ T cell–specific genes versus Treg-specific genes was significantly reduced in high versus low SCNA level tumors (Fig. 3C). A significant reduction was also found in the ratio between M1 macrophage (antitumorigenic)–specific genes versus M2 (protumorigenic) (44) macrophage–specific genes (Fig. 3D). These results suggest that the tumor immune microenvironment of high SCNA level tumors is more protumorigenic and immunosuppressive. Finally, we analyzed the ratio of expression of proinflammatory cytokines (IFN-γ, IL-1A, IL-1B, and IL-2), which are among the most important markers of immune stimulation, and immunosuppressive molecules [IL-4, IL-10, IL-11, and TGFB1 (transforming growth factor–β1); see Materials and methods]. This ratio was significantly decreased in high versus low SCNA level tumors, suggesting a relative reduction of proimmunogenic responses in high aneuploidy tumors (P = 5 × 10−12; Fig. 3B, see below).
These results from the pan-cancer analysis were recapitulated in several individual tumor types, with the exception of brain tumors (Figs. 2C and 3E; figs. S5, A and B, and S6; and table S4B). Overall, these data point to a reduction of immune-mediated cytotoxic and proinflammatory activities in the microenvironment of high aneuploidy tumors.
Different SCNA classes predict cell cycle and immune signatures
To determine which SCNA classes (chromosome, arm, and focal) best predict cell cycle and immune signatures, we defined a cell cycle gene signature [genes associated with proliferation not frequently amplified/deleted; table S5A; see Materials and methods (3, 45)] and a cytotoxic immune signature (genes specific for cytotoxic CD8+ T cells and NK cells; table S5B; see Materials and methods). For each tumor sample, we determined a cell cycle signature score and an immune signature score as the average expression level of the genes composing each signature (see Materials and methods). We found the cell cycle and immune signature scores had no significant correlation (average correlation coefficient among all tumor types is 0.08). We then used logistic regression to determine the contribution of focal SCNA level and arm/chromosome SCNA level in predicting the cell cycle and immune signature score (see Materials and methods). Arm and chromosome SCNAs were considered together as they each affect a larger number of genes (~10 to 20 times higher) than focal SCNAs.
In the pan-cancer analysis, both focal (β coefficient: β= 0.50, P = 1 × 10−18) and arm/chromosome SCNA levels (β= 0.30, P = 4 × 10−9) significantly predicted the cell cycle signature score, with focal SCNAs showing a stronger contribution to the prediction of this signature (Fig. 4A, fig. S7, and table S5C). In 11 of 12 tumor types, the focal SCNA level represented a positive predictor of the cell cycle signature, whereas the arm/chromosome SCNA level was a positive predictor in 8 tumor types (Fig. 4C).
Fig. 4. Relationship between arm/chromosome and focal SCNAs and the immune or cell cycle signature scores.
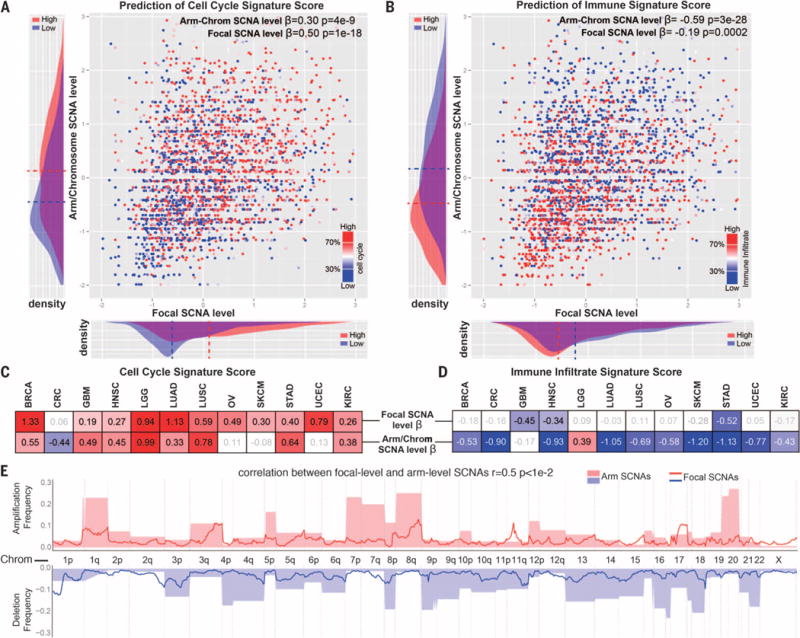
(A and B) Logistic regression was performed by comparing the tumor samples with high versus low cell cycle (A) or immune (B) signature scores and considering the arm/chromosome SCNA level and the focal SCNA level as predictors (both after standardization). The β coefficient and P value are indicated for each of the predictors. In addition, next to each of the axes on the graphs, the distribution of the corresponding parameter among tumors with high or low cell cycle (A) or immune infiltrate (B) signature is shown (dashed line indicates average value). (C and D) Logistic regression was performed as in (A) and (B) for individual tumor types. The β coefficient is indicated for each of the predictors for the indicated tumor type. A heat map is used to show the value of the β coefficients (for the coefficients with a P value of <0.1; see also table S5C). (E) Relationship between the focal and arm-level SCNAs. For each genome segment (807 genomic regions corresponding to cytogenetic bands), the frequency of amplification or deletion, for both focal and arm-level events in the pan-cancer data set, is shown (see Materials and methods and table S5D).
For the immune signature, the arm/chromosome SCNA level represented a stronger predictor (β = −0.59, P = 3 × 10−28; Fig. 4B) than the focal SCNA level (β= −0.19, P = 0.0002) in the pan-cancer analysis. Within three individual tumor types, the focal SCNA level represented a negative predictor of the immune signature (table S5C), whereas in 10 of 12 tumor types (all but gliomas), the arm/chromosome SCNA level represented a significant negative predictor (Fig. 4D) and, in each case, outperformed the focal SCNA level. Together, this analysis shows that the immune signature is more accurately predicted by arm and chromosome SCNA events than by focal SCNA events.
The differential role of arm/chromosome and focal SCNAs in predicting the cell cycle and the immune signature could be due to differences in the genes targeted or to other distinctive features of these SCNA classes. We found that focal and arm-level deletion/amplification frequencies were correlated along the genome (r = 0.5, P < 0.01; Fig. 4E and table S5D; see Materials and methods). This suggests that the focal and arm/chromosome SCNAs may be selected during tumor evolution in part due to the dosage alteration of overlapping gene sets that affect cell proliferation. However, it is a distinct property of arm/chromosome SCNAs that predicts the immune signature score (see Discussion).
Role of aneuploidy and mutations in predicting immune signatures
Tumor neoantigen and overall mutation burden have been posited to correlate with cytotoxic immune infiltrates (25, 26, 29, 46). We investigated the relative contribution of aneuploidy and mutation number in predicting the immune signature by comparing tumors with high and low arm/chromosome SCNA levels or point mutation burden. In CRC, lung adenocarcinoma (LUAD), and UCEC, tumors with high mutation burden showed an increased immune signature score, consistent with previous observations (Fig. 5, A and B) (29). In contrast, in all cancer types except gliomas, tumor samples with high arm/chromosome SCNA levels showed a significant decrease in the immune signature score (~50% difference on average) (Fig. 5, A and C, and fig. S8). This effect was particularly evident in head and neck squamous cell carcinoma (HNSC), skin cutaneous melanoma (SKCM), CRC, and stomach adenocarcinoma (STAD), tumor types with a high mutation burden (see Discussion). Compared to mutation number, the level of SCNAs showed a stronger correlation with the cytotoxic immune signature in most of the tumor types examined, even in those where mutation number positively correlated with the SCNA level (Fig. 1A). We also note that in breast cancer, the negative correlation between aneuploidy and the immune signature is stronger in estrogen receptor/progesterone receptor (ER/PR)–negative tumors, the subset of tumors with overall higher mutation burden and higher degree of immune infiltrate (47).
Fig. 5. Comparison between the number of mutations and SCNA level in the prediction of the immune signature.
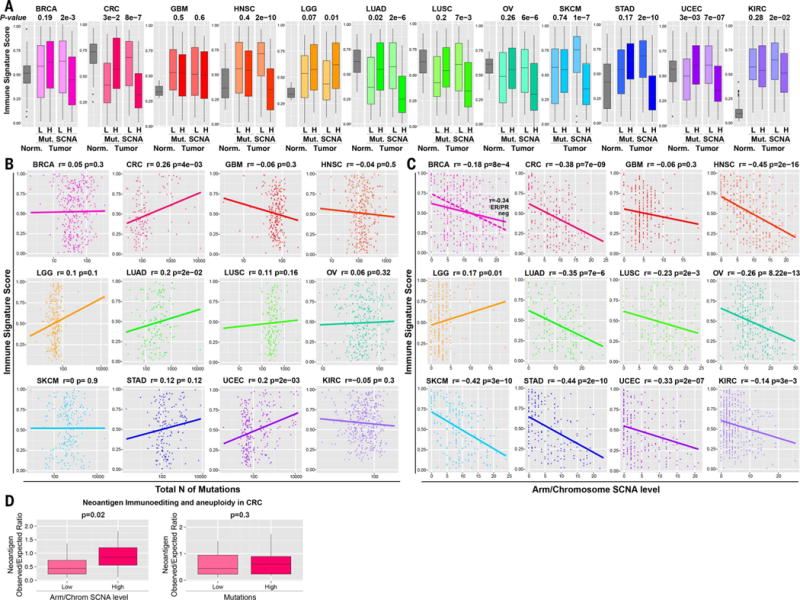
(A) Box plots illustrating the distribution of the immune signature among the normal tissue samples and the tumors with a low (L) or with a high (H) number of mutations or arm/chromosome SCNAs, as indicated. The P value from Wilcoxon test is shown. (B and C) For each tumor type, a plot is shown containing the immune signature score (y axis) versus the total number of (B) mutations (x axis) or (C) the arm/chromosome SCNA level in each tumor sample. The Spearman correlation coefficient and P value are shown. For BRCA, the relationship within the ER/PR-negative tumors only is shown separately as a dashed line. (D) The ratio between the number (n) of neoepitopes observed versus expected is shown in CRC for tumors with low and high SCNA level or mutations as indicated. The t test P value and percent increase in the observed versus expected neoepitope ratio are shown.
One of the mechanisms of cancer immune evasion is neoantigen editing or depletion, which occurs through the negative selection of tumor variants that carry and express immunoreactive neoantigens (48). Neoantigen depletion can be assessed by measuring the ratio between the number of observed neoantigens relative to the number of expected neoantigens, which is based on the number and spectrum of silent mutations (29). When the number of observed neoantigens is lower than expected, this suggests that neoantigen editing occurred during tumorigenesis. We studied the impact of SCNAs on neo-antigen editing in CRC, a tumor type where this phenomenon has been previously observed (29). We found that the ratio of observed to expected neoantigen numbers was significantly higher in tumors with high SCNA levels, indicating less neoantigen editing (Fig. 5D). In contrast, tumor mutational load had no significant correlation with neoantigen editing. Overall, these results are consistent with the hypothesis that aneuploidy inhibits cytotoxic immune activities during tumorigenesis, thus lowering the selective pressure against tumor variants containing neoantigens.
Prediction model for cell cycle and immune signatures
In addition to the total number of mutations, other molecular and clinical parameters may contribute to the prediction of the immune and cell cycle signatures. We used least absolute shrinkage and selection operator (lasso) (49) to determine the contribution of SCNA level, the total number of point mutations, TP53 mutations, patient age, patient gender, and tumor stage to both signatures. Some of these parameters have been suggested to play a role in the immune escape or cell cycle regulation of cancer cells (50–52). After splitting the data set into a training set (70% of the data set) and test set (remaining 30% of the data set), we applied lasso to select the best parameters predicting the cell cycle signature or immune signature by comparing the tumors with high versus low cell cycle or immune signature scores, respectively. The selected parameters were then used to refit a logistic regression model, and the final model was applied to the test set.
As expected (Fig. 4C), focal SCNA and arm/chromosome SCNA levels were selected by lasso to predict the cell cycle signature (Fig. 6A), as were mutations in TP53, a negative regulator of cell cycle entry (50, 51). Mutation number was also selected by lasso as a predictor of the cell cycle signature in several tumor types, possibly because cell proliferation drives mutation accumulation. For the immune signature, the arm/chromosome SCNA level was frequently selected by lasso across different tumor types (Fig. 6B), whereas the focal SCNA level was selected in only two tumor types. The total number of mutations was selected by lasso in three tumor types (CRC, LUAD, and UCEC) (Fig. 6B), but in these cases, the magnitude of the coefficients for arm/chromosome SCNAs was larger than that for mutation number. Overall, we achieved a good prediction of the cell cycle signature and the immune signature [area under the curve of the receiver-operating characteristic (ROC-AUC) ≥ 0.7] in 6 of 12 tumor types (Fig. 6, A and B). This analysis shows that, in several cancer types, aneuploidy and, in some of these cases, aneuploidy together with mutation number represent an important parameter for the predictor of the immune signature.
Fig. 6. Prediction of the cell cycle and immune signature score and survival analysis of melanoma patients after immunotherapy.
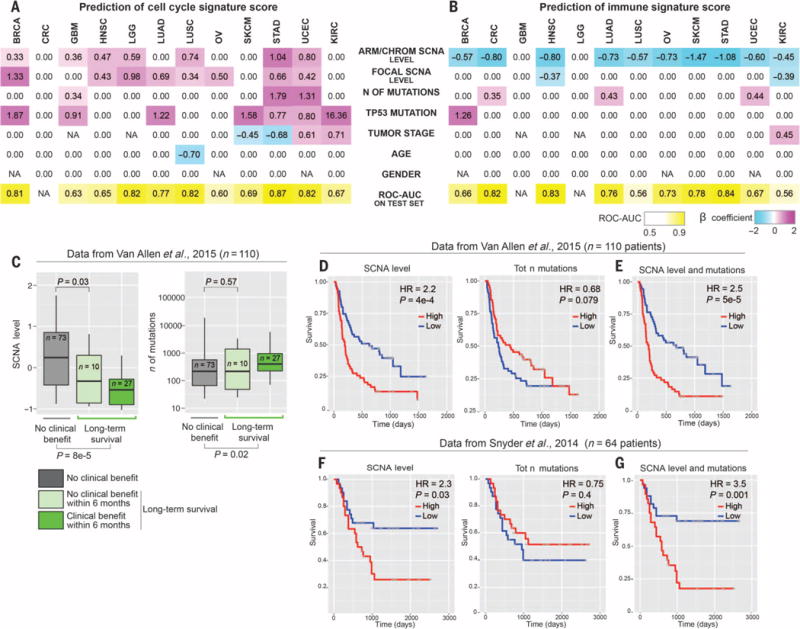
(A and B) For each tumor type, lasso was used to identify the best parameters predicting the cell cycle signature (A) or the immune signature score (B) on the training set. The selected parameters were used to refit a logistic regression model on the training set, and the corresponding β coefficients are shown for each indicated parameter and tumor type. A coefficient of 0.00 refers to parameters that were not selected. The resulting model was applied to the test set, and ROC-AUC is shown for each tumor type. The number of mutations was considered as log-transformed and standardized. TP53 mutation and gender were considered as binary parameters, and all the other parameters were standardized. NAs indicate that the corresponding parameters were not applicable (see also table S5, E and F). (C) Relationship between the SCNA level and the number of mutations with the response to anti–CTLA-4 immunotherapy. Data from the anti–CTLA-4 trial in melanoma patients described by Van Allen et al. (27) were used as described in Materials and methods. In the box plots, patients were divided into those who did achieve long-term survival (Long-term survival; different shades of green) and those who did not (No clinical benefit; gray). In addition, among the first group, a subset of 10 patients was further identified as patients with no clinical benefit within 6 months, as previously described (27). P values are shown for the comparison of the distribution of the number of mutations or the SCNA level between the indicated groups of patients (Wilcoxon test). (D and E) Survival analysis for the SCNA level, the number of mutations, and a combination of the two parameters. The number of mutations and the SCNA level were derived as in (C). In (D), the SCNA level (left) and the number of mutations (right) were used as individual predictors of survival in a univariate Cox proportional hazards models. In both cases, patients were stratified in two groups using the median value as a cutoff. HR and Wald test P values from the Cox proportional hazards model comparing the two groups of patients are shown. Kaplan-Meier survival curves are shown. In (E), multivariate Cox proportional hazards model was used including both the SCNA level and the number of mutations as predictors. On the basis of this multivariate model (table S6A), we derived a risk score for each patient and we stratified the patients in two groups using the median as a cutoff. HR and Wald test P values from the Cox proportional hazards model comparing the two groups of patients are shown. Kaplan-Meier survival curves for patients stratified in the two groups are also shown (table S6A). (F and G) The survival analyses presented were generated as in (D) and (E) with the indicated data set (table S6B).
SCNA levels predict survival of melanoma patients after immunotherapy
In light of the correlation between SCNA levels and immune activity in the tumor microenvironment, we next examined whether there was a correlation between SCNA levels and patient survival after immunotherapy. We analyzed a data set derived from a recent clinical trial of anti–CTLA-4 blockade in metastatic melanoma patients (27). We derived the SCNA level by applying VarScan 2 to whole-exome sequencing data (53) and determined the purity of each sample using AscatNGS (see Materials and methods and table S8A) (54). In addition, we considered the number of point mutations and neoantigens, which have been described as predictors of survival after immunotherapy (26, 27).
First, we assessed tumor SCNA levels and mutational load in patients who did or did not achieve long-term survival after treatment (27). Tumor SCNA levels in patients experiencing long-term survival were significantly lower (P = 8 × 10−5, Fig. 6C). As expected, tumor mutational load also distinguished these two classes of patients (P = 0.02, Fig. 6C) (27). An additional (n = 10) subgroup was defined for patients who experienced long-term survival (overall survival longer than 2 years) without apparent clinical benefit within 6 months after treatment (27). Tumor SCNA levels distinguished this subgroup from the nonresponders group (P = 0.03, Fig. 6C), whereas tumor mutational load did not distinguish between them (Fig. 6C).
Next, we examined survival after stratifying the patients into two equal groups (that is, upper and lower 50%) based on either the tumor SCNA level or mutational load. We then applied the Cox proportional hazards model to compare the survival of the two groups of patients (55). We found that higher SCNA levels correlated with poor survival [hazard ratio (HR) = 2.24; P = 4 × 10−4; Fig. 6D and table S6A]. A higher number of tumor mutations correlated with better survival (HR = 0.68, P = 0.079; Fig. 6, C to G, and table S6A); a similar result was found for the number of predicted neoantigens (table S6A). We next used a multivariable Cox proportional hazards model to include both SCNA levels and mutation number to derive a combined risk score. The HR for this combined risk score (binary classification of patients based on the median value of this score) was increased compared to the individual predictors (HR = 2.51, P = 5 × 10−5; Fig. 6E). In this multivariable model, the contribution of the SCNA level was significant (HR = 2.27, P = 3 × 10−4; table S6A) and similar to the univariate model that included only the SCNA level, demonstrating that the SCNA level is a predictor independent of the number of mutations. In addition, the number of mutations trended toward the prediction of better survival (HR = 0.85, P = 0.16). There was no significant correlation between the SCNA level and the number of mutations (r = −0.04, P = 0.63; also see Fig. 1A). As an alternative method, we performed survival analysis after considering each predictor as a continuous variable and we obtained similar results (see Materials and methods and table S6A) (56). Furthermore, we performed a similar analysis in another data set derived from an independent clinical trial of anti–CTLA-4 blockade in melanoma patients (Fig. 6, F and G, and tables S6B and S8B) (26) and obtained similar results.
As previous work has shown that high tumor aneuploidy correlates with poor survival (12–15), it is possible that the survival effects on patients undergoing immunotherapy might be explained by that general property of aneuploidy. Thus, we sought to determine whether the magnitude of this general effect explained the correlation we observed in the data from immunotherapy-treated patients. Therefore, we evaluated a TCGA data set of melanoma patients who were not treated with immunotherapy. In this data set, a higher number of tumor mutations predicted better survival (HR = 0.61, P = 0.039; table S6C and fig. S9). In the same data set, higher SCNA levels predicted poorer survival but, in contrast to its effect in the context of immunotherapy, the correlation did not reach statistical significance (HR = 1.53, P = 0.06; table S6C and fig. S9).
The correlation of both high mutation number and low SCNA level with better survival may be due to the role of an antitumor immune response in predicting a better outcome even in the absence of immunotherapy, an observation that has been previously described (57). The prediction of patient outcome by SCNA levels was more prominent in the immunotherapy context than in the nonimmunotherapy context, indicating that it likely affects immune blockade efficacy beyond its general role in patient survival (see also interaction analysis between SCNA level and immunotherapy in table S6D). Finally, a gene expression signature derived from the TCGA data set (23 genes; table S6E) and composed of genes that are positively correlated with aneuploidy and negatively correlated with the immune infiltrate was associated with worse survival in melanoma patients treated with immunotherapy (HR = 2.2; P = 0.05; fig. S9C and table S6E) (27). Overall, these data indicate that tumor SCNA levels and mutational load can be used together to predict patients’ survival after immunotherapy.
Discussion
A key unanswered question in cancer research is how tumors evolve and how aneuploidy shapes that evolution. Previous data from our laboratory and others indicated that cancer-associated SCNAs represent driver events during tumorigenesis (4–9, 58). Here, we report that, in human tumors, distinct types of SCNAs differentially predict gene expression signatures that are associated with two hallmarks of cancer: cell proliferation and immune evasion.
We uncovered relationships between the level of SCNAs and mutations in driver genes acting in specific cancer pathways that deepen our understanding of tumor evolution. Mutations in driver genes involved in the DNA damage pathway correlated positively with the overall level of SCNAs, consistent with a likely role for this pathway in preventing SCNAs (Fig. 1C and fig. S1B). Unexpectedly, the level of SCNAs negatively correlated with mutations in driver genes involved in the RTK pathway, such as EGFR, PIK3CA, KRAS, and BRAF. This negative correlation has important implications for the OG-induced genomic instability hypothesis (38). Current models posit that activation of OGs such as KRAS, BRAF, and HRAS leads to replication stress due to fork stalling, resulting in double-strand breaks in the DNA and consequent genomic instability that affects tumor evolution (32, 38). Our analysis suggests that mutated OGs (which may behave differently in vivo than in an experimental overexpression setting) may not represent a significant source of genomic instability and SCNAs in human tumors.
Consistent with previous observations, we found a positive correlation between high SCNA scores and the expression of genes involved in cell cycle regulation and proliferation. Whereas whole arm and chromosome aneuploidy contribute, focal SCNA levels play a more potent role in predicting the expression of a cell cycle and proliferation signature (Fig. 4, A and C). A plausible explanation for this observation is that frequently selected focal SCNAs contain the optimal chromosomal segments for promotion of cell proliferation; in contrast, arm-level events are less optimized with respect to which genes are altered, and the gene dosage changes of an entire arm or chromosome also create proteotoxic stress that is deleterious for cell proliferation (7, 19, 59, 60).
We uncovered a negative correlation between aneuploidy and the presence of a cytotoxic immune infiltrate (Figs. 5 and 6). We find that aneuploidy is a significant predictor of the immune signature score in 10 of 12 tumor types (Fig. 6, A and B). The two exceptions are brain tumors. The contribution of aneuploidy to the prediction of the immune signature score is independent of several clinical parameters including the total number of tumor mutations, which is known to positively correlate with the immune infiltrate in some tumor types (25, 29, 46).
Using published data from two independent clinical trials of immunotherapy (CTLA-4 blockade) in melanoma patients (26, 27), we found that high levels of tumor SCNAs predict poorer survival of patients. Because the number of mutations predicts patients’ survival independently of the SCNA level, combining the SCNA level with the number of mutations results in a further improvement of survival prediction.
In contrast to the cell proliferation signature, the immune signature score is predicted principally by arm and chromosome aneuploidy, with a minor role for focal SCNAs in some tumor types (Figs. 4, A to D, and 6, A and B). Because the genes altered in the focal and arm-level SCNAs significantly overlap (Fig. 4E), it is unlikely that the individual genes driving the selection of focal and arm-level SCNAs through dosage change are responsible for the reduction in the immune infiltrate. This suggests that differences in other features between these two types of SCNAs are responsible for the relationship. One property that distinguishes arm-level SCNAs from focal SCNAs is the number of gene products involved and the potential for proteotoxic stress. Thus, one hypothesis is that protein imbalance may impair a tumor signal needed for cytotoxic immune cell infiltration. Aneuploidy may weaken some aspects of antigen presentation on major histocompatibility complex (MHC), which is known to play an important role in tumor detection by the immune system (21). An alternative, speculative hypothesis involves the relative concentration of neoantigen peptides in high versus low aneuploidy tumors. Evidence suggests that aneuploidy evolves earlier than the bulk of mutations (61, 62). If, for the sake of argument, the typical tumor is roughly triploid, the average neoantigen would exist at a relative concentration of 33% lower than the same neoantigen in a near-diploid tumor. Furthermore, given the imbalanced stoichiometry of many complexes in aneuploid tumors, it is possible that the increased flux of unstable wild-type proteins through the proteasome generates more self peptides that would place the neoantigens at a further competitive disadvantage for loading onto limiting MHC protein, further limiting neoantigen presentation. These hypotheses also predict that the role of aneuploidy in promoting cancer immune escape is dependent on the presence of neoantigens, thus mutations. This is consistent with the fact that the tumor types, where aneuploidy has a more prominent role in predicting the immune signature, are the types with a high mutation load on average, such as HNSC, CRC, STAD, and SKCM (11).
In summary, our data suggest that assays of tumor SCNA levels might be useful, alone or in combination with estimates of tumor mutational load, for determining which patients are most likely to respond to therapies based on immune checkpoint blockade. Information on both point mutations and copy number changes can simultaneously be derived from sequencing performed on patient tumors. Furthermore, understanding the mechanism by which aneuploidy affects immune cell infiltration and responses to checkpoint blockade may provide an avenue for therapeutic intervention that could improve the efficacy of current checkpoint blockade immunotherapies.
Materials and methods
Data sets
We obtained the data sets for SCNAs (SNP-array based data), point mutations, RNA-seq, microarray expression data and clinical parameters from the TCGA database (https://tcga-data.nci.nih.gov) for the following tumor types: lower grade glioma (LGG), glioblastoma multiforme (GBM), skin cutaneous melanoma (SKCM), colorectal adenocarcinoma (CRC or COADREAD), stomach adenocarcinoma (STAD), lung squamous cell carcinoma (LUSC), head and neck squamous cell carcinoma (HNSC), ovarian adenocarcinoma (OV), uterine corpus endometrial carcinoma (UCEC), breast adenocarcinoma (BRCA), lung adenocarcinoma (LUAD), and kidney renal clear cell carcinoma (KIRC). See table S1A for the total number of samples in each tumor type. See table S7 for the data set of tumor samples with genomic and clinical parameters utilized in the analyses described below.
SCNA data processing and determination of the SCNA level
Determination of the noise threshold for SCNA calls based on the purity estimates
To determine SCNA calls, i.e., the presence or absence of amplifications or deletions, we considered different noise thresholds in different tumor types based on tumor purity (63). We estimated the threshold for the SCNA calls based on the following relationship between the purity and the relative copy number of a certain genomic region in tumor cells. For a tumor with purity p and mean ploidy T, the relative copy number R of a certain region, in relationship to the integer copy number of that region q in the tumor cells will be as follows (63):
Based on the Mitelman database (http://cgap.nci.nih.gov/Chromosomes/Mitelman), the average ploidy of the cells in a solid tumor is approximately 3N (corresponding to 69 chromosomes), thus we assume this average ploidy level in the tumors. In the ideal scenario of a tumor with purity of 100%, we would consider a noise threshold of ±0.35 for the log2-transformed copy number ratio (tumor versus normal), corresponding to a ~33% change of the copy number in the tumor cells. Based on the formula above between purity and relative copy number of a certain region, we adjusted the threshold for SCNA calls in each tumor type based on the estimated average purity level in that tumor type. The utilized threshold for the log2-transformed copy number ratio corresponds to a copy number change of ~33% within tumor cells.
An estimate of the average purity level within each tumor type was obtained from ABSOLUTE calls (3, 63), when available and, when not available, from the estimate of the purity based on the pathology report. The tumor types where ABSOLUTE estimate of the tumor purity was not available were LGG, STAD, and SKCM. For the pathology reports, we utilized the files of the type “nationwidechildrens.org_biospecimen_tumor_sample” and we considered the percent of tumor cells. For LGG, since this type of information was not available for most of the samples, we used the information on the files of the type “nationwidechildrens.org_biospecimen_slide” and we considered the tumor nuclei percent. To determine an estimate of the average purity in these three tumor types (STAD, SKCM, LGG), we considered the average purity by ABSOLUTE (3) of other tumor types (for which ABSOLUTE calls were available) with similar purity levels as estimated by the pathology report. The average estimated purity level for each tumor type and the corresponding noise threshold used for SCNA calls can be found in table S1A. This noise threshold was utilized to determine the SCNA level as described below. In addition, the analysis on the correlation between the SCNA level and the immune signature score was repeated by considering for each tumor sample a threshold level for SCNA calls determined from its purity level from the pathology report (table S5G).
Definition of SCNA events
Focal, arm, and chromosome SCNAs represent biologically distinct events, arising from different molecular mechanisms during tumorigenesis. We aimed to consider the contribution of these three distinct classes of SCNA events to different gene expression signatures as well as the relationship between focal and arm-level events. For the focal SCNAs (deletions or amplifications involving a region smaller than 50% of a chromosome arm), we applied GISTIC2 (30, 64) to the segmentation file (from all tumor samples) of the copy number data, after excluding the arm-level (and chromosome-level) copy number changes. The focal SCNA events (amplification and deletion) resulting from GISTIC2 analysis are listed in table S1, B and C. To distinguish between arm and chromosome events, among the arm-level SCNA events, all cases where both arms of a chromosome had the same copy number change (in value and sign) were considered as chromosome SCNA events, while all the others were considered as arm SCNA events (table S1D).
Calculation of the SCNA level
We first determined in each tumor sample whether each arm, chromosome or focal region (focal events listed in table S1, B and C) was amplified, highly amplified, deleted, or highly deleted. For each patient x with a tumor of type j with noise threshold t (explained above), each SCNA region i was assigned a copy number change C, depending on the sign and amplitude of its log2 copy number ratio c.
| (1) |
We determined a chromosome SCNA level, arm SCNA level, and focal SCNA level, representing the total level of deletions or amplifications at the chromosome, arm, and focal level, respectively. Each patient x was assigned the chromosome SCNA level ChromL, the arm SCNA level ArmL and the focal SCNA level FocL, based on the copy number changes Cxi associated with each of the chromosome events Chrom, arm events Arm and focal events Foc, respectively, as follows:
The ChromL, ArmL, and FocL values of each tumor sample were normalized to the mean and standard deviation calculated among the samples of the same tumor type. The SCNA level represents the sum of the normalized ChromL, ArmL, and FocL levels. The arm/chromosome SCNA level represents the sum of the normalized ChromL and ArmL.
In addition, we determined an additional SCNA level score for each tumor sample, which is proportional to the total extent of genomic regions affected by the SCNAs present in the sample. This score, called “SCNA level normalized by size” (utilized in fig. S8) represents the integrated level of SCNAs along the genome, weighted by the size of each SCNA event. To determine this “SCNA level normalized by size,” 800 genomic regions corresponding to cytogenetic bands were considered and for each region its copy number change (amplification/deletion) was determined based on Eq. 1. The “SCNA level normalized by size” represents the sum of the amplifications/deletions determined in the 800 genomic regions.
Mutation data processing
We utilized data available from TCGA on somatic point mutations (MAF files) from all tumor types and previously described lists of predicted TSGs and OGs (11) (table S2A). REACTOME and KEGG databases were utilized to classify the TSGs and OGs according to different cancer pathways (table S2B). For each TSG or OG, we defined a tumor sample as mutated in that predicted driver gene if the tumor contained at least one mutation predicted to be functionally relevant. For TSGs, functionally relevant mutations were defined as loss of function mutations (nonsense mutations or frameshift mutations) or missense mutations predicted as possibly or probably damaging by PolyPhen2 (65). For OGs, the functionally relevant mutations are represented by recurrent missense mutations. Specifically, for each OG, recurrent mutations were defined as all the missense mutations recurring in the same position (same amino-acid residue) with a frequency of 20% among all missense mutations (in the OG) or a frequency of 5% and a minimum total absolute number of 10 mutations among all tumor samples.
As shown in fig. S1A, for CRC, UCEC, STAD, and other tumor types, the distribution of the number of mutations across samples is bimodal, highlighting the presence of a subset of hypermutated tumors. The thresholds for the total number of mutations (in exons) per sample utilized to define the hypermutated samples in each tumor type were defined based on the distributions of mutations shown in fig. S1A and are listed in table S1A. When specified, the analyses considering the mutation data were performed after removing the hypermutated samples.
Gene expression analysis for the comparison of high versus low SCNA level tumors
We applied the generalized linear model (glm) for RNA-seq analysis using the EdgeR package (39, 66) to the TCGA pre-processed level-3 RNA-seq data sets (files of the type “uncv2.mRNA-seq_raw_counts”), containing the raw read counts mapped to genes. For STAD, “STAD.mRNA-seq_raw_counts.txt” file was utilized. To compare high SCNA level versus low SCNA level tumors, we considered the tumors having an SCNA level within the 30th percentile (low SCNA level or aneuploidy level) and the tumors having an SCNA level higher than the 70th percentile (high SCNA level or aneuploidy level).
For the pan-cancer analysis, we utilized the EdgeR glm model considering the tumor type as a covariate. To maintain an equal representation of all the tumor types in the data set analysis, we considered a randomly selected subset of 400 samples, for any of the tumor types represented by a higher number of samples. For the tumor-type specific analyses we included as a covariate the tumor histological type, when available. For BRCA, an equal representation of ER/PR positive and negative samples was considered. Gene Set Enrichment Analysis was performed by applying the GSEA (40) “weighted” enrichment statistics on a score for enrichment or depletion, defined as the negative log10 of the EdgeR analysis-derived FDR multiplied by the sign of the logFC (log fold change). For GSEA, we utilized pathways contained in REACTOME, KEGG, BIOCARTA, and PID databases, utilizing gene sets containing between 20 and 500 genes and 1000 permutations.
ImmGen-derived gene expression signatures
We utilized the ImmGen database (43) to derive a set of characteristic genes whose expression uniquely defines the following types of immune cells: CD8+ T cells, B cells, NK cells, Tregs, CD4+ T cells, dendritic cells, and macrophages. To derive these gene sets, we first identified gene sets distinguishing between pairs of immune cell types, utilizing the population comparison tool on ImmGen. Next, for each gene set we analyzed the top 20 genes and selected those that are characteristic of only one specific type of immune cell (about 5 to 10 per cell type, table S4D). To derive the gene sets specific for M1 and M2 macrophages, we used the data set from Newman et al. (67). GSEA analysis was performed on the output of the RNA-seq analysis (comparing high versus low SCNA level tumors) across all tumor types (pan-cancer) and in the tumor-type specific analyses using the gene sets specific for different immune cells (Fig. 3E).
To estimate the extent of the decrease in the expression of gene set specific for the different immune cell types (Fig. 3A), the average expression level of the gene set was determined by utilizing the RSEM (RNA-Seq by Expectation-Maximization) value of each gene after log transformation and normalization to the mean and standard deviation among the samples from each tumor type. The percentage decrease (% dec., Fig. 3A) in the expression of markers specific for each cell type between high and low SCNA level tumors was estimated by the percentage difference in the average expression level between high and low SCNA level tumors. For calculating the ratios of CD8+ T cell–specific versus Treg-specific genes and M1 macrophage-specific versus M2 macrophage-specific genes, for each tumor sample, we derived the ratio between the average expression level (log2 transformed RSEM value) of the gene set specific for the corresponding cell types (table S4D). For “pro to anti-inflammatory cytokine ratio,” the ratio of the average expression level (log2 transformed RSEM value) of pro-inflammatory cytokines (IFN-γ, IL-1, IL-2) versus anti-inflammatory cytokines (TGFB1, IL-10, IL-4, IL-11) was calculated in each tumor sample. Wilcoxon test was utilized to compare the distribution of the values between high and low SCNA level tumors. The calculated ratio for each tumor sample was standardized among the samples from the same tumor type.
Immune signature and cell cycle signature score
The gene sets utilized for the immune signature and the cell cycle signature score are presented in table S5, A and B. For the cytotoxic immune signature, we utilized a set of genes representing molecular markers of cytotoxic CD8+ T cells and NK cells, similar to the one utilized in Rooney et al. (29). For the list of genes utilized for the cell cycle signature, we considered a set of genes considered to be bona fide markers of cellular proliferation both by literature search and by identification in a specific study aimed to experimentally determine cell-cycle regulated genes (45). For both cell cycle and immune signatures, genes that are significantly amplified or deleted in a pan-cancer analysis (3) were excluded. To derive the cell cycle and immune signature score, we considered all the available gene expression data sets, i.e., RNA-seq and microarray gene expression data, relative to the tumor samples or the corresponding normal tissue (when available). For LGG, we considered the normal brain tissue corresponding to the GBM samples, as the normal tissue. For LUAD and LUSC, normal samples were pooled together as representative of normal lung tissue and used as normal tissue control. First, for each gene in the cell cycle or immune signatures, we considered the RSEM [or RPKM (reads per kilobase per million mapped reads)] normalized values or the gene level after LOWESS normalization for RNA-seq or microarray gene expression data, respectively. As an estimate of the expression level, we considered the rank position for each gene across samples within each data set (either RNA-seq or microarray gene expression data). As the cell cycle and immune signature scores, we considered the average of the expression value of all the genes in the corresponding signature. The presence of a subset of tumor samples in multiple gene expression data sets (RNA-seq or microarray) allowed us to confirm the expected correlation between the signature scores calculated on the basis of different expression data sets, i.e., RNA-seq and microarray.
Relationship between arm-level SCNAs and focal-level SCNAs
We aimed to determine the relationship between the frequency (across tumor samples) of focal and arm-level events along the genome. For each of 800 genomic regions, corresponding to cytogenetic bands, the corresponding copy number level (average copy number change along the region) at the arm-level or focal-level were considered (using GISTIC2). For each of these genomic regions, the frequency of amplification and deletion across samples within each tumor type was determined. To derive the correlation between focal-level and arm-level SCNAs along the genomic regions (in each tumor type), we considered the net frequency of copy number change in each region (difference between its frequency of amplification and its frequency of deletion) at the arm-level or focal-level. Both Pearson and Spearman correlation between the net frequencies of copy number changes at the focal-level versus arm-level across the 807 genomic regions are reported for each tumor type in table S5D. Additionally, for the correlation coefficient in the pan-cancer analysis shown in Fig. 4E, we considered the average value of the correlations obtained for all the individual tumor types (using the Fisher z transformation).
Logistic regression for the prediction of cell cycle and immune signature scores
To determine the role of the focal or arm/chromosome SCNAs in the prediction of the cell cycle signature or the immune signature scores, we utilized logistic regression to compare the tumors with low (lower than 30th percentile) and high (higher than 70th percentile) cell cycle or immune signature score. The focal SCNA level and arm/chromosome SCNA level was calculated for each tumor sample, as described above (normalized across the samples of the same tumor type). The results of the tumor-type analysis are shown in Fig. 4, C and D, and table S5C. In the tumor-type specific analyses, the SCNA scores were normalized within the samples of each specific histological subtype, when this information was available. For the pan-cancer analysis (Fig. 4, A and B), the cell cycle and immune signature score was normalized across the samples of the same tumor type.
Lasso classification method
To determine the parameters most predictive of the cell cycle signature or the immune signature scores, we utilized the statistical method lasso. We defined the tumors as having low or high cell cycle or immune signature scores using the 30th and 70th percentiles, as described above, and used a binomial model. In cases where the training set contained fewer than 150 tumor samples, we utilized the 40th and 60th percentiles as thresholds. We divided the data set into a training and test set, representing two thirds and the remaining one third of the data set, respectively, for each tumor type. We applied lasso to the training set using 10-fold cross validation. The total number (N) of mutations was log10 transformed and standardized. We then selected the parameters that had a β-coefficient different than zero in lasso, refit a logistic model on the training set. In the tumor-type specific analyses, for the tumor types for which tumor histological subtype was available, we normalized the SCNA levels within the tumors of each specific subtype. This model built on the training set was then applied to the test set, and the ROC-AUC (area under the curve of the receiver-operating characteristic) was calculated to assess the sensitivity and specificity of the model itself [using R package pROC (68)].
Immuno-editing signature
As described in Rooney et al. (29), we considered the ratio between the number of neoepitopes observed and the number of neoepitopes predicted based on the number and spectrum of silent mutations found in colorectal cancer samples. Only tumors with more than one predicted neoepitope were considered. As above, we considered the low and high SCNA level tumors based on the 30th and 70th percentile of the SCNA level or mutation number distribution.
Survival analysis of the immunotherapy trials
Number of mutations or number of neoantigens
For both the study from Van Allen et al. (27) and the study from Snyder et al. (26), we considered the number of mutations and/or the number of neoantigens as reported in the corresponding data sets. See table S8 for the data set of tumor samples with genomic and clinical parameters utilized in the analyses described below.
Calculation of the SCNA level
Sample files were filtered for duplicate sequences using Picard Tools (v1.14). The resulting filtered output files were then analyzed using BamTools to ascertain insert sizes and the total number of uniquely mapped reads present in each sample (69). This information was used to calculate a relative ratio between each pair of tumor and normal samples for use as an initial normalization to baseline (e.g. 2N) copy number between each pair. This is done to compensate for any systematic variance in total reads between samples (53). This ratio is typically defined as normal_unique basepairs/tumor_unique basepairs; unique basepairs are calculated as mapped nonduplicate reads * median read length (53).
Each pair of files was first converted into mpileup format using samtools (v1.3) and an hg19 (GRCh37) human reference (www.broadinstitute.org/ftp/pub/seq/references/Homo_sapiens_assembly19.fasta). The output of this conversion was piped directly into VarScan (v2.4.0) using its copy number mode, the –mpileup flag, and setting the –data-ratio variable to the custom ratio calculated for each sample pair (as described above). All other settings to run VarScan were default. Samples were assessed for improper baselining.
The output of this processing was then segmented using R and the DNAcopy library (70). Each file was smoothed using the default smooth. CNA process. Smoothed samples were then segmented using the segment command with a minimum lesion width of five and using undo. splits = “sdundo” (SD = 2). P values and lesion classifications were assessed using segments.p and this annotated output was used for all further analysis. Finally, the scored segmentation files were individually merged to coalesce multiple small segments into larger regions to correct for any over-segmentation in the data set. To this end, the VarScan accessory script mergeSegments.pl was employed, using chromosome arm sizes derived from the hg19 reference file used for the initial analysis. For the samples derived from Snyder et al. (26), the output of copy number analysis was kindly provided by Dr. Timothy Chan and Dr. Vladimir Makarov.
To calculate the level of SCNA for each sample, large-scale events were considered, i.e., events affecting at least 10% of a chromosome arm or 5% of a chromosome. Regions of deletions and amplifications were defined as described above in the section “Definition of SCNA events” (threshold of ±0.3 on the segment mean log2FC to define amplification/deletions; see also table S1A). The SCNA level was then calculated as the sum of deletions and amplifications, each weighted by its segment length. The SCNA level was then normalized to the mean and standard deviation of the distribution.
Cellularity analysis using AscatNGS and RNA-seq data
Deduplicated BAM files were processed with AscatNGS (https://github.com/cancerit/ascatNgs), a version of the Ascat algorithm (53) modified to work with NGS output. Gender information for each sample was provided to the algorithm and all 24 chromosomes included in the analysis. Variants were derived from the 1000 Genomes “multiple observations” data set and includes all observed SNVs with a Minor Allele Fraction of 0.05 or higher. The SNV panel was derived (example script available here: https://github.com/cancerit/ascatNgs/wiki/Human-reference-files-from-1000-genomes-VCFs) and a GC-correction table was calculated relative to the hg19 reference using the default parameters of the Ascat utility script ascatSnpPanelGcCorrections.pl. Across all alignment and analytical steps, a common hg19 reference file was used and is available at www.broadinstitute.org/ftp/pub/seq/references/Homo_sapiens_assembly19.fasta. AscatNGS ascertained cellularity and ploidy (and related data) for each sample. Samples that could not be modeled by the algorithm or that reported 100% purity were excluded from further analysis. We determined the role of purity in predicting survival. Purity did not represent a significant predictor of survival (HR = 1.16, p = 0.5; table S6A).
Aligned BAM files were assessed for quality and bias corrected using RNA-seQC (www.broadinstitute.org/cancer/cga/rna-seqc) as previously described (71) using default options, the common hg19 reference file, and a current known gene list for the hg19 build that was downloaded from the UCSC database. The output of this analysis includes an adjusted RPKM measurement for all observed genes in each sample, which was used for downstream analysis.
Survival analysis: Cox proportional hazards model
To perform survival analysis, we first applied univariate Cox proportional hazards model through the R package glmnet (55). We utilized the number of mutations (or neoantigens) and the SCNA level, as individual predictors (univariate model; Fig. 6, D and F, and table S6). The patients were divided in two groups based on whether the value associated to their tumors (e.g. mutation number) was higher or lower than the median level of the predictor, i.e., upper or lower 50%. The hazard ratio (HR) and the Wald test P value were determined. In addition, for each model, we calculated the C-statistics (in cross validation: cvK = 4, Rep = 10) using the R package SurvC1 (56) (table S6). For the N of mutations, an additional analysis was performed by using a specific threshold of 100 mutations to group the patients for survival analysis, as this threshold was utilized in the study by Snyder et al. (26) (table S6, A and B).
To determine the contribution of the SCNA level compared to the number of mutations as predictors of survival, both these parameters were combined in a multivariable Cox proportional hazards model (55). A combined risk score was derived from this multivariable model and the median of this combined score was utilized to divide the patients into two groups (based on whether the associated risk score was higher or lower than the median). The hazard ratio (HR) and the Wald test P value were then determined for this combined score (Fig. 6, E and G). In addition, the individual HR and P value for the two parameters (SCNA level and number of mutations) in the multivariable model were also determined (table S6). Patients who died for reasons other than melanoma were not excluded from the analysis but considered as censored at the time of death.
Analysis of TCGA data set of melanoma patients
We considered the TCGA data set of skin melanoma (SKCM) patients with available clinical information, copy number data and mutation data. Patients treated with ipilimumab (N = 2) or interferon (N = 10) were excluded as well as patients with early stage disease. The data set was composed of 159 patients including 78 events of death with a median survival time of 848 days. Stage and gender did not represent significant predictor of survival in this data set. The SCNA level was determined as described in the section “Calculation of the SCNA level.” Cox-proportional hazards model was applied as described in “Survival Analysis of the immunotherapy trials” (table S6C). To determine the interaction between the SCNA level and immunotherapy, the TCGA data set of melanoma patients not treated with immunotherapy and the data set of patients treated with anti-CTLA-4 from Van Allen et al. (27) and Snyder et al. (26) were combined together. Since the patients from TCGA were followed for a longer period of time, we considered only the patients followed for a similar time to the other data sets (up to 1700 days). Multivariable Cox proportional hazard model was applied considering the combination of low SCNA level (bottom 50%) and immunotherapy treatment and combination of high N of mutations and immunotherapy treatment as predictors, in addition to other covariates to control for differences between the three data sets (table S6D). A significant prediction of survival associated with the predictor representing the combination of low SCNA level and immunotherapy treatment indicated an interaction between the two parameters.
The TCGA data set was also utilized to derive a set of genes (23 genes, table S6E) that correlated with the SCNA level (positively) and with the immune infiltrate (negatively). The threshold used as cutoff for the correlation coefficient was 0.4. This gene set was utilized for survival analysis in the data set of melanoma patients treated with immunotherapy (27).
Supplementary Material
Acknowledgments
We thank K. Naxerova, P. Park, S. Sunyaev, and K. Wucherpfennig for critical reading of the manuscript and helpful advice. We thank L. F. de Andrade, L. Sironi, F. Ferrari, and members of the Elledge laboratory for helpful advice. We thank T. Chan, V. Makarov, L. Garraway, and E. V. Allen for providing sequencing data. This work was funded by a Department of Defense Breast Cancer Innovator Award, The Ludwig Foundation, NIH grant to S.J.E., and NIH Pathway to Independence Award (K99/R00) to T.D. S.J.E. is an investigator with the Howard Hughes Medical Institute.
Footnotes
REFERENCES AND NOTES
- 1.Holland AJ, Cleveland DW. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10:478–487. doi: 10.1038/nrm2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zack TI, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 5.Cahill DP, Kinzler KW, Vogelstein B, Lengauer C. Genetic instability and darwinian selection in tumours. Trends Cell Biol. 1999;9:M57–M60. doi: 10.1016/S0962-89249901661-X. [DOI] [PubMed] [Google Scholar]
- 6.Fujiwara T, et al. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–1047. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- 7.Weaver BAA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 8.Sotillo R, Schvartzman JM, Socci ND, Benezra R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature. 2010;464:436–440. doi: 10.1038/nature08803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duesberg P, Li R. Multistep carcinogenesis: A chain reaction of aneuploidizations. Cell Cycle. 2003;2:202–210. doi: 10.4161/cc.2.3.382. [DOI] [PubMed] [Google Scholar]
- 10.Solimini NL, et al. Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science. 2012;337:104–109. doi: 10.1126/science.1219580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davoli T, et al. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948–962. doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedlander ML, Hedley DW, Taylor IW. Clinical and biological significance of aneuploidy in human tumours. J Clin Pathol. 1984;37:961–974. doi: 10.1136/jcp.37.9.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark GM, et al. Prediction of relapse or survival in patients with node-negative breast cancer by DNA flow cytometry. N Engl J Med. 1989;320:627–633. doi: 10.1056/NEJM198903093201003. [DOI] [PubMed] [Google Scholar]
- 14.Kallioniemi OP, Koivula T, Punnonen R, Mattila J, Lehtinen M. Prognostic significance of DNA index, multiploidy, and S-phase fraction in ovarian cancer. Cancer. 1988;61:334–339. doi: 10.1002/1097-01421988011561:2<334∷AID-CNCR2820610224>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 15.Choma D, Daurès JP, Quantin X, Pujol JL. Aneuploidy and prognosis of non-small-cell lung cancer: A meta-analysis of published data. Br J Cancer. 2001;85:14–22. doi: 10.1054/bjoc.2001.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torres EM, et al. Identification of aneuploidy-tolerating mutations. Cell. 2010;143:71–83. doi: 10.1016/j.cell.2010.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheltzer JM, et al. Aneuploidy drives genomic instability in yeast. Science. 2011;333:1026–1030. doi: 10.1126/science.1206412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crasta K, et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–58. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oromendia AB, Dodgson SE, Amon A. Aneuploidy causes proteotoxic stress in yeast. Genes Dev. 2012;26:2696–2708. doi: 10.1101/gad.207407.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Torres EM, et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- 21.Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3:999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tumeh PC, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rizvi NA, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan TA, Wolchok JD, Snyder A. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2015;373:1984. doi: 10.1056/NEJMc1508163. [DOI] [PubMed] [Google Scholar]
- 27.Van Allen EM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGranahan N, et al. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med. 2015;7:283ra54. doi: 10.1126/scitranslmed.aaa1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mermel CH, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maser RS, DePinho RA. Connecting chromosomes, crisis, and cancer. Science. 2002;297:565–569. doi: 10.1126/science.297.5581.565. [DOI] [PubMed] [Google Scholar]
- 32.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—An evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 33.Ciriello G, et al. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–1133. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartkova J, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 35.Gorgoulis VG, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 36.Bartkova J, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 37.Di Micco R, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 38.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 39.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: A desktop application for gene set enrichment analysis. Bioinformatics. 2007;23:3251–3253. doi: 10.1093/bioinformatics/btm369. [DOI] [PubMed] [Google Scholar]
- 41.Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38:1043–1048. doi: 10.1038/ng1861. [DOI] [PubMed] [Google Scholar]
- 42.Sheltzer JM, Torres EM, Dunham MJ, Amon A. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci USA. 2012;109:12644–12649. doi: 10.1073/pnas.1209227109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heng TSP, et al. The immunological genome project: Networks of gene expression in immune cells. Nat Immunol. 2008;9:1091–1094. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- 44.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 45.Whitfield ML, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown SD, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;24:743–750. doi: 10.1101/gr.165985.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu F, et al. CD8+ cytotoxic T cell and FOXP3+ regulatory T cell infiltration in relation to breast cancer survival and molecular subtypes. Breast Cancer Res Treat. 2011;130:645–655. doi: 10.1007/s10549-011-1647-3. [DOI] [PubMed] [Google Scholar]
- 48.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: From immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 49.Tibshirani R. Regression selection and shrinkage via the lasso. J R Statist Soc B. 1994;58:267–288. doi: 10.2307/2346178. [DOI] [Google Scholar]
- 50.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/S0092-86740081871-1. [DOI] [PubMed] [Google Scholar]
- 51.Allred DC, et al. Association of p53 protein expression with tumor cell proliferation rate and clinical outcome in node-negative breast cancer. J Natl Cancer Inst. 1993;85:200–206. doi: 10.1093/jnci/85.3.200. [DOI] [PubMed] [Google Scholar]
- 52.Beer DG, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 53.Koboldt DC, et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Loo P, et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci USA. 2010;107:16910–16915. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33:1–22. doi: 10.18637/jss.v033.i01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uno H, Cai T, Pencina MJ, D’Agostino RB, Wei LJ. On the C-statistics for evaluating overall adequacy of risk prediction procedures with censored survival data. Stat Med. 2011;30:1105–1117. doi: 10.1002/sim.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Angell H, Galon J. From the immune contexture to the immunoscore: The role of prognostic and predictive immune markers in cancer. Curr Opin Immunol. 2013;25:261–267. doi: 10.1016/j.coi.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 58.Davoli T, de Lange T. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell. 2012;21:765–776. doi: 10.1016/j.ccr.2012.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Storchová Z, et al. Genome-wide genetic analysis of polyploidy in yeast. Nature. 2006;443:541–547. doi: 10.1038/nature05178. [DOI] [PubMed] [Google Scholar]
- 60.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carter SL, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reich M, et al. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 65.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Newman AM, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robin X, et al. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barnett DW, Garrison EK, Quinlan AR, Strömberg MP, Marth GT. Bamtools: A C++ API and toolkit for analyzing and managing BAM files. Bioinformatics. 2011;27:1691–1692. doi: 10.1093/bioinformatics/btr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seshan VE, Olshen AB. DNAcopy: A Package for Analyzing DNA Copy Data (Bioconductor) 2014 [Google Scholar]
- 71.Deluca DS, et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012;28:1530–1532. doi: 10.1093/bioinformatics/bts196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.